")
Back to Journals » OncoTargets and Therapy » Volume 13
Identification of P-Rex1 in the Regulation of Liver Cancer Cell Proliferation and Migration via HGF/c-Met/Akt Pathway
Authors Qiu W, Chang Y, Liu J, Yang X, Yu Y, Li J, Liang Q, Sun G
Received 10 June 2020
Accepted for publication 21 August 2020
Published 24 September 2020 Volume 2020:13 Pages 9481—9495
DOI https://doi.org/10.2147/OTT.S265592
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Carlos E Vigil
Wancheng Qiu,1 Yanhua Chang,2 Jing Liu,1 Xu Yang,1 Yan Yu,1 Jiajia Li,1 Qing Liang,1 Guangchun Sun1
1Department of Pharmacy, Shanghai Fifth People’s Hospital, Fudan University, Shanghai, People’s Republic of China; 2Department of Pathology, The Affiliated Wuxi People’s Hospital of Nanjing Medical University, Wuxi, Jiangsu Province, People’s Republic of China
Correspondence: Guangchun Sun; Qing Liang Email [email protected]; [email protected]
Background: Rho-GTPases and their activators, guanine nucleotide exchange factors (GEFs), are increasingly being recognized as essential mediators of oncogenic signaling. Although it is known that P-Rex1, a member of the Dbl family of GEFs for the Rac small GTPase, contributes to the migration of cancer cells, its exact role in liver cancer and the underlying mechanisms remain unclear.
Materials and Methods: Public datasets from the Gene Expression Omnibus database (GEO) and clinical liver cancer samples were analyzed to explore the expression of P-Rex1. P-Rex1 knockdown and overexpression cell lines were established using a recombinant lentiviral transfection system. BrdU and colony formation assays were performed to determine cell viability. Migratory capacity was analyzed using a transwell migration assay and an in vitro wound-healing assay. Nude mice bearing subcutaneous xenograft tumors were established to determine the effects of P-Rex1 on tumorigenesis in vivo. The role of P-Rex1 in hepatocarcinogenesis was determined through Western blot and co-immunoprecipitation.
Results: Induced expression of endogenous P-Rex1 was identified in liver cancer tumors when compared with adjacent nonmalignant tissues from clinical data. In response to HGF treatment, P-Rex1-knockdown cells displayed reduced proliferation and migration in vitro as well as reduced xenograft tumor growth in vivo. Overexpression of P-Rex1 promoted liver cancer cell proliferation and migration. P-Rex1 primarily acts as a downstream effector of GPCR signaling. This study demonstrated that downregulation of P-Rex1 led to a significant decrease in the phosphorylation of Akt and Erk1/2 by reducing the phosphorylation of the tyrosine kinase receptor c-Met. Furthermore, a physical association between P-Rex1 and c-Met was observed after HGF treatment, suggesting that P-Rex1 may be involved in the HGF/c-Met signaling pathway.
Conclusion: These results support the role of P-Rex1 as a novel player in liver cancer, which suggest that targeting P-Rex1 may provide a potential strategy for liver cancer treatment.
Keywords: liver cancer, P-Rex1, c-Met, proliferation, migration
Introduction
Liver cancer is one of the most common causes of cancer-related death worldwide, making it an increasing public health concern. One of the limiting factors in liver cancer treatment is late diagnosis that renders surgical resection or radiofrequency ablation, otherwise potentially curative treatment modalities, inapplicable.1,2 Sorafenib, a commonly used oral drug, is a standard systemic treatment for patients with advanced liver cancer, but resistance usually takes less than six months to develop. Although lenvatinib was approved by the Food and Drug Administration as first-line treatment for patients with unresectable liver cancer in 2018, acquired drug resistance remains inevitable.3 Thus, the need for novel treatment strategies has become increasingly urgent. The MET oncogene, which encodes the tyrosine kinase receptor for hepatocyte growth factor (HGF), has been observed to play a pivotal role in the promotion of tumor growth and metastasis, including liver cancer.4,5 Aberrant c-Met activity occurs in various cancer types and is often associated with a poor prognosis. Binding of HGF to c-Met activates multiple intracellular pathways such as mitogen-activated protein kinase (MAPK) and phosphatidylinositol 3-kinase (PI3K) via phosphorylation of the receptor tyrosine kinase domain. Several studies have demonstrated the effects of HGF on phenotypic changes in liver cancer, including epithelial-mesenchymal transition (EMT), migration, and invasion. Knockdown of c-Met impairs liver cancer cell proliferation, colony formation, and migration in vitro as well as tumor growth in vivo.6–8 In addition, HGF has been shown to induce resistance to multiple kinase inhibitors by activating phosphorylated c-Met, a process that is dependent on the Akt/Erk1/2-EGR1 pathway.9 Although it is widely believed that c-Met is one of the most promising therapeutic targets for liver cancer treatment, none of the clinical trials of nonselective inhibitors have shown the desired effects.
Rho-GTPase family proteins are key molecular switches in whole-intracellular signal transduction, cycling between the inactive GDP-bound and active GTP-bound states. The exchange of GDP for GTP and the subsequent activation of Rho-GTPases are mediated by GEFs, whose activation often occurs downstream of receptor signaling.10,11 Rho-GTPases and Rho-GEFs play central roles in controlling intracellular processes such as cytoskeleton regulation, cell polarity, reactive oxygen species (ROS) formation, and gene transcription.12 Overexpression of Rho-GEFs contributes to cancer progression and metastasis in various cancer types, including breast cancer and glioblastoma, mainly through the promotion of tumor cell migration and invasion.13–15 PtdIns(3,4,5)P3-dependent Rac exchanger 1 (P-Rex1), a member of the Dbl family of Rho-GEFs, is synergistically activated by PtdIns(3,4,5)P3 and the βγ subunits of G-protein-coupled receptors (GPCR).16,17 The PREX1 gene region is frequently deleted or amplified in cancer, suggesting the role of PREX1 as a putative oncogene in human cancers.18–20 In addition to the P-Rex1 pathway’s extensive involvement in the inflammatory response, neurite differentiation,21 lung endothelial permeability,22,23 and pulmonary fibrosis,24 its involvement in cancer has only recently been studied.17,25,26 However, the role of P-Rex1 in liver cancer remains elusive. The present work indicates that P-Rex1 functions in liver cancer cell proliferation, migration, and xenograft tumor growth by acting as a downstream effector of HGF signaling. Knockdown of P-Rex1 suppressed Akt and Erk1/2 phosphorylation by reducing the phosphorylation of the tyrosine kinase receptor c-Met. A physical association between P-Rex1 and c-Met was observed after HGF treatment. Collectively, these findings suggest the possibility of targeting P-Rex1 for liver cancer therapy based on its dual function in liver cancer pathogenesis and inflammation.27
Materials and Methods
Immunohistochemistry
Five paraffin sections of liver cancer tumors and paired adjacent non-tumor tissues were obtained from the Shanghai Fifth People’s Hospital repository and subjected to deparaffinization and antigen retrieval. All human-sample experimental procedures were approved by the Research Ethics Committee of Shanghai Fifth People’s Hospital, Fudan University (2016EC119) and conducted in accordance with the Code of Ethics of the World Medical Association (Declaration of Helsinki); the donor was informed completely and provided consent for the experiment. P-Rex1 was detected by incubating the sections with primary antibodies at 4°C overnight. Coloration was developed using a DAB chromogenic kit (Boster, Wuhan, China). The immunohistochemical score was measured based on the staining intensity standard.
Analysis of the Liver Cancer Database
GSE121248 and GSE112790 were obtained from the NCBI Gene Expression Omnibus (GEO) (http://www.ncbi.nlm.nih.gov/geo). Prior to downstream analysis, the array probes were mapped to the respective gene symbols using array annotations. The average expression values of genes matching multiple probes were calculated as the expression value of genes. A t-test was used to identify significant differences between two groups. Correlation between the mRNA expression of P-Rex1 and liver cancer prognosis was evaluated using the online Kaplan-Meier plotter database. The overall survival (OS) of liver cancer patients with vascular invasion was analyzed.
Construction of the P-Rex1 Knockdown and Overexpression System
Human liver cancer cell lines (SMMC7721, HepG2, Huh7, PLC/PRF/5, and SK-Hep1) were obtained from the cell bank of the Chinese Academy of Science (Shanghai, China). Cells were cultured in DMEM supplemented with 10% fetal bovine serum (FBS, Gibco, USA) at 37°C in a humidified 5% CO2 atmosphere. Three small hairpin RNAs (shRNAs) specifically targeting P-Rex1 (target sequence-1, 5ʹ-GCAACGACTTCAAGCTGGTGGAGAA-3ʹ, target sequence-2, 5ʹ-GACTCCTACAGCGAGTGTAAC-3ʹ, target sequence-3, 5ʹ-GCATCAAGAAGGTGTGCTTCA −3ʹ) and one scrambled shRNA (5ʹ-AGTACTGCTTACGATACGGTT-3ʹ) were designed and cloned into the pLKO.1-puromycin lentiviral vector for construction of the P-Rex1 knockdown system. Full-length human P-Rex1 coding sequences were kindly provided by Richard D. Ye and cloned into the pCDH-CMV-MCS-EF1-Puro lentiviral vector to construct a P-Rex1 overexpression system. The cloned plasmids as well as the packaging plasmids (psPAX2 and pVSVG) were transfected into 293T cells to produce the lentiviral particles used to infect liver cancer cells. At about two and three days post-transfection, supernatants were collected and filtered using 0.45-μm cellulose acetate filters. The stably transfected cells were screened by puromycin 48 h after viral infection, and then P-Rex1 expression was verified using Western blotting.
BrdU Assay
Cell proliferation ELISA was performed using the BrdU assay kit (Roche, Switzerland) following the manufacturer’s instructions. Briefly, shP-Rex1 or PROE cells as well as control cells were seeded in 96-well plates (4000/well) and cultured with DMEM/FBS containing HGF (20 ng/mL, PeproTech, USA) or buffer control. Cells were incubated with BrdU for 4 h and fixed using FixDenat. Anti-BrdU antibody was used to detect BrdU incorporation.
Colony Formation Assay
Scrambled control or shP-Rex1 cells were diluted in culture medium and seeded into six-well plates at a density of 500 per well, in triplicate. Cells were treated with HGF (20 ng/mL) or buffer control for 14 days until optimal clones were formed. The clones were fixed with methanol, stained with 0.1% crystal violet, and then photographed. Colony formation was evaluated using NIH Image J software.
Cell Migration Assay
Cells were serum-deprived 12 h before the transwell migration assay. A total of 5×104 cells were seeded in the upper chamber (8 μM pore size, Corning) with 100 μL serum-free DMEM containing 20 ng/mL HGF. Fetal bovine serum (10%) was placed in the wells of the lower chamber to induce migration. The chamber was incubated in a humidified environment with 5% CO2 at 37°C for 24 h. The cells that migrated on the membrane underside were counted and photographed in five random fields.
For the wound-healing assay, scrambled control or shP-Rex1 cells were seeded into six-well plates and left overnight. At confluence, the cells were scratched using a standard 1-mL pipette tip to create a cell-free strip. The cells were maintained for a total period of 32 h in DMEM containing 2% FBS with HGF (20 ng/mL) or buffer control. Cell migration was evaluated by measuring the reduced wound area versus total area throughout the entire observation period, and photographs were taken 0, 16, and 32 h after scratching.
Co-Immunoprecipitation (Co-IP)
293T cells were either co-transfected with an HA-tagged human c-Met plasmid and an AU5-tagged human P-Rex1 plasmid or transfected separately with one or the other. At about 48 h post-transfection, the cells were lysed on ice with cold RIPA lysis buffer. For endogenous immunoprecipitation, SK-Hep1 cells were stimulated with HGF (20 ng/mL) or buffer control for the indicated times. Cell lysates were cleared and incubated with 3 μg anti-HA or c-Met antibodies (Cell Signaling Technology, USA) overnight at 4°C. Protein A/G PLUS-Agarose (Santa Cruz, USA) was added to the lysates and incubated by rotation at 4°C for 5 h. The immunoprecipitates were collected and subjected to Western blotting using different antibodies, as indicated in the figures.
Western Blot Analysis
Total protein of the cells or tumor tissues was extracted in RIPA lysis buffer containing protease and phosphatase inhibitor cocktails (Sigma-Aldrich, USA). Protein samples were separated by SDS-PAGE and transferred to PVDF membranes. The membranes were blocked with 5% skim milk for 1.5 h at room temperature and incubated overnight at 4°C with primary antibodies, including P-Rex1, phospho-c-Met, c-Met, phospho-Akt (473), Akt, phospho-Erk1/2, Erk1/2, and β-actin. All antibodies were purchased from Cell Signaling Technology, and the dilution ratio was 1:1000. Protein bands were detected using the Enhanced Chemiluminescence Substrate Kit and visualized using the FluorChem E system.
In vivo Tumor Xenograft Model
Male nude mice (BALB/c-nu, five weeks) were purchased from Shanghai SLAC Laboratory Animal Co. Ltd. (Shanghai, China) and housed under specific pathogen-free conditions. A total of 5×106 SK-Hep1 (scrambled-shRNA or P-Rex1-shRNA) cells in a volume of 100 μL PBS were inoculated subcutaneously in the right oxter of each mouse. Tumor volumes were measured and recorded every three days after inoculation using the formula V= (L×W2)/2, where the length and width of tumors were determined using Vernier calipers. The mice were sacrificed after a one-month period of observation. Solid tumors were surgically resected and weighed. Animal’s welfare and use adhered to the guideline for ethical review of laboratory animal welfare (China National Standard GB/T 35892–2018) following a protocol approved by the Institutional Animal Care and Use Committee (IACUC) at Shanghai Jiaotong University.
Statistical Analysis
Quantitative data are expressed as the means ± SEM obtained from at least three independent experiments. Statistically significant differences between samples were determined by a one-way ANOVA (Bonferroni test). The Wilcoxon signed-rank test was used for the statistical analysis of tumor growth. Statistical significance was defined as P < 0.05. Analysis and graphing were performed using Prism software.
Results
Induced Expression of P-Rex1 in Liver Cancer
To determine whether P-Rex1 is involved in the pathogenesis of liver cancer, human tissues obtained from the Shanghai Fifth People’s Hospital repository were analyzed for P-Rex1 expression. The results confirmed that P-Rex1 expression in tumors was markedly upregulated compared with that in normal tissues (Figure 1A). Liver cancer patient data from public datasets of the Gene Expression Omnibus database (GEO) showed that P-Rex1 was presented at a significantly higher expression level in tumors than in adjacent nonmalignant tissues (GSE121248 and GSE112790). P-Rex1 was originally purified from leukocyte cytosol and was found to be highly expressed in neutrophils. The expression of the neutrophil marker myeloperoxidase (MPO) showed no significant difference between groups, indicating that the high P-Rex1 levels were not due to infiltrating neutrophils (Figure 1B and C). The overall survival (OS) of liver cancer patients was evaluated using the online Kaplan-Meier plotter database. Liver cancer patients with vascular invasion in high P-Rex1 expression group experienced shorter survival compared with patients with low P-Rex1 expression group (Figure 1D). These results demonstrate that P-Rex1 may be involved in the pathogenesis of liver cancer and its overexpression indicates a worse clinical outcome.
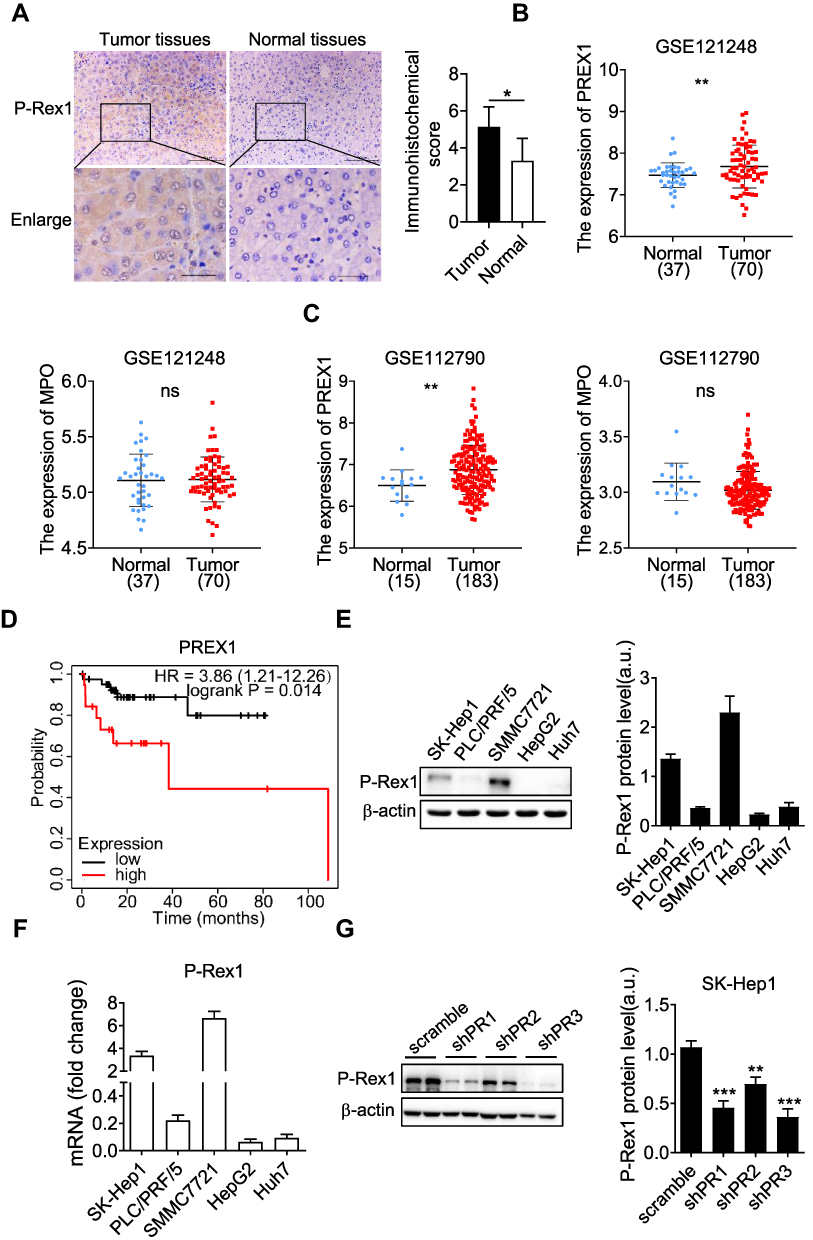 |
Figure 1 P-Rex1 expression in liver cancer. (A) Immunohistochemical staining of P-Rex1 in tumor and paired adjacent non-tumor (normal) tissues from patients (original magnification: scale bars = 100 μm, inset: scale bars = 25 μm), n=5. (B and C) P-Rex1 and MPO mRNA expression was analyzed in malignant liver tissues and adjacent non-malignant tissues using the GEO datasets (GSE121248 and GSE112790). (D) The Kaplan-Meier overall survival (OS) curves were presented. Liver cancer patients with vascular invasion were divided into a high P-Rex1 expression group and a low P-Rex1 expression group. (E and F) The protein and mRNA levels of P-Rex1 in five liver cancer cell lines. (G) The stable scrambled control clone and P-Rex1 knockdown clones (shPR1, shPR2, and shPR3) were constructed using recombinant lentiviral transfection. Representative Western blots and bar charts are shown. Quantitative data are shown as means ± SEM from three independent experiments. (*P < 0.05, **P < 0.01, and ***P < 0.001). |
Establishment of the P-Rex1 Knockdown SK-Hep1 Cell Line
Expression of P-Rex1 was detected in five common liver cancer cell lines (SK-Hep1, PLC/PRF/5, SMMC7721, HepG2, and Huh7). Western blot results showed that the expression of P-Rex1 was high in the highly metastatic cell lines, especially SMMC7721 and SK-Hep1. HepG2 cells expressed low levels of P-Rex1 (Figure 1E and F). To evaluate the role of P-Rex1 in liver cancer, a P-Rex1 knockdown SK-Hep1 cell line was established using a recombinant lentiviral transfection system. Three shRNA sequences were designed and constructed using lentivirus plasmids. Group shPR3, which showed maximum efficiency in silencing P-Rex1 protein expression, was selected for further investigation into the role of P-Rex1 in proliferation and migration (Figure 1G).
P-Rex1 Knockdown Inhibits Progression into the G2/M Phase in SK-Hep1 Cells
To explore the role of P-Rex1 in the cell cycle, flow cytometry was performed to detect cell cycle distribution. The cell phase distribution was found to be 77.40 ± 0.90% vs 71.23 ± 0.84% in G0/G1 phase, 2.74 ± 0.10% vs 4.70 ± 0.13% in S phase (P < 0.001), and 9.75 ± 0.98% vs 5.45 ± 0.18% in G2/M phase (P < 0.05), before vs after P-Rex1 knockdown in SK-Hep1 cells, respectively (Figure 2A). The fraction of cells in S phase was significantly increased, while the G2/M phase fraction was decreased after P-Rex1 knockdown, suggesting that P-Rex1 depletion induces S-phase arrest and inhibits progression into the G2/M phase. Moreover, p21 was reported to inhibit progression into G2 and through G2/M via inhibition of the CDK1-cyclinA and CDK1-cyclinB1 complexes.28 The protein level of p21 in the P-Rex1 knockdown group was significantly increased (P < 0.05) 6 h after HGF stimulation (Figure 2B), further demonstrating the inhibitory effect of P-Rex1 knockdown on the cell cycle.
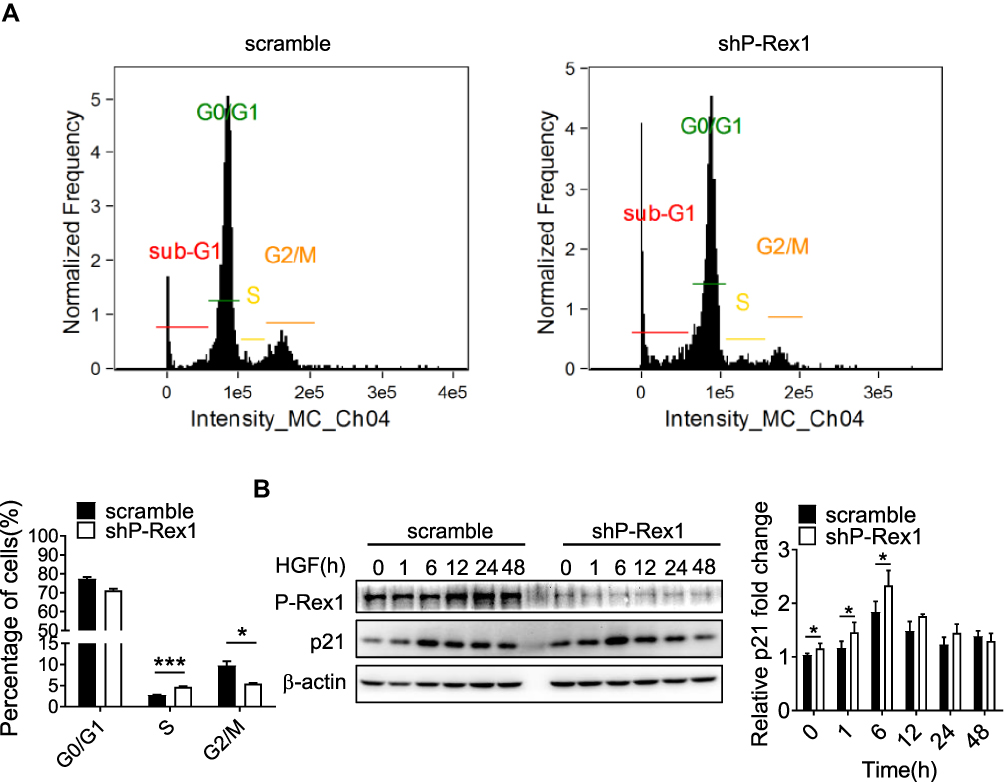 |
Figure 2 P-Rex1 regulates cell cycle transition in SK-Hep1 cells. (A) Cell phase distribution in scrambled control and P-Rex1 knockdown (shP-Rex1) cells. The ratio is presented as a bar diagram. (B) The expression of p21 protein after HGF treatment for the indicated time (h) in the control and shP-Rex1 groups of SK-Hep1 cells. Each experiment was repeated three times. The data are presented as means ± SEM. (*P < 0.05 and ***P < 0.001). |
Downregulation of P-Rex1 Reduces Proliferation and Migration of SK-Hep1 Cells
To evaluate the effect of P-Rex1 on liver cancer cell proliferation, a BrdU assay was performed to determine cell viability in serum-containing medium supplemented with HGF (20 ng/mL) or buffer control. The 48-h cell proliferation rates were significantly reduced after P-Rex1 depletion in both the normal (P < 0.05) and HGF-stimulated conditions (P < 0.05, Figure 3A). Moreover, a colony formation assay was conducted to assess the effect of P-Rex1 on colonogenicity. ShP-Rex1 cells formed fewer and smaller colonies than control cells did in response to HGF (P < 0.001, Figure 3B and C).
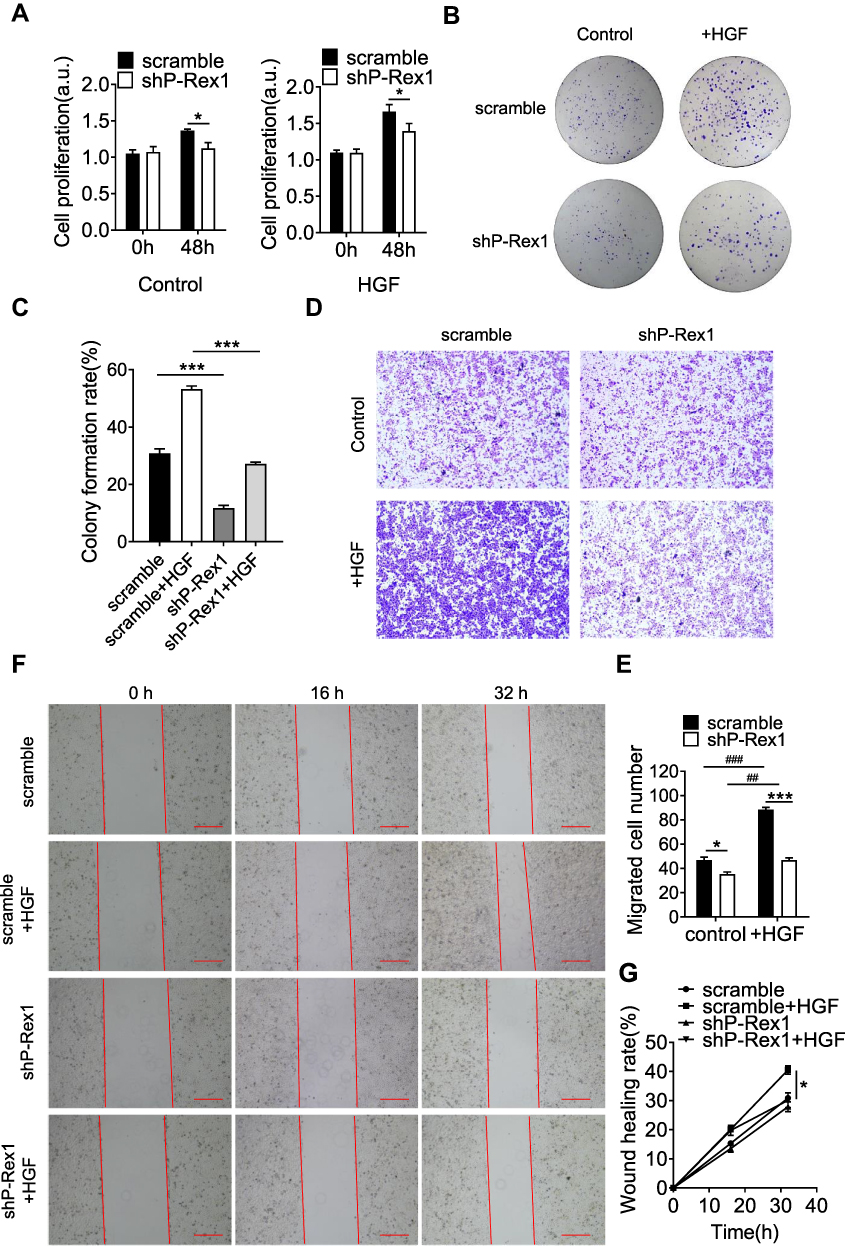 |
Figure 3 Downregulation of P-Rex1 attenuates proliferation and migration in SK-Hep1 cells. (A) A BrdU assay was performed to determine the proliferation of scrambled control and P-Rex1 knockdown (shP-Rex1) cells in culture medium treated with or without HGF (20 ng/mL) for 48 h. (B and C) Scrambled control or shP-Rex1 cells were diluted and seeded into six-well plates to determine their colony forming capabilities. (D and E) A transwell migration assay was performed to evaluate the migratory behavior of scrambled control or shP-Rex1 cells treated with HGF (20 ng/mL). Representative images of migrated cells are shown, scale bars = 200 μm. (F and G) Wound closure with scrambled control or shP-Rex1 cells treated with HGF (20 ng/mL) or buffer control for a total of 32 h. Microphotographs were taken at 0, 16, and 32 h. Scale bars = 500 μm. Each experiment was repeated three times. The data are presented as means ± SEM. *P < 0.05 and ***P < 0.001 comparing shP-Rex1 cells with scrambled control cells; ##P < 0.01 and ###P < 0.001 comparing HGF-treated samples with untreated samples in the same group. |
Migratory capacity was analyzed using a transwell migration assay and an in vitro wound-healing assay. HGF induced a higher rate of migration in the SK-Hep1 cells. Compared with that of the scrambled controls, the increased number of migrated cells was diminished in P-Rex1-knockdown cells (Figure 3D and E). In agreement with the results from the transwell assay, treatment of SK-Hep1 cells with HGF increased wound healing rates significantly 32 h after scratching (P < 0.05). However, much slower migration was observed in P-Rex1-knockdown cells (Figure 3F and G). These results demonstrate that P-Rex1 is involved in liver cancer cell proliferation and migration.
Silencing P-Rex1 Suppresses Xenograft Tumor Growth in vivo
Nude mice bearing subcutaneous xenograft tumors were established to determine the effects of P-Rex1 on tumorigenesis in vivo. SK-Hep1 cells expressing scrambled-shRNA or P-Rex1-shRNA were inoculated into nude mice subcutaneously, and then tumor growth was observed. The P-Rex1-knockdown group exhibited a significant decrease in tumor volume compared with the scrambled control group (914.77 ± 114.99 mm3 vs 1483.70 ± 186.90 mm3, respectively, P < 0.01) (Figure 4A). The average tumor weights of the P-Rex1-knockdown group and the control group were 0.70 ± 0.09 g and 1.20 ± 0.13 g, respectively, 29 days after infusion with SK-Hep1 cells (P < 0.01) (Figure 4B). Additionally, a Western blot analysis of tumor tissue homogenates showed that phosphorylated Akt (p-Akt) and Erk1/2 (p-Erk1/2) levels were attenuated in the P-Rex1-knockdown group. Significantly lower levels of the HGF receptor c-Met were observed in P-Rex1-knockdown tumor tissues compared with those in control tissues (Figure 4C). All of these molecules are closely related to tumorigenesis, tumor cell growth, and apoptosis. Collectively, these findings suggest that P-Rex1 knockdown downregulates liver cancer cell ectopic tumor formation.
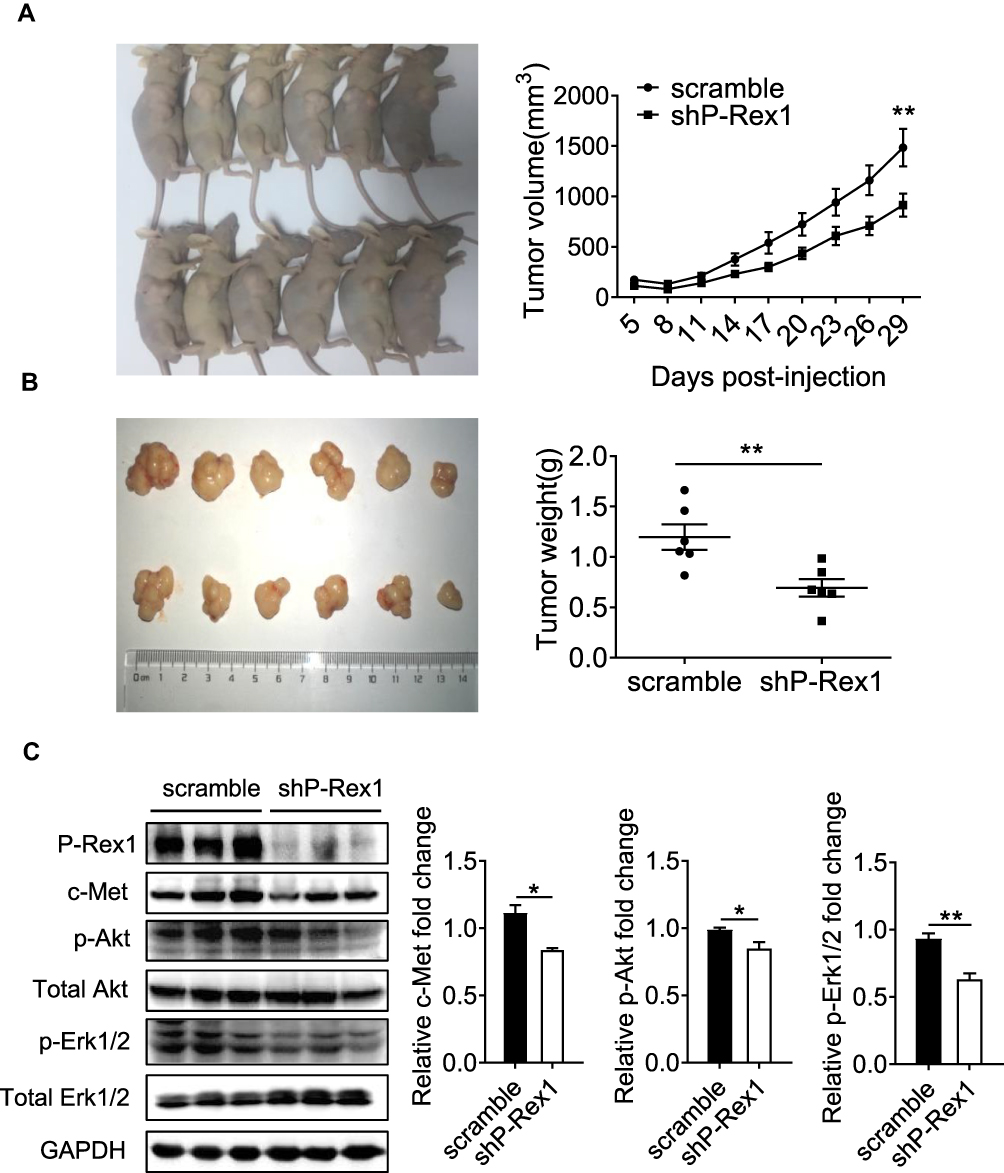 |
Figure 4 P-Rex1 downregulation inhibits xenograft tumor growth in vivo. (A) BALB/c-nu mice (male) were subcutaneously transplanted with SK-Hep1 cells transfected with scrambled-shRNA or P-Rex1-shRNA (n=6). The volumes of the tumors were measured and presented as the mean ± SEM. The tumors were removed for weight analysis at 29 days post-inoculation (B). (C) The expression of c-Met, p-Akt, and p-Erk1/2 in tumor tissue homogenates. The data are shown above as means ± SEM from three independent experiments. (*P < 0.05 and **P < 0.01). |
P-Rex1 Knockdown Inhibits Akt and Erk1/2 Phosphorylation by Targeting c-Met
Having established a role for P-Rex1 in tumorigenesis and metastasis, the subsequent experiment was focused on the signaling mediated by P-Rex1 in vitro. HGF/c-Met has been implicated in liver cancer tumor proliferation, motility, and invasion. The phosphorylation levels of c-Met (p-c-Met) were significantly elevated after HGF stimulation. The upregulation of p-c-Met was less pronounced in P-Rex1-knockdown cells, suggesting a correlation between P-Rex1 and the HGF/c-Met signaling pathway (P<0.05, Figure 5A and B). Moreover, P-Rex1-knockdown cells showed much less phosphorylation of downstream signaling molecules Akt and Erk1/2 compared with control cells (P < 0.05, Figure 5A, C, and D). Furthermore, an empty vector or P-Rex1-containing plasmid was transfected into shP-Rex1 cells to investigate whether re-expression of the P-Rex1 protein could rescue c-Met phosphorylation. In contrast with the shP-Rex1 group, a small but significant increase in p-c-Met protein levels was observed in the P-Rex1 re-expression group following HGF stimulation (P < 0.05, Figure 5E and F). These data demonstrate that P-Rex1 is involved in HGF/c-Met signaling in SK-Hep1 cells.
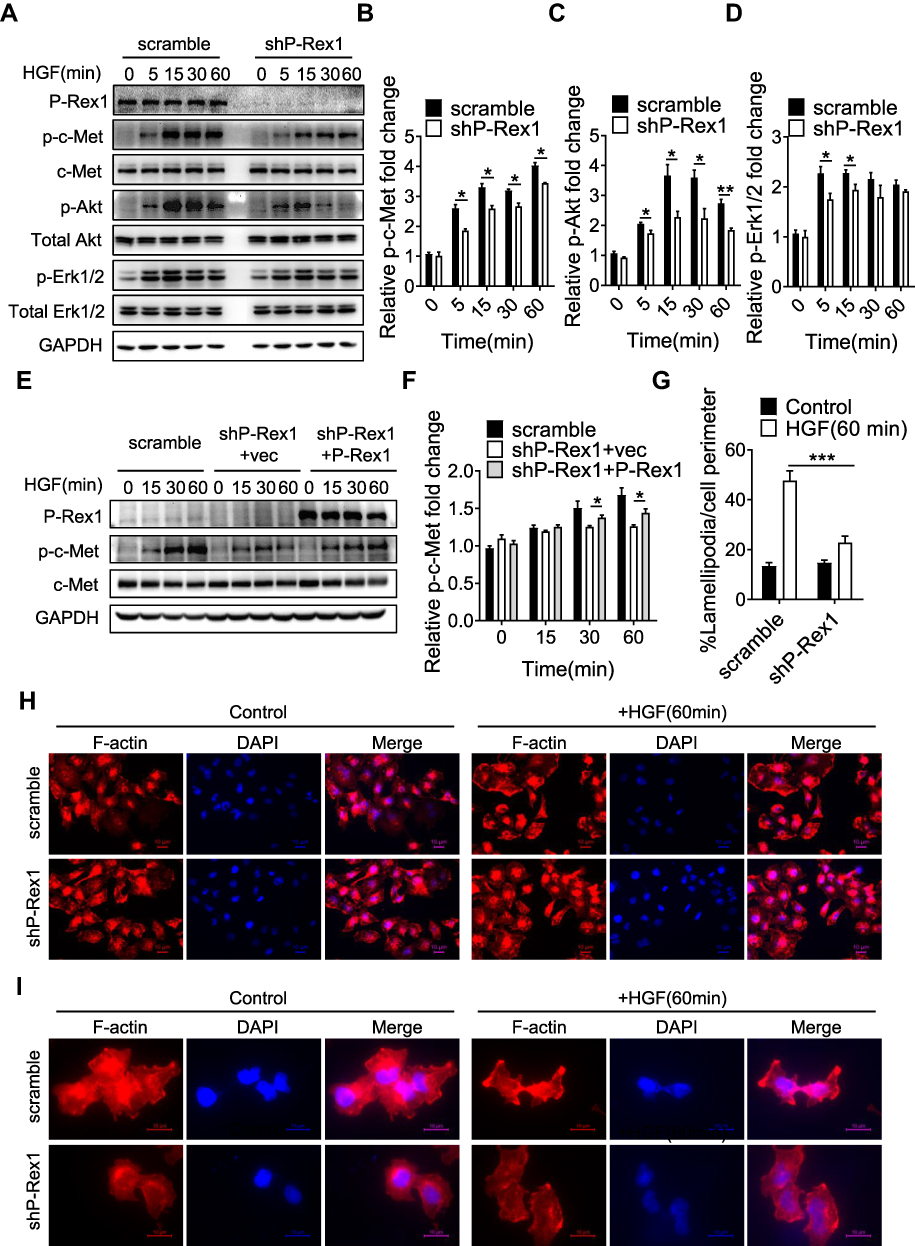 |
Figure 5 P-Rex1 is involved in HGF/c-Met signaling in SK-Hep1 cells. (A–D) Levels of phosphorylation of c-Met, Akt, and Erk1/2 after HGF stimulation for the indicated time (min) in scrambled control and P-Rex1 knockdown (shP-Rex1) cells. (E and F) ShP-Rex1 cells were transiently transfected with a P-Rex1-containing plasmid to restore its function. Western blotting for p-c-Met and c-Met was performed. Data quantification is shown below. (G–I) Control and P-Rex1 knockdown cells were stained with Rhodamine-conjugated phalloidin after HGF stimulation for 60 min, and signal was quantified. Different power images were included. Scale bars =10 μm. The data are shown above as means ± SEM from three independent experiments. (*P < 0.05, **P < 0.01, and ***P < 0.001). |
As P-Rex1 was primarily identified as a GEF for the activation of Rac1, a key element in controlling the formation of lamellipodia,29 examinations were conducted to reveal whether Rac1 is involved in P-Rex1-mediated oncogenic signaling. Immunofluorescence showed that P-Rex1 depletion significantly decreased HGF-induced lamellipodia formation in SK-Hep1 cells, supporting a Rac1-dependent role (P < 0.001, Figure 5G, H, and I). According to published reports, c-Met participates in cytoskeletal reorganization via stimulation of Rac1 activation in different endosomal pathways.30 These results not only indicate the formation of a P-Rex1-Rac1-c-Met positive feedback loop but also confirm the essential role of P-Rex1 in the migration and spread of liver cancer cells.
P-Rex1 Overexpression Facilitates Proliferation and Migration by Promoting the HGF/c-Met/Akt Pathway in HepG2 Cells
To further validate the role of P-Rex1 in liver cancer, stable overexpression of P-Rex1 in HCC cells was constructed using recombinant lentiviral transfection. HepG2 cells, which expressed the lowest levels of endogenous P-Rex1, were used to generate the P-Rex1 stable overexpression cell line. One vector control clone (VEC) and one P-Rex1 overexpression clone (PROE) were obtained. Western blot analysis and RT-qPCR showed that recombinant P-Rex1 was expressed in the PROE clone but not in the control VEC clone, suggesting stable overexpression of P-Rex1 in the HepG2 cell line (Figure 6A and B). P-Rex1 expression significantly promoted HepG2 cell proliferation after HGF (20 ng/mL) treatment (Figure 6C). As shown in Figure 6D and E, HepG2 cells displayed relatively low metastasis. A much higher migration level, induced by 10% fetal bovine serum, was observed in PROE cells than in VEC cells. Treatment of HepG2 cells with human HGF prior to 10% fetal bovine serum increased the migration abilities of both VEC and PROE clones. Furthermore, HGF induced an increase in the phosphorylation of c-Met as well as downstream Akt and Erk1/2. The increase was more significant in PROE cells than in VEC cells (Figure 6F-I). These findings demonstrate that P-Rex1 facilitates the motility and migration of HepG2 cells through the HGF/c-Met/Akt pathway.
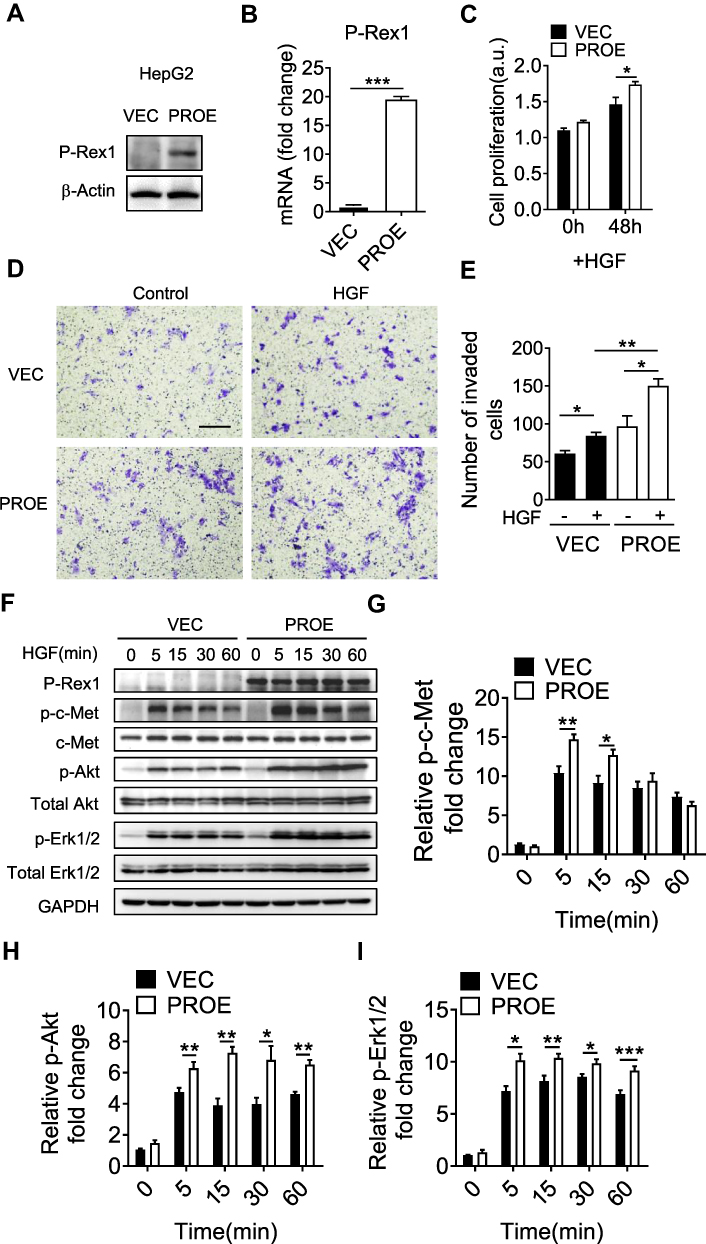 |
Figure 6 P-Rex1 expression promotes proliferation and migration in HepG2 cells. Stable vector control clone (VEC) and P-Rex1 overexpression clone (PROE) were constructed using recombinant lentiviral transfection. A representative Western blot (A) and RT-qPCR (B) are shown. (C) A BrdU assay was performed to determine the proliferation of VEC and PROE cells in culture medium treated with HGF (20 ng/mL) for 48 h. (D and E) A transwell assay was performed to evaluate the invasive behavior of VEC and PROE cells treated with HGF (20 ng/mL). Representative images of migrated cells are shown, scale bars = 250 μm. (F–I) Levels of phosphorylation of c-Met, Akt, and Erk1/2 after HGF stimulation in VEC and PROE cells. The data are shown as mean ± SEM from three independent experiments. (*P < 0.05, **P < 0.01, and ***P < 0.001). |
P-Rex1 and c-Met Form a Protein Complex
Given the requirement for P-Rex1 in c-Met-promoted HCC, this study analyzed whether P-Rex1 interacts with c-Met through immunoprecipitation. HA-tagged c-Met and AU5-tagged P-Rex1 plasmids were co-transfected into 293T cells to construct a heterologous expression system. Co-transfection with P-Rex1 and c-Met increased the co-immunoprecipitation of P-Rex1 with c-Met more than transfection with P-Rex1 alone (Figure 7A). More importantly, a markedly improved interaction between endogenous P-Rex1 and c-Met was observed after HGF treatment for 30 min (P < 0.001, Figure 7B-D). A schematic drawing depicting P-Rex1’s involvement in the HGF/c-Met/Akt pathway illustrates these interactions (Figure 7E). Taken together, these data suggest a physical association between P-Rex1 and c-Met.
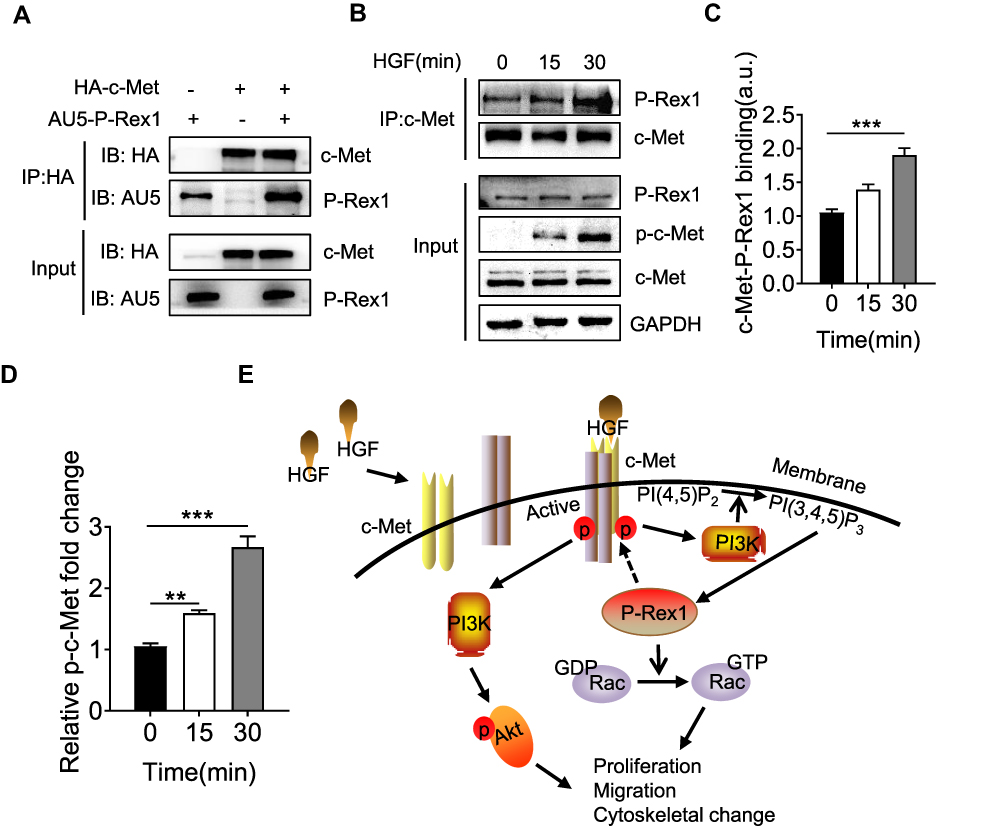 |
Figure 7 P-Rex1 and c-Met form a protein complex. (A) 293T cells were co-transfected with both HA-tagged human c-Met plasmid and AU5-tagged human P-Rex1 plasmid or were transfected separately. Cell lysates were subjected to immunoprecipitation with anti-HA antibody followed by Western blot analysis with an anti-AU5 antibody. (B and C) SK-Hep1 cells were treated with HGF (20 ng/mL) or buffer control for the indicated times. Co-immunoprecipitation was performed using anti-c-Met antibody. P-Rex1 expression was determined through Western blotting. (D) Levels of phosphorylation of c-Met. (E) Schematic drawing depicting P-Rex1’s involvement in the HGF/c-Met/Akt pathway. The data are shown above as means ± SEM from three independent experiments. (**P < 0.01 and ***P < 0.001). |
Discussion
P-Rex1, the first member of the P-Rex family, was originally purified from leukocyte cytosol prepared from pig blood.16 The family includes three members: P-Rex1, P-Rex2a, and P-Rex2b. The sequences of P-Rex2a and P-Rex2b are homologous with those of P-Rex1.31,32 Despite various studies pointing out the oncogenicity of guanine nucleotide exchange factors (GEFs) in multiple cancer types, especially melanoma, breast cancer, glioblastoma, and prostate cancer, the present work unveiled a specific new function of P-Rex1 in the pathogenesis of liver cancer. P-Rex1 knockdown attenuated liver cancer cell proliferation, migration, and xenograft tumor growth by mediating the activation of the HGF signaling pathway. Silencing P-Rex1 suppressed Akt and Erk1/2 phosphorylation by targeting c-Met. Additionally, the present data demonstrated that P-Rex1 and c-Met could form a protein complex after HGF treatment, suggesting that P-Rex1 might be an attractive therapeutic target in liver cancer therapy.
P-Rex1 primarily acts as a downstream effector of GPCR signaling.31,33 There is compelling evidence that PIP3 positively regulates the activation of P-Rex1, which directly activates Rac1.16 More recent studies have focused on the crosstalk between the P-Rex1/Rac1 pathway and other classical oncogenic signaling pathways, such as the ErbB pathway in breast cancer26 and the VEGF/VEGFR pathway in prostate cancer, and considered P-Rex1 as an essential mediator in cancer progression. In liver cancer, aberrant c-Met activity causes c-Met monomers to recruit other receptor monomers to form heterodimers of receptor proteins, leading to increased tumor cell proliferation and drug resistance. The present results indicated crosstalk between the P-Rex1/Rac1 pathway and the HGF/c-Met pathway. Downregulation of P-Rex1 diminished HGF-induced c-Met phosphorylation as well as phosphorylation of the downstream signaling molecules Akt and Erk1/2. Recombination and endogenous co-immunoprecipitation assays identified an interaction between P-Rex1 and c-Met, suggesting that P-Rex1 may directly or indirectly contribute to the HGF/c-Met pathway. The interaction between GEFs and tyrosine kinase receptors has rarely been reported. P-Rex1 was found to be associated with platelet-derived growth factor receptors (PDGFRβ), driving alterations in cell shape and invasion.34 Li et al reported that c-Met, AXL, ELMO2, and DOCK180 could form a complex to induce cytoskeletal reorganization after HGF treatment.35 DOCK180 is a guanine nucleotide exchange factor for Rac1. Thus, the present findings show for the first time that HGF promotes the recruitment of P-Rex1 to c-Met, independent of GPCRs. However, understanding the mode of interaction (direct or indirect) and the exact mechanism underlying P-Rex1-mediated receptor activation requires further investigation.
HGF-induced P-Rex1/Rac1 activation was supported by the observation that 1) P-Rex1 depletion significantly decreased HGF-induced lamellipodia formation in SK-Hep1 cells, which is suggestive of a Rac1-dependent mechanism; 2) a much slower migration in P-Rex1-knockdown cells implies the involvement of Rac1 in HGF signaling. Activated Rac1 binds and activates the PI3K/p110β subunit to increase PIP3 production, while the phosphorylated tyrosine kinase receptor c-Met recruits more PI3K/p85 subunits.36,37 More importantly, detection of the P-Rex1/c-Met interaction in a co-immunoprecipitation assay supports the role of P-Rex1 in HGF/c-Met signaling. These findings placed P-Rex1 downstream of the HGF signaling pathway as a component of the positive feedback loop. Recent evidence has suggested that activated c-Met is associated with poor outcomes of sorafenib therapy in HCC, leading to sorafenib resistance via reactivation of the Akt/Erk1/2-EGR1 signaling pathway.9,38 The HGF/c-Met axis offers a potential target for drug therapy in liver cancer. Of note, the combination of c-Met inhibitors with other related molecule inhibitors would be a promising strategy due to the low sensitivity and effectiveness of single-agent regimens in preclinical and clinical trials.39,40 More recent studies have identified a set of small molecules against P-Rex1 by differential scanning fluorimetry, but inhibition of P-Rex1-related cell function needs to be further confirmed.41
Liver cancer is an extraordinarily heterogeneous malignancy with a poor prognosis and a lack of curative treatments that requires continued research efforts. This study demonstrated that P-Rex1 is involved in liver cancer cell proliferation, migration, and tumor growth by modulating Akt and Erk1/2 phosphorylation via c-Met targeting. Identification of P-Rex1 as a key molecule in HGF/c-Met signaling may shed light on the pathogenesis of liver cancer. P-Rex1 could be a potential molecular drug target in the design of novel therapeutic approaches for liver cancer treatment, but further investigation is required.
Disclosure
The authors declare no competing financial or non-financial interests for this work.
References
1. Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65(2):87–108. doi:10.3322/caac.21262
2. Eggert T, Greten TF. Current standard and future perspectives in non-surgical therapy for hepatocellular carcinoma. Digestion. 2017;96(1):1–4. doi:10.1159/000464282
3. Liang Q, Shen X, Sun G. Precision medicine: update on diagnosis and therapeutic strategies of hepatocellular carcinoma. Curr Med Chem. 2018;25(17):1999–2008. doi:10.2174/0929867325666180117101532
4. Birchmeier C, Birchmeier W, Gherardi E, Vande Woude GF. Met, metastasis, motility and more. Nat Rev Mol Cell Biol. 2003;4(12):915–925. doi:10.1038/nrm1261
5. Trusolino L, Bertotti A, Comoglio PM. MET signalling: principles and functions in development, organ regeneration and cancer. Nat Rev Mol Cell Biol. 2010;11(12):834–848. doi:10.1038/nrm3012
6. Garcia-Vilas JA, Medina MA. Updates on the hepatocyte growth factor/c-Met axis in hepatocellular carcinoma and its therapeutic implications. World J Gastroenterol. 2018;24(33):3695–3708. doi:10.3748/wjg.v24.i33.3695
7. Hung TH, Li YH, Tseng CP, et al. Knockdown of c-MET induced apoptosis in ABCB1-overexpressed multidrug-resistance cancer cell lines. Cancer Gene Ther. 2015;22(5):262–270. doi:10.1038/cgt.2015.15
8. Bouattour M, Raymond E, Qin S, et al. Recent developments of c-Met as a therapeutic target in hepatocellular carcinoma. Hepatology (Baltimore, Md). 2018;67(3):1132–1149. doi:10.1002/hep.29496
9. Xiang QF, Zhan MX, Li Y, et al. Activation of MET promotes resistance to sorafenib in hepatocellular carcinoma cells via the AKT/ERK1/2-EGR1 pathway. Artif Cells Nanomed Biotechnol. 2019;47(1):83–89. doi:10.1080/21691401.2018.1543195
10. Schmidt A. Guanine nucleotide exchange factors for Rho GTPases: turning on the switch. Genes Dev. 2002;16(13):1587–1609. doi:10.1101/gad.1003302
11. Bustelo XR, Sauzeau V, Berenjeno IM. GTP-binding proteins of the Rho/Rac family: regulation, effectors and functions in vivo. Bioessays. 2007;29(4):356–370. doi:10.1002/bies.20558
12. Heasman SJ, Ridley AJ. Mammalian Rho GTPases: new insights into their functions from in vivo studies. Nat Rev Mol Cell Biol. 2008;9(9):690–701. doi:10.1038/nrm2476
13. Fernandez-Zapico ME, Gonzalez-Paz NC, Weiss E, et al. Ectopic expression of VAV1 reveals an unexpected role in pancreatic cancer tumorigenesis. Cancer Cell. 2005;7(1):39–49. doi:10.1016/j.ccr.2004.11.024
14. Minard ME, Kim LS, Price JE, Gallick GE. The role of the guanine nucleotide exchange factor Tiam1 in cellular migration, invasion, adhesion and tumor progression. Breast Cancer Res Treat. 2004;84(1):21–32. doi:10.1023/B:BREA.0000018421.31632.e6
15. Murray DW, Didier S, Chan A, et al. Guanine nucleotide exchange factor Dock7 mediates HGF-induced glioblastoma cell invasion via Rac activation. Br J Cancer. 2014;110(5):1307–1315. doi:10.1038/bjc.2014.39
16. Welch HC, Coadwell WJ, Ellson CD, et al. P-Rex1, a PtdIns(3,4,5)P3- and Gbetagamma-regulated guanine-nucleotide exchange factor for Rac. Cell. 2002;108(6):809–821. doi:10.1016/S0092-8674(02)00663-3
17. Srijakotre N, Man J, Ooms LM, Lucato CM, Ellisdon AM, Mitchell CA. P-Rex1 and P-Rex2 RacGEFs and cancer. Biochem Soc Trans. 2017;45(4):963–977. doi:10.1042/BST20160269
18. Lindsay CR, Lawn S, Campbell AD, et al. P-Rex1 is required for efficient melanoblast migration and melanoma metastasis. Nat Commun. 2011;2:555. doi:10.1038/ncomms1560
19. Barrio-Real L, Kazanietz MG, Rho GE. Fs and cancer: linking gene expression and metastatic dissemination. Sci Signal. 2012;5(244):pe43. doi:10.1126/scisignal.2003543
20. Barrio-Real L, Lopez-Haber C, Casado-Medrano V, et al. P-Rex1 is dispensable for Erk activation and mitogenesis in breast cancer. Oncotarget. 2018;9(47):28612–28624. doi:10.18632/oncotarget.25584
21. Pantarelli C, Welch HCE. Rac-GTPases and Rac-GEFs in neutrophil adhesion, migration and recruitment. Eur J Clin Invest. 2018;48(Suppl 2):e12939. doi:10.1111/eci.12939
22. Huang Y, Xie Y, Jiang H, et al. Upregulated P-Rex1 exacerbates human airway smooth muscle hyperplasia in asthma. J Allergy Clin Immunol. 2019;143(2):778–781 e775. doi:10.1016/j.jaci.2018.09.020
23. Naikawadi RP, Cheng N, Vogel SM, et al. A critical role for phosphatidylinositol (3,4,5)-trisphosphate-dependent Rac exchanger 1 in endothelial junction disruption and vascular hyperpermeability. Circ Res. 2012;111(12):1517–1527. doi:10.1161/CIRCRESAHA.112.273078
24. Liang Q, Cheng N, Zhang G, et al. Identification of P-Rex1 as an anti-inflammatory and anti-fibrogenic target for pulmonary fibrosis. Sci Rep. 2016;6:25785. doi:10.1038/srep25785
25. Liu HJ, Ooms LM, Srijakotre N, et al. PtdIns(3,4,5)P3-dependent Rac Exchanger 1 (PREX1) Rac-guanine nucleotide exchange factor (GEF) activity promotes breast cancer cell proliferation and tumor growth via activation of extracellular signal-regulated Kinase 1/2 (ERK1/2) Signaling. J Biol Chem. 2016;291(33):17258–17270. doi:10.1074/jbc.M116.743401
26. Sosa MS, Lopez-Haber C, Yang C, et al. Identification of the Rac-GEF P-Rex1 as an essential mediator of ErbB signaling in breast cancer. Mol Cell. 2010;40(6):877–892. doi:10.1016/j.molcel.2010.11.029
27. Ye RD. The Rho guanine nucleotide exchange factor P-Rex1 as a potential drug target for cancer metastasis and inflammatory diseases. Pharmacol Res. 2020;153:104676. doi:10.1016/j.phrs.2020.104676
28. Abbas T, Dutta A. p21 in cancer: intricate networks and multiple activities. Nat Rev Cancer. 2009;9(6):400–414.
29. Ridley AJ, Paterson HF, Johnston CL, Diekmann D, Hall A. The small GTP-binding protein rac regulates growth factor-induced membrane ruffling. Cell. 1992;70(3):401–410. doi:10.1016/0092-8674(92)90164-8
30. Menard L, Parker PJ, Kermorgant S. Receptor tyrosine kinase c-Met controls the cytoskeleton from different endosomes via different pathways. Nat Commun. 2014;5:3907. doi:10.1038/ncomms4907
31. Welch HC. Regulation and function of P-Rex family Rac-GEFs. Small GTPases. 2015;6(2):49–70. doi:10.4161/21541248.2014.973770
32. He S, Lin J, Yu S, Sun S. Upregulation of PREX2 promotes the proliferation and migration of hepatocellular carcinoma cells via PTEN-AKT signaling. Oncol Lett. 2016;11(3):2223–2228. doi:10.3892/ol.2016.4164
33. Cervantes-Villagrana RD, Adame-Garcia SR, Garcia-Jimenez I, et al. Gbetagamma signaling to the chemotactic effector P-REX1 and mammalian cell migration is directly regulated by Galphaq and Galpha13 proteins. The. J Biologica Chem. 2019;294(2):531–546. doi:10.1074/jbc.RA118.006254
34. Campbell AD, Lawn S, McGarry LC, Welch HC, Ozanne BW, Norman JC. P-Rex1 cooperates with PDGFRbeta to drive cellular migration in 3D microenvironments. PLoS One. 2013;8(1):e53982. doi:10.1371/journal.pone.0053982
35. Li W, Xiong X, Abdalla A, et al. HGF-induced formation of the MET-AXL-ELMO2-DOCK180 complex promotes RAC1 activation, receptor clustering, and cancer cell migration and invasion. J Biol Chem. 2018;293(40):15397–15418. doi:10.1074/jbc.RA118.003063
36. Yang HW, Shin MG, Lee S, et al. Cooperative activation of PI3K by Ras and Rho family small GTPases. Mol Cell. 2012;47(2):281–290. doi:10.1016/j.molcel.2012.05.007
37. Fritsch R, de Krijger I, Fritsch K, et al. RAS and RHO families of GTPases directly regulate distinct phosphoinositide 3-kinase isoforms. Cell. 2013;153(5):1050–1063. doi:10.1016/j.cell.2013.04.031
38. Xiang Q, Chen W, Ren M, et al. Cabozantinib suppresses tumor growth and metastasis in hepatocellular carcinoma by a dual blockade of VEGFR2 and MET. Clin Cancer Res. 2014;20(11):2959–2970. doi:10.1158/1078-0432.CCR-13-2620
39. Liu Y, Tan J, Ou S, Chen J, Chen L. MicroRNA-101-3p suppresses proliferation and migration in hepatocellular carcinoma by targeting the HGF/c-Met pathway. Invest New Drugs. 2019.
40. Luo T, Zhang SG, Zhu LF, et al. A selective c-Met and Trks inhibitor Indo5 suppresses hepatocellular carcinoma growth. J Exp Clin Cancer Res. 2019;38(1):130. doi:10.1186/s13046-019-1104-4
41. Cash JN, Chandan NR, Hsu AY, et al. Discovery of small molecules that target the Phosphatidylinositol (3,4,5) Trisphosphate (PIP3)-Dependent Rac Exchanger 1 (P-Rex1) PIP3 -Binding Site and Inhibit P-Rex1–dependent functions in neutrophils. Mol Pharmacol. 2020;97(3):226–236. doi:10.1124/mol.119.117556
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.