")
Back to Journals » Drug Design, Development and Therapy » Volume 13
Identification Of Natural Compound Derivative For Inhibition Of XLF And Overcoming Chemoresistance In Colorectal Cancer Cells
Authors Liu Z, Yu M, Fei B, Sun J, Wang D
Received 16 May 2019
Accepted for publication 27 September 2019
Published 6 November 2019 Volume 2019:13 Pages 3823—3834
DOI https://doi.org/10.2147/DDDT.S215967
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Cristiana Tanase
Zhuo Liu,1 Miao Yu,1 Bingyuan Fei,1 Jing Sun,2 Dongxin Wang3
1Department of Gastrointestinal Colorectal and Anal Surgery, China-Japan Union Hospital of Jilin University, Changchun, Jilin, People’s Republic of China; 2Department of Biochemistry and Molecular Medicine, The George Washington University, Washington, DC, USA; 3Department of Anesthesiology, Jilin Cancer Hospital, Jilin, People’s Republic of China
Correspondence: Dongxin Wang
Department of Anesthesiology, Jilin Cancer Hospital, 1018 Huguang Road, Changchun, Jilin 130031, People’s Republic of China
Tel +86 139 4487 2227
Email [email protected]
Purpose: A previous study has identified that XRCC4-like factor (XLF) is a potential target to overcome resistance to 5-fluorouracil (5-Fu) and oxaliplatin (OXA) in colorectal cancer (CRC). The purpose of this study is to develop potent XLF inhibitors to chemoresistance in CRC.
Methods: Virtual screening was adopted to identify novel XLF-binding compounds by initially testing 6800 molecules in Chemical Entities of Biological Interest library. Hit compounds were further validated by Western blot assay. Cell sensitivity to 5-Fu and OXA was measured using sulforhodamine B assay. The effect of XLF inhibitor on DNA repair efficiency was evaluated by comet assay, fluorescent-based nonhomologous end joining (NHEJ) and homologous recombination (HR) reporter assays. DNA-binding activity of NHEJ key factors was examined by chromatin fractionation assay.
Results: We identified G3, a novel and potent XLF inhibitor (IC50 0.47±0.02 μM). G3 induced XLF protein degradation in CRC cells. Significantly, G3 improved cell sensitivity to 5-Fu and OXA in chemoresistant CRC cell lines. Mechanistically, G3 depleted XLF expression, severely compromised NHEJ efficiency by up to 65% and inhibited NHEJ key factor assembly on DNA. G3 also inhibited HR efficiency in a time-dependent manner.
Conclusion: These results suggest that G3 overcomes 5-Fu and OXA resistance in CRC cells by inhibiting XLF expression. Thus, XLF is a promising target and its inhibitor G3 is a potential candidate for treatment of chemoresistant CRC patients.
Keywords: virtual screening, XLF inhibitor, chemoresistance, colorectal cancer
Introduction
Colorectal cancer (CRC) is the third most commonly diagnosed cancer affecting both men and women and the second common cause of cancer-related death worldwide.1 There are one million new cases diagnosed per year, and the death rate is around 0.6 million.2 Unfortunately, a significant amount of patients diagnosed with CRC are at advanced stage who will require chemotherapy.3 5-fluorouracil (5-Fu) has been the mainstay of chemotherapy for CRC since the 1950s.4 5-Fu is a synthetic fluorinated pyrimidine analog that generates replication stress by replacing thymidine with fluorinated nucleotides, hereby leading to DNA damage and cell death. Oxaliplatin (OXA) is a platinum-based drug that also serves as one of the first-line chemotherapy drugs approved for CRC treatment.5 However, a majority of patients develop chemoresistance or multidrug resistance eventually. Therefore, it is critical to identify new targets or new regimens to overcome resistance in CRC.
Both 5-Fu and OXA are DNA-damaging agents that cause replication stress, genome instability, and cell death. Therefore, DNA repair mechanisms would be expected to participate in chemoresistance. DNA double-strand breaks (DSBs) are the most toxic DNA lesions that could be generated by chemotherapeutic drugs.6,7 A single unrepaired DSB is sufficient to induce cell death.8 There are two major DSB repair pathways in human cells: homologous recombination (HR) and nonhomologous end joining (NHEJ).9,10 NHEJ is the major DSB repair pathway that can be used throughout the whole cell cycle stages.11 While HR predominantly incorporates sister chromatid as a template to repair DSB, its function is limited in late S and G2 phase where sister chromatid is available.12 NHEJ is initiated by binding ring-shaped Ku 70/80 (Ku) heterodimer to DSB. Ku is an abundant NHEJ factor that has extremely high affinity for DSB that results in Ku-DNA complex rapidly. This complex recruits DNA-PK catalytic subunit (DNA-PKcs) to active DNA-PK activity.13 Other NHEJ essential key factors, including X-ray cross-complementing 4 (XRCC4), XRCC4-like factor (XLF), and ligase IV, are also recruited by Ku to bind DSB.14 NHEJ also requires DNA end-processing factors to remove adducts or abnormal damages from the ends of DSB, such as Exo1, Artemis, and TDP1.15–18 HR, on the other hand, uses undamaged DNA sequence from sister chromatid as a template to repair DSB. Therefore, HR is considered as an error-free pathway as compared to NHEJ, which generally causes insertions and deletions.12 The initiation step of HR is the generation of a long 3ʹ single-strand overhang by CtIP/MRE11-RAD50-NBS1 (MRN) complex.19 Subsequently, replication protein A (RPA) binds to single-strand DNA (ssDNA) to prevent secondary structure. RAD51 is then loaded to the ssDNA via displacing RPA and promotes strand invasion to seek for template in sister chromatid that results in Holliday junction.20 HR will be finally repaired by resolving Holliday junction followed by ligation of DNA ends.21
Our recent study found that XLF, which is an essential NHEJ factor, contributes to chemoresistance to 5-Fu and OXA in CRC cells. Deficiency of XLF significantly sensitizes CRC cells to these DNA-damaging agents.22 Therefore, in this study, we incorporated virtual screen and experimental validation to identify natural compounds for XLF inhibition. We found a bibenzyl compound gigantol, which was originally extracted from the stem of Dendrobium aurantiacum,23 binds to XLF, and inhibits its expression. Gigantol has been described as an antitumor compound against lung cancer24,25 and liver cancer.26 However, the potency of gigantol to inhibit XLF or CRC cell growth is not ideal. Here, we optimized the structure of gigantol and identified its derivative G3 as a very potent XLF inhibitor. Significantly, G3 sensitizes chemoresistant CRC cells to both 5-Fu and OXA, suggesting G3 is a promising compound to improve chemotherapy in chemoresistant CRC patients.
Methods And Materials
Virtual Screening
XLF protein (1–233 homodimer at 2.3 A resolution) crystal structure was downloaded from PubMed.27 The small molecule library (6800 compounds) was acquired from Chemical Entities of Biological Interest (ChEBI). Protein–ligand binding was performed by using LigPrep (Schrödinger). The top 10% (652 compounds) compounds, which showed low free energy of binding, were picked for further study. The natural compounds (8 compounds) from the hits were manually picked. The picked natural compounds were experimentally validated using Western blotting assay.
Docking
Before docking, the LigPrep model was used to refine ligands. Ionization states within pH=7.0±1.5 were selected for docking. XLF crystal structure, which represents homodimeric XLF ranging from amino acids 1 to 230, was downloaded from protein databank (PDB ID: 2R9A, http://www.rcsb.org/). XLF structure was prepared using Protein Preparation Wizard to correct metal ionization states, enumerate bond orders, and remove co-crystallized water molecules. For docking, the induced fit methodology was used, and a 10 Å side length of the cubic grid was allowed during docking. Residues within 4.5. Å of ligand poses were refined.
Cell Lines And Cell Cultures
HCT116 (ATCC, CCL-247) and LS174T cells (ATCC, CL-188) were cultured at 37°C in 5% CO2 atmosphere in McCoy’s 5a medium (ATCC 30–2007) and Eagle’s minimum essential medium (ATCC 30–2003), respectively, with 10% FBS for less than 6 months. Resistant cells were generated according to previous studies.28 Briefly, HCT116 and LS174T 5-Fu-resistant cells were generated by incubating with 5 µM and 10 µM 5-Fu, respectively, for 3 months. HCT116 and LS174T OXA-resistant cells were generated by incubating with 7 µM and 15 µM OXA, respectively, for 3 months.
Comet Assay
HCT116 cells were treated with 60 µM of OXA for 2 hrs to introduce DNA-damaged agents. 5 µM G3 was added after OXA for another 2 hrs. Cells were recovered in drug-free medium for 24 hrs and subjected to alkaline comet assay. The comet assay was performed according to the manufacturer’s instruction (Trevigen, #4250-050-K). Briefly, the cells were resuspended in molten LMAgarose at 3x105 cells/mL. 50 µl of the agarose was pipetted and spread onto the Comet Slide®. The slides were dried at 4°C in a dark room for 10 mins and incubated in cold lysis solution for 30 mins. The slides were then immersed in freshly prepared alkaline unwinding solution (pH>13) for 20 mins and placed in an electrophoresis slide tray. The DNA was separated at 21 V for 30 mins. The slides were washed twice in dH2O for 5 mins followed by 70% ethanol incubation for 5 mins. The slides were dried at 37°C for 10–15 mins. DNA was stained using SYBR Gold and the signal was viewed by epifluorescence microscopy (excitation/emission is 496 nm/522 nm)
HR And NHEJ Reporter Assay
10 μg of NHEJ reporter plasmid (addgene 44026) was linearized with 50 U of NheI in 50 μL reaction for 6 hrs at 37°C. Linearized DNA was purified using Qiagen gel extraction kit. 1 μg linearized DNA was transfected into HCT116 and LS174T cells using Lipofectamine 3000 (ThermoFisher) according to the manufacturer’s instruction. Cells with chromosomally integrated reporter constructs were selected by 1 mg/mL geneticin 24 hrs after transfection for 2 weeks. Plasmid-integrated cells were seeded at 3x105 cells/mL in a 6-well plate and cultured for 24 hrs to allow adhere. 2 µg/well of I-SceI plasmid was transfected into the cell by lipofectamine 3000 to generate DSB and incubated for 48 hrs to allow repair. Cells were harvested by using trypsin and resuspended in PBS by pipetting. GFP-positive cells were counted by flow cytometry (Beckman Coulter). For experiments with G3 treatment, I-Scel was transfected 24 hrs after 5 μM of G3 treatment. HR reporter assay was performed similarly.
Cell Viability Assay
Cells were seeded at 3x103 cells/well and cultured for overnight in a 96-well plate. Cells were incubated with the drug for 72 hrs, and the cell viability was detected by using sulforhodamine B (SRB) assay. Cells were then fixed by 100 µL/well of 10% trichloroacetic acid (TCA) at 4°C for 1 hr. TCA was washed with 200 μL ddH2O for 4 times and air-dried for 20 mins in fume hood. Cells were stained by 100 µL/well SRB solution (0.02% SRB in 1% acetate acid) for 1 hr at room temperature. Plates were washed for 3 times with 200 µL/well 1% acetate acid and air-dried. 200 µL/well of 10 mM tris-HCl, pH 10.5, was added in each well to extract SRB for 1 hr. The absorbance was measured at 510 nm by a microplate reader (BioTek). In drug synergy assay, cells were incubated with G3 for 24 hrs followed by 5-Fu or OXA treatment for 72 hrs.
Chromatin Fractionation Assay
5×105 cells/mL cells were seeded in a 6-well plate and incubated for 24 hrs to allow attachment. G3 was added with different concentrations and incubated for 48 hrs. Cells were then washed in PBS before resuspension in CSK/Triton buffer (10 mM PIPES pH6.8, 100 mM NaCl, 1 mM EGTA, 300 mM sucrose, 3 mM MgCl2, 1 mM DTT, 0.5% Triton-X100, protease inhibitor cocktail) to extract the cytoplasmic and soluble nuclear fraction by centrifugation (5 mins, 1300 x g at 4°C). The remaining pellet was washed in CSK/Triton buffer and resuspended in CSK buffer (without Triton-X100) with the addition of 25 U of RNase-free DNase I to incubate at 37°C for 30 mins. 250 mM of (NH4)2SO4 was added to disrupt the nuclear membrane and extract the chromatin-bound fraction. After centrifugation, the pellet was washed in CSK buffer and resuspended in CSK/NaCl buffer (CSK buffer with 2M NaCl) to extract the histone-containing fraction. The remaining insoluble fraction, which contains nuclear matrix-bound material along with any precipitated proteins, was washed twice in CSK/NaCl buffer and resuspended in Western blot 2x SDS loading buffer.
Western Blot Assay
Protein samples were denatured using a 2x SDS-PAGE sample buffer and boiled for 5 mins. The samples were then loaded and separated on an 7% polyacrylamide gel (29:1) (BIO-RAD, 1610156) at 120 V for 1.5 hrs on an electrophoresis apparatus (BioRad). Separated samples were transferred to nitrocellulose membrane at 100v at 4°C for 1 hr. Membrane was blocked by blocking buffer (3% BSA, 0.1% Tween 20 and 1x PBS) and probed by a relevant antibody. The membrane was washed 3 times by washing buffer (0.1% Tween 20 and 1x PBS) followed by binding of HRP-conjugated rabbit secondary antibody. The protein signal was developed by SuperSignalTM west pico PLUS Chemiluminescent Substrate (ThermoFisher Scientific #34580) and detected by ChemiDocTM (BioRad).
Statistical Analysis
Statistical analysis was done using GraphPad Prism version 5.01 (Graphpad Software Inc., USA). Results are expressed as the mean ± SD from 3 independent experiments. Student’s t-test was incorporated to analyze differences among the groups. P < 0.05 was considered statistically significant.
Results
Identification Of XLF Inhibitor G3
Our previous work found that NHEJ key factor XLF is responsible for 5-Fu and OXA resistance in CRC.22 Since XLF enzymatic activity has not yet been found, we used visual screening to identify XLF-binding small molecules that may potentially inhibit XLF. We are particularly interested in natural compounds; therefore, we selected ChEBI drug-like small-molecule database, which contains 3500 natural compounds. By docking 6800 small molecules to the XLF crystal structure, we acquired XLF-binding affinities of each compound. We picked 8 natural compounds (hits) from the top 10% compounds according to their XLF-binding activity, including oleanolic acid, betulinic acid, pomiferin, edgeworin, vincristine, resveratrol, stigmasterol, and gigantol (Figure 1A). To further determine whether these candidates interact with XLF in cells, we evaluated XLF stability in the presence of each candidate using a thermoshift assay. We picked the top 3 hits (gigantol, vincristine, and resveratrol) that interacted with XLF. To evaluate the effect of these hits on XLF in CRC, we treated HCT116 cells with 0–500 µM hits and subsequently analyzed XLF protein expression by Western blot assay. Among these hits, gigantol showed the best IC50 to inhibit XLF expression (38.20 µM) (Table 1) (Supplementary Figure 1A). Gigantol is a natural compound that provides antitumor potential in liver and lung cancer.23,26,29
 |
Table 1 Inhibitory Effect Of Gigantol And Its Derivatives On XLF Expression In HCT116 Cells |
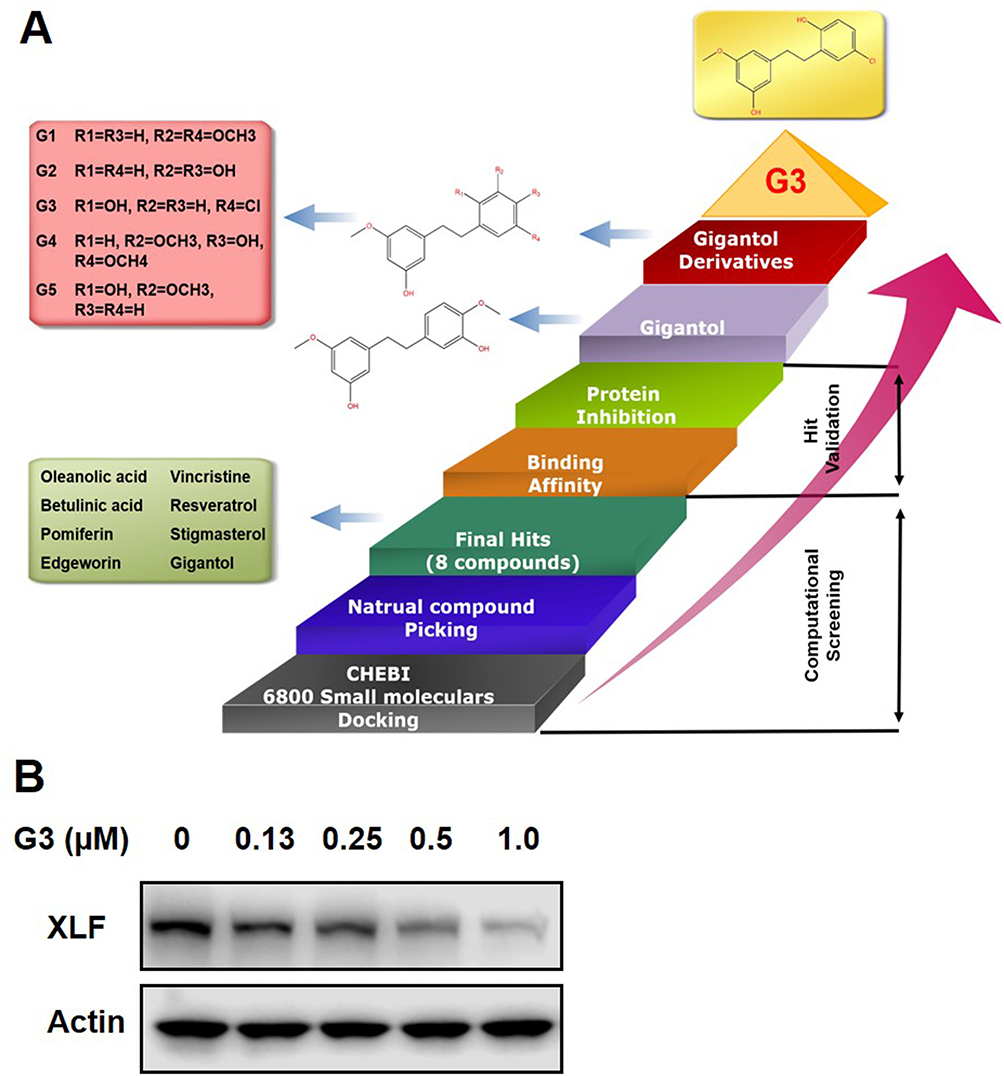 |
Figure 1 Identification of XLF inhibitor G3. Notes: (A) Virtual screening and experimental validation of XLF inhibitors. (B) G3 inhibits XLF protein expression in HCT116 cells. XLF is visualized using Western blot assay. |
To develop a more potent XLF inhibitor, we optimized gigantol and evaluated the XLF protein level in HCT116 cells treated with five gigantol derivatives (Supplementary Figure 1B). We identified the most potent derivative G3 (Supplementary Figure 1C; Table 1) that inhibited XLF protein expression, with IC50 of 0.47 µM (Figure 1B). We will use G3 to further evaluate its anticancer activity in CRC cells.
G3 Sensitizes Chemoresistant Cells To 5-Fu And OXA
We developed 5-Fu and OXA-resistant HCT116 and LS174T cell lines and validated their IC50 against 5-Fu and OXA in our previous study (Supplementary Figure 2).22 To determine whether G3 inhibits chemoresistant CRC cells, we treated 5-Fu-resistant HCT116 or LS174T cells (HCT116 Fu-R or LS174T Fu-R) with G3 for 24 hrs, 48 hrs, or 72 hrs and measured cell growth inhibition using SRB assay. We found that G3 inhibits 5-Fu CRC cells in a dose- and time-dependent manner (Figure 2A and B). Similarly, G3 had an increasing antitumor effect in HCT116 OXA-R or LS174T OXA-R in a dose- and time-dependent manner (Figure 2C and D). These data suggest that G3 has the potential, as an alternative regimen, to benefit CRC patient who has already developed chemoresistance to 5-Fu or OXA.
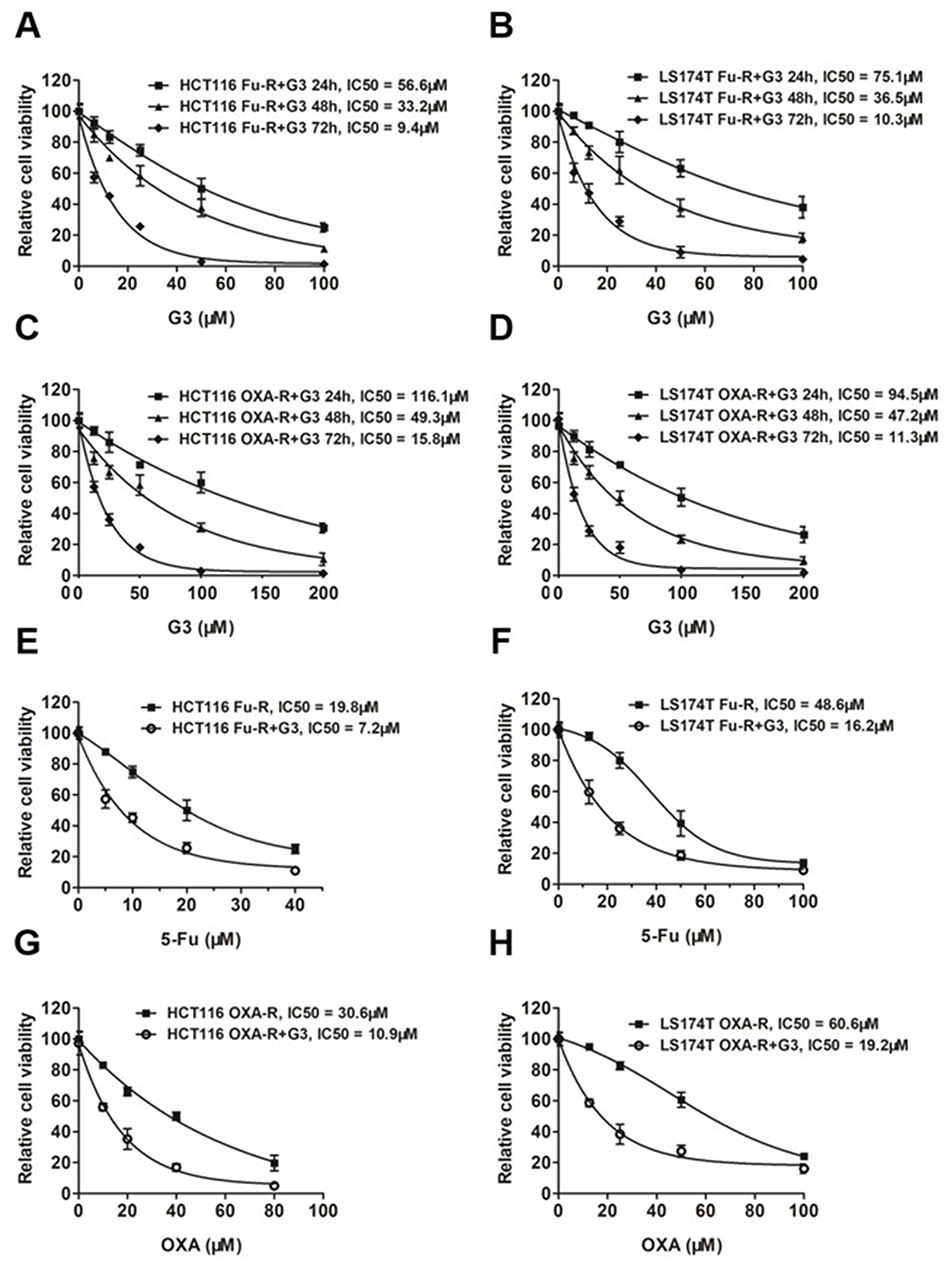 |
Figure 2 G3 sensitizes chemoresistant cells to 5-Fu and OXA. Notes: (A) Cell survival of HCT116-Fu-R and (B) LS174T Fu-R cell lines treated with G3 for 24 hrs, 48 hrs, and 72 hrs. IC50s were indicated. (C) Cell survival of HCT116-OXA-R and (D) LS174T OXA-R cell lines treated with G3 for 24 hrs, 48 hrs, and 72 hrs. IC50s were indicated. (E) Cell survival of HCT116-Fu-R and (F) LS174T Fu-R cell lines treated with G3 followed by 5-Fu. For G3 treatment: cells were treated with 5 μM of G3 for 24 hrs before 5-Fu treatment for another 72 hrs. IC50s were indicated. (G) Cell survival of HCT116-OXA-R (H) LS174T OXA-R cell lines treated with G3 followed by OXA. For G3 treatment, cells were treated with 5 μM of G3 for 24 hrs before OXA treatment for another 72 hrs. IC50s were indicated. |
To validate the synergy of G3/5-Fu or G3/OXA, we treated the HCT116 Fu-R cells with 5 μM of G3 for 24 hrs followed by exposure to increasing concentrations of 5-Fu or OXA for 72 hrs and evaluated the viability of the cells. As we expected, G3 sensitized HCT116 Fu-R and LS174T Fu-R to 5-Fu by 2.75-folds and 3-folds, respectively (Figure 2E and F). Consistently, G3 also decreased IC50 of OXA by 2.81-folds and 3.16-folds in HCT116 OXA-R and LS174T OXA-R cells, respectively (Figure 2G and H), suggesting G3 does not specifically affect resistant mechanisms of either 5-Fu or OXA. Since G3 degraded XLF in CRC cells, we hypothesize that G3 inhibits the DSB repair mechanism, which contributes to chemoresistance in a variety of cancer types.
G3 Inhibits NHEJ Efficiency In Chemoresistant CRC Cells
To examine the role of G3 on NHEJ in chemoresistant cells, we first stably transfected classic NHEJ reporter assay30 in HCT116 Fu-R, LS174T Fu-R, HCT116 OXA-R, and LS174T OXA-R cell lines. This assay uses endonuclease I-SceI to generate complementary DSB, which impedes green fluorescent protein (GFP) gene expression. Successful repair by NHEJ results in the expression of GFP gene, which is detected by fluorescence-activated cell sorting (FACS) to provide quantification of NHEJ efficiency. Consistent with our previous results,22 NHEJ was significantly upregulated in 5-Fu- and OXA-resistant CRC cells (Figure 3A and B). Upon treatment of 5 μM G3 before NHEJ assay, HCT116 5-Fu-R and HCT116 OXA-R exhibited NHEJ efficiency almost equivalent to that in HCT116 WT (Figure 3A), suggesting G3 sensitizes 5-Fu- and OXA-resistant HCT116 cells via NHEJ inhibition. Consistently, G3 decreased NHEJ efficiency in LS174T 5-Fu-R and LS174T OXA-R cells (Figure 3B).
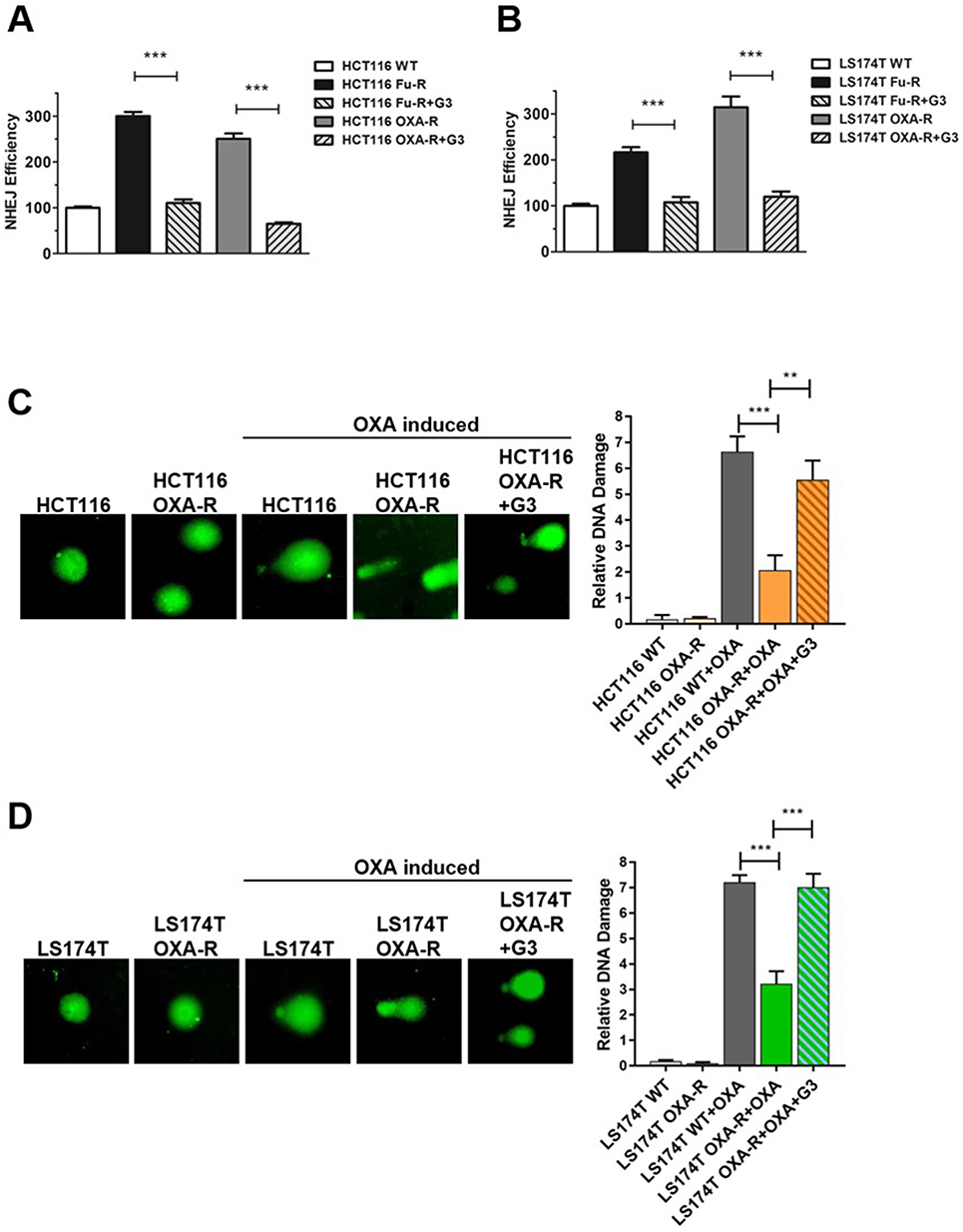 |
Figure 3 G3 inhibits NHEJ efficiency in chemoresistant CRC cells. Notes: (A) Quantification of GFP events generated by NHEJ in HCT116 WT, HCT116 Fu-R, HCT116-Fu-R+G3, HCT116 OXA-R, and HCT116-OXA-R+G3 cell lines. The GFP events were normalized to that in HCT116 WT cell line. Each result represents 3 independent experiments, ***P<0.001. (B) Quantification of GFP events generated by NHEJ in LS174T WT, LS174T Fu-R and LS174T-Fu-R+G3, LS174T OXA-R and LS174T-OXA-R+G3 cell lines. The GFP events were normalized to that in LS174T WT cell line. Each result represents 3 independent experiments, ***P<0.001. (C) XLF inhibitor G3 induces DNA damage in OXA-resistant HCT116 and (D) LS174T cell lines. Left, representative comet assay images. Right, relative DNA damage scaled by tail length 1–5 from 3 independent experiments, ***P<0.001, **P<0.01. |
To evaluate the effect of G3 on DNA damage repair with single-cell resolution, we treated CRC cells with OXA to introduce DNA damage and then examined DNA damage after 24 hrs recovery by comet assay. As shown in Figure 3C, compared to HCT116 WT cells treated with control (diluted DMSO), HCT116 WT cells treated with OXA generated a significant amount of DNA damage. Importantly, HCT116 OXA-R cells exhibited less DNA damage as compared to HCT116 WT, indicating DNA repair efficiency indeed increases in OXA-resistant cells (Figure 3C). HCT116 OXA-R cells treated with OXA and G3 restored DNA damage to levels similar to those in HCT116 WT cells treated with OXA, indicating G3 inhibits DNA repair in OXA-resistant HCT116 cells (Figure 3C). Similar results were found using LS174T cell lines (Figure 3D). Together, these results suggest that G3 inhibits DSB repair via NHEJ suppression, which contributes to sensitizing chemoresistant CRC cells.
G3 Dissembles NHEJ Factors From DNA
To elucidate the consequence of G3-XLF interaction in NHEJ, we evaluated the kinase activity of DNA-PK and ligase activity of ligase IV in vitro. However, G3 did not affect their enzymatic activity, indicating G3 may affect the scaffolding of the NHEJ apparatus. NHEJ is a compact process requiring the cooperation of all the key factors.10 We hypothesized that G3 may interrupt protein–protein interaction between XLF and other NHEJ factors. Therefore, we used Schrödinger software to dock G3 with XLF homodimer and determined the interaction sites. As shown in Figure 4A and Table 2, G3 interacts with the N-terminal region of XLF. XLF consists of a globular head domain, an elongated coil-coil domain, and a disordered C-terminal region.31 The globular head domain of XLF (N-terminal) is responsible for the interaction between XLF and XRCC4. Therefore, G3 may inhibit NHEJ via competing with XRCC4 to bind XLF.
 |
Table 2 Interaction Types And Binding Residues Between XLF And G3 |
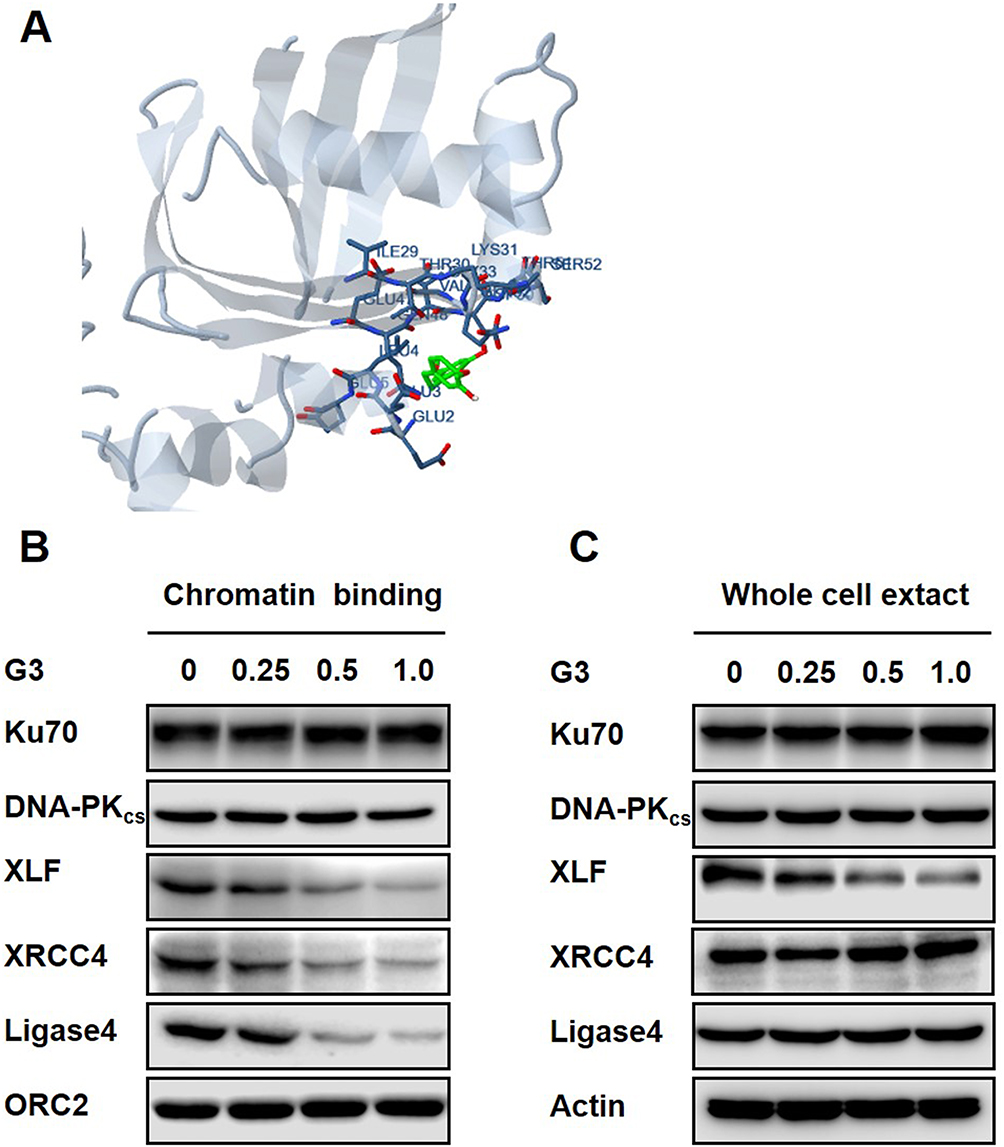 |
Figure 4 G3 dissembles NHEJ factors from DNA. Notes: (A) Docked complexes for XLF (PDB ID: 2R9A) complex with G3. G3 is colored in green, and the interface is colored in sky gray. (B) Western blot against NHEJ key factors in chromatin fraction from cells treated with G3. G3 concentrations: 0 µM, 0.25 µM, 0.5 µM, and 1 µM. (C) Western blot against NHEJ key factors in the whole extract from cells treated with G3. G3 concentrations: 0 µM, 0.25 µM, 0.5 µM, and 1 µM. |
XLF and XRCC4 interact and form positively charged filament to tether DNA.32 Therefore, G3 may disrupt the DNA binding of NHEJ key factors. We then used chromatin fractionation assay to evaluate the DNA-binding ability of NHEJ key factors after G3 treatment. We found that, as compared to that in HCT116 5-Fu-R cell without G3 treatment, G3 significantly decreased DNA binding of XLF, XRCC4, and Ligase IV in a dose-dependent manner (Figure 4B). However, decreased DNA binding of XLF resulted from a lack of XLF expression in the presence of G3 (Figure 4C). To exclude the possibility that G3 may degrade the protein expression of other NHEJ factors, we also evaluated the protein expression of Ku70, DNA-PKcs, XLF, XRCC4, and Ligase IV in the whole-cell extractions. We found that G3 did not change the protein levels of all the NHEJ key factors except XLF (Figure 4C), suggesting G3 inhibits NHEJ apparatus assembly after early-stage binding of Ku and DNA-PKcs.
G3 Inhibits HR Efficiency In Chemoresistant HCT116 Cells
XLF and proteins encoded by XRCC genes share similar structures in human cells.33 G3, as an XLF-binding molecule, could also interact with XLF like proteins, such as XRCC2, which participates in HR by forming a complex with Rad51.34 Therefore, we hypothesize that the inhibitory effect of G3 is not limited to NHEJ. We then used the HR reporter assay to monitor HR efficiency in HCT116 5-Fu-R cells. As shown in Figure 5A, HR efficiency decreased with 5 µM of G3 treatment in a time-dependent manner, suggesting G3 also inhibits HR in 5-Fu-resistant HCT116 cells. Similar results were observed in LS174T Fu-R, HCT116 OXA-R, and LS174T OXA-R cells (Figure 5B–D), indicating G3 inhibits both DSB repair pathways in 5-Fu- and OXA-resistant CRC cells.
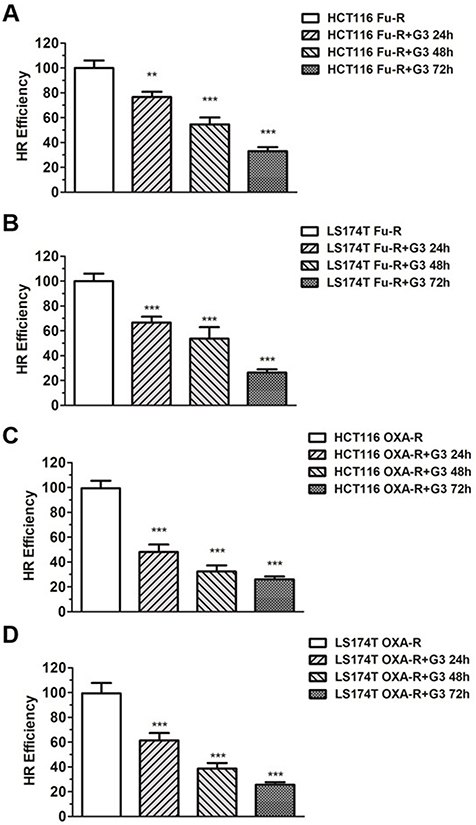 |
Figure 5 G3 inhibits HR efficiency in chemoresistant HCT116 cells. Notes: (A) Quantification of GFP events generated by HR in HCT116 Fu-R, HCT116 Fu-R+G3 and (B) LS174T Fu-R, LS174T Fu-R+G3 for 24 hr, 48 hr, and 72 hr cell lines. The GFP events were normalized to that in cells without G3 treatment. Each result represents 3 independent experiments, **P<0.01, ***P<0.001. (C) Quantification of GFP events generated by HR in HCT116 OXA-R, HCT116-OXA-R+G3 and (D) LS174T OXA-R, LS174T OXA-R+G3 for 24 hr, 48 hr and 72 hr cell lines. The GFP events were normalized to that in cells without G3 treatment. Each result represents 3 independent experiments, ***P<0.001. |
Discussion
Multidrug resistance has been one of the obstacles of CRC treatment for decades and is the major cause of death in metastatic or recurrent CRC patients.35 Therefore, it is urgent to develop a new regimen and combination chemotherapy to inhibit cell growth.
It has been reported that gigantol, which is a natural compound extracted from Dendrobium draconis, has a variety of bioactivities as well as antitumor effects in liver and lung cancers.25,26,29,36,37 To our knowledge, it has not been used in anti-CRC therapy by itself or as a sensitizer for existing anticancer drugs. Moreover, the IC50 of gigantol is not optimal to be clinically relevant. Therefore, it is necessary to develop gigantol derivatives that can inhibit cell growth and overcome drug resistance with better potency. Our study, for the first time, demonstrated that G3 has the ability to inhibit 5-Fu- and OXA-resistant CRC cell growth. Furthermore, G3 can sensitize chemoresistant cells to 5-Fu or OXA by around 3-folds.
XLF and XRCC4 belong to a homologous superfamily with a very similar N-terminal structure.33 However, G3, which interacts with the N-terminal region of XLF and inhibits XLF expression, does not affect the protein level of XRCC4 (Figure 4C). Therefore, G3 mediates XLF degradation in a mechanism distinct from other structure-related proteins. We also found that G3 dissembles NHEJ factors from DNA in a dose-dependent manner (Figure 4B). Hence, we speculated that G3 instantly interacts with XLF and disrupts its interaction with XRCC4 to compromise the assembly of NHEJ machinery. In addition, G3 can further degrade XLF to deplete NHEJ subsequently.
Interestingly, 5-Fu and OXA does not generate DSB at first but creates replication stress via adding DNA adducts.38,39 Therefore, it is not surprising that chemoresistant mechanisms of 5-Fu or OXA often related to thymidine phosphorylase or NER, respectively.40 Our research linked DSB repair mechanisms to chemoresistance generated by 5-Fu and OXA, indicating that preventing DSB repair is also necessary in anticancer therapy to keep persistence of DSB that may result from DNA single-strand breaks.41–44 Since DSB is the most toxic DNA lesion and the consequence of the collapse of DNA single-strand break, compromising repair of DSB could serve as the final and essential step to maintain DNA damage in cancer cells. Therefore, our study provides a novel and potent DSB repair inhibitor G3 that may overcome chemoresistance induced by a variety of DNA damaging agents, such as platinum-based drugs, etoposide, camptothecin, and hydroxyurea.
Conclusion
This study has demonstrated that natural compound derivative G3 is a potent XLF inhibitor. G3 sensitizes chemoresistant HCT116 and LS174T cells to 5-Fu and OXA. Mechanistically, G3 decreases NHEJ and HR efficiency, which are upregulated in chemoresistant cells. We also elucidated that G3 inhibits NHEJ by disassembly of key NHEJ factors on DNA. Our study supports further optimization of G3 as a leading anticancer compound in chemotherapy.
Abbreviations
CRC, colorectal cancer; 5-Fu, 5-florouracil; OXA, oxaliplatin; NHEJ, nonhomologous end joining; HR, homologous recombination; DSB, DNA double-strand break; Ku, Ku70/80 heterodimer; WT, wild type; DNA-PKcs, DNA-PK catalytic subunit; MRN, CtIP/MRE11-RAD50-NBS1; RPA, replication protein A; ssDNA, single-strand DNA; XRCC4, X-ray cross-complementing 4; XLF, XRCC4-like factor.
Disclosure
This work was supported by funding from Jilin Province Science and Technology Institute (20160101034JC and 20170204050SF to Zhuo Liu and Dongxin Wang, respectively). The authors report no conflicts of interest in this work.
References
1. Siegel RL, Miller KD, Fedewa SA, et al. Colorectal cancer statistics, 2017. CA Cancer J Clin. 2017;67:177–193. doi:10.3322/caac.21395
2. Brenner H, Kloor M, Pox CP. Colorectal cancer. Lancet. 2014;383:1490–1502. doi:10.1016/S0140-6736(13)61649-9
3. Sugihara K. Overview of treatment strategy of Stage IV colorectal cancer. Nihon Shokakibyo Gakkai Zasshi. 2017;114:1195–1200. doi:10.11405/nisshoshi.114.1195
4. Salonga D, Danenberg KD, Johnson M, et al. Colorectal tumors responding to 5-fluorouracil have low gene expression levels of dihydropyrimidine dehydrogenase, thymidylate synthase, and thymidine phosphorylase. Clin Cancer Res. 2000;6:1322–1327.
5. Peinert S, Grothe W, Stein A, et al. Safety and efficacy of weekly 5-fluorouracil/folinic acid/oxaliplatin/irinotecan in the first-line treatment of gastrointestinal cancer. Ther Adv Med Oncol. 2010;2:161–174. doi:10.1177/1758834010365061
6. Tomicic MT, Kaina B. Topoisomerase degradation, DSB repair, p53 and IAPs in cancer cell resistance to camptothecin-like topoisomerase I inhibitors. Biochim Biophys Acta. 2013;1835:11–27. doi:10.1016/j.bbcan.2012.09.002
7. Britten RA, Kuny S, Perdue S. Modification of non-conservative double-strand break (DSB) rejoining activity after the induction of cisplatin resistance in human tumour cells. Br J Cancer. 1999;79:843–849. doi:10.1038/sj.bjc.6690135
8. Bennett CB, Lewis AL, Baldwin KK, Resnick MA. Lethality induced by a single site-specific double-strand break in a dispensable yeast plasmid. Proc Natl Acad Sci U S A. 1993;90:5613–5617. doi:10.1073/pnas.90.12.5613
9. Kim JS, Krasieva TB, Kurumizaka H, Chen DJ, Taylor AMR, Yokomori K. Independent and sequential recruitment of NHEJ and HR factors to DNA damage sites in mammalian cells. J Cell Biol. 2005;170:341–347. doi:10.1083/jcb.200411083
10. Kakarougkas A, Jeggo PA. DNA DSB repair pathway choice: an orchestrated handover mechanism. Br J Radiol. 2014;87:20130685. doi:10.1259/bjr.20130685
11. Arnoult N, Correia A, Ma J, et al. Regulation of DNA repair pathway choice in S and G2 phases by the NHEJ inhibitor CYREN. Nature. 2017;549:548–552. doi:10.1038/nature24023
12. San Filippo J, Sung P, Klein H. Mechanism of eukaryotic homologous recombination. Annu Rev Biochem. 2008;77:229–257. doi:10.1146/annurev.biochem.77.061306.125255
13. Gottlieb TM, Jackson SP. The DNA-dependent protein kinase: requirement for DNA ends and association with Ku antigen. Cell. 1993;72:131–142. doi:10.1016/0092-8674(93)90057-w
14. Britton S, Coates J, Jackson SP. A new method for high-resolution imaging of Ku foci to decipher mechanisms of DNA double-strand break repair. J Cell Biol. 2013;202:579–595. doi:10.1083/jcb.201303073
15. Riballo E, Kühne M, Rief N, et al. A pathway of double-strand break rejoining dependent upon ATM, Artemis, and proteins locating to gamma-H2AX foci. Mol Cell. 2004;16:715–724. doi:10.1016/j.molcel.2004.10.029
16. Bahmed K, Seth A, Nitiss KC, Nitiss JL. End-processing during non-homologous end-joining: a role for exonuclease 1. Nucleic Acids Res. 2011;39:970–978. doi:10.1093/nar/gkq886
17. Li J, Summerlin M, Nitiss KC, Nitiss JL, Hanakahi LA. TDP1 is required for efficient non-homologous end joining in human cells. DNA Repair (Amst). 2017;60:40–49. doi:10.1016/j.dnarep.2017.10.003
18. Heo J, Li J, Summerlin M, et al. TDP1 promotes assembly of non-homologous end joining protein complexes on DNA. DNA Repair (Amst). 2015;30:28–37. doi:10.1016/j.dnarep.2015.03.003
19. Symington LS, Gautier J. Double-strand break end resection and repair pathway choice. Annu Rev Genet. 2011;45:247–271. doi:10.1146/annurev-genet-110410-132435
20. Johnson RD, Jasin M. Sister chromatid gene conversion is a prominent double-strand break repair pathway in mammalian cells. Embo J. 2000;19:3398–3407. doi:10.1093/emboj/19.13.3398
21. Aylon Y, Liefshitz B, Kupiec M. The CDK regulates repair of double-strand breaks by homologous recombination during the cell cycle. Embo J. 2004;23:4868–4875. doi:10.1038/sj.emboj.7600469
22. Liu Z, Yu M, Fei B, Sun J, Wang D. Nonhomologous end joining key factor XLF enhances both 5-florouracil and oxaliplatin resistance in colorectal cancer. Onco Targets Ther. 2019;12:2095–2104. doi:10.2147/OTT.S192923
23. Charoenrungruang S, Chanvorachote P, Sritularak B, Pongrakhananon V. Gigantol, a bibenzyl from Dendrobium draconis, inhibits the migratory behavior of non-small cell lung cancer cells. J Nat Prod. 2014;77:1359–1366. doi:10.1021/np500015v
24. Bhummaphan N, Chanvorachote P. Gigantol suppresses cancer stem cell-like phenotypes in lung cancer cells. Evid Based Complement Alternat Med. 2015;2015:836564. doi:10.1155/2015/836564
25. Unahabhokha T, Chanvorachote P, Pongrakhananon V. The attenuation of epithelial to mesenchymal transition and induction of anoikis by gigantol in human lung cancer H460 cells. Tumour Biol. 2016;37:8633–8641. doi:10.1007/s13277-015-4717-z
26. Chen H, Huang Y, Huang J, Lin L, Wei G. Gigantol attenuates the proliferation of human liver cancer HepG2 cells through the PI3K/Akt/NF-kappaB signaling pathway. Oncol Rep. 2017;37:865–870. doi:10.3892/or.2016.5299
27. Li Y, Chirgadze DY, Bolanos-Garcia VM, et al. Crystal structure of human XLF/Cernunnos reveals unexpected differences from XRCC4 with implications for NHEJ. Embo J. 2008;27:290–300. doi:10.1038/sj.emboj.7601942
28. Meng Y, Chen C-W, Yung MMH, et al. DUOXA1-mediated ROS production promotes cisplatin resistance by activating ATR-Chk1 pathway in ovarian cancer. Cancer Lett. 2018;428:104–116. doi:10.1016/j.canlet.2018.04.029
29. Unahabhokha T, Chanvorachote P, Sritularak B, Kitsongsermthon J, Pongrakhananon V. Gigantol inhibits epithelial to mesenchymal process in human lung cancer cells. Evid Based Complement Alternat Med. 2016;2016:4561674. doi:10.1155/2016/5040528
30. Seluanov A, Mao Z, Gorbunova V. Analysis of DNA double-strand break (DSB) repair in mammalian cells. J Vis Exp. 2010. doi:10.3791/2002
31. Andres SN, Modesti M, Tsai CJ, Chu G, Junop MS. Crystal structure of human XLF: a twist in nonhomologous DNA end-joining. Mol Cell. 2007;28:1093–1101. doi:10.1016/j.molcel.2007.10.024
32. Hammel M, Rey M, Yu Y, et al. XRCC4 protein interactions with XRCC4-like factor (XLF) create an extended grooved scaffold for DNA ligation and double strand break repair. J Biol Chem. 2011;286:32638–32650. doi:10.1074/jbc.M111.272641
33. Ochi T, Blackford AN, Coates J, et al. DNA repair. PAXX, a paralog of XRCC4 and XLF, interacts with Ku to promote DNA double-strand break repair. Science. 2015;347:185–188. doi:10.1126/science.1261971
34. Thacker J, Zdzienicka MZ. The mammalian XRCC genes: their roles in DNA repair and genetic stability. DNA Repair (Amst). 2003;2:655–672.
35. Jensen NF, Stenvang J, Beck MK, et al. Establishment and characterization of models of chemotherapy resistance in colorectal cancer: towards a predictive signature of chemoresistance. Mol Oncol. 2015;9:1169–1185. doi:10.1016/j.molonc.2015.02.008
36. Wu J, Li X, Wan W, et al. Gigantol from Dendrobium chrysotoxum Lindl. binds and inhibits aldose reductase gene to exert its anti-cataract activity: an in vitro mechanistic study. J Ethnopharmacol. 2017;198:255–261. doi:10.1016/j.jep.2017.01.026
37. Wu J, Li X, Fang H, et al. Investigation of synergistic mechanism and identification of interaction site of aldose reductase with the combination of gigantol and syringic acid for prevention of diabetic cataract. BMC Complement Altern Med. 2016;16:286. doi:10.1186/s12906-016-1251-5
38. Nakamura H, Yu-Qin W, Miyauchi S, et al. [Studies on the mechanism of antitumor activity of 5-FU and its derivatives–relationship between the inhibition of tumor growth and the inhibition of thymidylate synthetase in vivo]. Gan To Kagaku Ryoho. 1984;11:1049–1055.
39. Raymond E, Faivre S, Woynarowski JM, Chaney SG. Oxaliplatin: mechanism of action and antineoplastic activity. Semin Oncol. 1998;25:4–12.
40. Van der Jeught K, Xu HC, Li YJ, Lu XB, Ji G. Drug resistance and new therapies in colorectal cancer. World J Gastroenterol. 2018;24:3834–3848. doi:10.3748/wjg.v24.i34.3834
41. Yajima H, Lee KJ, Zhang S, Kobayashi J, Chen BP. DNA double-strand break formation upon UV-induced replication stress activates ATM and DNA-PKcs kinases. J Mol Biol. 2009;385:800–810. doi:10.1016/j.jmb.2008.11.036
42. Ho EL, Parent M, Satoh MS. Induction of base damages representing a high risk site for double-strand DNA break formation in genomic DNA by exposure of cells to DNA damaging agents. J Biol Chem. 2007;282:21913–21923. doi:10.1074/jbc.M610651200
43. MacLean HE, Favaloro JM, Warne GL, Zajac JD. Double-strand DNA break repair with replication slippage on two strands: a novel mechanism of deletion formation. Hum Mutat. 2006;27:483–489. doi:10.1002/humu.20327
44. Nowosielska A, Marinus MG. Cisplatin induces DNA double-strand break formation in Escherichia coli dam mutants. DNA Repair (Amst). 2005;4:773–781. doi:10.1016/j.dnarep.2005.03.006
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.