")
Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 13
Identification of novel target genes in human lung tissue involved in chronic obstructive pulmonary disease
Authors Heinbockel L, Marwitz S, Schromm AB, Watz H, Kugler C, Ammerpohl O, Schnepf K , Rabe KF , Droemann D, Goldmann T
Received 8 January 2018
Accepted for publication 26 April 2018
Published 26 July 2018 Volume 2018:13 Pages 2255—2259
DOI https://doi.org/10.2147/COPD.S161958
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Richard Russell
Lena Heinbockel,1,2 Sebastian Marwitz,1,2 Andra B Schromm,3 Henrik Watz,2,4 Christian Kugler,2,5 Ole Ammerpohl,2,6 Karoline Schnepf,7 Klaus F Rabe,2,5 Daniel Droemann,2,7 Torsten Goldmann1,2
1Pathology of the University Medical Center Schleswig-Holstein (UKSH), Campus Luebeck and Research Center Borstel, Borstel, Germany; 2Airway Research Center North (ARCN), German Center for Lung Research (DZL), Großhansdorf, Germany; 3Immunobiophysics, Research Center Borstel, Borstel, Germany; 4Pulmonary Research Institute at LungenClinic Grosshansdorf, Grosshansdorf, Germany; 5LungenClinic Grosshansdorf, Grosshansdorf, Germany; 6Institute of Human Genetics, University Medical Center Ulm, Ulm, Germany; 7Medical Clinic III, Pulmonology/Infectious Diseases, University Hospital Schleswig-Holstein, Campus Luebeck, Luebeck, Germany
Introduction: As part of a study aimed at illuminating at least some of the complex molecular events taking place in COPD, we screened tissues by means of transcriptome analyses.
Materials and methods: Tissues were subjected to transcriptome analysis. Candidate genes were identified and validated by immunohistochemistry. Primary human lung cells were subjected to stimulation with cigarette smoke extract for further validation by real time PCR.
Results: Six candidate genes were selected for further investigations: Aquaporin 3 (AQP3), extracellular matrix protein 1 (ECM1), four and a half LIM domain 1 (FHL1), milk fat globule epidermal growth factor 8 (MFGE8, lactadherin), phosphodiesterase 4D-interacting protein (PDE4DIP), and creatine transporter SLC6A8. All six proteins were allocated to distinct cell types by immunohistochemistry. Upon stimulation with cigarette smoke extract, human type II pneumocytes showed a dose-dependent down-regulation of MFGE8, while ECM1 and FHL1 also tended to be down-regulated. Although present, none of the candidates was regulated by cigarette smoke extract in primary human macrophages.
Discussion: MFGE8 turned out to be an interesting new candidate gene in COPD deserving further studies.
Keywords: COPD, transcriptome, MFGE8, CSE, cigarette smoke extract, ETS2
Introduction
The complex disease mechanisms resulting in COPD are poorly understood. We investigated COPD-related changes of the gene expression in tumor-free lung tissues of COPD patients by transcriptome analysis.
Materials and methods
Lung tissue
Tumor-free lung tissue obtained from lobectomy or pneumectomy specimens, with a minimum distance to the tumor of 10 cm, of 19 lung cancer patients, was fixed using 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid [HEPES]-glutamic acid buffer-mediated organic solvent protection effect (HOPE) and embedded in paraffin according to DCS Innovative Diagnostik-Systeme protocol (Hamburg, Germany). Fourteen patients (mean age 66 years; 11 males; eight ex-smokers and six current smokers) had concomitant COPD; five patients served as control (mean age 69.5 years, all female and never-smokers). Detailed information is deposited at Gene Expression Omnibus (GSE106986).
This study was conducted using material from non-small-cell lung cancer resections and performed anonymously. Patient tissue was used and all experimental procedures were performed with the permission of the local ethical committee at the University of Lübeck (AZ12-220).
Transcriptome analysis
Microarray analysis was performed using extracted RNA of human lung tissues. RNA quality was determined with the Agilent Bioanalyzer. The Low Input Quick Amp Labeling Kit (Agilent Technologies, Santa Clara, CA, USA) was used according to the manufacturer’s protocol for cDNA synthesis, amplification, and labeling with Cyanine 3-CTP (Cy3) during reverse transcription by T7 RNA polymerase. Cy3-labeled cRNA was purified using the RNeasy Mini Kit (Qiagen NV, Venlo, the Netherlands) and the specific activity was calculated using a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA). Labeled cRNA was hybridized to Agilent Human Whole Genome Gene Expression 4×44 k V2 arrays and scanned with a SureScan microarray scanner (Agilent Technologies) at a resolution of 5 μm. GeneSpring software version 13 (one-color gene expression protocol) was used for quantile normalization and statistical analyses (one-way ANOVA and Benjamini–Hochberg multiple testing correction, cutoff p-value <0.05). The complete gene list is deposited at Gene Expression Omnibus (GSE106986).
Histologic analyses
A tissue microarray of HOPE-fixed, paraffin-embedded tissues of 20 patients was used for analysis. The samples were dewaxed in 100% isopropanol for 10 min at 60°C. Sections were air dried, rehydrated in 70% acetone for 10 min at 4°C, and transferred into distilled water for 10 min at 4°C and 5 min at room temperature. Endogenous peroxidases were blocked by incubation in 3% H2O2 for 10 min. The polymer blocking reagent (Zytomed, Bargteheide, Germany) was incubated 5 min before the first antibodies were applied. The first antibodies were used in concentrations of anti-AQP3 ab125219 5 μg/mL (Abcam, Cambridge, United Kingdom); anti-ECM1 ab3924 1 μg/mL (Abcam); anti-FHL-1 ab58067 1 μg/mL (Abcam); anti-MFGE8 MAB12684 1 μg/mL (Abnova, Heidelberg, Germany); anti-PDE4DIP ab56655 4 μg/mL (Abcam); anti-SLC6A8 ab62196 1 μg/mL (Abcam) in antibody diluent (Zytomed) for 60 min at room temperature. The CytoChem Plus HRP Polymer Kit (Zytomed) was used for detection. The tissue slides were counterstained with Mayer’s hemalum solution (Merck).
Cigarette smoke extract (CSE) stimulation of human primary alveolar pneumocytes type 2 and macrophages
Tumor-free lung tissue (around 30 g) was rinsed with phosphate-buffered saline and cut into pieces of 3 mm size. The tissue pieces were rinsed with cell buffer (pH 6.64, 10 mM HEPES, 1.9 mM CaCl2, 1.3 mM MgSO4, 137 mM NaCl, 5.4 mM KCl, 6.2 mM glucose, 6.2 mM Na2HPO4) and incubated for 60 min at 400 rpm, at 37°C in 5% CO2 in cell buffer supplemented with 200 mg Dispase II, 500 μg DNase I, and 100 μL Accutase for the enzymatic dissociation of the cells. The cells were separated by density centrifugation, resulting in single-cell suspensions of epithelial cells and immune cells. Cells were seeded on cell culture flasks to separate fast-adhering immune cells from slow-adhering epithelial cells. P2 cells were separated by negative selection of CD45. Therefore, CD45 MicroBeads (human, Miltenyi, Bergisch Gladbach, Germany) were incubated for 15 min at 4°C and the cell-bound beads were captured in an LD column (Miltenyi, Bergisch Gladbach, Germany) with the Multi-MACS magnet. The eluted CD45-negative P2 cells were seeded on 48-well plates at low density (4×105 cells/well) in RPMI (10% fetal calf serum, penicillin/streptomycin, and L-Glutamine) for 16 h.
CSE was obtained using Tobacco-Health Research cigarettes (University of Kentucky) by drawing smoke of one cigarette slowly through a water pump into a tube containing 10 mL of ddH2O (100% CSE). One percent and 5% CSE in RPMI (with penicillin/streptomycin and L-Glutamine) were used for 16 h cell stimulation. Cells were harvested and used for RNA isolation.
Quantitative real-time polymerase chain reaction of stimulated human primary alveolar pneumocytes type 2 and macrophages
RNA was isolated using the RNeasy Plus Mini Kit (Qiagen) and quantified with the Agilent Bioanalyzer. The RevertAid First Strand cDNA Synthesis Kit (Thermo Fisher Scientific) was used for cDNA synthesis. Quantitative polymerase chain reaction was implemented with universal Probe Library equipment (Roche) and the following primers for AQP3 (for: cgcctttttacagcccttg/rev: caggagtggggacacgat), ECM1 (for: gctggagagggagttccag/rev: ggtcacagtatttgtcaagggtatc), FHL1 (for: gcgtggattgctacaagaact/rev: accacactggagcctttacc), IL8 (for: gagcactccataaggcacaaa/rev: atggttccttccggtggt), MFGE8 (for: tttgagctactgggctgtga/rev: ggccgtgatctgcttgtc), PDE4DIP (for: agccactgtctttgctcagg/rev: gcttcaccaaggattcacct), SLC6A8 (for: tgggaggtgaccctttgtc/rev: cgtaggggaatgtagcagtga) in the LightCycler 480 (Roche) instrument.
Results
After quantile normalization and ANOVA, 3,974 genes were found to be differently regulated in COPD patients compared to non-COPD patients (Figure 1A). Six candidate genes, which have not been previously evaluated in COPD, were selected for further investigations. 1) Aquaporin 3 (AQP3), a water/glycerol channel, which is upregulated in murine asthma models and involved in the chemokine production of alveolar macrophages.1 2) Extracellular matrix protein 1 (ECM1), a secreted glycoprotein that is expected to be involved in the development of various epithelial tumors.2 3) Four and a half LIM domain 1 (FHL1), a protein that is highly expressed in muscle disorders with the development of respiratory failure.3 4) Milk fat globule epidermal growth factor 8 (MFGE8, lactadherin), a secreted glycoprotein that opsonizes apoptotic cells4 but also collagen for phagocytes. Furthermore, it counteracts airway smooth muscle hypercontractility through binding to integrin receptors on the muscles in an ovalbumin challenge model of asthma.5 5) Phosphodiesterase 4D-interacting protein (PDE4DIP), a protein that is a major component of skeletal muscles interacting with phosphodiesterase PDE4D. 6) Creatine transporter SLC6A8, a plasma membrane protein that transports creatine required for rapid ATP supply into tissues such as muscle and brain.
 | Figure 1 Transcriptome analyses and selected protein candidates in tissue microarrays. |
The corresponding proteins were assigned to distinct cell types by immunohistochemistry of tissue from COPD patients (Figure 1B). AQP3 was detected in type I pneumocytes (P1), type II pneumocytes (P2), macrophages, and bronchi; ECM1 in P2, macrophages, bronchi, and extracellular region; FHL1 in P2, macrophages, bronchi, and capillaries; MFGE8 was present in P1, P2, and bronchi; PDE4DIP in macrophages, bronchi smooth muscles, and capillaries; SLC6A8 in macrophages, bronchi, and capillaries.
In a Gene Set Enrichment Analysis (MSigDB) after Baran et al, we found a potential regulation of three of the six genes (AQP3, ECM1, and FHL1) by the transcription factor ETS proto-oncogene 2 (ETS2). ETS2 was described to be substantially involved in the pathogenesis of pulmonary fibrosis in its phosphorylated form,6 suggesting that ETS2 might also have a role in peribronchiolar fibrosis in COPD.
We further investigated the possible regulation of the selected genes in a CSE model of primary human lung cells (Figure 2). After CSE stimulation primary human type II pneumocytes exhibited a significant, dose-dependent regulation on the RNA level for one (MFGE8) of the six studied candidates, while ECM1 and FHL1 showed a tendency to be downregulated. Despite the presence of five candidate proteins in alveolar macrophages of COPD patients (all except MFGE8, Figure 1B), none of them were found to be regulated upon CSE stimulation in human primary macrophages. Thus, the protein expression of the chosen candidates in alveolar macrophages appears to be independent of CSE stimulation.
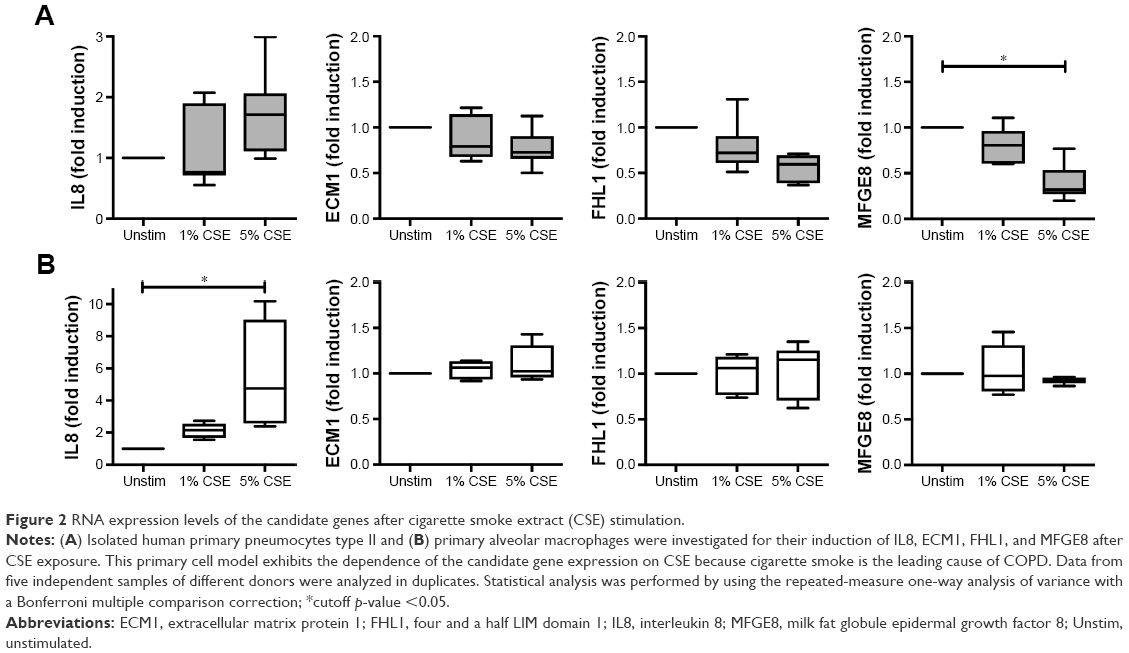 | Figure 2 RNA expression levels of the candidate genes after cigarette smoke extract (CSE) stimulation. |
MFGE8 is described as an inhibitor of the inflammasome-induced interleukin 1β (IL1β) production and thus a potential candidate for reducing the inflammatory cytokines with simultaneous clearance of apoptotic cells.4 Inflammatory mediators such as IL1β, which are increased in lungs and bronchoalveolar lavage fluids of COPD patients, might be promising targets in therapeutic studies.7
Particularly the CSE-inducible genes are valuable subjects for further investigations. In conclusion, we found an interesting protein, MFGE8, which is present in type II cells of human lungs and significantly downregulated after CSE stimulation, a possible contribution to the inflammatory manifestations of COPD. Thus, we will investigate in further studies MFGE8 as a potential inhibitor for CSE-induced inflammatory effects on human lungs.
Acknowledgments
The study was funded by the German Center for Lung Research (DZL), disease area COPD, Gen.2. Patient tissues were provided by the BioMaterialBank North, which is funded in part by the Airway Research Center North (ARCN), a member of the German Center for Lung Research (DZL), and is a member of popgen 2.0 network (P2N), which is supported by a grant from the German Ministry for Education and Research (01EY1103). The authors thank Bettina Baron-Lühr, Patricia Prilla, Stefanie Fox, Jasmin Tiebach, Kristin Wiczkowski, and Maria Lammers for their excellent technical assistance as well as Dr Klaas F. Franzen for providing the CSE.
Disclosure
The author Lena Heinbockel is supported by the Deutsche Forschungsgemeinschaft (DFG, project DR797/3-1 611672). The authors report no other conflicts of interest in this work.
References
Ikezoe K, Oga T, Honda T, et al. Aquaporin-3 potentiates allergic airway inflammation in ovalbumin-induced murine asthma. Sci Rep. 2016;6:25781. | ||
Wang L, Yu J, Ni J, et al. Extracellular matrix protein 1 (ECM1) is over-expressed in malignant epithelial tumors. Cancer Lett. 2003;200(1):57–67. | ||
Pfeffer G, Povitz M, Gibson GJ, Chinnery PF. Diagnosis of muscle diseases presenting with early respiratory failure. J Neurol. 2015;262(5):1101–1114. | ||
Hanayama R, Tanaka M, Miwa K, Shinohara A, Iwamatsu A, Nagata S. Identification of a factor that links apoptotic cells to phagocytes. Nature. 2002;417(6885):182–187. | ||
Kudo M, Khalifeh Soltani SM, Sakuma SA, et al. Mfge8 suppresses airway hyperresponsiveness in asthma by regulating smooth muscle contraction. Proc Natl Acad Sci U S A. 2013;110(2):660–665. | ||
Baran CP, Fischer SN, Nuovo GJ, et al. Transcription factor ets-2 plays an important role in the pathogenesis of pulmonary fibrosis. Am J Respir Cell Mol Biol. 2011;45(5):999–1006. | ||
Barnes PJ. Identifying molecular targets for new drug development for chronic obstructive pulmonary disease: what does the future hold? Semin Respir Crit Care Med. 2015;36(4):508–522. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.