")
Back to Journals » Advances and Applications in Bioinformatics and Chemistry » Volume 8
Identification of potential drug targets by subtractive genome analysis of Escherichia coli O157:H7: an in silico approach
Authors Mondal SI, Ferdous S, Jewel NA , Akter A, Mahmud Z, Islam MM, Afrin T, Karim N
Received 13 May 2015
Accepted for publication 27 October 2015
Published 8 December 2015 Volume 2015:8 Pages 49—63
DOI https://doi.org/10.2147/AABC.S88522
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Juan Fernandez-Recio
Shakhinur Islam Mondal,1,6,* Sabiha Ferdous,1,* Nurnabi Azad Jewel,1 Arzuba Akter,2,6 Zabed Mahmud,1 Md Muzahidul Islam,1 Tanzila Afrin,3 Nurul Karim4,5
1Genetic Engineering and Biotechnology Department, Shahjalal University of Science and Technology, Sylhet, Bangladesh; 2Biochemistry and Molecular Biology Department, Shahjalal University of Science and Technology, Sylhet, Bangladesh; 3Department of Pharmacy, East West University, Aftabnagar, Bangladesh; 4Biochemistry and Molecular Biology Department, Jahangirnagar University, Savar, Bangladesh; 5Division of Parasitology, 6Division of Microbiology, Department of Infectious Diseases, Faculty of Medicine, University of Miyazaki, Miyazaki, Japan
*These authors contributed equally to this work
Abstract: Bacterial enteric infections resulting in diarrhea, dysentery, or enteric fever constitute a huge public health problem, with more than a billion episodes of disease annually in developing and developed countries. In this study, the deadly agent of hemorrhagic diarrhea and hemolytic uremic syndrome, Escherichia coli O157:H7 was investigated with extensive computational approaches aimed at identifying novel and broad-spectrum antibiotic targets. A systematic in silico workflow consisting of comparative genomics, metabolic pathways analysis, and additional drug prioritizing parameters was used to identify novel drug targets that were essential for the pathogen’s survival but absent in its human host. Comparative genomic analysis of Kyoto Encyclopedia of Genes and Genomes annotated metabolic pathways identified 350 putative target proteins in E. coli O157:H7 which showed no similarity to human proteins. Further bioinformatic approaches including prediction of subcellular localization, calculation of molecular weight, and web-based investigation of 3D structural characteristics greatly aided in filtering the potential drug targets from 350 to 120. Ultimately, 44 non-homologous essential proteins of E. coli O157:H7 were prioritized and proved to have the eligibility to become novel broad-spectrum antibiotic targets and DNA polymerase III alpha (dnaE) was the top-ranked among these targets. Moreover, druggability of each of the identified drug targets was evaluated by the DrugBank database. In addition, 3D structure of the dnaE was modeled and explored further for in silico docking with ligands having potential druggability. Finally, we confirmed that the compounds N-coeleneterazine and N-(1,4-dihydro-5H-tetrazol-5-ylidene)-9-oxo-9H-xanthene-2-sulfonamide were the most suitable ligands of dnaE and hence proposed as the potential inhibitors of this target protein. The results of this study could facilitate the discovery and release of new and effective drugs against E. coli O157:H7 and other deadly human bacterial pathogens.
Keywords: E. coli O157:H7, KEGG metabolic pathways, novel and broad-spectrum antibiotic targets, DNA polymerase III alpha, homology modeling
Introduction
Enteropathogenic Escherichia coli and enterohemorrhagic E. coli (EHEC) infections in humans are a major source of morbidity and mortality in both developing and developed countries.1 Among various pathogenic E. coli strains that cause intestinal or extra-intestinal diseases in humans, the most devastating are shiga toxins producing EHEC strains, because they cause not only diarrhea and hemorrhagic colitis but also life-threatening hemolytic uremic syndrome and encephalopathy.2 Over 100 serotypes of shiga toxin-producing E. coli have been associated with human infections and the most common serotype is E. coli O157:H7. Several deadly outbreaks of E. coli O157:H7 were reported in Canada, United States, Great Britain, and Japan.3–5 However, the massive outbreak in Sakai city, Japan, in 1996 is of great concern as a number of deaths from the infection were reported.6
The genome of EHEC O157:H7 Sakai strain was sequenced in 2001.7 The sequence analysis revealed that this strain contains 18 prophages (Sp1 to Sp18), six prophage-like elements (SpLE1 to SpLE6), and two plasmids (pO157 and pOSAKl). To know the mechanism underlying pathogenicity of this bacterium, a substantial number of virulence-related genes or functions associated with various stages of infection have been identified.7 However, lack of details of functional annotations often limit the possibility to use them as targets for designing new drugs against this pathogen.
The treatment of E. coli O157:H7 mostly relies on conventional antibiotic therapy although some studies have highlighted that there is no evidence that this improves the course of disease and antibiotic treatment of patients with E. coli O157:H7 infection increases the risk of hemolytic–uremic syndrome.8,9 Moreover, an increase of antibiotic resistance has been reported in E. coli O157:H7 over the last 30 years which is also alarming.10–14 The accumulated results strongly suggest that there is an urgent and continuing need to find new drug and vaccine candidates to tackle this deadly pathogen.
Drug target identification is the first step in the drug discovery process.15 However, traditional drug discovery methods are time-consuming, expensive, and often yield few drug targets. In contrast, advances in complete genome sequencing, bioinformatics, and cheminformatics represent an attractive alternative approach to identify drug targets worthy of experimental follow-up. Because of the availability of both pathogen and host–genome sequences, it has become easier to identify drug targets at the genomic level for any given pathogen.16,17 In recent years, computational methods have been used widely for the identification of potential drug and vaccine targets in different pathogenic microorganisms.18–21 Subtractive and comparative genomics approach combined with metabolic pathway analysis was found to be an efficient way to identify the protein-set essential for the pathogen’s survival but absent in the host.22 Subtraction of the host genome from essential genes of pathogens helps in searching for non-human homologous targets which ensures no interaction of drugs with human targets. On the other hand, comparative genomics method emphasizes the selection of conserved proteins amongst several species as most favorable targets.23–26 The use of advanced bioinformatics tools with integrated genomics, proteomics, and metabolomics may ensure the discovery of potential drug targets for most of the infectious diseases. Once the target(s) have been identified, the in silico virtual screening of different chemical databases could provide unprecedented opportunity to select and design the best possible inhibitor(s).27
In this study, we took an in-depth in silico approach to identify novel therapeutic targets in E. coli O157:H7 Sakai strain by combining analysis of metabolomics and genomics data. Instead of analysis of whole genome, we particularly considered the key essential or survival proteins of the pathogen which are non-homologous to the host. We elucidated a good number of novel targets in E. coli O157:H7 to design effective drugs against broad-spectrum pathogenic bacteria. Moreover, we provided a modeled 3D structure of DNA polymerase III alpha (dnaE) which was selected as the best possible target for inhibition and designing potential drugs. To the best of our knowledge this was the first in silico identification of drug targets in E. coli O157:H7.
Materials and methods
Pathway analysis and protein retrieval
Figure 1 showed the strategies for identification of suitable drug targets and prediction of inhibitor used. The Kyoto Encyclopedia of Genes and Genomes (KEGG) database28,29 was searched for metabolic pathways for both human genomes and E. coli O157:H7 Sakai strain. The identification numbers of all pathways from both organisms were listed. A manual comparison was made, and pathways that did not appear in the human genomes but were present in the pathogen, according to the KEGG database annotations, were selected as unique to E. coli O157:H7, while the remaining pathways were listed as common. Amino acid sequences of proteins from common and unique pathways were obtained from UniprotKB.30
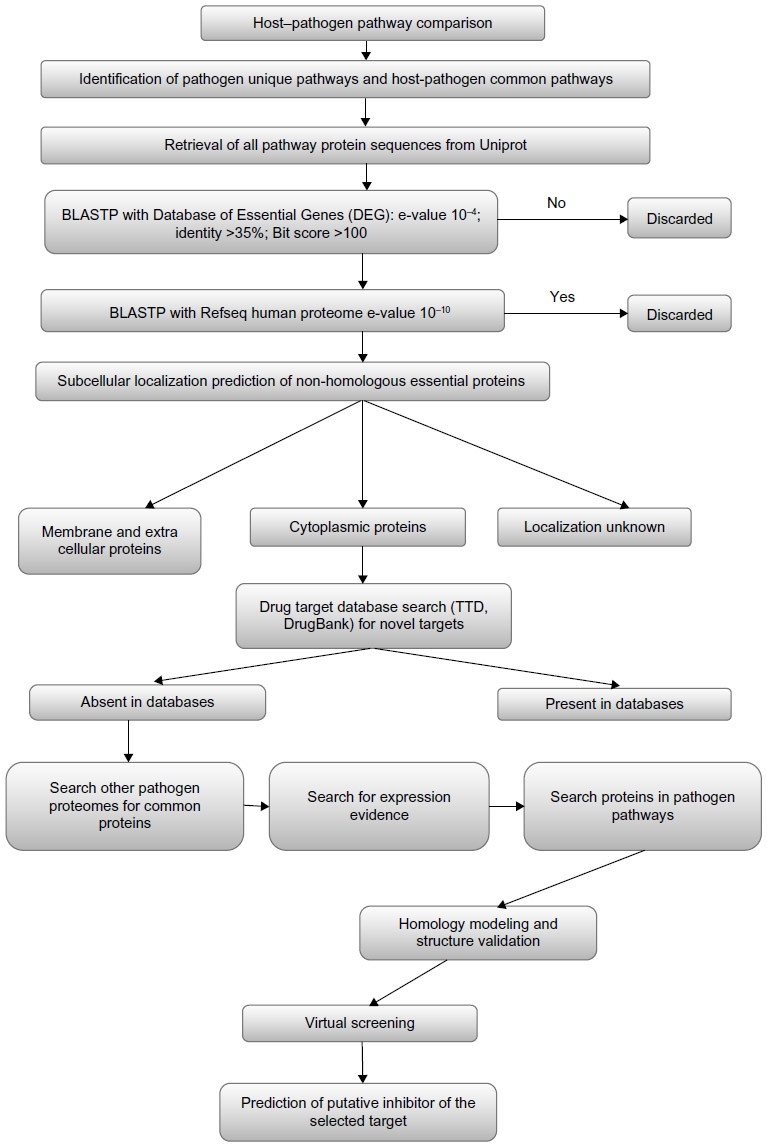 | Figure 1 A schematic representation of the workflow of computational drug target identification and prediction of putative inhibitors of the selected target |
Identification of essential proteins and non-homologous proteins in humans
Database of Essential Genes (DEG)31 was used to identify the essential proteins involved in host-pathogen pathways. The DEG 6.8 database was retrieved from http://www.tubic.tju.edu.cn/deg/. The BioEdit Sequence Alignment Editor (version 7.1.3) was used for Protein Basic Local Alignment Search Tool (BLASTP) search to screen for and eliminate the probable essential proteins of the organism setting e-value cut off 10−4, sequence identity >35%, bit score >100 and others as default.
All human protein sequences were retrieved from Refseq database ftp site (ftp://ftp.ncbi.nlm.nih.gov/genomes/H_sapiens/protein/) and essential proteins were subjected to BLASTP search against the human proteins with BioEdit. Only the non-hit proteins at e-value cut off 10−10, were selected as non-homologous proteins to avoid any functional similarity with host proteome.
Subcellular localization prediction and targets’ prioritization
Subcellular localization prediction of the essential non-human proteins was done by PSORTb version 3.0.2,32 which predicts three types of localization such as: cytoplasmic, membrane, and extracellular proteins for Gram-negative bacteria. The potential drug targets were evaluated by several molecular and structural criteria33 for prioritizing suitable drug targets. Drug targets’ prioritization involved calculation of molecular weight (MW) using computational tools and drug targets associated literature available at Swiss-Prot database. Protein Data Bank and ModBase (http://www.salilab.org/modbase) databases were searched for identifying experimentally and computationally solved 3D structures respectively.34 The selected protein was searched for any structural identity with the 3D ligand binding site of any human protein structure on the web server SMAP-WS at a cut off value of 30% sequence identity.35
Moreover, druggability is another important prioritization criterion for therapeutic targets; that is defined as the likelihood of being able to modulate the activity of the therapeutic target protein with a small-molecule drug.36,37 The druggability of identified drug targets was measured by mining DrugBank contents. BLASTP with default parameters was performed against the list of targets of compounds found within DrugBank to align the potential drug targets from E. coli O157:H7. Alignments with e-values less significant than 10−25 were removed as described previously as selection criteria for filtering BLAST results in identifying drug targets of bacterial genomes.38
Identification of novel targets and searching for common proteins
To identify novel targets among the potential targets, databases DrugBank, SuperTarget, and Therapeutic Target Database, were searched for similarity with the cytoplasmic proteins.39–41 Parameters were set as e-value <10−5, sequence identity >35%, and bit score >100. The non-hit proteins at the threshold value were selected as novel drug targets. To search for the common proteins amongst pathogenic bacteria, all protein sequences of 73 different strains of pathogenic bacteria were retrieved from PATRIC database.42 The novel targets were subjected to BLASTP against these proteomes at e-value cut off 10−5, sequence identity >35%, bit score >100 with BioEdit software. The proteins that were found to be common in at least 40 pathogenic strain proteomes were listed as broad-spectrum targets. Different bacterial species were used as references.
Homology modeling
As no exact protein data bank (PDB) structure was available for dnaE in PDB, it was subjected to BLAST search against PDB structures using 0.001 e-value cut off. The template for homology modeling was chosen considering X-ray diffraction resolution and highest sequence similarity. Homology modeling was done on ESyPred3D server.43
Structure validation and active site prediction
The modeled structure was assessed through SWISS-MODEL structure assessment tool44 and ANOLEA (atomic non-local environment assessment)45 assessing the packing quality of the models. PROCHECK suite of programs46 checked the stereochemical quality of protein structures. Energy minimization was carried out by GROMOS96 with default parameters implemented in Swiss PDB Viewer (version 4.0.4).47 Active site of the modeled structure was determined by CASTp server.48
Virtual screening, drug likeliness, and toxicity analysis
For visual analysis and comparison of the active site interaction with the ligands, 24 ligand molecules of E. coli K-12 dnaE subunit were extracted from BindingDB database and docked with the subject receptor.49 Virtual screening was done with a total 6,460 molecules, 5,040 experimental and 1,447 approved molecules deposited in DrugBank, based on selected active sites into the dnaE. Virtual screening was performed on Linux (Ubuntu 10.04) based cluster with 32 core systems. The top 100 molecules were selected based on lowest binding energy after being docked several times. The selected molecules were analyzed by Lipinski’s rule of five. The ligand interaction analysis and visualization was done with the help of Pymol and Discovery Studio (Accelrys, San Diego, CA, USA). Absorption, distribution, metabolism, excretion, and toxicity (ADMET) prediction was carried out with PreADMET server. PreADMET predicts mutagenicity and carcinogenicity of a compound and helps to avoid toxic compound. Oral bioavailability was predicted with FAF-Drugs2 program of Mobyle@RPBS server.50
Results and discussion
Identification of pathogen-specific pathways
Here we report the first computational comparative and subtractive genomics analysis of different metabolic pathways from E. coli O157:H7, for the identification of potential drug targets. A systematic workflow was defined involving several bioinformatics tools, databases, and drug target prioritization parameters (Figure 1), with the goal of obtaining information about proteins that were involved in various metabolic pathways of E. coli O157:H7, but absent in its host, therefore avoiding any potential side effects. When we searched the KEGG database for pathogen metabolic pathways, a total of 105 different pathways appeared. In order to identify drug targets involved in pathogen-specific metabolic pathways, comparative analysis of the metabolic pathways of the host and pathogen was performed. Detailed pathways analyses revealed that a total of 35 pathways were present only in the pathogen and termed as pathogen-specific pathways, and the remaining 70 pathogen pathways were defined as common host-pathogen pathways as listed in Table 1.
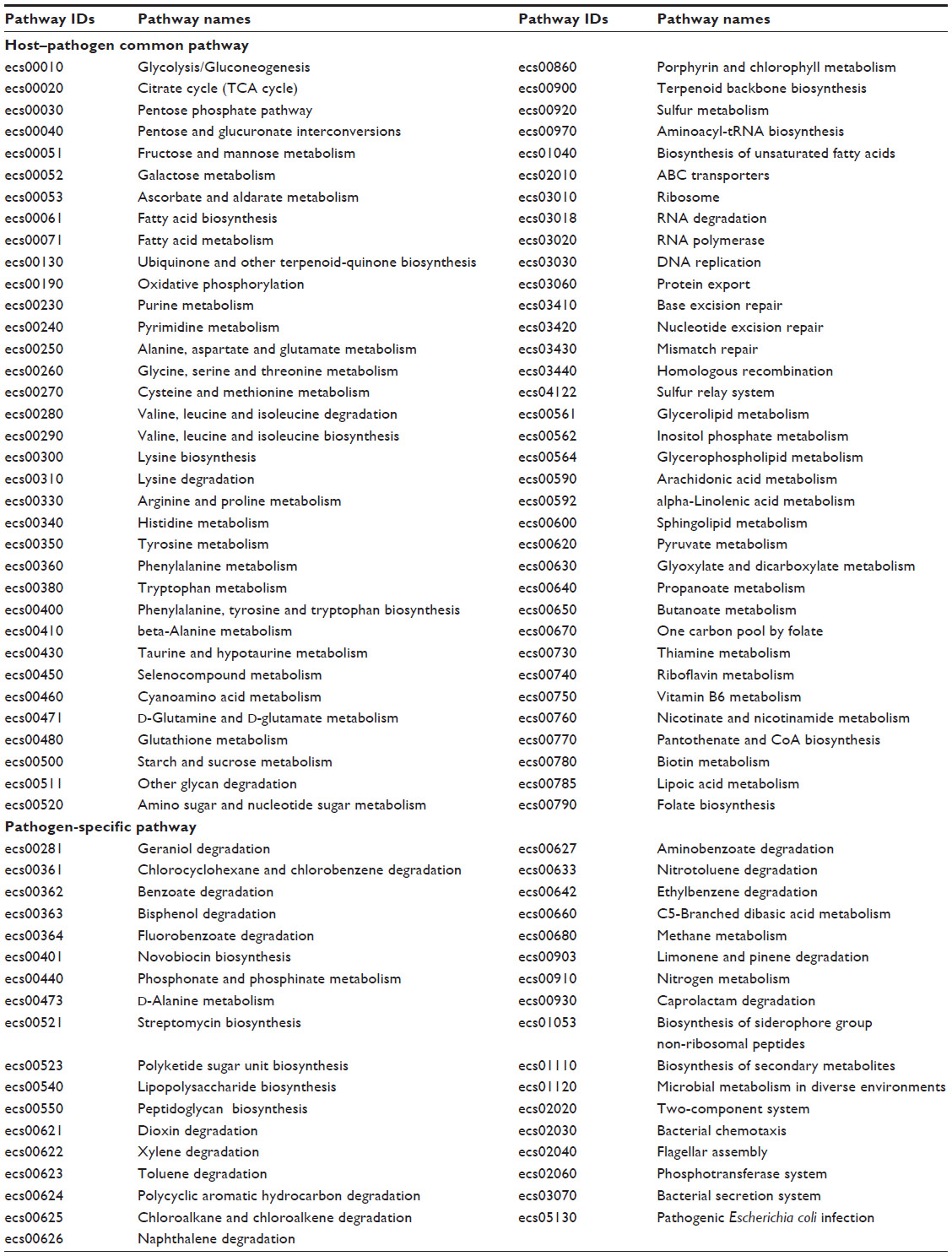 | Table 1 Host–pathogen common and pathogen-specific pathways from KEGG database |
Identification of non-homologous essential proteins
To be an effective drug target, the protein should be crucial for the survival of pathogen in the host body but non-homologous to human proteins and this criterion is a prerequisite for avoiding cross binding of drugs with human proteins, and drug side effect probability.51 Unique pathways are those that are specific to the pathogen but absent in its host. Proteins in these pathways can also be considered as unique to the pathogen and might serve as potential drug and vaccine targets.34 Moreover, several unique proteins are known to be present in common pathways as identified during our analysis of E. coli O157:H7 (data not shown) and in several previous studies on other bacteria.19,20,52
Additionally, we also identified that a single unique protein can also take part in multiple pathways. Proteins that are involved in more than one pathway could be more effective drug targets when, in addition, they are non-homologous proteins. Nevertheless, being unique or non-human and involved in metabolic pathways are not the sole criteria for selecting favorable drug targets. It is possible that a bacterial protein showing no similarity to host proteins might be involved in multiple metabolic pathways, but its disruption might be of no therapeutic benefit. The reasons may include presence of paralogs, isoenzymes, and most importantly, being non-essential for the pathogen’s survival. Again, not all essential proteins are non-homologous in nature. Therefore, pathogen proteins that fulfill the criteria of being unique and essential at the same time represent more attractive drug targets.34
Based on the criteria discussed above, we identified 780 probable essential proteins from host–pathogen common pathways, and 234 from pathogen unique pathways (Supplementary materials). These proteins showed good similarity with the experimentally proven essential proteins recorded in DEG database. BLASTP search against human proteome narrowed down the target proteome to only 220 and 130 proteins from common pathways and unique pathways respectively resulting in 350 proteins which were essential for the pathogen’s survival and non-homologous to the host (Supplementary materials).
Subcellular localization and prediction of drug target prioritization
Localization of the proteins in the cell is an important factor for identification of suitable and effective drug targets. Membrane localized proteins are difficult to purify and assay53 and therefore, cytoplasmic proteins are more favorable as drug targets. Other major factors are: accessibility value of a target protein; preferably low MW (<100 kDa); whether a potential drug is a transmembrane protein; and availability of 3D structural information.20 Based on these essential features, the identified non-homologous essential proteins of E. coli O157:H7 were further characterized. Most of the proteins had MW less than 100 kDa indicating the possibility to experimentally study these proteins for drug development.
From the common pathways, 152 proteins were found to be cytoplasmic, 54 proteins to be membrane localized, and the other 14 proteins to be of unknown localization. From pathogen unique pathways, 70, 52, and eight proteins were found to be cytoplasmic, membrane localized, and of unknown localization respectively (Supplementary materials). Based on these results, 152 and 70 cytoplasmic proteins from common pathways and unique pathways respectively were considered for further analysis to identify suitable drug targets (Figure 2).
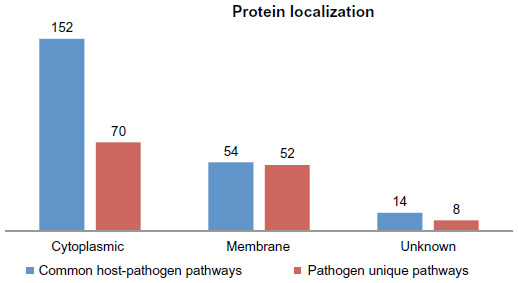 | Figure 2 Comparative subcellular localization of proteins from the common host-pathogen pathways and pathogen-specific pathways. |
The DrugBank is a unique bioinformatics and cheminformatics resource which combines detailed drug data with comprehensive information about drug targets. By utilizing the DrugBank database, druggability of non-homologous essential proteins of E. coli O157:H7 was measured by sequence similarity to the targets of small-molecule drugs and BLASTP search was performed to align the non-homologous essential proteins to the list of drug-targeted proteins from DrugBank. A total of 129 proteins of E. coli O157:H7 showed high similarities with the binding partners of US Food and Drug Administration (FDA)-approved drugs, experimental small-molecule compounds, or nutraceutical compounds supporting the potential of comparative genomics in drug discovery. Among these, 80 proteins and 49 proteins were from common and unique pathways respectively (Supplementary materials).
By this comparison with drug-targeted proteins additionally a list of approved drug and drug-like compounds was identified that bind to proteins with similar sequences to those of E. coli O157:H7. It is reasonable that careful filtering of this set could reveal a number of potential compounds that were primed for optimization and derivatization using traditional medicinal chemistry although protein sequence similarity does not guarantee identical structures or binding pockets.38 We searched the presence of 3D structures of the non-host essential proteins of E. coli O157:H7. Such information could greatly facilitate a structure-based drug design, including homology modeling, docking, virtual screening or pharmacophore-based screening.54 The PDB and ModBase were used as sources for the 3D structural information. Out of 350 non-host essential proteins, ten proteins were identified as having experimentally determined 3D structures in PDB and 312 were found to have 3D models in ModBase (Supplementary materials).
Novel targets’ identification
Proteins which showed significant similarity with the databases were discarded and the remaining protein sequences were taken as novel targets. Forty-four proteins from pathogen unique pathways and 76 proteins from common pathways totaling 120 proteins were defined as novel proteins (Supplementary materials). Metabolic pathway analysis indicated that these 120 proteins were involved in 12 biological processes unique to pathogens and 49 biological processes that were common in both host and pathogen. Moreover, all of these 61 biological processes were classified into 12 classes: amino acid metabolism, carbohydrate metabolism, energy metabolism, glycan biosynthesis and metabolism, lipid metabolism, metabolism of cofactors and vitamins, metabolism of other amino acids, nucleotide metabolism, genetic information processing, environmental information processing, cellular processes, and others (Supplementary materials). Figure 3 showed the percentage distribution of novel drug targets involved in different biological process.
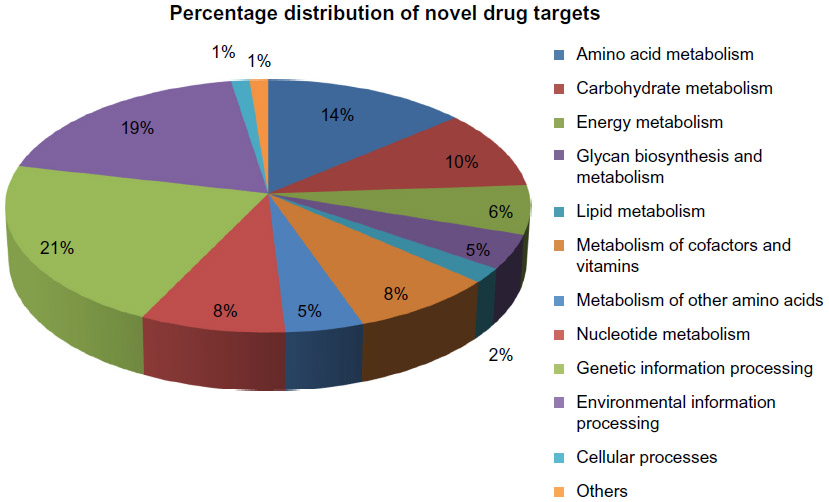 | Figure 3 Percentage distribution of novel drug targets involved in different metabolic pathways or biological processes. |
Novel targets in pathogens’ unique pathways
Our study revealed that 44 proteins were uniquely involved in pathogen-specific 12 unique pathways and these were lipopolysaccharide (LPS) biosynthesis, peptidoglycan biosynthesis, methane metabolism, C5-branched dibasic acid metabolism, nitrogen metabolism, phosphonate and phosphinate metabolism, bacterial secretion system, phosphotransferase system (PTS), flagellar assembly, two-component system, biosynthesis of siderophore group non-ribosomal peptides, and bacterial chemotaxis.
Three enzymes of LPS core biosynthesis and three enzymes of lipid A pathway were found uniquely present in LPS biosynthesis pathway (KEGG Pathway: map00540). In LPS of the Enterobacteriaceae, the core oligosaccharide is responsible for many of the biological properties of the antigenic O-polysaccharide.55 On the other hand, enzymes of lipid A pathway required for bacterial growth could be excellent targets for the development of new antibiotics.56 Moreover, murF; EC 6.3.2.10 was the only enzyme present in peptidoglycan biosynthesis (KEGG Pathway: map00550). In Gram-positive bacteria, the cell wall composed of peptidoglycan macromolecules and many surface proteins of Gram-positive bacteria is thought to be important for survival within an infected host.57
In methane metabolism (KEGG Pathway: map00680), AckA; EC 2.7.2.1 was found to be uniquely present. Methanotrophs involved in the global methane cycle consume methane as their sole source of carbon and energy for growing58 whereas methanogens can obtain energy for growth by converting a limited number of substrates to methane.59 The protein ilvH, acetolactate synthase III small subunit, is a unique protein present in C5-branched dibasic acid metabolism (KEGG Pathway: map00660) which provides alternative sources of carbon and energy.60
There was a unique presence of four proteins in nitrogen metabolism (KEEG Pathway: map00910). In the nitrogen cycle, different reductive or oxidative reactions are utilized by prokaryotes for energy conservation.61 In case of oxygen deprived growth conditions for E. coli, anaerobic respiration nitrate is the preferred electron acceptor.62 The putative resistance protein was identified as unique protein present in phosphonate and phosphinate metabolism (KEGG Pathway: map00440). Natural products containing carbon-phosphorous bonds, so-called C-P compounds have been found in many organisms, but only protists and bacteria, mostly actinobacteria, have biosynthetic capacity. Moreover, the secB; protein-export protein secB and etpN; type 4 prepilin-like proteins leader peptide-processing enzyme, that are the parts of bacterial secretion system (KEGG Pathway: map03070), were found as unique. Many proteins implicated in efflux of different toxins and drugs; virulence and biogenesis of different organelles (pili and flagella) are secreted.63 The secB is involved in efficient export of proteins across the cytoplasmic membrane in E. coli.64 In PTS (KEGG Pathway: map02060), n9 proteins were found to be uniquely present. It has been known that PTS is involved in transportation of more than 20 carbohydrates in bacteria and plays a major role in phosphorylation and uptake of carbohydrates and controlling their metabolism.65,66 Flagellar FliJ protein which is part of flagellar assembly (KEGG Pathway: map02040) as well as a putative general chaperone and cytoplasmic protein67 was identified as unique in the pathogen. The bacterial flagellum extending from the cytoplasm to the cell exterior serves as both a motor organelle and a protein export/assembly apparatus.68 A total of 19 proteins were identified as unique in the pathogen-specific pathway in a two-component system (KEGG Pathway: map02020). Bacterial two-component system is required for adaptation to external stimuli and can affect changes in cellular physiology.69 A single protein was identified to be involved in biosynthesis of siderophore group non-ribosomal peptides pathway. Polyketide synthases and non-ribosomal polypeptide synthetases are known to be responsible for the biosynthesis of several siderophores such as enterobactin in E. coli.70 Two proteins were detected in case of bacterial chemotaxis. In chemotaxis, bacteria sense chemical gradients in the environment and move toward favorable conditions. The pathway is arguably best characterized in the case of E. coli.71
Novel targets in common host-pathogen pathways
We also identified 76 proteins in 49 host–pathogen common metabolic pathways as novel targets. The pathways were grouped as metabolism of amino acid, carbohydrate, energy, lipid, other amino acids, nucleotide, cofactors and vitamins, and genetic information processing, environmental information processing, cellular processes, and others (Supplementary materials).
Identification of broad-spectrum targets
Common proteins among several species would be well broad-spectrum antibiotic targets.72 We used 73 species as reference and proteins which were common in at least 40 different species were listed as broad-spectrum targets (Supplementary materials). Forty-four proteins were identified as broad-spectrum targets (Supplementary materials). In addition, broad-spectrum proteins involved in multiple pathways would be better targets as their inhibition of activity will hamper more than one system in the pathogen.73 dnaE subunit and AckA were involved in a maximum number of pathways. AckA catalyzes the reversible reaction of formation of acetyl phosphate from acetate and ATP. Whereas dnaE participates in some critical pathways of the pathogen like purine metabolism, pyrimidine metabolism, DNA replication, mismatch repair, and homologous recombination. There is no resolved X-ray crystallography structure for both dnaE (E. coli O157:H7 Sakai strand) and AckA (E. coli O157:H7 Sakai strand) as we intended to do homology modeling. However, dnaE subunit (Uniprot ID: Q8X8X5) was preferred over AckA (Uniprot ID: P0A6A5) based on suitability of homology modeling and docking studies. During the BLAST search against PDB structures (threshold e-value <0.001), dnaE subunit showed 98% sequence identity with E. coli replicative dnaE subunit (PDB ID: 2HNH), whereas 94% for AckA with Salmonella enterica subspecies enterica serovar Typhimurium AckA (PDB ID: 3SK3 and 3SLC) (data not shown). Furthermore, an inter-domain motion during ligand binding as well as a ligand binding pocket located at the dimeric interface of form-II AckA has been reported74 that is very hard to address through in silico docking software. In case of dnaE subunit, literature searching helped us to identify the pocket for ligand binding as well as important and conserved residues within the pocket that are important for catalytic activity.75
Moreover, we conducted BLAST searching for dnaE (Uniprot ID: Q8X8X5) with e-value cut off of 0.001 against UniprotKB. We selected the organisms whose proteins showed at least 75% sequence identity with dnaE. The organisms were searched in PATRIC database to check their host and pathogenicity. Only the human hosts were considered and predicted to be pathogenic if involved in disease(s) according to PATRIC database.42 We found that all the organisms are pathogenic except Lelliottia, Kluyvera, Hafnia, Ewingella, Cedecea, and Yokenella. However, literature searching helped us to conclude that Kluyvera, Yokenella, Ewingella, Hafnia, and Cedecea are also involved in diseases.76–81 We did not find sufficient information about Lelliottia. Thus it is clearly demonstrated that E. coli O157:H7 dnaE does not share any sequence similarity with non-pathological bacterial dnaE. Therefore, due to the above advantages, dnaE was selected for homology modeling and subsequent structure-based drug designing.
Homology modeling
DNA polymerase III holo-enzyme has ten different peptides arranged in an asymmetric dimer and contains a 3′-5′ exonuclease activity. The alpha subunit is at the core enzyme and mainly functions in the polymerase activity. The best hit of similarity search identified the crystal structure of the catalytic alpha subunit of E. coli replicative dnaE DNA polymerase III (PDB ID: 2HNH).82 It showed 99.98% sequence similarity with our target sequence and was used as the template for homology modeling. The modeled structure was shown in Figure 4.
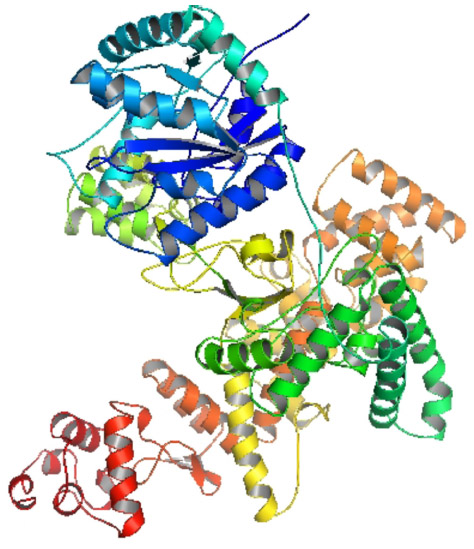 | Figure 4 Homology modeled structure of the Escherichia coli O157:H7 Sakai strand DNA polymerase III alpha, modeled with ESyPred3D server. |
Structure validation and energy minimization
Advancing toward the way of in silico drug design with protein models depends largely on the quality of the models. Inspection of the Psi/Phi Ramachandran plot analysis showed that the model built by ESyPred3D has residues in most favored regions 92.4%, residues in additional allowed regions 7.1%, and residues in generously allowed regions 0.3%. A good quality model would be expected to have over 90% in the most favored regions (Figure 5A).
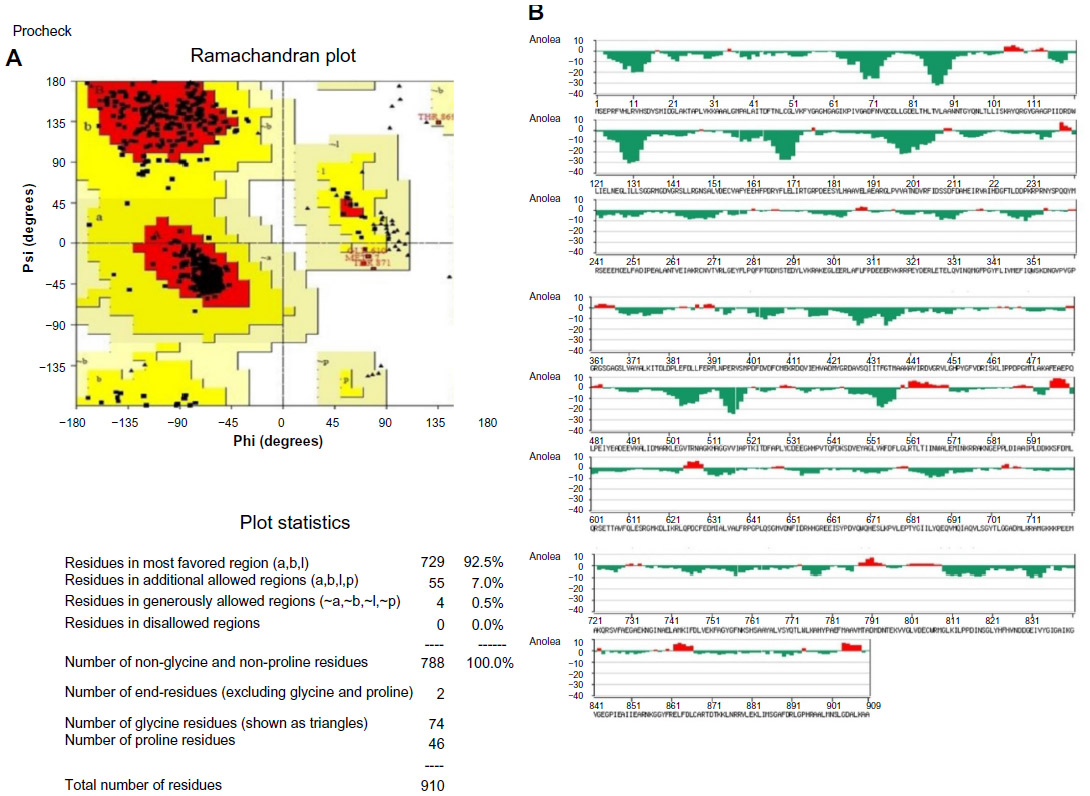 | Figure 5 Structure validation and energy minimization. |
This structure was also verified with ANOLEA. The y-axis of the plot represents the energy for each amino acid of the protein chain and it showed that maximum residues are in favorable energy environment (Figure 5B). As a result the structure modeled was considered as a good quality model for further analysis. To obtain a better refined model energy minimization was done using Swiss PDB viewer. It minimizes energy using GROMOS96. The force-field energies of the overall structure before and after minimization were −13,816.546 KJ/mol and −36,224.391 KJ/mol respectively.
Active site analysis
Active site is the region on the surface of an enzyme to which a specific substrate (ligand) or set of substrates (ligands) binds. The properties of the active site are determined by the sequences of amino acids and the 3D arrangement of the polypeptide chains of the enzyme. Identification of the active site of E. coli O157:H7 Sakai strain dnaE was done by CASTp server. This server calculates the surface area and volume of the pocket of the given structure. In addition, it also shows the active site residues. The active site residues with a volume of 17,116 were shown in Figure 6. Literature searching helped us to identify the important residues within the pocket. The structure of catalytic alpha subunit of E. coli is known to have three conserved residues of aspartate (Asp 402, Asp 404, and Asp 556 in our model, ensured by aligning with the reference structure, data not shown) and four conserved positive residues (Arg 391, Arg 397, Arg 710, and Arg 711) in this structure (Arg 390, Arg 396, Arg 709, and Arg 710 in the reference structure) that are important in catalysis83 and proposed to interact with the negatively charged triphosphate tail of the incoming nucleotide respectively. Moreover, two aromatic residues, Tyr 754 and Phe 756, that could potentially interact with the nascent base pair resembles Tyr 755 and Phe 757 in our modeled structure. These residues were therefore considered as important in inhibition of DNA binding and catalysis of the dnaE subunit.
 | Figure 6 Active site residues (shown in green) of the Escherichia coli O157:H7 Sakai strand DNA polymerase III alpha. |
Virtual screening, drug likeliness, and toxicity analysis
Twenty-four ligand molecules of E. coli K-12 obtained from BindingDB database were docked on the receptor. Molecules showing lowest binding energy were listed in Table 2. The 251D molecule is a potent inhibitor of the bacterial replicative dnaE.84 Information on these molecules’ binding interaction was taken as reference to predict convenient inhibitors of dnaE of E. coli O157:H7 Sakai strand from the molecules of DrugBank (Figure 7A). The top 100 hits from DrugBank showing lower binding energies after being docked five times were filtered for Lipinski’s rule of five85 that reduced the compounds to 59. To avoid off target binding, the compounds having human targets were excluded. The top two hits showed stable or nearly stable binding energy and good clustering performances from the docking results (Table 3) and structures are shown in Figure 8. Among the final high affinity binding molecules, DB04118 (N-coeleneterazine) and DB04698 (N-(1,4-dihydro-5H-tetrazol-5-ylidene)-9-oxo-9H-xanthene-2-sulfonamide) were found to interact with important residues (Figure 7B) required for DNA binding and catalysis. According to DrugBank database, both molecules are experimental drugs. DB04118 has no specific target yet. However, 3-dehydroquinate dehydratase of Helicobacter pylori (American Type Culture Collection strain 700392/26695), that catalyzes a trans-dehydration via an enolate intermediate, is the target for DB04698. These molecules could be proposed as potential inhibitors of the E. coli O157:H7 Sakai strand dnaE. For the identification of potential drug candidates human intestinal absorption is imperative.86 By using PreADMET we found human intestinal absorption was 95.36% and 88.10% for DB04118 and DB04698 respectively, indicating well-absorbed compounds (70%~100% according to PreADMET) that is desirable for drug candidates. As only the unbound drug is necessary for diffusion or transport across the cell membranes and target-drug interaction, we predicted percent drug bound in plasma protein. Plasma protein binding prediction results showed 99.28% and 100.00% plasma protein binding for DB04118 and DB04698 respectively, indicating strongly bound chemicals which are not desirable. Caco-2 cell model serves as a reliable in vitro model for the prediction of oral drug absorption. PreADMET predicted 17.45 (nm/second) and 0.36 (nm/second) Caco-2 cell permeability for DB04118 and DB04698 respectively that are considered middle and low permeability respectively. Both drugs exhibit good oral bioavailability and mostly negative result in Ames test as predicted by FAF-Drugs2 and PreADMET toxicity prediction (data not shown) and no evidence of carcinogenic activity for only DB04698 as predicted by PreADMET rodent carcinogenicity prediction. We also analyzed other ADMET properties like blood–brain barrier penetration, skin permeability, MDCK cell, and P-gp inhibition, and in all of these cases results are positive for our proposed compounds (data not shown).
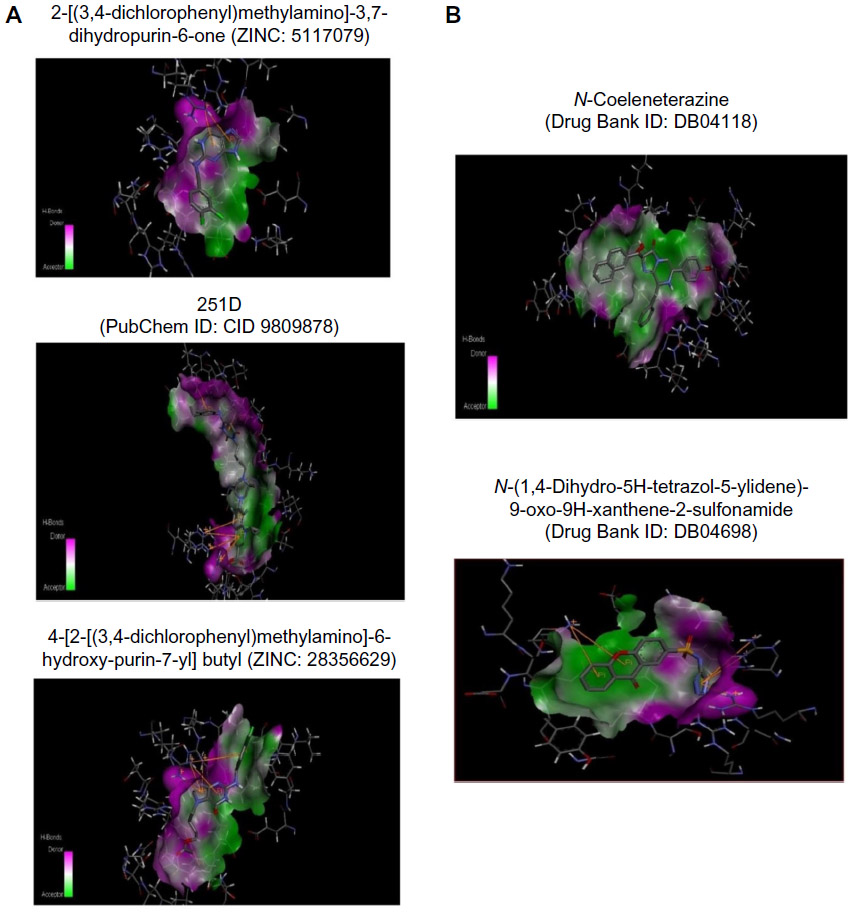 | Figure 7 Three-dimensional representation. |
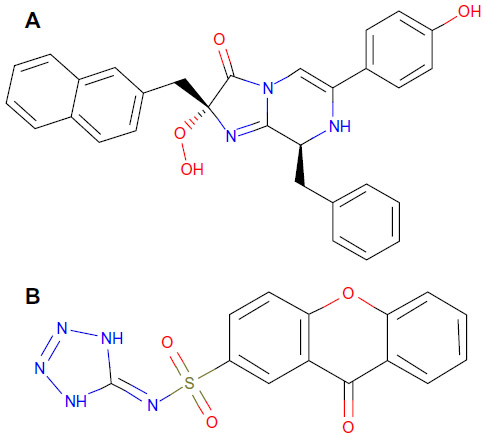 | Figure 8 Structure of top hit compounds by in silico screening. |
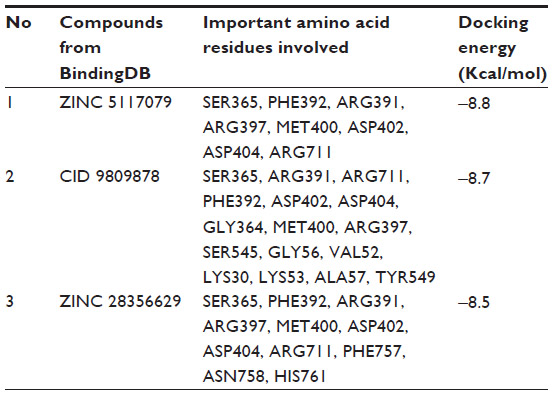 | Table 2 Lowest docking energies and important residues of the binding site observed to be interactive with the ligands from BindingDB database |
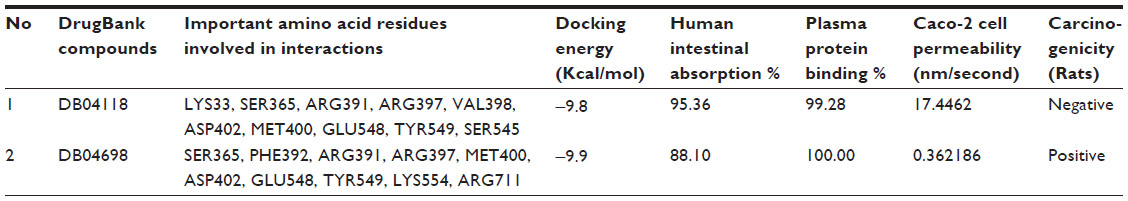 | Table 3 Lowest docking energies, important residues of the binding site observed to be interactive with the ligands from DrugBank, percentage of human intestinal absorption and plasma protein binding, Caco-2 cell permeability and carcinogenicity in rats |
Conclusion
The overall picture emerging from this study was the identification of broad-spectrum antibiotic target as well as prediction of potential inhibitors of DNA III alpha of deadly pathogen E.coli 0157:H7 using extensive in silico tools. Bacterial replicative dnaE is a member of C family of polymerases87,88 that are unique in terms of sequence. Bacterial replicative DNA polymerase III share no sequence similarity and is strikingly different from canonical DNA polymerases including those of eukaryotic replicative polymerases.89 It has been reported that DNA polymerases are responsible for pathogen survival and drug resistance so they have been considered to be a drug target in a broad group of Gram-positive pathogens such as Staphylococcus, Streptococcus, Enterococcus, and Mycoplasma.90 It has also been reported that DNA polymerase III inactivation would impede survival of Mycobacterium tuberculosis within the host.91
Since bacterial evolution aims to acquire resistance against single or multiple antibiotics to ensure their survival in the environment, the development, not only of new conventional antibiotics but also of novel compounds and alternative strategies for the battle against bacterial infections, is becoming a topical and widely recognized need. In this study, a number of criteria such as essentiality of proteins, dissimilarity with host, conservation among pathogens, availability in drug databases, virtual screening etc, were utilized to explore potential drug targets as well as to predict a drug that can block dnaE of deadly E coli O157:H7 strain. This in silico strategy can be used for screening novel and alternative targets in a way to design and develop new drugs against other emerging human pathogens.
Acknowledgments
We acknowledge Professor Dr Muhammed Zafar Iqbal for his cooperation and support throughout the research. We would like to thank the Department of Genetic Engineering and Biotechnology, and the Department of Computer Science and Engineering, Shahjalal University of Science and Technology, Sylhet, Bangladesh for providing us research facilities.
Disclosure
The authors report no conflicts of interest in this work.
References
Kaper JB, Nataro JP, Mobley HL. Pathogenic Escherichia coli. Nat Rev Microbiol. 2004;2(2):123–140. | |
Law D. Virulence factors of Escherichia coli O157 and other Shiga toxin-producing E. coli. J Appl Microbiol. 2000;88(5):729–745. | |
Ostroff SM, Griffin PM, Tauxe RV, et al. A statewide outbreak of Escherichia coli O157:H7 infections in Washington State. Am J Epidemiol. 1990;132(2):239–247. | |
Griffin PM, Tauxe RV. The epidemiology of infections caused by Escherichia coli O157:H7, other enterohemorrhagic E. coli, and the associated hemolytic uremic syndrome. Epidemiol Rev. 1991;13:60–98. | |
Besser RE, Lett SM, Weber JT, et al. An outbreak of diarrhea and hemolytic uremic syndrome from Escherichia coli O157:H7 in fresh-pressed apple cider. JAMA. 1993;269(17):2217–2220. | |
Michino H, Araki K, Minami S, et al. Massive outbreak of Escherichia coli O157:H7 infection in schoolchildren in Sakai City, Japan, associated with consumption of white radish sprouts. Am J Epidemiol. 1999; 150(8):787–796. | |
Hayashi T, Makino K, Ohnishi M, et al. Complete genome sequence of enterohemorrhagic Escherichia coli O157:H7 and genomic comparison with a laboratory strain K-12. DNA Res. 2001;8(1):11–22. | |
Walterspiel JN, Ashkenazi S, Morrow AL, Cleary TG. Effect of subinhibitory concentrations of antibiotics on extracellular Shiga-like toxin I. Infection. 1992;20(1):25–29. | |
Wong CS, Jelacic S, Habeeb RL, Watkins SL, Tarr PI. The risk of the hemolytic-uremic syndrome after antibiotic treatment of Escherichia coli O157:H7 infections. N Engl J Med. 2000;342(26):1930–1936. | |
Mora A, Blanco JE, Blanco M, et al. Antimicrobial resistance of Shiga toxin (verotoxin)-producing Escherichia coli O157:H7 and non-O157 strains isolated from humans, cattle, sheep and food in Spain. Res Microbiol. 2005;156(7):793–806. | |
Meng J, Zhao S, Doyle MP, Joseph SW. Antibiotic resistance of Escherichia coli O157:H7 and O157:NM isolated from animals, food, and humans. J Food Prot. 1998;61(11):1511–1514. | |
Wilkerson C, Samadpour M, van Kirk N, Roberts MC. Antibiotic resistance and distribution of tetracycline resistance genes in Escherichia coli O157:H7 isolates from humans and bovines. Antimicrob Agents Chemother. 2004;48(3):1066–1067. | |
Kim HH, Samadpour M, Grimm L, et al. Characteristics of antibiotic-resistant Escherichia coli O157:H7 in Washington State, 1984–1991. J Infect Dis. 1994;170(6):1606–1609. | |
Schroeder CM, Zhao C, DebRoy C, et al. Antimicrobial resistance of Escherichia coli O157 isolated from humans, cattle, swine, and food. Appl Environ Microbiol. 2002;68(2):576–581. | |
Chan JN, Nislow C, Emili A. Recent advances and method development for drug target identification. Trends Pharmacol Sci. 2010;31(2):82–88. | |
Allsop AE. Bacterial genome sequencing and drug discovery. Curr Opin Biotechnol. 1998;9(6):637–642. | |
Stumm G, Russ A, Nehls M. Deductive genomics: a functional approach to identify innovative drug targets in the post-genome era. Am J Pharmacogenomics. 2002;2(4):263–271. | |
Damte D, Suh JW, Lee SJ, Yohannes SB, Hossain MA, Park SC. Putative drug and vaccine target protein identification using comparative genomic analysis of KEGG annotated metabolic pathways of Mycoplasma hyopneumoniae. Genomics. 2013;102(1):47–56. | |
Amineni U, Pradhan D, Marisetty H. In silico identification of common putative drug targets in Leptospira interrogans. J Chem Biol. 2010;3(4):165–173. | |
Barh D, Kumar A. In silico identification of candidate drug and vaccine targets from various pathways in Neisseria gonorrhoeae. In Silico Biol. 2009;9(4):225–231. | |
Oany AR, Emran AA, Jyoti TP. Design of an epitope-based peptide vaccine against spike protein of human coronavirus: an in silico approach. Drug Des Devel Ther. 2014;8:1139–1149. | |
Rahman MA, Noore MS, Hasan MA, et al. Identification of potential drug targets by subtractive genome analysis of Bacillus anthracis A0248: An in silico approach. Comput Biol Chem. 2014;52:66–72. | |
Galperin MY, Koonin EV. Searching for drug targets in microbial genomes. Curr Opin Biotechnol. 1999;10(6):571–578. | |
Tatusov RL, Koonin EV, Lipman DJ. A genomic perspective on protein families. Science. 1997;278(5338):631–637. | |
Koonin EV, Tatusov RL, Galperin MY. Beyond complete genomes: from sequence to structure and function. Curr Opin Struct Biol. 1998; 8(3):355–363. | |
Kobayashi K, Ehrlich SD, Albertini A, et al. Essential Bacillus subtilis genes. Proc Natl Acad Sci U S A. 2003;100(8):4678–4683. | |
Lavecchia A, Di Giovanni C. Virtual screening strategies in drug discovery: a critical review. Curr Med Chem. 2013;20(23):2839–2860. | |
Kanehisa M, Goto S, Hattori M, et al. From genomics to chemical genomics: new developments in KEGG. Nucleic Acids Res. 2006; 34(Database issue):D354–D357. | |
Kanehisa M, Goto S, Furumichi M, Tanabe M, Hirakawa M. KEGG for representation and analysis of molecular networks involving diseases and drugs. Nucleic Acids Res. 2010;38(Database issue): D355–D360. | |
Magrane M, Consortium U. UniProt Knowledgebase: a hub of integrated protein data. Database (Oxford). 2011;2011:bar009. | |
Zhang R, Lin Y. DEG 5.0, a database of essential genes in both prokaryotes and eukaryotes. Nucleic Acids Res. 2009;37(Database issue): D455–D458. | |
Yu NY Wagner JR, Laird MR, et al. PSORTb 3.0: improved protein subcellular localization prediction with refined localization subcategories and predictive capabilities for all prokaryotes. Bioinformatics. 2010;26(13):1608–1615. | |
Aguero F, Al-Lazikani B, Aslett M, et al. Genomic-scale prioritization of drug targets: the TDR Targets database. Nat Rev Drug Discov. 2008;7(11):900–907. | |
Butt AM, Nasrullah I, Tahir S, Tong Y. Comparative genomics analysis of Mycobacterium ulcerans for the identification of putative essential genes and therapeutic candidates. PLoS One. 2012;7(8):e43080. | |
Ren J, Xie L, Li WW, Bourne PE. SMAP-WS: a parallel web service for structural proteome-wide ligand-binding site comparison. Nucleic Acids Res. 2010;38(Web Server issue):W441–W444. | |
Cheng AC, Coleman RG, Smyth KT, et al. Structure-based maximal affinity model predicts small-molecule druggability. Nat Biotechnol. 2007;25(1):71–75. | |
Keller TH, Pichota A, Yin Z. A practical view of ‘druggability’. Curr Opin Chem Biol. 2006;10(4):357–361. | |
Holman AG, Davis PJ, Foster JM, Carlow CK, Kumar S. Computational prediction of essential genes in an unculturable endosymbiotic bacterium, Wolbachia of Brugia malayi. BMC Microbiol. 2009;9:243. | |
Knox C, Law V, Jewison T, et al. DrugBank 3.0: a comprehensive resource for ‘omics’ research on drugs. Nucleic Acids Res. 2011; 39(Database issue):D1035–D1041. | |
Hecker N, Ahmed J, von Eichborn J, et al. SuperTarget goes quantitative: update on drug-target interactions. Nucleic Acids Res. 2012;40(Database issue):D1113–D1117. | |
Zhu F, Shi Z, Qin C, et al. Therapeutic target database update 2012: a resource for facilitating target-oriented drug discovery. Nucleic Acids Res. 2012;40(Database issue):D1128–D1136. | |
Gillespie JJ, Wattam AR, Cammer SA, et al. PATRIC: the comprehensive bacterial bioinformatics resource with a focus on human pathogenic species. Infect Immun. 2011;79(11):4286–4298. | |
Lambert C, Leonard N, De Bolle X, Depiereux E. ESyPred3D: Prediction of proteins 3D structures. Bioinformatics. 2002;18(9):1250–1256. | |
Arnold K, Bordoli L, Kopp J, Schwede T. The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics. 2006;22(2):195–201. | |
Melo F, Feytmans E. Assessing protein structures with a non-local atomic interaction energy. J Mol Biol. 1998;277(5):1141–1152. | |
Laskowski RA MM, Moss DS, Thornton JM. PROCHECK: A program to check the stereochemical quality of protein structures. J Appl Cryst. 1993;26:283–291. | |
Guex N, Peitsch MC. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis. 1997;18(15):2714–2723. | |
Binkowski TA, Naghibzadeh S, Liang J. CASTp: Computed Atlas of Surface Topography of proteins. Nucleic Acids Res. 2003;31(13):3352–3355. | |
Liu T, Lin Y, Wen X, Jorissen RN, Gilson MK. BindingDB: a web-accessible database of experimentally determined protein-ligand binding affinities. Nucleic Acids Res. 2007;35(Database issue):D198–D201. | |
Alland C, Moreews F, Boens D, et al. RPBS: a web resource for structural bioinformatics. Nucleic Acids Res. 2005;33(Web Server issue):W44–W49. | |
Sakharkar KR, Sakharkar MK, Chow VT. A novel genomics approach for the identification of drug targets in pathogens, with special reference to Pseudomonas aeruginosa. In silico Biol. 2004;4(3):355–360. | |
Chong CE, Lim BS, Nathan S, Mohamed R. In silico analysis of Burkholderia pseudomallei genome sequence for potential drug targets. In Silico Biol. 2006;6(4):341–346. | |
Duffield M, Cooper I, McAlister E, Bayliss M, Ford D, Oyston P. Predicting conserved essential genes in bacteria: in silico identification of putative drug targets. Mol Biosyst. 2010;6(12):2482–2489. | |
Caffrey CR, Rohwer A, Oellien F, et al. A comparative chemogenomics strategy to predict potential drug targets in the metazoan pathogen, Schistosoma mansoni. PLoS One. 2009;4(2):e4413. | |
Heinrichs DE, Yethon JA, Whitfield C. Molecular basis for structural diversity in the core regions of the lipopolysaccharides of Escherichia coli and Salmonella enterica. Mol Microbiol. 1998;30(2):221–232. | |
Raetz CR, Guan Z, Ingram BO, et al. Discovery of new biosynthetic pathways: the lipid A story. J Lipid Res. 2009;50 Suppl:S103–S108. | |
Navarre WW, Schneewind O. Surface proteins of gram-positive bacteria and mechanisms of their targeting to the cell wall envelope. Microbiol Mol Biol Rev. 1999;63(1):174–229. | |
Theisen AR, Murrell JC. Facultative methanotrophs revisited. J Bacteriol. 2005;187(13):4303–4305. | |
Welander PV, Metcalf WW. Loss of the mtr operon in Methanosarcina blocks growth on methanol, but not methanogenesis, and reveals an unknown methanogenic pathway. Proc Natl Acad Sci U S A. 2005; 102(30):10664–10669. | |
Peregrin-Alvarez JM, Sanford C, Parkinson J. The conservation and evolutionary modularity of metabolism. Genome Biol. 2009;10(6):R63. | |
Cabello P, Roldan MD, Moreno-Vivian C. Nitrate reduction and the nitrogen cycle in archaea. Microbiology. 2004;150(Pt 11):3527–3546. | |
Brondijk TH, Nilavongse A, Filenko N, Richardson DJ, Cole JA. NapGH components of the periplasmic nitrate reductase of Escherichia coli K-12: location, topology and physiological roles in quinol oxidation and redox balancing. Biochem J. 2004;379(Pt 1):47–55. | |
Kostakioti M, Newman CL, Thanassi DG, Stathopoulos C. Mechanisms of protein export across the bacterial outer membrane. J Bacteriol. 2005;187(13):4306–4314. | |
Kumamoto CA. Escherichia coli SecB protein associates with exported protein precursors in vivo. Proc Natl Acad Sci U S A. 1989;86(14):5320–5324. | |
Dahl U, Jaeger T, Nguyen BT, Sattler JM, Mayer C. Identification of a phosphotransferase system of Escherichia coli required for growth on N-acetylmuramic acid. J Bacteriol. 2004;186(8):2385–2392. | |
Kotrba P, Inui M, Yukawa H. Bacterial phosphotransferase system (PTS) in carbohydrate uptake and control of carbon metabolism. J Biosci Bioeng. 2001;92(6):502–517. | |
Van Arnam JS, McMurry JL, Kihara M, Macnab RM. Analysis of an engineered Salmonella flagellar fusion protein, FliR-FlhB. J Bacteriol. 2004;186(8):2495–2498. | |
Macnab RM. How bacteria assemble flagella. Annu Rev Microbiol. 2003;57:77–100. | |
Yamamoto K, Hirao K, Oshima T, Aiba H, Utsumi R, Ishihama A. Functional characterization in vitro of all two-component signal transduction systems from Escherichia coli. J Biol Chem. 2005;280(2):1448–1456. | |
Silakowski B, Kunze B, Nordsiek G, Blocker H, Hofle G, Muller R. The myxochelin iron transport regulon of the myxobacterium Stigmatella aurantiaca Sg a15. Eur J Biochem. 2000;267(21):6476–6485. | |
Rao CV, Ordal GW. The molecular basis of excitation and adaptation during chemotactic sensory transduction in bacteria. Contrib Microbiol. 2009;16:33–64. | |
White TA, Kell DB. Comparative genomic assessment of novel broad-spectrum targets for antibacterial drugs. Comp Funct Genomics. 2004;5(4):304–327. | |
Barh D TS, Jain N, Ali A, et al. In Silico Subtractive Genomics for Target Identification in Human Bacterial Pathogens. Drug Development Research. 2011;72:162–177. | |
Chittori S, Savithri HS, Murthy MR. Structural and mechanistic investigations on Salmonella typhimurium acetate kinase (AckA): identification of a putative ligand binding pocket at the dimeric interface. BMC Struct Biol. 2012;12:24. | |
Lamers MH, Georgescu RE, Lee SG, O’Donnell M, Kuriyan J. Crystal structure of the catalytic α subunit of E. coli replicative DNA polymerase III. Cell. 2006;126(5):881–892. | |
Jain S, Gaind R, Gupta KB, et al. Yokenella regensburgei infection in India mimicking enteric fever. J Med Microbiol. 2013;62(Pt 6):935–939. | |
Sarria JC, Vidal AM, Kimbrough RC. Infections caused by Kluyvera species in humans. Clin Infect Dis. 2001;33(7):E69–E74. | |
Ridell J, Siitonen A, Paulin L, Mattila L, Korkeala H, Albert MJ. Hafnia alvei in stool specimens from patients with diarrhea and healthy controls. J Clin Microbiol. 1994;32(9):2335–2337. | |
Palaniswamy C, Selvaraj DR, Selvaraj T. Gangrenous cholecystitis caused by Hafnia alvei: a case report and review of literature. J Am Med Dir Assoc. 2009;10(5):361–363. | |
Hassan S, Amer S, Mittal C, Sharma R. Ewingella americana: an emerging true pathogen. Case Rep Infect Dis. 2012;2012:730720. | |
Bayir O, Yildirim GA, Saylam G, Yuksel E, Ozdek A, Korkmaz MH. Atrophic rhinitis caused by Cedecea davisae with accompanying mucocele. Kulak Burun Bogaz Ihtis Derg. 2015;25(4):249–253. | |
Lamers MH, Georgescu RE, Lee SG, O’Donnell M, Kuriyan J. Crystal structure of the catalytic alpha subunit of E. coli replicative DNA polymerase III. Cell. 2006;126(5):881–892. | |
Pritchard AE, McHenry CS. Identification of the acidic residues in the active site of DNA polymerase III. J Mol Biol. 1999;285(3):1067–1080. | |
Butler MM, Lamarr WA, Foster KA, et al. Antibacterial activity and mechanism of action of a novel anilinouracil-fluoroquinolone hybrid compound. Antimicrob Agents Chemother. 2007;51(1):119–127. | |
Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 2001;46(1–3):3–26. | |
Zhao YH, Le J, Abraham MH, et al. Evaluation of human intestinal absorption data and subsequent derivation of a quantitative structure–activity relationship (QSAR) with the Abraham descriptors. J Pharm Sci. 2001;90(6):749–784. | |
Rothwell PJ, Waksman G. Structure and mechanism of DNA polymerases. Adv Protein Chem. 2005;71:401–440. | |
Kelman Z, O’Donnell M. DNA polymerase III holoenzyme: structure and function of a chromosomal replicating machine. Annu Rev Biochem. 1995;64(1):171–200. | |
McCauley MJ, Shokri L, Sefcikova J, Venclovas C, Beuning PJ, Williams MC. Distinct double-and single-stranded DNA binding of E. coli replicative DNA polymerase III α subunit. ACS Chem Biol. 2008;3(9):577–587. | |
Barnes MH, Leo CJ, Brown NC. DNA polymerase III of Gram-positive eubacteria is a zinc metalloprotein conserving an essential finger-like domain. Biochemistry. 1998;37(44):15254–15260. | |
Jadaun A, Sudhakar DR, Subbarao N, Dixit A. In Silico Screening for Novel Inhibitors of DNA Polymerase III Alpha Subunit of Mycobacterium tuberculosis (Mtb DnaE2, H 37 R v). 2015;10(3):e0119760. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.