")
Back to Journals » International Journal of Nanomedicine » Volume 16
Identification of Novel Ligands for Targeted Antifibrotic Therapy of Chronic Pancreatitis
Authors Hung J , Awasthi R , Klibanov AL, Kelly KA
Received 4 May 2021
Accepted for publication 15 July 2021
Published 14 August 2021 Volume 2021:16 Pages 5495—5512
DOI https://doi.org/10.2147/IJN.S318331
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Farooq A. Shiekh
Jessica Hung,1 Rohni Awasthi,1 Alexander L Klibanov,2– 5 Kimberly A Kelly2
1Department of Biomedical Engineering, School of Engineering, University of Virginia, Charlottesville, Virginia, 22908, USA; 2Department of Biomedical Engineering, School of Medicine, University of Virginia, Charlottesville, Virginia, 22908, USA; 3Division of Cardiovascular Medicine, University of Virginia, Charlottesville, Virginia, 22908, USA; 4Robert M. Berne Cardiovascular Research Center, University of Virginia, Charlottesville, Virginia, 22908, USA; 5Department of Radiology, University of Virginia, Charlottesville, Virginia, 22908, USA
Correspondence: Kimberly A Kelly
Department of Biomedical Engineering, School of Medicine, University of Virginia, Box 800759, Charlottesville, Virginia, 22908, USA
Tel +1 434-243-9352
Fax +1 434-982-3870
Email [email protected]
Purpose: Chronic pancreatitis (CP) is an inflammatory disorder of the pancreas that leads to impaired pancreatic function. The limited therapeutic options and the lack of molecular targeting ligands or non-serum-based biomarkers hinder the development of target-specific drugs. Thus, there is a need for an unbiased, comprehensive discovery and evaluation of pancreatitis-specific ligands.
Methods: This study utilized a computational-guided in vivo phage display approach to select peptide ligands selective for cellular components in the caerulein-induced mouse model of CP. The identified peptides were conjugated to pegylated DOPC liposomes via the reverse-phase evaporation method, and the in vivo specificity and pharmacokinetics were determined. As proof of concept, CP-targeted liposomes were used to deliver an antifibrotic small molecular drug, apigenin. Antifibrotic effects determined by pancreatic histology, fibronectin expression, and collagen deposition were evaluated.
Results: We have identified five peptides specific for chronic pancreatitis and demonstrated selectivity to activated pancreatic stellate cells, acinar cells, macrophages, and extracellular matrix, respectively. MDLSLKP-conjugated liposomes demonstrated an increased particle accumulation by 1.3-fold in the inflamed pancreas compared to the control liposomes. We also observed that targeted delivery of apigenin resulted in improved acini preservation, a 37.2% and 33.1% respective reduction in collagen and fibronectin expression compared to mice receiving the free drug, and reduced oxidative stress in the liver.
Conclusion: In summary, we have developed a systematic approach to profile peptide ligands selective for cellular components of complex disease models and demonstrated the biomedical applications of the identified peptides to improve tissue remodeling in the inflamed pancreas.
Keywords: phage display, next-generation sequencing, peptide ligands, targeted liposomes, drug delivery
Introduction
Chronic pancreatitis (CP) is an inflammatory disorder that causes irreversible damage in the pancreas and induces long-standing sequelae encompassing recurrent severe pain, fibrosis, duct distortion, parenchymal calcification, and loss of exocrine and endocrine function at the advanced stage.1,2 In addition to physical debilities, patients with CP typically struggle with psychological and financial challenges, resulting in a significantly impaired quality of life.3–5 Patients with a history of CP have an increased risk of developing pulmonary diseases, diabetes mellitus, and pancreatic cancer.6 The annual incidence of CP worldwide ranges between 4.4 and 14 per 100,000 population, with an approximate prevalence of 36.9–52.4 per 100,000.7 Despite low prevalence, the frequent demands of pain management and necessary procedures directly or indirectly caused by CP lead to a disproportional high cost of medical care, resulting in a high socioeconomic burden on diseased individuals and the health care system in the United States.8–10
Fibrosis is a characteristic feature of CP, which is not only an outcome of recurrent pancreatic parenchymal cell necrosis but is also responsible for the post-injury reactions that induce a cascade of events and signify molecular and cellular damage. Various cellular components and molecular crosstalk are involved in fibrogenesis, with each of the components contributing to the clinical outcome of pancreatic remodeling. However, no FDA-approved drug is available to address the fundamental causes of inflammation and fibrogenesis and to halt and reverse the damage of pancreatitis. In fact, current therapeutic strategies for CP are limited to palliative care and pain alleviation and these approaches fail at the advanced stage when invasive surgical procedures are the only available options. Thus, addressing fibrotic conditions has become one of the main areas to improve pancreatitis outcomes. Apigenin, a natural, small molecule compound, has been shown preclinically to have antifibrotic, anti-inflammatory, antioxidant, and proapoptotic properties in CP and various cancers in vitro and in vivo.11–13 Despite these promising features, apigenin suffers from low aqueous solubility, metabolic instability, and off-target effects;14 therefore, no approved clinical applications of apigenin are available.
Liposome-based drug delivery has been clinically proven in the cancer setting to successfully encapsulate small molecule drugs for enhanced bioavailability, prolong drug circulation half-life, and improve patient outcomes.15,16 Surface modifications with targeting ligands such as antibodies, antibody fragments, and peptides enable specific binding at the diseased sites while minimizing undesirable side effects. In the tissue remodeling process of CP, various cellular and molecular components are involved and contribute to the progression, which make them potential targets. There are, however, no molecular targeting ligands or non-serum-based biomarkers available, limiting the development of imaging agents and therapeutics for pancreatitis. To fill that void, we used phage display and our innovative and computationally guided target selection approach, PHASTpep,17 to identify peptides specific for key cellular components involved in fibrogenesis. The cellular selectivity of the identified CP-specific clones was evaluated using fluorescent microscopy and colocalization analysis with common cell types present in the CP microenvironment. As proof of concept, we developed an apigenin-encapsulated liposomal formulation surface modified with the identified CP-targeted peptides. The pharmacokinetic profile of targeted liposomes was examined in vivo and ex vivo after intravenous injection in a mouse model of CP. Pancreata from the CP mice receiving apigenin-encapsulated, targeted liposomes were evaluated for antifibrotic effects based on histology compared to mice receiving vehicle, free drug, or non-targeted liposome treatments. Through a high-throughput ligand identification process, we aim to demonstrate the application of targeting peptides in drug delivery to mediate the development of target-specific interventions for CP.
Materials and Methods
Lipids for Liposome Preparation
1.2-Dioleoyl-sn-glycerol-3-phosphocholine (DOPC), and 1.2-distearoyl-sn-glycero-3- phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] (DSPE-PEG2000) were purchased from Avanti Polar Lipids, Alabaster, AL; DSPE-PEG3400-maleimide was purchased from Laysan Bio Inc., Arab, AL; 1.1′-dioctadecyl-3,3,3′,3′- tetramethylindodicarbocyanine, 4-chlorobenzenesulfonate salt (DiD) was purchased from Biotium Inc., Hayward, CA; cholesterol was purchased from Millipore Sigma, Burlington, MA.
Caerulein-Induced Pancreatitis in Mice
All animal experiments were approved by the Animal Care and Use Committee at the University of Virginia and conformed to the NIH “Guide for the Care and Use of Laboratory Animals in Research.”
C57BL/6J mice (6–12 week-old, female) were used for the in vivo phage screening. For chronic pancreatitis, caerulein (Bachem, Torrance, CA) was dissolved in sterile saline and administrated via intraperitoneal injection twice a day, 8 hours apart for 14 consecutive days, at a concentration of 250 μg/kg body weight18 as shown in Figure S1A. During the 14-day course, intraperitoneal injections of 100 μg/kg buprenorphine were given every 3 days to minimize the induced pain. For healthy controls, c57BL/6J mice were injected intraperitoneally with an equal volume of sterile saline following the same schedule as in the chronic pancreatitis models. The inflammatory status of the pancreas was confirmed at the end of caerulein treatments via immunohistochemistry using hematoxylin-eosin (H&E) staining (Figure S1B). Picrosirius red (Polysciences, Inc., Warrington, PA) was used to visualize collagen in paraffin-embedded pancreas sections (Figure S1B).
In vivo Phage Screening
1x1012 plaque-forming units (pfu) of the Ph.D.TM-7 Phage Display Peptide Library (New England Biolabs, Ipswich, MA) were injected intravenously via tail vein into CP and healthy c57BL/6J mice (N=3, each). Phage were allowed to circulate in the blood for 4h post-injection to allow extravasation out of the bloodstream and into tissues to facilitate cellular binding before the mice were euthanized. Various organs, including heart, liver, spleen, pancreas, kidneys, and skeletal muscle, were harvested, weighed, and homogenized in lysis buffer: 1x EDTA and 1x protease inhibitor cocktail (Fisher Scientific, Hampton, NH) in Dulbecco’s phosphate-buffered saline (DPBS, HyClone, Logan, UT). Phage titers of the tissues were determined by bacteriophage plaque assay following the manufacturer’s instructions (Ph.D.TM Phage Display Libraries Instruction Manual, NEB) and calculated in percent injected dose (%ID) per organ-weight. Phage recovered from the pancreas pool were amplified in Escherichia coli strain ER2738 at the early-log phase in LB media for 5h at 37°C. Bacterial debris was centrifuged at 12,000 rpm, 10 min, and phage in the supernatant were purified via PEG precipitation (PEG/NaCl: 20% w/v polyethylene glycol-8000, 2.5mM NaCl) overnight at 4°C. The amplified phage were then washed with DPBS, precipitated again with PEG/NaCl at 4°C for 30 min, centrifuged, and resuspended in DPBS for the next round of biopanning. Three rounds of biopanning were performed in both CP and healthy mice.
Phage DNA Sequencing
Thirty phage plaques from the pancreas of round-3 were randomly selected for DNA sequencing. The insert oligo in the integrated section of the phage was amplified by polymerase chain reaction (PCR) using the forward primer, 5ʹ-CCTTTAGTGGTACCTTTCTAT-3ʹ, and the reverse primer, 5ʹ-GCCCTCATAGTTAGCGTAACG-3′, and then Sanger sequenced (Eurofins). For next-generation sequencing, phage DNA of the pancreas pooled at all rounds was extracted via sodium iodide precipitation, followed by PCR amplification using previously published primer sets17 with KAPA HiFi PCR kit (Fisher Scientific, Hampton, NH). The PCR cycles are initialized with one step of 95°C for 1min, followed by 17 cycles of 95°C for 30 s, 60°C for 30 s, and 72°C for 30 s. PCR purification was performed using a QIAquick PCR purification kit (Qiagen, Hilden, Germany) following the manufacturer’s instructions. Phage DNA was sent to the UVA Biomolecular Research Core Facility for single-ended Next-generation sequencing (NGS) on an Illumina Miseq Sequencer. FASTA files generated from the Illumina sequencing were processed by PHASTpep software.17
Specificity of Phage Clones
Eighteen candidate clones were grouped (4–5 clones per group, 4 groups total) based on sequence similarity using GibbsCluster Server 2.0.16 by the Technical University of Denmark (Figure S2A). Phage clones were pooled in equimolar amounts and labeled with fluorophore VivoTag 680 (VT680, PerkinElmer, Waltham, MA). Wild type M13Ke phage, used as a negative control, was labeled with VivoTag S-750 (VT750, PerkinElmer, Waltham, MA). For each group, the fluorescently labeled candidate and WT phage were mixed in equal number and co-injected into CP and healthy mice (n = 5). Phage accumulation was determined by measuring the ex vivo fluorescent intensity of the pancreas at 20h post-injection via the IVIS Spectrum Series (Perkin Elmer, Waltham, MA) using the excitation and emission wavelength at 675nm/720nm for VT680 and 745nm/800nm for VT750. To account for variations of fluorescent labeling efficiency, the raw readouts of VT680 and VT750 were normalized to the dye-per-phage ratio (Figure S2B). The specificity ratio was calculated as %ID/g of the normalized VT680 divided by the %ID/g of the normalized VT750 (Figure S2C). A similar procedure was applied to determine the specificity of individual phage clones in group 2 and 3. Fluorescent intensity was measured at 20h post-injection of candidate phage (VT680) and WT phage (VT750). Fold change of specificity ratio was calculated as the specificity ratio of CP mice divided by the specificity ratio of the healthy mouse (Table S1).
Immunofluorescence (IF)
Post ex vivo imaging, mouse pancreata were submerged in Neg-50 Frozen Section Medium (Thermo Scientific, Waltham, MA) and snap-frozen over liquid nitrogen vapor. The embedded tissues were cut into 5 μm sections using a cryostat (Leica Microsystems Inc., Buffalo Grove, IL) for subsequent imaging with ZEISS LSM-880 Confocal Laser Scanning Microscope (Carl Zeiss Meditec, Inc., Jena, Germany) at the Advanced Microscopy Facility at the University of Virginia. Cell types of interest in the pancreatitis microenvironment were identified by IF analysis using the following antibodies: rat anti-mouse CD31 at 1:200 dilution (endothelial markers) (BD Biosciences, San Jose, CA), rabbit anti-mouse cytokeratin 7 at 1:3000 dilution (CK-7, epithelial markers) (Abcam, Cambridge, MA), rabbit anti-mouse collagen IIIa at 1:200 dilution (ECM markers) (Abcam, Cambridge, MA), goat anti-mouse carboxypeptidase A1 at 1:200 dilution (CPA1, acini markers) (R&D Systems, Inc., Minneapolis, MN), rat anti-mouse F4/80 at 1:500 (macrophage markers) (Bio-Rad, Hercules, CA), and FITC-conjugated, mouse anti-mouse α-SMA at 1:200 dilution (Sigma, St. Louis, MO). Alexa Fluor 488 (Abcam, Cambridge, MA) antibodies from appropriate species, including donkey anti-goat, donkey anti-rat, donkey anti-rabbit at 1:500 dilution, were used as secondary antibodies. IF-stained pancreatic sections were mounted with ProLong Gold Antifade Mountant with DAPI (Thermo Fisher Scientific Inc, Waltham, MA) for nucleus visualization.
Preparation and Characterization of Liposomes
Peptide 7-mers identified in the phage screen were chemically synthesized to include a C terminal addition of the amino acid linker, GGSK(FAM)C, by the Tufts University Peptide Synthesis Core Facility using standard FMOC chemistry and Rink-Amide resin (Figure S7). Liposome preparation was carried out as previously described with minor modifications.19 In brief, 4mg of peptides were first conjugated to 9.5mg of DSPE-PEG3400-maleimide in 1mL of 0.5mM EDTA/PBS under argon to prepare the aqueous micellar solution. The micelle mixture was left 1h at room temperature, followed by overnight incubation at 4°C. Overnight dialysis was performed in PBS and then in MilliQ H2O twice to remove free peptides and salts from the conjugated micelles. The purified DSPE-PEG3400-peptide was then lyophilized and ready for use in liposome preparations. Liposomes were prepared by hydration of lipid film composed of the following reagents: DOPC (9.5mg), cholesterol (4.5mg), DSPE-PEG2000 (4.5mg), DSPE-PEG3400-peptide (1mg), and DiD (0.5mg). DiD was incorporated into the lipid bilayer as a non-exchangeable near-infrared lipid dye, allowing in vivo detection of liposomes by IVIS (PerkinElmer, Waltham, MA). The lipid contents were mixed by sonication in 1 mL of chloroform, 1 mL of saline, and 3 mL of ether followed by placing on a rotary evaporator overnight to remove residual organic solvents. The lipid mixtures were then size-extruded 21 times through a syringe extruder with a 0.2 μm Nuclepore filter (Thermo Fisher Scientific, Inc., Waltham, MA). The size-extruded liposomes were centrifuged on A-100/18 Fixed-Angle Rotor (Beckman Coulter, Brea, CA) by Airfuge Air-Driven Ultracentrifuge (Beckman Coulter, Brea, CA) at 20 psig for 1h to separate micelles and unattached lipids from liposomes. The resulting liposomal pellets were resuspended in saline and characterized by NanoSight NS300 (Malvern Instruments Ltd., Worcestershire, UK) to determine particle size and concentration (Figure 1 and S3). The settings of NanoSight are as follows, camera level = 11; slider shutter = 600; slider gain = 300; FPS = 25.0; number of frames = 749; temperature = 20.7–20.9°C; viscosity = (water) 0.979–0.982 cP. The absorbance of FAM at 495nm was used to determine the number of peptides incorporated in each liposome formulation.
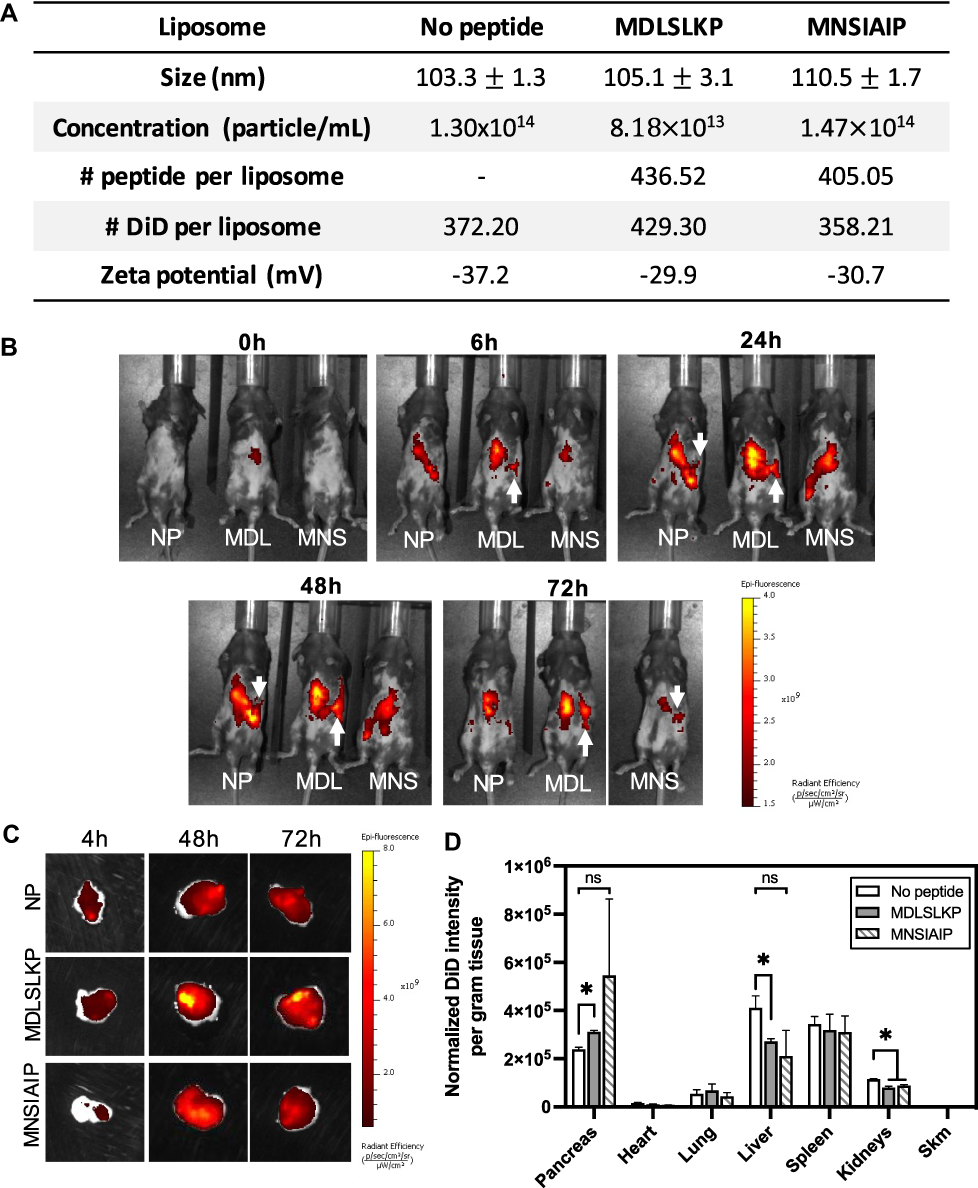 |
Figure 1 Pharmacokinetics of peptide-modified liposomes. (A) Table showing characteristic features of surface-modified liposomes with peptides identified to target collagen IIIa+ and acinar cells. (B) In vivo IVIS images of CP mice over 72h time course post-injection of peptide-modified liposomes. The white arrow indicates fluorescent signals detected at the pancreas region. (C) Ex vivo IVIS images of the pancreas at 4, 48, and 72h post liposome injection. (D) Biodistribution of DiD-labeled liposomes in CP mice at 48h post injection. Fluorescent intensity is normalized to the number of particles injected, the number of DiD per liposome, and the mass of the pancreas. N = 3. Student’s t-test was used to compare the targeted liposomes to the no peptide liposomes. *p < 0.05. Abbreviations: ns, not statistically significant; Skm, skeletal muscle. |
Specificity and Selectivity of Peptide-Conjugated Liposomes
Peptide-conjugated liposomes (150 μL containing 5×1011 particles) were injected via tail vein in CP mice (N=3) to determine the pharmacokinetic properties using IVIS. No peptide liposomes were used as negative controls. Mice hair was shaved and removed by depilatory creams prior to imaging. In vivo imaging via IVIS at 0, 6, 24, 48 and 72h post injections was performed. 4, 48, and 72h post injection, animals were perfused with saline, and organs were harvested for ex vivo imaging on IVIS using the Ex/Em 640/680 nm filter sets to detect DiD accumulation in the pancreas and other organs. The cellular targets of targeting liposomes were determined by IF using antibodies against the acinar cell marker (CPA1), aPSC marker (αSMA), ECM marker (collagen IIIa), epithelial marker (CK7), endothelial marker (CD31), and macrophage marker (F4/80). Colocalization analysis was performed using the JACoP plugin of the ImageJ software (National Institute of Health, Bethesda, MD). Mander’s colocalization coefficient (MCC), which represents the percentage of liposomes overlapping with cell markers, was used as an indicator to quantitate the extent of colocalization of liposomes with each cell type.20
Apigenin Drug Loading and Release Kinetics
The lipid mixture, containing DOPC (9.5mg), cholesterol (4.5mg), DSPE-PEG2000 (4.5mg), DSPE-PEG3400-peptide (1mg), and 2mg apigenin (Sigma, St. Louis, MO), was pre-dissolved in 100uL ethanol, respectively. The drug and lipid mixture was mixed and added to 1mL PBS at 55℃, 1h. Liposomes were prepared by passing through a 0.2 μm Nucleopore filter using a syringe extruder. The free drug was removed by Zeba Spin Desalting Column, 40K MWCO (Thermo Scientific, Waltham, MA). The resulting liposomes were characterized by Nanosight NS300. The drug loading per liposome was determined by Ultrospec 3000 UV/visible spectrophotometer (Pharmacia Biotech, Sweden). The extinction coefficient of apigenin in saline is 664.02 (M−1cm−1) at 337nm, which was determined using Beer’s law on serial-diluted samples of known concentrations. To determine the degree of apigenin remaining encapsulated in the purified liposomes at storage conditions (PBS, 4℃), a release kinetic study was performed for 14 days. The purified liposomes were centrifuged in Pierce Protein Concentrators PES, 10K MWCO (Thermo Scientific, Waltham, MA) on day 0, 9, and 14 at storage conditions. The free drug concentration in the filtrate was determined by UV spectrometer to calculate the amount of free apigenin released from the purified liposomes. In vitro release kinetics were determined by placing drug-loaded liposomes into the cartridge of Slide-A-Lyzer Mini Dialysis devices, 10K MWCO (Thermo Fisher Scientific Inc., Waltham, MA), with the conical tube filled with 50% FBS/saline at 37℃. The FBS buffer was used as a blank to set up UV spectrometer readouts. Free drug released into FBS buffer was collected at day 0, 1, 3, and 4 post incubation and determined by UV spectrometer.
Pharmacodynamic Measurement and Evaluation of Therapeutic Efficacy of Targeted Delivery of Apigenin
Six-week old C57bl/6J mice were given intraperitoneal injections of caerulein (125 μg/kg, twice daily, 5 weeks). Following 2 weeks of caerulein treatment, animals were randomly divided into 4 groups, N = 5 per group: 1) intravenous injections of empty (drug-free) MDLSLKP-conjugated liposomes (ECM Lip), twice-weekly 2) free drug – oral gavage of apigenin, 2mg/kg, 6 days per week,11 3) Api-NP Lip – intravenous injections of apigenin encapsulated no peptide liposomes, 6mg/kg, twice per week, and 4) Api-ECM Lip – intravenous injections of apigenin encapsulated, MDLSLKP-conjugated liposomes, 6mg/kg, twice per week. Apigenin treatments lasted for 3 weeks for the remaining 3 weeks of caerulein induction. At the end of week 5, mice were euthanatized and serum harvested through cardiac puncture. Pancreata were perfused, harvested and paraffin-embedded for staining. Livers were perfused, harvested, and partially paraffin-embedded for staining. The remaining of the liver tissue was homogenized in lysis buffer (200mM HEPES, pH7.5, 10mM KCl, 1.5mM MgCl2, 1mM EDTA, 1mM EGTA, 2mM PMSF, 1mM DTT, and 1x Protease Inhibitor Cocktail (Thermo Fisher Scientific Inc, Waltham, MA)).21 Pancreata sectioned at 5 μm were stained with H&E, picrosirius red, and fibronectin (1:50, Abcam, Cambridge, MA). Visualization of fibronectin was performed with DAB (Acros Organics, Fair Lawn, NJ) and the tissue counter-stained using Hematoxylin 1 (Richard Allen Scientific, San Diego, CA). The quantification of picrosirius red was determined by Image J, using a threshold applied on the red composites of the RGB images. In the analysis, a total of 60 images per group (12 images per animal) were used for picrosirius red quantification. Fibronectin expression was quantified using the positive pixel count function on QuPath.22 Acinar atrophy was determined by counting the number of damaged acinus within a 256 μm x 256 μm region of interest in the pancreas H&E images. Five images/animal, 3 animals/group were analyzed. Hepatotoxicity induced by the treatments was evaluated by Western blot of the liver lysates probing against SOD1 (rabbit anti-mouse 1:1000, Cell Signaling, Danvers, MA) and β-actin (mouse anti-mouse 1:1000, Cell Signaling, Danvers, MA). Serum alanine aminotransferase (ALT) and blood urea nitrogen (BUN) were determined using the VetTest 8008 Chemistry Analyzer (IDEXX, Westbrook, ME). Liver, heart, and lung from all treatment groups were sectioned and stained with fibronectin to evaluate treatments in tissues other than the pancreas (Figure S6).
Statistical Analysis
Statistical analysis of the data was performed by Student’s t-test, one-way analysis of variance (ANOVA) and Tukey–Kramer test. All data presented are expressed as mean ± standard error of at least three independent measurements. For all comparisons, p-value <0.05 was considered significant.
Results
Enrichment of Phage Clones Specific to the Inflamed Pancreas
The Kelly laboratory has pioneered methods to identify novel ligands and targets for various diseases.17,19,23,24 To select phage clones that bind specifically to cells in the complex CP microenvironment, an in vivo phage display screen was performed by injecting the phage library into the caerulein-induced CP mouse model (Figure 2A). The PhD.7 phage library (1×1012 pfu per mouse) was injected via tail vein in CP mice and allowed to circulate for 4h before animals were euthanized to allow extravasation out of the bloodstream and into tissues. Phage harvested from the pancreas were amplified and re-injected into CP animals for a total of 3 rounds of biopanning. For each round, tissues other than pancreas were also harvested and phage titered to determine the selectivity of the phage pool for CP pancreas. After three rounds of selection, the phage titer in the CP pancreas showed a statistically significant increase from round 1 (0.89% ID/g) and round 2 (0.39% ID/g) to round 3 (14.38% ID/g) (Figure 2B). Phage titers in the clearance organs, including liver, spleen, and kidneys, decreased over rounds, implying the selectivity of phage pools shifted towards the inflamed tissues and away from the clearance organs over the course of the selection process. At the end of round 3, we Sanger (30 clones per animal, N = 3) and deep sequenced phage clones isolated from pancreata to identify CP-specific peptides using an in silico selection approach from the enriched phage pools.
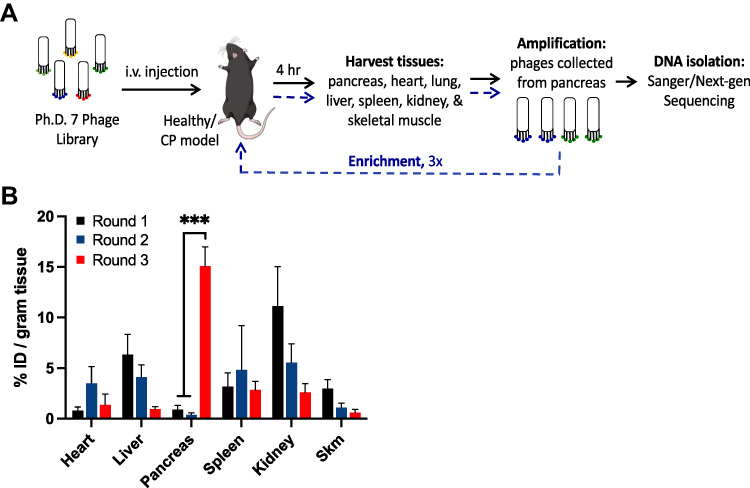 |
Figure 2 In vivo phage screening in the chronic pancreatitis mouse model. (A) A schematic of the in vivo phage biopanning process to screen for clones specific for CP pancreas. (B) Phage titering of in vivo phage screening in caerulein-induced CP mice. Phage pools (% injected dose per gram tissue) were recovered from the pancreas and various organs in 3 rounds of the biopanning process. One-way ANOVA and Tukey–Kramer tests were used to compare round 3 vs round 1, and round 3 vs round 2. N = 3; ***p<0.0001. Abbreviation: Skm: Skeletal muscle. |
In silico Selections of CP-Specific Candidate Clones
The Phage Analysis for Selective Targeted PEPtides (PHASTpep) software has been previously used to identify target-specific phage clones in screening against recombinant proteins and cells in culture.17 The process involves phage display selection, then Next-Generation Sequencing (NGS) to identify the peptide sequences that bind the target. From there, peptide sequences are subjected to the selectivity algorithm, which searches through the Kelly laboratory database of all previous phage display experiments. This step functions as an in silico negative selection to remove any phage clone that appears in multiple screens and would therefore, be non-specific or non-selective and capable of binding to multiple targets. To ensure selectivity for the inflamed pancreas, in addition to the caerulein-treated mice, we screened healthy mice of the same mouse strain and used the outputs from these screens in our algorithms to in silico counter-select clones that also bind to the healthy pancreas. PHASTpep applied normalization algorithms to remove non-specific binders and to ensure selectivity.17 However, the algorithms were used for in vitro selections with only one cell type present. In this work, four different methods to analyze the data and choose CP-specific peptide ligands were developed to account for a more complex system that involves thousands of available targets expressed in the multiple cell types present in an in vivo screen.
PHASTpep for Clone Selection
Conventional clone picking suffers from high false-positive rates and lacks a robust approach to select target-specific candidate clones. To address this issue, we used the NGS frequency counts from round 3 to allow quantitative sorting. To ensure specificity, each individual clone’s frequency count in the target screens was normalized to that in the naïve library screen (normalized frequency) to account for amplification and library biases.17 To ensure selectivity for CP pancreas over the healthy pancreas, normalized frequency of clones from CP pancreas screen was compared with pancreas from healthy animals. Of the 90 clones that Sanger sequencing methods alone would have identified, only 6 clones were selected as meeting the criteria of high normalized frequency counts in the CP pancreas (>50) but low accumulation in the healthy pancreas (<10) (Figure 3A). These selection criteria ensured a 5~10-fold higher expression of phage clones in the inflamed over the benign pancreas. It also removed a non-specific phage clone, ADARYKS,25,26 from the candidate list, which would have been selected using Sanger sequencing alone since it was the most abundant clone among clones picked (10%).
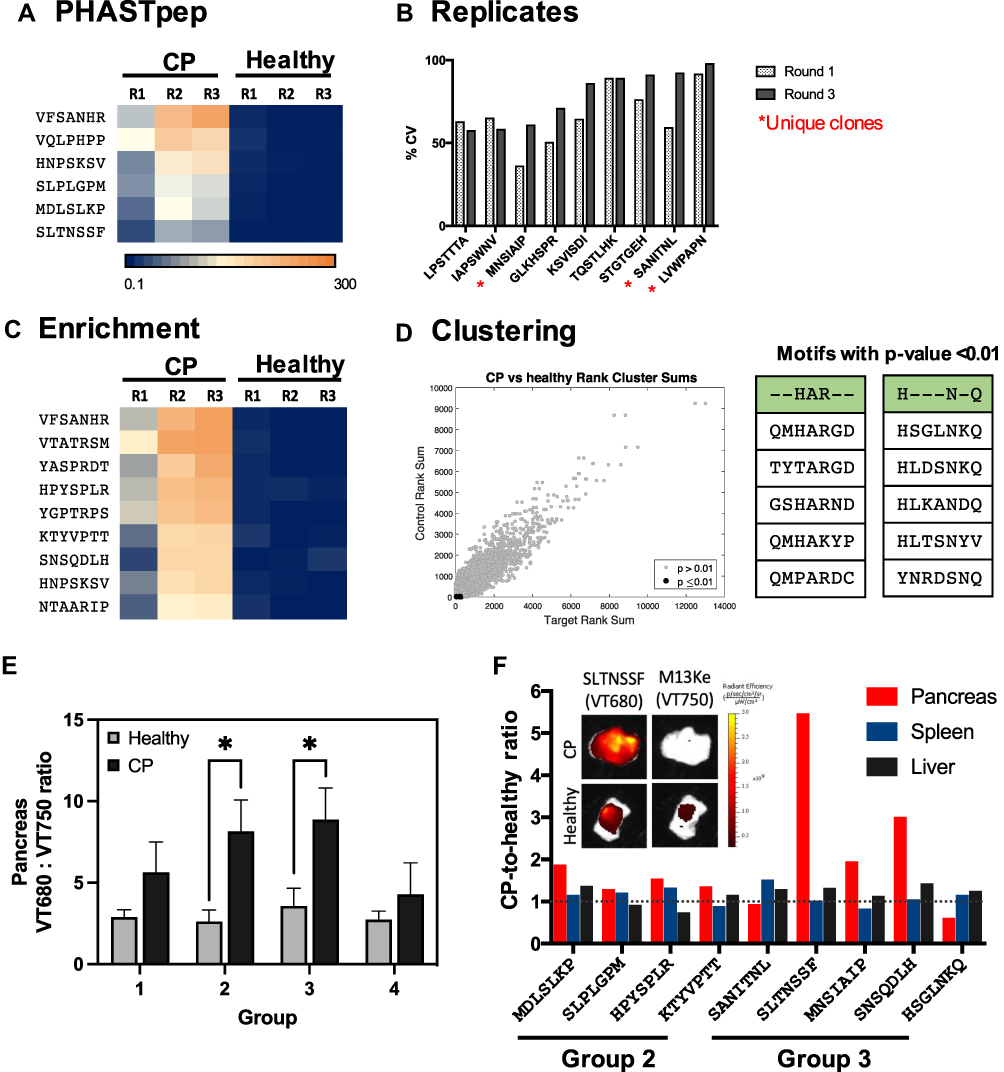 |
Figure 3 In silico selection and in vivo validation of CP targeting clones. CP targeting candidate clones were selected based on (A) PHASTpep, (B) replicability between 3 mice (asterisk indicates clones that have not been identified as ligands for known targets), (C) clone enrichment between 3 rounds of biopanning, and (D) motif clustering analysis. The normalized frequency count of each clone is represented in heatmaps. (E) Phage clone specificity validation by homology groups revealed preferential bindings of Group 2 and 3 targeting clones to CP pancreas over the healthy pancreas. N = 5. Mean ± SEM. A Student’s t-test was used to compare the targeting phage (VT680)-to-wild type phage (VT750) ratio in CP versus the same ratio in the healthy pancreas. *p-value < 0.05 (p = 0.0275 for Group 2; p = 0.0443 for Group 3). (F) Phage clones from Group 2 and 3 were validated individually in both healthy and CP mice. Fold change represents the ratio of targeting-to-wild type ratio in CP over healthy mice. 7/9 clones showed higher phage accumulation in CP over healthy pancreas (fold change >1). |
Replicability Between Libraries
Lot-to-lot variations of naïve libraries may induce amino acid distribution bias and skew the screening results.27 Therefore, we developed a method to take the variability into account and ensure the utility of the data to facilitate comparability of the sequences from every screen. This allows better selectivity as we can rapidly remove phage clones that are present across multiple selections. To elucidate the variability, we calculated the mean and standard deviation of the normalized frequency of a clone across two lots of phage display libraries (n=3 animals) and selected those clones with coefficient variants (CV) <1 and that have a minimum of 5-fold increase expression in CP over healthy pancreas (Figure 3B). Of 9 clones that meet these criteria, 3 unique clones, LVWPAPN, MNSIAIP & SANITNL, were selected.
Clone Enrichment Over Rounds
During biopanning, high affinity binders and/or clones binding to targets with high expression tend to remain in the eluates and be enriched after amplification. This process is indicated by a clone’s increasing frequency over rounds. As limited amounts (<0.0001%) of clones can be sequenced in the traditional clone picking process, the loss of potential candidate clones occurs early in the biopanning process, making direct comparisons of individual clone’s frequency change between rounds not as informative. With NGS, several orders of magnitude in the quantity of sequences in the library are obtained, enabling the calculation of a more reflective growth rate for each clone in silico. Therefore, we can utilize the growth rate (GR), determined by the ratio of the numbers of clones present between rounds, to select high affinity clones. In notation, GR2(3)1 = frequency in round 2(3)/frequency in round 1. Of the populations with the top 60 frequency counts in rounds 2 and 3, we selected 9 clones that show consistent positive GR (GR21 and GR31 >1), ensuring the analysis was not skewed towards burst growth in a single selection (Figure 3C). Using this method, we identified clones that would have been discarded using conventional clone picking alone.
Homologous Motif Identification Using Clustering Analysis
As protein interactions are often determined by a few amino acids, motifs conferring phage binding to its target can be repeatedly seen among different clones in the same screen. Recognizing target-specific homology families can offer insights to the libraries that would have been missed when evaluating each sequence as an independent read. Therefore, we clustered phage pools using the Hobohm algorithm28 to reveal homologous motifs in the CP screens. The frequency ranks of the identified homologies in the CP screens were then compared to the ranks in the healthy pancreas. Two clones, QMHARGD and HSGLNKQ, from the two statistically significant (p-value < 0.01) motif families were selected based on their target selectivity and growth rate (Figure 3D).
After combining the 4 analysis methods and determining selectivity using the Kelly laboratory database of phage screens that contains tissue-specific phage clones,17 we selected 18 candidate clones for further validation.
In vivo Validation of Phage Clones That Show Selectivity to Cellular Components in the Inflamed Pancreas
To efficiently evaluate 18 candidate clones, we divided them into 4 groups (Figure S2A) based on sequence similarity using GibbsCluster Server 2.0.29 Phage in the selected group and the wildtype M13Ke phage were fluorescently conjugated to fluorophore VT680 and VT750, respectively, pooled then were intravenously injected via tail vein into healthy or CP mice. At 20h post-injection, ex vivo ratiometric imaging of targeted-to-M13Ke phage analysis revealed a significantly higher accumulation of targeting phage clones from group 2 and 3 in the CP pancreas when compared with pancreas from healthy mice (p<0.05, Figure 3E). A total number of 9 individual clones gathered from group 2 and 3 were subsequently screened individually for their specificity for the inflamed pancreas. Seven out of nine clones (MDLSLKP, SLPLGPM, HPYSPLR, KTYVPTT, SLTNSSF, MNSIAIP, and SNSQDLH) showed increased specificity for CP over healthy pancreas (Figure 3F). From the validation results, we concluded that PHASTpep-guided selection, replicates and enrichment algorithm can reveal sequences specific for CP pancreas from an in vivo screen. That none of the clones identified from clustering analysis showed specificity to CP could be a result of the diverse available targets present in tissues, increasing the difficulty to converge valid motifs using the Hobohm algorithm.
To identify the cell types that the CP targeting clones were binding, we performed immunofluorescent analysis on common cellular components in the CP microenvironment in the inflamed pancreas sections following in vivo validation of individual clones (Figure 4A). Mander’s correlation coefficient (MCC) analysis was performed to determine colocalization of the targeting phage to the cell markers. Among each clone, we compared the MCC value of each cell marker to the rest of the markers using the Tukey–Kramer test (Table S2). Five clones were identified to demonstrate statistically significant colocalization to one single cell type; thus, an indication for cellular selectivity. Through this analysis, we revealed that KTYVPTT was selective for αSMA+ cells (MCC = 0.521 ± 0.067), MDLSLKP for collagen IIIa+ cells (MCC = 0.828 ± 0.089), MNSIAIP for CPA-1+ cells (MCC = 0.633 ± 0.179), and SLTNSSF and SNSQDLH for F4/80+ cells (MCC = 0.804 ± 0.090 and 0.800 ± 0.197, respectively) (Figure 4B). Phage clones and the associated cellular components in the inflamed pancreas are summarized in Figure 4C.
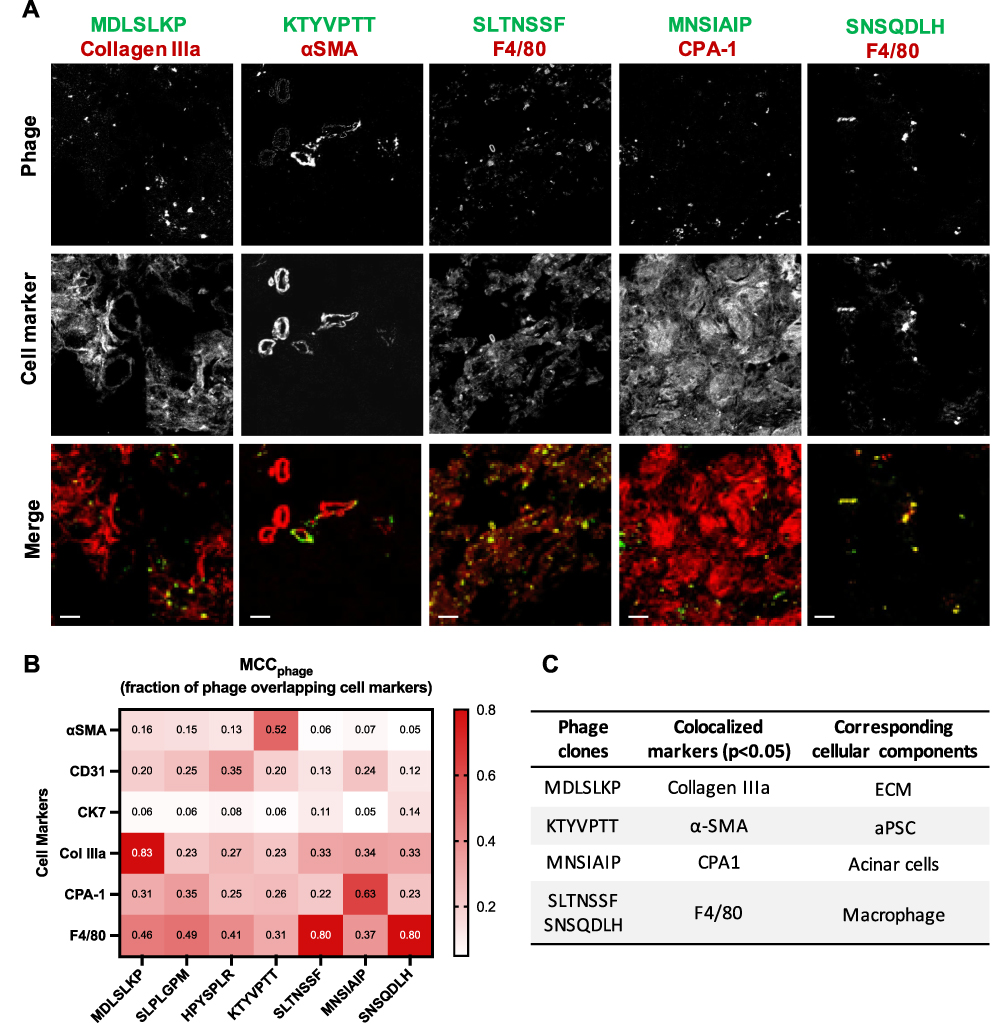 |
Figure 4 CP-homing phage clones show selectivity for cellular components in the CP pancreas. (A) Immunofluorescence images of VT680-labeled phage colocalized with cell markers in the inflamed pancreas. Six cell markers were stained to represent six common cellular components in CP: αSMA (activated PSC), CD31 (endothelium), CK7 (epithelium), Collagen IIIa (ECM), CPA-1 (acinar cells), and F4/80 (macrophages). Colors in the merged images represent phage (green) and cell markers (red). Scale bar: 20 μm. (B) Heatmap of mean Manders’ correlation coefficient (MCC) representing the fraction of phage overlapping with the cell markers. Manders’ colocalization analysis was performed using the ImageJ plug-in JACoP. N = 10–12 images per marker, per clone. Col IIIa: collagen IIIa. (C) A table summarizing phage clones shown statistically significant selectivity for a single cellular component in CP. One-way ANOVA and Tukey–Kramer tests were used to compare the MCC of all cell markers for each clone. The result was considered significant if the p-value ≤ 0.05. |
Peptide-Conjugated Liposomes Altered Nanoparticle Pharmacokinetics and Showed Cellular Selectivity in the Inflamed Pancreas
The extracellular matrix and acinar cells are highly abundant cells in CP; therefore, we chose peptides MDLSKLP and MNSIAIP to develop targeted liposomes. Despite demonstrating the highest in vivo ratio for specificity between the inflamed and healthy pancreas, we did not select the macrophage-targeting peptides for this proof-of-concept drug delivery system because macrophages present in a wide spectrum of activated phenotypes in inflammatory and fibrotic diseases and are subject to change in response to microenvironmental stimuli.30,31 Better understanding of the roles of macrophages in CP and further characterization of the targeting specificity of SLTNSSF and SNSQDLH for macrophage subsets will be needed. Peptide-conjugated liposomes were prepared by the reverse-phase evaporation method with an average size of 90–110 nm in diameters, and the number of peptides displayed on the surface ranged from 400 to 450 per liposome (Figure 1A). All peptides used in the study have a net charge of +1 at pH 7.0. A non-exchangeable lipid dye, DiD, was incorporated into the lipid formula at an average of 350–450 dye per liposome to allow particle tracking by noninvasive fluorescent imaging (Figure 1A). Liposomes without surface modification (no-peptide liposomes, NP) were used as negative controls in the pharmacokinetics studies as liposomes are readily taken up by the abundant phagocytic cells present in CP. 5×1011 liposomes were injected into CP mice via tail vein, and the animals were imaged at 0, 6, 24, 48, and 72h post injection using IVIS. Starting at 6h and lasting until the 72h timepoint, fluorescent accumulation was observed in the MDLSLKP liposome-injected mice in the left abdomen, consistent with the location of the pancreas (Figure 1B). At 48h post injection, a 1.3-fold increase of MDLSLKP liposomal accumulation was detected in the inflamed pancreas compared to the NP liposomes (Figure 1C and D). As expected, liposome accumulation was also observed in the clearance organs (liver, spleen, and kidney) as previously reported.32,33 A significant reduction in liver and kidney accumulation of the MDLSLKP liposomes was observed, suggesting the addition of targeting ligands altered the particle distribution away from the clearance organs to the inflamed pancreas. In contrast to MDLSLKP targeted liposomes, there was not a significant difference in pancreas and liver accumulation of the MNSIAIP liposome compared to the NP liposome.
The cellular selectivity of the targeting liposomes in the inflamed pancreas at 48h-post injection was systematically validated via colocalization analyses on IF-stained tissue sections stained for 6 cell markers (Figure 5A). As expected, NP liposomes were taken up and colocalize with macrophages (MCC = 0.534 ± 0.178) as liposomes are readily phagocytosed by macrophages, which are abundant in the inflamed pancreas.34,35 The addition of peptide ligands shifted the cellular targets of the MDLSLKP liposome to collagen IIIa+ expressing cells (MCC = 0.493 ± 0.142), demonstrating the preservation of similar ECM selectivity as it was observed by the phage (Figure 5B). The MNSIAIP liposome, however, did not show statistically significant selectivity towards CPA-1+ cells, suggesting that this peptide lost its selectivity once conjugated to a liposome. The loss of cell selectivity may explain the result of the in vivo studies where increased pancreas accumulation was observed in the phage form but not in the liposomal form. Combining the pharmacokinetics and cell colocalization results, we concluded that MDLSLKP peptide improved liposome targeting to collagen IIIa+ expressing cells by 1.5–3 fold as compared with other cell types, resulting in increased pancreas accumulation when compared with non-targeted liposomes. The targeting ligands identified shift liposomes away from macrophage uptake and towards target cells as supported by the Tukey–Kramer test of MCCs of all cell types in the NP and MDLSLKP liposomes (Table S3).
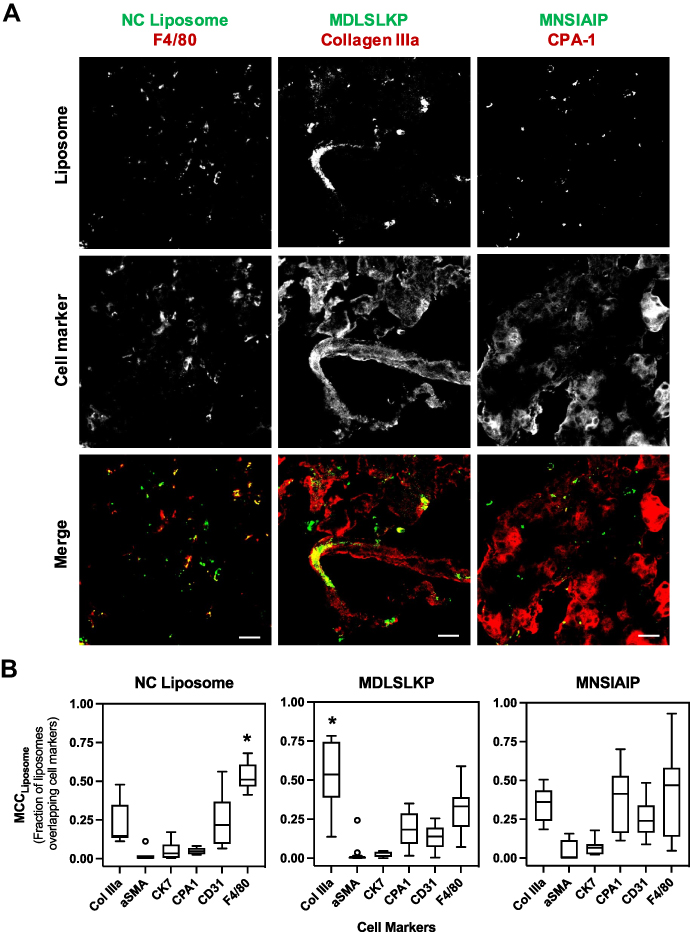 |
Figure 5 Immunofluorescence of peptide-conjugated liposomes in CP pancreas. (A) NC liposomes were non-specifically taken up by macrophages present in the inflamed pancreas. MDLSLKP liposomes colocalized with extracellular matrix (collagen IIIa+ cells). Color code: green for liposome (DiD) and red for cell markers. Scale bar: 20 μm. (B) Box-and-whisker plot of MCC values of liposomes overlapping cell markers. Liposome selectivity for the corresponding cell types was analyzed using the ImageJ plug-in JACoP. N = 7~12 images per group. One-way ANOVA and Tukey–Kramer tests were used to compare the MCC of all cell markers for each liposome. *p < 0.05. No statistically significant difference was observed in spatial localization of MNSIAIP liposomes with any stained cell types. |
Targeted Delivery of Apigenin Enhances Antifibrotic Effects in CP Mice
Apigenin is a small molecule natural compound, that has been demonstrated preclinically to have antifibrotic and anti-inflammatory properties in CP.11,12 Apigenin, however, suffers from low aqueous solubility, metabolic instability, and off-target effects and there are no approved clinical applications of apigenin available.21,36,37 Therefore, we used apigenin as a proof of concept and directly loaded it into the MDLSLKP-conjugated liposomes (ECM liposome). Physicochemical properties of apigenin are summarized in Figure S4A and B. Free apigenin was removed from liposomes by size exclusion chromatography. The drug loading was determined using UV spectrometer and showed a 40–50% encapsulation rate in both NP and ECM liposomes. The final drug-to-lipid ratio was estimated at an average of 80–100 ug apigenin per mg of lipid, and each liposome contains 28,000~34,000 drug per particle (Figure S4C). Both apigenin-loaded NP and ECM liposomes had a diameter of 90–100 nm measured by NanoSight. We also determined the shelf-life of the drug-encapsulated liposomes and showed >95% of the encapsulated apigenin remained in liposomes under storage conditions (PBS, 4℃) over 14 days (Figure S4D). Hydrophobic molecules are usually encapsulated in the lipid layer of liposomes and may burst release from the particles in vivo.38 To test whether apigenin will burst release in our system, we performed an in vitro release study in 50% FBS/PBS at 37℃. We did not see burst release of apigenin in the first couple of hours upon placing liposomes in 50% FBS. Instead, 14.02% of the encapsulated apigenin molecules were released by day 1 and 52.69% by day 4 (Figure S4E).
To test the antifibrotic efficacy of targeted liposomes, we evaluated the pharmacodynamics of apigenin in the free drug form, encapsulated in non-targeted liposomes and in ECM liposomes (Figure 6A). ECM liposomes without drug loading were included in the study as vehicle control. C57BL/6J mice were injected with caerulein 14 days before treatment started to establish inflammation in the pancreas. Apigenin, in the free drug or liposomal form, was given in the remaining 3-week course along with caerulein. As expected, increased acini atrophy and cellular heterogeneity in size and shape, in addition to increased interstitial space, fibrosis, and collagen deposition (34.83% area) were observed in the control group (Figure 6B–D).11 Compared to free drug and NP liposomes, targeted delivery of apigenin resulted in enhanced preservation of acini units with a respective 4-fold and 2-fold reduction of acinar atrophy counts observed in the pancreas and the decrease of interstitial space between acinus (Figure 6B and E). Although NP liposomes have substantial macrophage uptake, the enhanced pharmacodynamic activity of apigenin loaded into ECM targeted liposomes demonstrated the importance of cell-specific targeting to drug activity. Using Picrosirius red to stain for collagen, we found that collagen deposition was reduced from 24.90% area to 15.63% in the free apigenin versus targeted liposomal form, respectively (p-value < 0.0001, Figure 6D and G). Apigenin loaded in NP liposomes reduced collagen to 19.18% area of the inflamed tissue, suggesting that liposomal formulation of apigenin alone resulted in better therapeutic efficacy than free drug but was not as effective as targeted delivery to the ECM. Targeted liposomes resulted in 33.1% better reduction in fibronectin expression when compared to free drug alone and 41.0% reduction compared to NP liposomes (p-value < 0.05. Figure 6C and F). In addition to therapeutic efficacy, we also evaluated hepatotoxicity induced by apigenin21 in free drug and liposomal form and showed a 1.2-fold increase of SOD1 expression in targeted liposomes, indicating reduction of oxidative stress in the liver (Figure S5C), matching the reduced liver accumulation found via liposome biodistribution. Liposome-based antifibrotic therapies have been evaluated in many pre-clinical studies.39–41 Lacking a selective and specific targeting ligand, however, has limited the clinical implementation as the non-targeted liposomes are taken up readily by macrophages. The 7-mer peptides we identified from the inflamed pancreas can be applied to other cell type-selective therapeutic molecules to potentially improve tissue remodeling and reduce fibrosis and inflammation in CP.
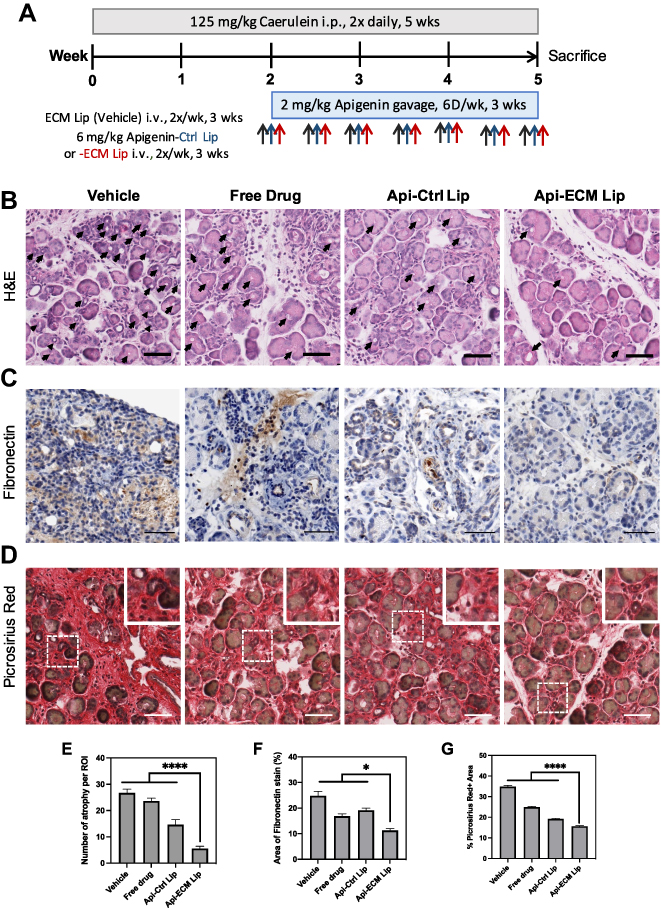 |
Figure 6 Targeted delivery of Apigenin reduces fibrosis. (A) Schematic of CP mouse model followed by 3-week treatments of either empty ECM liposome (vehicle), free Apigenin (free drug), Apigenin-encapsulated naked liposomes (Api-Naked Lip), or Apigenin-encapsulated MDLSLKP liposomes (Api-ECM Lip) (N=5). (B) H&E staining of pancreas by the end of 3-week treatments. Reduced interstitial space and acinar atrophy (indicated by arrows) were observed in the pancreas treated by targeted liposomes compared to free drug and the control liposomes. Scale bar: 50 μm. (C) Pancreas immuno-stained for fibronectin demonstrated targeted delivery of Apigenin significantly decreased fibronectin expression. Scale bar: 100 μm. (D) Picrosirius red staining of the pancreas. Scale bar: 50 μm. (E) Number of acini atrophy found in a 256 μm x 256 μm image. N = 5 images/animal, 3 animals/group. (F) Quantification of fibronectin-positive area. N = 8 images/animal, 5 animals/group. (G) Quantification of Picrosirius red-positive area. N = 12 images/animal, 5 animals/group. In all images, ANOVA and Tukey’s test were used to compare Api-ECM Lip to the rest of the treatment groups. *p<0.05, ****p<0.0001. |
Discussion
Chronic pancreatitis is a complex inflammatory pancreatic disease that remains incurable.42–44 Current treatments for CP are limited to palliative care and pain alleviation, and these approaches fail at the advanced stage when invasive surgical procedures such as endoscopic intervention, bypass, and total pancreatectomy are the only available options.45 The pancreatic community has recently reached a consensus that precision medicine can provide a more sophisticated approach for complex disorders like CP to assist the development of target-specific interventions.44 Despite omics-based technology being widely used to profile disease-specific biomarkers and therapeutic targets in many diseases,46,47 transcriptomics reveals little about CP-specific pathways due to the universal genetic backgrounds shared between pancreatitis, pancreatic cancer, and the benign pancreas.48 Looking only in epithelial cells, Sanh et al showed differential expression in 34 proteins in malignant and pancreatitis pancreas compared to the benign tissue, but were not able to distinguish pancreatitis from pancreatic cancer.49 Considering the heterogeneity of cellular components involved in the disease progression of CP, there is a definite need to provide an unbiased, comprehensive evaluation of pancreatitis-associated proteomes.
In this paper, we utilized a computational-guided in vivo phage display approach to profile 7-mer peptide ligands selective for cellular components in the caerulein-induced CP mouse pancreas. In contrast to indirect proteomic techniques, phage display allows probing of proteins in their native context during biopanning, thus increasing the clinical relevance of the identified targeting agents.50 Additionally, in vivo screening ensures identifying targeting agents with high selectivity as subtractions for all other tissues are carried out while enrichment occurs in the target tissue.19 By comparing the in vivo screens against CP, benign pancreas, and pancreatic cancer using our database, we were able to ensure peptide selectivity to the diseased pancreas by not choosing peptides in any condition but CP. Phage display combined with Illumina NGS overcomes conventional biopanning limitations, including high false-positive rates and lack of a robust analytical target selection method.51 Using the PHASTpep-guided approach, our team has successfully identified peptides specific for pancreatic cancer-associated fibroblasts in vitro.17 Here, we expanded the application in analyzing in vivo phage screens and assessed the identified phage clones with live animal imaging modalities and fluorescent microscopy to show peptide specificity towards inflamed pancreas and selectivity towards potential cellular target of interest, including activated PSC, acinar cells, macrophages, and extracellular matrix.
To explore the potential of the CP targeting peptides in cell type-specific drug delivery, we conjugated the peptides to pegylated DOPC liposomes and characterized the pharmacokinetics using fluorescent-based imaging modalities. Although in vivo and ex vivo imaging has lower sensitivity compared to direct quantification of liposome lipid markers in tissues using high-performance liquid chromatography, imaging modalities provide a noninvasive approach to allow serial evaluation in the same animals on particle distribution. A hallmark of CP is the presence of large numbers of phagocytic cells, and indeed, NP liposomes were readily taken up by the macrophage cell population. The addition of targeting ligands, however, shifted the cellular targets of the MDLSLKP-conjugated liposomes away from macrophages and demonstrated selectivity to collagen IIIa+ expressing cells, consistent with its phage clone. A shift in organ accumulation from the clearance organs to the inflamed pancreas was also achieved when injecting liposomes displaying MDLSLKP peptides. Disappointingly, cellular selectivity of MNSIAIP to CPA-1-positive cells was not observed after conjugation to liposomes, suggesting that the current orientation of MNSIAIP displayed on the liposome may have hindered the binding of the peptide to its target. The lack of selectivity could explain the no statistically significant difference observed in particle accumulation of the MNSIAIP liposome compared to the NP liposome. This finding emphasizes the importance of characterizing targeted liposome’s cellular targets.
The improvement in increasing the ratio of on-target to off-target effects could address the side effects induced by antifibrotic drugs that act on canonical extracellular factors, including growth factors, cytokines, and MMPs. For example, TGF-β inhibitors are amongst the majority of approved or investigational anti-fibrosis drug families and have demonstrated efficacy in reducing cardiac, liver, and kidney fibrosis.52 However, galnisertib, a TGF-β R1 kinase inhibitor, caused cardiac toxicity, bone development abnormality and induced irregular inflammatory responses in skin and gut at long-term use, which ended with termination on Phase II clinical trial (NCT0113801).53 The ability to selectively target multiple different cell types in CP can open a new avenue for therapeutic strategies that address the crosstalk between ECM components and aPSCs, which initiates multiple cascades of events in fibrogenesis and inflammation in CP.12,54 Indeed, we demonstrated that targeting apigenin to the ECM demonstrated enhanced pharmacodynamic effects beyond that of targeting macrophages alone with NP liposomes, underscoring the importance of targeting. Apigenin is a small molecule drug that has been shown to have antifibrotic, anti-inflammatory, and proapoptotic effects in cancer and chronic inflammatory diseases in vitro and in vivo.11–13,55,56 The clinical use of apigenin, however, is limited by low aqueous solubility, high metabolic instability, and potential hepatotoxicity at acute use.21,36 As proof of concept, we encapsulated apigenin in liposomes and evaluated the antifibrotic effects through cellular component-specific delivery to the ECM in CP pancreas. After 3 consecutive weeks of treatments, mice receiving drug-encapsulated, ECM-targeted liposomes showed the best tissue remodeling effects, including acini unit preservation and stroma reduction, compared to free drug and non-targeted liposomes. In addition, targeted delivery reduced off-target effects as mice receiving apigenin encapsulated in targeted liposomes showed reduced ALT and liver SOD1 expression and preserved liver histology compared to the non-targeted delivery (Figure S5A and B). The results of our work demonstrated that cell type-specific targeting of small molecule drugs can improve pharmacodynamics and alter the anatomical endpoint (fibrosis) in the inflamed pancreas.
Conclusion
This study successfully established a high-throughput approach to guide ligand selection for cellular targets in a diseased mouse model. Our findings 1) revealed five heptapeptides specific to CP, 2) demonstrated that the conjugation of CP-specific peptides to pegylated DOPC liposomes is capable of increasing particle accumulation in the inflamed pancreas, and 3) showed that targeted delivery of apigenin in a mouse model of CP enhanced tissue remodeling, attenuated pancreatic fibrosis, and reduced liver toxicity. To our knowledge, this is the first study to identify a non-serum-based molecular ligand specific for CP. The ligand identification process is robust and requires no prior knowledge of the target, offering the potential to be easily applied to other disease models. Our study also provides opportunities for future applications of nanomedicine for the targeting of chronic inflammatory diseases.
Acknowledgments
The authors would like to thank Y. Bao of the UVA Biomolecular Research Core Facility for NGS; M. Bevard of the UVA Cardiovascular Research Center histology core for paraffin embedding and IHC staining. The work was supported by NIH (R21 CA231196-01, R01 CA233716-01 A1) and NCI (P30 CA044579-26).
Author Contributions
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agreed to be accountable for all aspects of the work.
Disclosure
KAK is the CEO and Founder of ZielBio, Inc. ZielBio played no role in funding this work. KAK and JH report a patent HIGH-THROUGHPUT ANALYTIC SELECTIONS OF NOVEL TARGETING LIGANDS FOR CHRONIC PANCREATITIS AND DEVELOPMENT OF TARGET-SPECIFIC LIPOSOMES FOR ANTIFIBROTIC THERAPY pending to United States Patent and Trademark Office. The application was filed on 2/5/2021. ALK is a subcontract of an NIH grant to SoundPipe Therapeutics, a company in the area of microbubble-assisted drug delivery. The authors report no other conflicts of interest in this work.
References
1. Gardner TB, Adler DG, Forsmark CE, Sauer BG, Taylor JR, Whitcomb DC. ACG clinical guideline: chronic pancreatitis. Off J Am Coll Gastroenterol ACG. 2020;115(3):322–339. doi:10.14309/ajg.0000000000000535
2. Beyer G, Habtezion A, Werner J, Lerch MM, Mayerle J. Chronic pancreatitis. Lancet Lond Engl. 2020;396(10249):499–512. doi:10.1016/S0140-6736(20)31318-0
3. Morgan K, Owczarski SM, Borckardt J, et al. Quality of life after pancreatectomy with islet autotransplantation for chronic pancreatitis. J Gastrointest Surg. 2012;16(1):129–134. doi:10.1007/s11605-011-1744-y
4. Machicado JD, Amann ST, Anderson MA, et al. Quality of life in chronic pancreatitis is determined by constant pain, disability/ unemployment, current smoking, and associated co-morbidities. Am J Gastroenterol. 2017;112(4):633–642. doi:10.1038/ajg.2017.42
5. Han S, Patel B, Min M, et al. Quality of life comparison between smokers and non-smokers with chronic pancreatitis. Pancreatol off J Int Assoc Pancreatol IAP Al. 2018;18(3):269–274. doi:10.1016/j.pan.2018.02.012
6. Bang UC, Benfield T, Hyldstrup L, Bendtsen F, Beck Jensen J-E. Mortality, cancer, and comorbidities associated with chronic pancreatitis: a Danish nationwide matched-cohort study. Gastroenterology. 2014;146(4):989–994. doi:10.1053/j.gastro.2013.12.033
7. Kichler A, Jang S. Chronic pancreatitis: epidemiology, diagnosis, and management updates. Drugs. 2020;80(12):1155–1168. doi:10.1007/s40265-020-01360-6
8. Hall TC, Garcea G, Webb MA, Al-Leswas D, Metcalfe MS, Dennison AR. The socio-economic impact of chronic pancreatitis: a systematic review. J Eval Clin Pract. 2014;20(3):203–207. doi:10.1111/jep.12117
9. Ting J, Wilson L, Schwarzenberg SJ, et al. Direct costs of acute recurrent and chronic pancreatitis in children in the insppire registry. J Pediatr Gastroenterol Nutr. 2016;62(3):443–449. doi:10.1097/MPG.0000000000001057
10. Machicado JD, Dudekula A, Tang G, et al. Period prevalence of chronic pancreatitis diagnosis from 2001–2013 in the commercially insured population of the United States. Pancreatology. 2019;19(6):813–818. doi:10.1016/j.pan.2019.07.003
11. Mrazek AA, Porro LJ, Bhatia V, et al. Apigenin inhibits pancreatic stellate cell activity in pancreatitis. J Surg Res. 2015;196(1):8–16. doi:10.1016/j.jss.2015.02.032
12. Mrazek AA, Bhatia V, Falzon M, Spratt H, Chao C, Hellmich MR. Apigenin decreases acinar cell damage in pancreatitis. Pancreas. 2019;48(5):711. doi:10.1097/MPA.0000000000001310
13. Ahmed SA, Parama D, Daimari E, et al. Rationalizing the therapeutic potential of apigenin against cancer. Life Sci. 2021;267:118814. doi:10.1016/j.lfs.2020.118814
14. Chen H, Mrazek AA, Wang X, et al. Design, synthesis, and characterization of novel apigenin analogues that suppress pancreatic stellate cell proliferation in vitro and associated pancreatic fibrosis in vivo. Bioorg Med Chem. 2014;22(13):3393–3404. doi:10.1016/j.bmc.2014.04.043
15. Gabizon AA, Patil Y, La-beck NM. New insights and evolving role of pegylated liposomal doxorubicin in cancer therapy. Drug Resist Updat Rev Comment Antimicrob Anticancer Chemother. 2016;29:90–106. doi:10.1016/j.drup.2016.10.003
16. Alavi M, Hamidi M. Passive and active targeting in cancer therapy by liposomes and lipid nanoparticles. Drug Metab Pers Ther. 2019;34(1). doi:10.1515/dmpt-2018-0032
17. Brinton LT, Bauknight DK, Dasa SSK, Kelly KA. PHASTpep: analysis software for discovery of cell-selective peptides via phage display and next-generation sequencing. PLoS One. 2016;11(5):e0155244. doi:10.1371/journal.pone.0155244
18. Halbrook CJ, Wen H-J, Ruggeri JM, et al. Mitogen-activated protein kinase kinase activity maintains acinar-to-ductal metaplasia and is required for organ regeneration in pancreatitis. Cell Mol Gastroenterol Hepatol. 2017;3(1):99–118. doi:10.1016/j.jcmgh.2016.09.009
19. Dasa SSK, Suzuki R, Gutknecht M, et al. Development of target-specific liposomes for delivering small molecule drugs after reperfused myocardial infarction. J Control Release off J Control Release Soc. 2015;220(Pt A):556–567. doi:10.1016/j.jconrel.2015.06.017
20. Manders EMM, Verbeek FJ, Aten JA. Measurement of co-localization of objects in dual-colour confocal images. J Microsc. 1993;169(3):375–382. doi:10.1111/j.1365-2818.1993.tb03313.x
21. Singh P, Mishra SK, Noel S, Sharma S, Rath SK. Acute exposure of apigenin induces hepatotoxicity in Swiss mice. PLoS One. 2012;7(2):e31964. doi:10.1371/journal.pone.0031964
22. Bankhead P, Loughrey MB, Fernández JA, et al. QuPath: open source software for digital pathology image analysis. Sci Rep. 2017;7(1):16878. doi:10.1038/s41598-017-17204-5
23. Bausch D, Thomas S, Mino-Kenudson M, et al. Plectin-1 as a novel biomarker for pancreatic cancer. Clin Cancer Res off J Am Assoc Cancer Res. 2011;17(2):302–309. doi:10.1158/1078-0432.CCR-10-0999
24. Gutknecht MF, Seaman ME, Ning B, et al. Identification of the S100 fused-type protein hornerin as a regulator of tumor vascularity. Nat Commun. 2017;8(1):552. doi:10.1038/s41467-017-00488-6
25. Kroiss D, Aramini JM, McPhee SA, Tuttle T, Ulijn RV. Unbiased discovery of dynamic peptide-ATP complexes. ChemSystemsChem. 2019;1(1––2):7–11. doi:10.1002/syst.201900013
26. Part:BBaK2043011: Design. Available from: http://parts.igem.org/Part:BBa_K2043011:Design.
27. Ryvkin A, Ashkenazy H, Weiss-Ottolenghi Y, Piller C, Pupko T, Gershoni JM. Phage display peptide libraries: deviations from randomness and correctives. Nucleic Acids Res. 2018;46(9):e52. doi:10.1093/nar/gky077
28. String::Cluster::Hobohm- Cluster strings using the Hobohm algorithm. Available from: https://metacpan.org/pod/String::Cluster::Hobohm.
29. Andreatta M, Alvarez B, Nielsen M. GibbsCluster: unsupervised clustering and alignment of peptide sequences. Nucleic Acids Res. 2017;45(W1):W458–W463. doi:10.1093/nar/gkx248
30. Hu F, Lou N, Jiao J, Guo F, Xiang H, Shang D. Macrophages in pancreatitis: mechanisms and therapeutic potential. Biomed Pharmacother Biomed Pharmacother. 2020;131:110693. doi:10.1016/j.biopha.2020.110693
31. Yang S, Yuan H-Q, Hao Y-M, et al. Macrophage polarization in atherosclerosis. Clin Chim Acta Int J Clin Chem. 2020;501:142–146. doi:10.1016/j.cca.2019.10.034
32. Koning GA, Kamps JAAM, Scherphof GL. Interference of macrophages with immunotargeting of liposomes. J Liposome Res. 2002;12(1–2):107–119. doi:10.1081/lpr-120004782
33. Immordino ML, Dosio F, Cattel L. Stealth liposomes: review of the basic science, rationale, and clinical applications, existing and potential. Int J Nanomedicine. 2006;1(3):297–315.
34. Vonarbourg A, Passirani C, Saulnier P, Simard P, Leroux JC, Benoit JP. Evaluation of pegylated lipid nanocapsules versus complement system activation and macrophage uptake. J Biomed Mater Res A. 2006;78A(3):620–628. doi:10.1002/jbm.a.30711
35. Zahednezhad F, Saadat M, Valizadeh H, Zakeri-Milani P, Baradaran B. Liposome and immune system interplay: challenges and potentials. J Control Release off J Control Release Soc. 2019;305:194–209. doi:10.1016/j.jconrel.2019.05.030
36. Zhang J, Liu D, Huang Y, Gao Y, Qian S. Biopharmaceutics classification and intestinal absorption study of apigenin. Int J Pharm. 2012;436(1–2):311–317. doi:10.1016/j.ijpharm.2012.07.002
37. Srivastava JK, Gupta S. Extraction, characterization, stability and biological activity of flavonoids isolated from chamomile flowers. Mol Cell Pharmacol. 2009;1(3):138. doi:10.4255/mcpharmacol.09.18
38. Gubernator J. Active methods of drug loading into liposomes: recent strategies for stable drug entrapment and increased in vivo activity. Expert Opin Drug Deliv. 2011;8(5):565–580. doi:10.1517/17425247.2011.566552
39. Sung SA, Jo SK, Cho WY, Won NH, Kim HK. Reduction of renal fibrosis as a result of liposome encapsulated clodronate induced macrophage depletion after unilateral ureteral obstruction in rats. Nephron Exp Nephrol. 2007;105(1):e1–9. doi:10.1159/000096859
40. Jose A, Mandapalli PK, Venuganti VVK. Liposomal hydrogel formulation for transdermal delivery of pirfenidone. J Liposome Res. 2016;26(2):139–147. doi:10.3109/08982104.2015.1060611
41. Khaja F, Jayawardena D, Kuzmis A, Önyüksel H. Targeted sterically stabilized phospholipid siRNA nanomedicine for hepatic and renal fibrosis. Nanomaterials. 2016;6(1). doi:10.3390/nano6010008
42. Whitcomb DC. Peering into the “black box” of the complex chronic pancreatitis syndrome. Pancreas. 2016;45(10):1361–1364. doi:10.1097/MPA.0000000000000715
43. Whitcomb DC, Shimosegawa T, Chari ST, et al. International consensus statements on early chronic Pancreatitis. Recommendations from the working group for the international consensus guidelines for chronic pancreatitis in collaboration with The International Association of Pancreatology, American Pancreatic Association, Japan Pancreas Society, PancreasFest Working Group and European Pancreatic Club. Pancreatol off J Int Assoc Pancreatol IAP Al. 2018;18:516–527. doi:10.1016/j.pan.2018.05.008
44. Lowe ME, Andersen DK, Caprioli RM, et al. Precision medicine in pancreatic disease-knowledge gaps and research opportunities: summary of a national institute of diabetes and digestive and kidney diseases workshop. Pancreas. 2019;48(10):1250–1258. doi:10.1097/MPA.0000000000001412
45. Bouwense SAW, Kempeneers MA, van Santvoort HC, Boermeester MA, van Goor H, Besselink MG. Surgery in chronic pancreatitis: indication, timing and procedures. Visc Med. 2019;35(2):110–118. doi:10.1159/000499612
46. Matthews H, Hanison J, Nirmalan N. “Omics”-informed drug and biomarker discovery: opportunities, challenges and future perspectives. Proteomes. 2016;4(3). doi:10.3390/proteomes4030028
47. Hasin Y, Seldin M, Lusis A. Multi-omics approaches to disease. Genome Biol. 2017;18(1):83. doi:10.1186/s13059-017-1215-1
48. Li S, Li R, Wang H, Li L, Li H, Li Y. The key genes of chronic pancreatitis which bridge chronic pancreatitis and pancreatic cancer can be therapeutic targets. Pathol Oncol Res POR. 2018;24(2):215–222. doi:10.1007/s12253-017-0217-3
49. Sanh N, Fadul H, Hussein N, et al. Proteomics profiling of pancreatic cancer and pancreatitis for biomarkers discovery. J Cell Sci Ther. 2018;9(4). doi:10.4172/2157-7013.1000287
50. Onogi Y, Khalil AEMM, Ussar S. Identification and characterization of adipose surface epitopes. Biochem J. 2020;477(13):2509–2541. doi:10.1042/BCJ20190462
51. Juds C, Schmidt J, Weller MG, et al. Combining phage display and next-generation sequencing for materials sciences: a case study on probing polypropylene surfaces. J Am Chem Soc. 2020;142(24):10624–10628. doi:10.1021/jacs.0c03482
52. Li X, Zhu L, Wang B, Yuan M, Zhu R. Drugs and targets in fibrosis. Front Pharmacol. 2017;8:855. doi:10.3389/fphar.2017.00855
53. Herbertz S, Sawyer JS, Stauber AJ, et al. Clinical development of galunisertib (LY2157299 monohydrate), a small molecule inhibitor of transforming growth factor-beta signaling pathway. Drug Des Devel Ther. 2015;9:4479–4499. doi:10.2147/DDDT.S86621
54. Xiao W, Jiang W, Shen J, et al. Retinoic acid ameliorates pancreatic fibrosis and inhibits the activation of pancreatic stellate cells in mice with experimental chronic pancreatitis via suppressing the wnt/β-catenin signaling pathway. PLoS One. 2015;10(11):e0141462. doi:10.1371/journal.pone.0141462
55. Zhang J, Chao L, Liu X, et al. The potential application of strategic released apigenin from polymeric carrier in pulmonary fibrosis. Exp Lung Res. 2017;43(9–10):359–369. doi:10.1080/01902148.2017.1380086
56. Chen L, Zhao W. Apigenin protects against bleomycin-induced lung fibrosis in rats. Exp Ther Med. 2016;11(1):230–234. doi:10.3892/etm.2015.2885
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.