")
Back to Journals » OncoTargets and Therapy » Volume 10
Identification of key pathways and genes with aberrant methylation in prostate cancer using bioinformatics analysis
Received 23 June 2017
Accepted for publication 20 August 2017
Published 10 October 2017 Volume 2017:10 Pages 4925—4933
DOI https://doi.org/10.2147/OTT.S144725
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Ingrid Espinoza
Anshika N Singh, Neeti Sharma
Symbiosis School of Biological Sciences, Symbiosis International University, Gram – Lavale, Taluka – Mulshi, Pune, India
Abstract: Prostate cancer (PCa), a multifocal clinically heterogeneous disease, is the most commonly diagnosed non-cutaneous neoplasm in men worldwide. The epigenome of PCa is a typical representation of catastrophic model of epigenetic alterations during tumorigenesis and its progression. Alterations in methylation patterns in tumor suppressors, cell cycle, oncogenes and metabolism-related genes are the most commonly observed epigenetic alterations in PCa. In this study, we have developed a computational strategy to identify methylated biomarker signature panels as potential targets of PCa by screening >160 genes reported to be epigenetically dysregulated, and shortlisted 26 differentially methylated genes (DMGs) that significantly contribute to oncogenesis. The gene ontology and functional enrichment analysis were performed, which showed that identified DMGs contribute to cellular and metabolic processes such as cell communication, cell cycle, response to drugs, apoptosis and p53 signaling. The top hub genes AR, CDH13, CDKN2A, DAPK1, GSTP1, CD44 and RASSF1 identified from protein–protein interaction network construction using Search Tool for the Retrieval of Interacting Genes contributed to hormonal response, inflammatory response, cell cycle, reactive oxygen species activity and receptor kinase activity, which are related to hallmarks of cancer as revealed by their functional enrichment analysis by Cytoscape. These genes were further scrutinized for CpG islands, transcription start sites and positions of methylated cytosines to study their methylation profiles. Our analysis revealed high negative correlation values between methylation frequencies and gene expressions of the hub genes, namely, AR, CDH13, CDKN2A, DAPK1, CD44, GSTP1 and RASSF1, which can be used as potential diagnostic biomarkers for PCa.
Keywords: prostate cancer, CpG islands, epigenetic modification, DNA methylation
Introduction
Prostate cancer (PCa) accounts for being the most commonly diagnosed non-cutaneous neoplasm and third leading cause of cancer mortalities in men worldwide.1 Reports suggest that men aged >40 years are ~30-fold more susceptible to developing PCa.2 Although a number of genetic, surface and intracellular markers have been identified to study cancer progression such as CD133, CD49f, Trop2, p63 and PSA, PSA is the only established biomarker for the detection of PCa.3 However, PSA lacks in specificity and sensitivity;3 thus, there exists an urgent need to identify biomarkers that are more specific.
Epigenetic alterations in the form of DNA methylation and histone modifications have emerged as a major focus area for cancer treatment considering their contribution in modulation of crucial gene expressions in various malignancies.4 Tumor suppressor genes and oncogenes play crucial roles in cancer by regulating functions such as cell cycle control, DNA repair, cell adhesion and apoptosis and are known to be differentially methylated in their promoter regions in several types of cancers.5,6 However till date, there is a limited knowledge of the specific functional mechanisms at the genome level that are being altered by epigenetic dysregulation during tumor progression in PCa.7 Given their key role in various stages of carcinogenesis by causing abnormal gene expression of tumor suppressor genes and oncogenes, histone modifications and hypermethylation status of genes are being explored as potential biomarkers of cancer progression and prognosis.8 Nowadays with the advancement of chromatin immunoprecipitation (ChIP) technology along with microarray profiling, scientists can screen thousands of differentially expressed genes (DEGs), analyze their mRNA profiles and methylation profiles computationally and subsequently scrutinize their involvement in various molecular and biological regulatory functions during various stages of tumor initiation and progression.9
The present study aims to explore the contribution of epigenetic modulation of genes involved in progression of PCa using computational approach. We screened >160 differentially methylated genes (DMGs), which are reported to be epigenetically dysregulated, and identified 26 genes that significantly contribute to oncogenesis in prostate gland. This was followed by screening the identified DMGs for gene ontology (GO) and their pathway enrichment. The protein–protein interaction (PPI) and functional enrichment analysis further shed light on the crucial roles played by the identified DMGs during tumor initiation and progression. Our analysis showed a high negative correlation value between methylation frequencies and gene expressions of identified hub genes, which have the potential to act as early prognostic and diagnostic biomarkers of PCa.
Methods
Identification of DMGs
A comprehensive literature survey was performed using PubMed and Google Scholar. The keywords used were “Prostate cancer” and “DNA methylation”. The literature survey was limited to full text articles available in English. A preliminary abstract review was performed to decide relevance of these research articles to our methylation study. Once the literature survey was updated, an extensive data extraction exercise was carried out to retrieve genes that are differentially methylated in PCa using online databases. The Cancer Genome Atlas (TCGA) (cancergenome.nih.gov/),10 Cancer Genetics Web (www.cancerindex.org/geneweb/)11 and MethyCancer (methycancer.psych.ac.cn/)12 were used for identifying the DMGs in PCa.
GO and functional enrichment analysis
Databases such as GeneCodis13 and Panther14 were used for functional enrichment analysis. GO of identified DMGs included categories such as cellular component (CC), biological process (BP) and molecular function (MF) terms. The parameters were set as p-value <0.01 for considering the results that were statistically significant. The enriched pathways were identified using Kyoto Encyclopedia of Genes and Genomes (KEGG)15 analysis. To further analyze the interrelationship of identified hub genes in regulating cellular pathways, gene set enrichment analysis module of Cytoscape software16 was used.
PPI network construction by search tool for the retrieval of interacting genes (STRING)
The interactive relationships between DMGs were identified using STRING database.17 A combined score of >0.7 (high) of only experimentally validated interactions was considered to be statistically significant.
Epigenetic analysis
The FASTA sequences of identified differentially methylated hub genes in PCa were retrieved from nucleotide database of National Center for Biotechnology Information (www.ncbi.nlm.nih.gov/). CpGProD (doua.prabi.fr/software/cpgprod)18 and EMBOSS CpGPlot (www.ebi.ac.uk/Tools/seqstats/emboss_cpgplot)19 were used to monitor the presence of CpG islands if any. Information related to GC%, G + C skew, A + T skew, etc. was also retrieved from these online programs. Methylation and Expression database of Normal and Tumor tissues (MENT; mgrc.kribb.re.kr:8080/MENT/)20 is an online integrative database to study DNA methylation and its correlation with gene expression in paired samples gathered from TCGA and Gene Expression Omnibus (GEO). MENT was used to identify the effect of methylation on the gene expression of the identified DMGs in PCa. Analysis of DNA methylation with exon–intron structures, genomic loci of epigenetic changes, positions of methylated cytosines, CpG islands and transcription start sites (TSS) using a graphical description was carried out using Prostate Epigenetic Database (PEpiD; wukong.tongji.edu.cn/pepid).21 The identified hub genes after epigenetic analysis were further classified using extensive literature survey based on their specificity and sensitivity for determining their performance as prognostic and diagnostic biomarkers for PCa. The literature survey covered published reports in which the specificity and sensitivity of DMGs were determined in cell lines and also bodily samples such as serum, ejaculates, post-massage urine and biopsy samples using methylation-specific polymerase chain reaction (MSP-PCR), ChIP and microarray analysis.22–45
Results
Identification of DMGs involved in PCa initiation and progression
A consolidated list of >160 genes from literature survey and online databases with evidences of differential methylation during PCa progression was prepared (Table S1). We shortlisted 26 candidate DMGs that were associated with metastatic and recurrent stages of the disease with at least two publications and were used for further analysis (Table 1).
 | Table 1 Differentially methylated genes (DMGs) shortlisted for analysis |
Functional annotation and pathway enrichment analysis
The altered biological function and MF of the identified DMGs were then analyzed through GO using GeneCodis and Panther. A p-value of <0.01 was considered as significant during the analysis. The enrichment GO terms were categorized into MF, BP and CC (Table 2). During the MF analysis, we found that majority of DMGs contributed to protein binding (16 genes), zinc ion binding (seven genes), metal ion binding (eight genes), DNA binding (six genes) and receptor activity (six genes). Other regulated MFs included enzyme binding, sequence-specific DNA binding and calcium ion binding. The CC analysis revealed that majority of differentially methylated candidate genes were associated with cytoplasm (20 genes), nucleus (16 genes), plasma membrane (10 genes), nucleoplasm (seven genes) and cytosol (six genes). In the BP ontology, the most significantly regulated processes included response to drugs (six genes), negative regulation of cell proliferation (six genes), signal transduction (seven genes) and negative regulation of apoptotic process (five genes). The other regulated processes included response to estradiol stimulus, cell cycle arrest and negative regulation of MAPK cascade. Thus, the most significantly enriched pathways identified using KEGG analysis include pathways in cancer (10 genes), cell cycle (five genes), p53 signaling (three genes) and PCa (three genes; Table 2). Functional gene set enrichment analysis using gene set enrichment analysis (GSEA) module of Cytoscape showed that the identified DMGs most commonly contributed in hormonal response, inflammatory response, cell cycle, reactive oxygen species (ROS) activity and receptor kinase activity, which are all related to hallmarks of oncogenesis (Table S2).
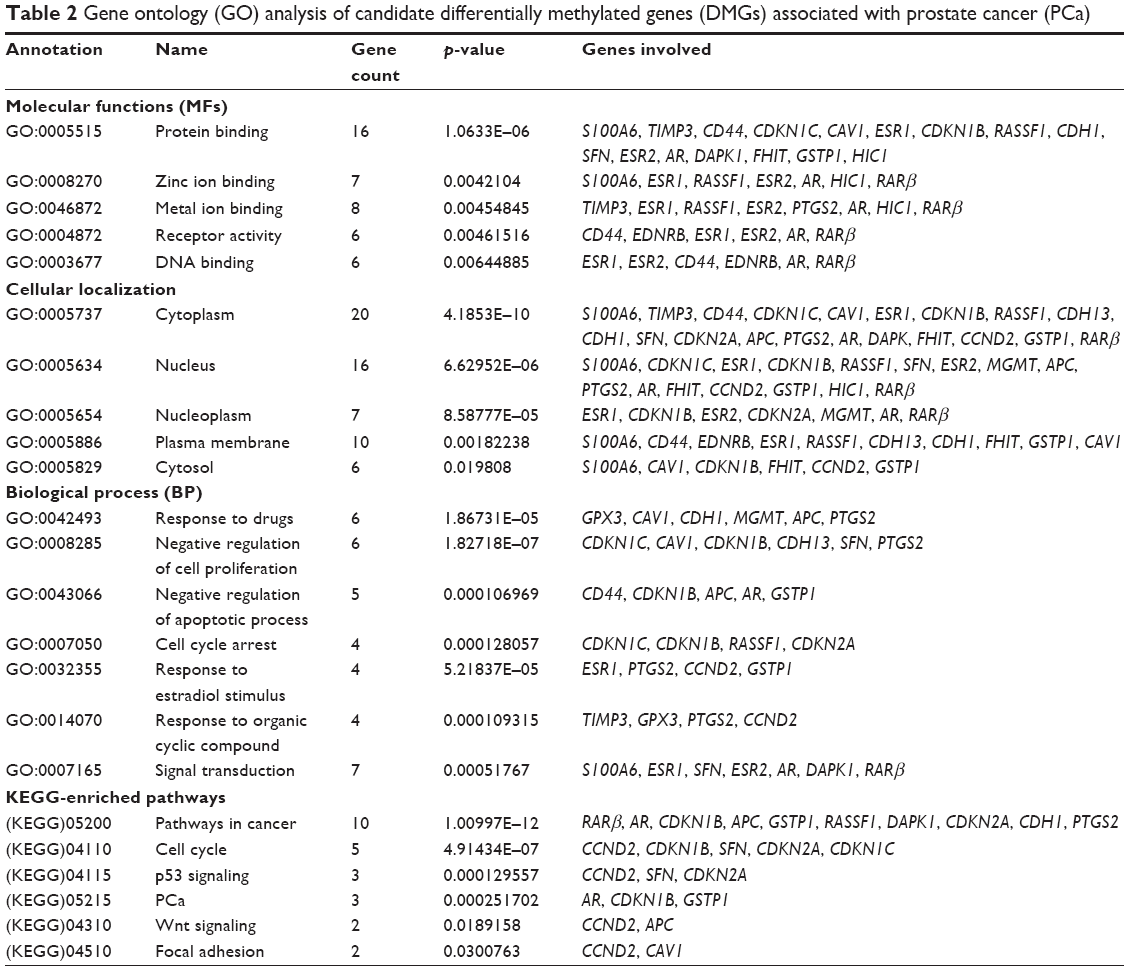 | Table 2 Gene ontology (GO) analysis of candidate differentially methylated genes (DMGs) associated with prostate cancer (PCa) |
PPI network
The PPI networks between DMGs were investigated using STRING server. In total, 26 nodes with 90 edges and 6.92 as average node degree were analyzed (Figure 1 and Table 3). In the PPI network, seven node proteins, namely, GSTP1, CDH13, CDKN2A, RASFF1, CD44, AR and DAPK1, showed maximum degree of association with the other proteins (>5), thus indicating that these genes (proteins) have higher hub degrees. Thus, these hub genes (proteins) may play a crucial role in PCa initiation and progression.
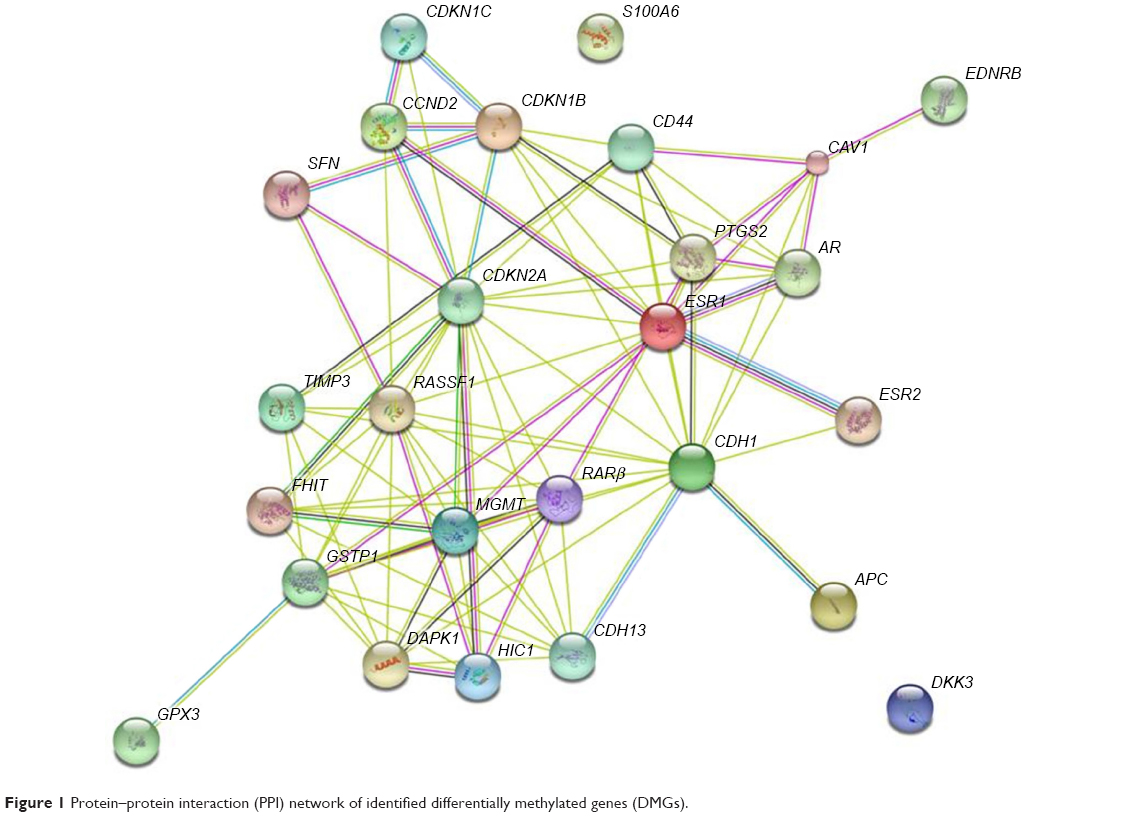 | Figure 1 Protein–protein interaction (PPI) network of identified differentially methylated genes (DMGs). |
 | Table 3 Associated genes of identified seven differentially methylated hub genes |
Epigenetic analysis
Evidences have shown that the cytosines in the CpG dinucleotides are often methylated to 5-methylcytosines leading to change in gene expression.46 Almost 70%–80% cytosines in CpG mammals are seen to be methylated in mammals and are often located near the promoter regions.47 CpGProd and EMBOSS CpGPlot were used to analyze positions of CpG islands, CpGo/e ratio, G + C skew and A + T skew (Table S3). The positions of methylated cytosines along with positions of TSS of the identified hub DMGs were also retrieved using PEpID (a prostate epigenetic database in mammals; Table S4). Once the CpG islands and the methylated cytosines positions were retrieved, MENT database was used to scrutinize the correlation between methylation frequencies and gene expressions. The MENT launched in 2012 is an integrated database of DNA methylation and gene expressions. Datasets containing information regarding DNA methylation and gene expression in paired samples were retrieved from TCGA and GEO. We opted for gene search tool to predict the correlation between methylation values and gene expressions of our hub genes. The Illumina methylation datasets consists of Human Methylation27 and Golden Gate Methylation Cancer Panel I, which is composed of 125 prostate tumor samples and 92 normal prostate samples for analysis. Based on the correlation values between mean methylation frequencies and gene expression, it was observed that expressions of hub genes GSTP1, CDH13, CDKN2A, RASFF1, AR, CD44 and DAPK1 were seen to be significantly affected by their hypermethylation during PCa progression due to an observed negative correlation of as high as −0.7 as shown in Table 4.
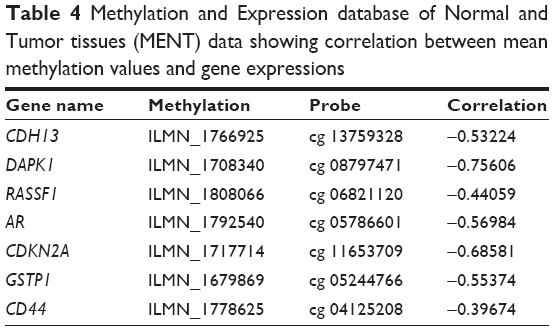 | Table 4 Methylation and Expression database of Normal and Tumor tissues (MENT) data showing correlation between mean methylation values and gene expressions |
These identified hub genes, ie, AR, CDH13, CDKN2A, DAPK1, GSTP1, CD44 and RASSF1, have been reported by various other research groups to show high specificity and sensitivity as independent biomarkers in prostatic intraepithelial neoplasia and different stages of PCa, including primary and metastatic tumors (Table 5). These genes have been reported to be differentially regulated due to hypermethylation in PCa cells, ie, PC3, DU145 and LNCaP, as well as bodily samples, including serum, ejaculates, post-massage urine and biopsy samples.22–45
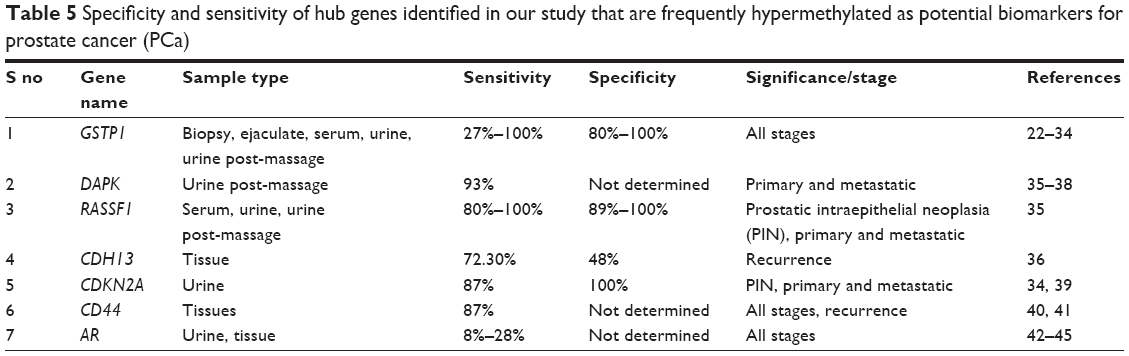 | Table 5 Specificity and sensitivity of hub genes identified in our study that are frequently hypermethylated as potential biomarkers for prostate cancer (PCa) |
Discussion
Alterations in methylation patterns in tumor suppressor genes, cell cycle genes, oncogenes and metabolism-related genes are the most commonly observed epigenetic alterations in PCa. With the advancement in high-throughput technologies, including microarray and ChIP array, it has become feasible to analyze not only the expression levels of several genes simultaneously but also their methylation frequencies that can eventually lead to discovery of genes that can act as potential diagnostic, prognostic and therapeutic biomarkers. In the present study, we evaluated a panel of 26 DMGs using computational approaches to study their differential methylation and gene annotation profiles that are regulated during different stages of PCa. Consequently, their interrelationships were analyzed using PPI server.
Using thorough literature survey and online databases such as TCGA, Cancer Genetics Web and MethyCancer, we identified 26 DMGs in PCa. Here, we hypothesized that DMGs may act as potential diagnostic and prognostic biomarkers for PCa. However, to better understand the interrelationship and contribution of DMGs in cancer progression, we further performed GO and KEGG pathway enrichment analysis. The GO analysis showed that the DMGs significantly contributed in MFs such as protein binding, DNA binding, zinc ion binding and metal ion binding, which may contribute to carcinogenesis. Our hypothesis was further strengthened by the BP analysis which showed that these 26 DMGs significantly contributed to signal transduction, negative regulation of cell proliferation and negative regulation of apoptosis. These identified BPs pointed toward the involvement of the identified DMGs in major hallmarks of cancer. CDKN2A, CDKN1B and CDKN1C among the identified genes are known to be critical regulators of cell cycle, acting as checkpoints at various stages of cell cycle. Aberrantly, methylated cyclin-dependent kinase genes lead to carcinogenesis and progression of cancer due to uncontrolled cell growth.48 CDKN2A has been seen to be frequently methylated in PCa progression.48–50 CCND2 is another important identified cell cycle regulator required for cell cycle G1/S transition.51 Other cell cycle genes such as CD44,52 HIC153 and PTGS254 identified in our analysis have been reported to be frequently methylated during PCa progression. Another important DMG identified was stratifin, which is known to inhibit the cyclin B1–CD2 complex from entering the nucleus by enforcing a G2/M arrest. The loss of stratifin due to promoter hypermethylation during tumor progression has been seen in various cancers, including PCa.55 Death-associated protein kinase (DAPK) and fragile histidine triad (FHIT) among the identified DMGs are involved in apoptosis and metastasis and are reported to be frequently hypermethylated in various cancers, including PCa.56,57 Our analysis also identified endothelin B receptor type B, protein encoded by the EDNRB gene, which is known to induce apoptosis and inhibit tumor progression by interacting with endothelins. Several studies have reported promoter hypermethylation of EDNRB in higher frequencies in prostate tumors as compared to normal tissues.2,58
KEGG analysis showed that the major enriched pathways included pathways in cancer, cell cycle, p53 signaling, focal adhesion and Wnt signaling (Table 2). Wnt signaling is reported to play integral roles in cell fate decisions, proliferation, neural patterning, migration, differentiation, tumor aggression and stem cell renewal during many malignancies including PCa.59,60 In addition, differential regulation of p53 signaling as regulated by CCND2, SFN and CDKN2A has been reported in various cases of PCa.61 Reports have suggested that expressions of AR and p53, which are balanced during androgen-dependent stages of PCa, eventually get disturbed during the progression of the disease.62 p53 signaling also plays a pivotal role in cell cycle progression by regulating G1/S-, S- and G2/M-phase checkpoints and also transcription-dependent and -independent cell death program, DNA repair and apoptosis.63 The functional enrichment analysis demonstrated that the identified genes contributed to hormonal response, cell cycle, ROS and inflammatory responses that are directly involved with cancer initiation and progression (Table S2). This further supported our hypothesis that the identified 26 DMGs played crucial roles in PCa development.
The PPI network constructed with identified DMGs led to the identification of high-degree hub genes (Table 3 and Figure 1) in the local network, which included AR, CDH13, CDKN2A, DAPK1, CD44, GSTP1 and RASSF1 that exhibited the highest degree of connectivity. Androgens that are responsible for stimulating growth of PCa cells mediate their effect through androgen receptors (ARs). Silencing of AR can lead to decreased growth and induce apoptosis.42–45 AR promoter hypermethylation has been reported by various groups to be between 8% and 39% in PCa tumor tissues.2 The results of our analysis were in concordance with several published research papers that have reported the downregulated expression of AR due to hypermethylation during progression of PCa from androgen-dependent to androgen-independent stages.42–45,64–69 Thereby, this has led to emergence of androgen deprivation as one of the most effective treatment for advanced PCa, in addition to prostatectomy and radiation therapy.42–45 T-cadherin (CDH13), another identified hub gene located on 16q24, is a tumor suppressor gene known to play a critical role in cell–cell adhesion.70 CDH13 expression can restrain the invasive potential and proliferation rate of tumor cells. Anomalous expression of CDH13 due to promoter methylation has been reported in various malignancies, including breast cancer, lung cancer and bladder cancer.70 Yet, another gene identified in our analysis was glutathione S-transferase Pi 1 (GSTP1), which is known to be the most frequently epigenetically altered metabolic gene in PCa. DNA hypermethylation of GSTP1 is reported to be as high as 90% in prostate cancerous samples and 70% in prostatic interepithelial neoplasia.22–34 GSTP1 located on chromosome 11q3 eliminates foreign chemicals and detoxifies electrophilic toxic carcinogens and oxidants. Methylation in GSTP1 has been detected with high frequencies in PCa cell lines, urine samples, patient ejaculates and blood samples as compared to low or no methylation detected in healthy control patients.71 Ras association domain family protein 1 isoform A (RASSF1A), another gene identified, was found to be associated with more than eight identified DMGs. RASSF1A is a tumor suppressor gene usually associated with DNA repair and apoptosis.72 Evidences have shown that aberrant methylation of RASSF1A may hamper cell cycle control and DNA repair in tumors.72 Silencing of RASSF1A has been observed in various cancers such as non-small-cell lung cancer, colorectal cancer, breast cancer and PCa. In fact, some researchers have shown the frequency of hypermethylation of RASSF1A to be correlated with PCa progression and aggressiveness.72
Conclusion
We identified seven DMGs, namely, AR, CDH13, CDKN2A, DAPK1, GSTP1, CD44 and RASSF1, with high negative correlation (~0.7), which depicts that the increase in methylation directly leads to decrease in the expression of these genes. Biological and functional annotation showed that this panel of selected candidate genes played a crucial part in cell proliferation, binding, Wnt regulation, enzyme regulator activities and drug response. Although nonspecific epigenetic targeting might have short-term undesirable effects, gene-specific demethylation approaches offer promising prognostic and therapeutic targets for PCa diagnosis. Thus, results of the present study may be used for basic and translational treatment; however, further in vitro and in vivo investigations are needed to confirm our hypothesis.
Acknowledgments
This work was supported by grants from the Symbiosis Centre for Research and Innovation (SCRI) and Symbiosis School of Biological Sciences (SSBS); Symbiosis International University (SIU), Lavale, Pune, India and Junior Research INSPIRE Fellowship (JRF) from the Department of Science and Technology, Government of India, to Miss Anshika Nikita Singh.
Disclosure
The authors report no conflicts of interest in this work.
References
Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65(2):87–108. | ||
Yang M, Park JY. DNA methylation in promoter region as biomarkers in prostate cancer. Methods Mol Biol. 2012;863:67–109. | ||
Velonas VM, Woo HH, dos Remedios CG, Assinder SJ. Current status of biomarkers for prostate cancer. Int J Mol Sci. 2013;14(6):11034–11060. | ||
Sarkar S, Horn G, Moulton K, et al. Cancer development, progression, and therapy: an epigenetic overview. Int J Mol Sci. 2013;14(10):21087–21113. | ||
Sharma S, Kelly TK, Jones PA. Epigenetics in cancer. Carcinogenesis. 2009;31(1):27–36. | ||
Tam WL, Weinberg RA. The epigenetics of epithelial-mesenchymal plasticity in cancer. Nat Med. 2008;144(5):724–732. | ||
Feinberg AP, Koldobskiy MA, Göndör A. Epigenetic modulators, modifiers and mediators in cancer aetiology and progression. Nat Rev Genet. 2016;17(5):284–299. | ||
Jones PA, Issa J-PJ, Baylin S. Targeting the cancer epigenome for therapy. Nat Rev Genet. 2016;17(10):630–641. | ||
Zhang L, Huang Y, Zhuo W, Zhu Y, Zhu B, Chen Z. Identification and characterization of biomarkers and their functions for Lapatinib-resistant breast cancer. Med Oncol. 2017;34(5). | ||
Weinstein JN, Collisson EA, Mills GB, et al. The cancer genome Atlas Pan-Cancer Analysis Project John. Nat Genet. 2014;45(10): 1113–1120. | ||
Cotterill SJ [homepage on the Internet]. Cancer Genetics Web. Available from: http://www.cancer-genetics.org/index.htm. Accessed October 8, 2016. | ||
He X, Chang S, Zhang J, et al. MethyCancer: the database of human DNA methylation and cancer. Nucleic Acids Res. 2008;36(suppl 1):836–841. | ||
Tabas-madrid D, Nogales-cadenas R, Pascual-montano A. GeneCodis3: a non-redundant and modular enrichment analysis tool for functional genomics. Nucleic Acids Res. 2012;40:478–483. | ||
Mi H, Poudel S, Muruganujan A, Casagrande JT, Thomas PD. PANTHER version 10: expanded protein families and functions, and analysis tools. Nucleic Acids Res. 2016;44(D1):D336–D342. | ||
Kanehisa M, Goto S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000;28(1):27–30. | ||
Lopes CT, Franz M, Kazi F, Donaldson SL, Morris Q, Bader GD. Cytoscape Web: an interactive web-based network browser. Bioinformatics. 2011;27(13):2347–2348. | ||
Jensen LJ, Kuhn M, Stark M, et al. STRING 8 – a global view on proteins and their functional interactions in 630 organisms. Nucleic Acids Res. 2009;37(suppl 1):412–416. | ||
Ponger L, Mouchiroud D. CpGProD: identifying CpG islands associated with transcription start sites in large genomic mammalian sequences. Bioinformatics. 2002;18(4):631–633. | ||
Li W, Cowley A, Uludag M, et al. The EMBL-EBI bioinformatics web and programmatic tools framework. Nucleic Acids Res. 2015;43:580–584. | ||
Baek S, Yang S, Kang T, Park S, Sung Y, Kim S. MENT: methylation and expression database of normal and tumor tissues. Gene. 2013;518(1):194–200. | ||
Shi J, Hu J, Zhou Q, Du Y, Jiang C. PEpiD: a prostate epigenetic database in mammals. PLoS One. 2013;8(5):e64289. | ||
Ahmed H, Cappello F, Rodolico V. Evidence of heavy methylation in the galectin 3 promoter in early stages of prostate adenocarcinoma: development and validation of a methylated marker for early diagnosis of prostate cancer 1. Transl Oncol. 2009;2(3):146–156. | ||
Suh CI, Shanafelt T, May DJ, et al. Comparison of telomerase activity and GSTP1 promoter methylation in ejaculate as potential screening tests for prostate cancer. Mol Cell Probes. 2000;14(4):211–217. | ||
Goessl C, Krause H, Mu M, Schrader M, Sachsinger J. Advances in brief fluorescent methylation-specific polymerase chain reaction for DNA-based detection of prostate cancer in bodily fluids. Cancer Res. 2000;1(5):5941–5945. | ||
Harden SV, Guo Z, Epstein JI, Sidransky D. Quantitative GSTP1 methylation clearly distinguishes benign prostatic tissue and limited prostate adenocarcinoma. J Urol. 2003;169(3):1138–1142. | ||
Harden SV, Goodman SN, Partin AAW, Patrick C, Epstein JI. Quantitative GSTP1 methylation and the detection of prostate adenocarcinoma in sextant biopsies. J Natl Cancer Inst. 2003;95(21):1634–1637. | ||
Goessl C, Mu M, Krause H, Schostak M, Straub B. Methylation-specific PCR for detection of neoplastic DNA in biopsy washings. J Pathol. 2002;196(3):331–334. | ||
Tokumaru Y, Harden SV, Sun D, Yamashita K, Epstein JI, Sidransky D. Optimal use of a panel of methylation markers with GSTP1 hypermethylation in the diagnosis of prostate adenocarcinoma. Clin Cancer Res. 2004;10:5518–5522. | ||
Cairns P, Esteller M, Herman JG, et al. Molecular detection of prostate cancer in urine by GSTP1 hypermethylation molecular detection of prostate cancer in urine by GSTP1. Clin Cancer Res. 2001;7(9):2727–2730. | ||
Ellinger J, Bastian PJ, Jurgan T, et al. CpG island hypermethylation at multiple gene sites in diagnosis and prognosis of prostate cancer. Urology. 2008;71(1):161–167. | ||
Sunami E, Shinozaki M, Higano CS, et al. Multimarker circulating DNA assay for assessing blood of prostate cancer patients methods. Clin Chem. 2009;567:559–567. | ||
Gonzalgo ML, Pavlovich CP, Lee SM, Nelson WG, James T, Brady B. Prostate cancer detection by GSTP1 methylation analysis of postbiopsy urine specimens. Clin Cancer Res. 2003;9(7):2673–2677. | ||
Rogers CG, Gonzalgo ML, Yan G, et al. High concordance of gene methylation in post-digital rectal examination and post-biopsy urine samples for prostate cancer detection. J Urol. 2006;176:2280–2284. | ||
Hoque MO, Topaloglu O, Begum S, Henrique R, Rosenbaum E. Quantitative methylation-specific polymerase chain reaction gene patterns in urine sediment distinguish prostate cancer patients from control subjects. J Clin Oncol. 2015;23(27):6569–6575. | ||
Rouprêt M, Hupertan V, Yates DR, et al. Molecular detection of localized prostate cancer using quantitative methylation-specific PCR on urinary cells obtained following prostate massage. Clin Cancer Res. 2007;13(6):1720–1725. | ||
Maruyama R, Toyooka S, Toyooka KO, et al. Aberrant promoter methylation profile of prostate cancers and its relationship to clinicopathological features aberrant promoter methylation profile of prostate cancers and its relationship to clinicopathological features. Clin Cancer Res. 2002;8(2):514–519. | ||
Kang GH, Lee S, Lee HJ, Hwang KS. Aberrant CpG island hypermethylation of multiple genes in prostate cancer and prostatic intraepithelial neoplasia. J Pathol. 2004;202:233–240. | ||
Yamanaka M, Watanabe M, Yamada Y, et al. Altered methylation of multiple genes in carcinogenesis of the prostate. Int J Cancer. 2003;106(3):382–387. | ||
Ginsburg GS, Williard HF. Genomic and Personalized Medicine. Amsterdam: Academia Press; 2012. | ||
Jan G. Decreased expression Of CD44 in metastatic prostate cancer. Int J Cancer. 1999;483:478–483. | ||
Alumkal JJ, Zhang Z, Humphreys EB, et al. Biomarkers and risk factors effect of DNA methylation on identification of aggressive prostate cancer. Urology. 2008;72(6):1234–1239. | ||
Jarrard DF, Kinoshita H, Shi Y, et al. Methylation of the androgen receptor promoter CpG island is associated with loss of androgen receptor expression in prostate cancer cells. Cancer Res. 1998;58(23):5310–5315. | ||
Kinoshita H, Shi Y, Sandefur C, et al. Methylation of the androgen receptor minimal promoter silences transcription in human prostate cancer. Cancer Res. 2000;60(13):3623–3630. | ||
Nakayama T, Watanabe M, Suzuki H, et al. Epigenetic regulation of androgen receptor gene expression in human prostate cancers. Lab Invest. 2000;80(12):1789–1796. | ||
Sasaki M, Tanaka Y, Perinchery G, et al. Methylation and inactivation of estrogen, progesterone, and androgen receptors in prostate cancer. J Natl Cancer Inst. 2002;94(5):384–390. | ||
Jin B, Li Y, Robertson KD. DNA methylation: superior or subordinate in the epigenetic hierarchy? Genes Cancer. 2011;2(6):607–617. | ||
Law JA, Jacobsen SE. Establishing, maintaining and modifying DNA methylation patterns in plants and animals Julie. Nat Rev Genet. 2011;11(3):204–220. | ||
Bonelli P, Tuccillo FM, Borrelli A, Schiattarella A, Buonaguro FM. CDK/CCN and CDKI alterations for cancer prognosis and therapeutic predictivity. Biomed Res Int. 2014;2014:361020. | ||
Nguyen TT, Nguyen CT, Gonzales FA, Nichols PW, Yu MC, Jones PA. Analysis of cyclin-dependent kinase inhibitor expression and methylation patterns in human prostate cancers. Prostate. 2000;43(3):233–242. | ||
Herman G, Merlo A, Mao L, et al. Inactivation of the CDKN2/p16/MTSJ gene is frequently associated with aberrant DNA methylation in all common human cancers’. Cancer Res. 1995;7(1):4525–4530. | ||
Musgrove EA, Caldon CE, Barraclough J, Stone A, Sutherland RL. Cyclin D as a therapeutic target in cancer. Nat Rev Cancer. 2011;11(8):558–572. | ||
Verkaik NS, van Steenbrugge GJ, van Weerden WM, Bussemakers MJ, van der Kwast TH. Silencing of CD44 expression in prostate cancer by hypermethylation of the CD44 promoter region. Lab Invest. 2000;80(8):1291–1298. | ||
Zheng J, Xiong D, Sun X, et al. Signification of hypermethylated in cancer 1 (HIC1) as tumor suppressor gene in tumor progression. Cancer Microenviron. 2012;1:285–293. | ||
Woodson K, O’Reilly KJ, Ward DE, et al. CD44 and PTGS2 methylation are independent prognostic markers for biochemical recurrence among prostate cancer patients with clinically localized disease. Epigenetics. 2006;1(4):183–186. | ||
Lodygin D, Hermeking H. The role of epigenetic inactivation of 14-3-3 σ in human cancer. Cell Res. 2005;15(4):237–246. | ||
Michie AM, Mccaig AM, Nakagawa R, Vukovic M. Death-associated protein kinase (DAPK) and signal transduction: regulation in cancer. FEBS J. 2010;277(1):74–80. | ||
Fournier G, Cussenot O, Pesche S, Latil A, Bie I, Lidereau R. Molecular analysis of the FHIT gene in human prostate cancer. Oncogene. 1998;16(14):1863–1868. | ||
Pao MM, Tsutsumi M, Liang G, Uzvolgyi E, Gonzales FA, Jones PA. The endothelin receptor B (EDNRB) promoter displays heterogeneous, site specific methylation patterns in normal and tumor cells. Hum Mol Genet. 2001;10(9):903–910. | ||
Mohammadi N, Babashah S. Interplay between microRNAs and WNT/b -catenin signalling pathway regulates epithelial – mesenchymal transition in cancer. Eur J Cancer. 2015;51(12):1638–1649. | ||
Zhan T, Rindtorff N, Boutros M. Wnt signaling in cancer. Oncogene. 2017;36(11):1461–1473. | ||
Osman I, Drobnjak M, Fazzari M, Ferrara J, Scher HI, Cordon-Cardo C. Inactivation of the p53 pathway in prostate cancer: impact on tumor progression. Clin Cancer Res. 1999;5(8):2082–2088. | ||
Cronauer MV, Schulz WA, Burchardt T, Ackermann R, Burchardt M. Inhibition of p53 function diminishes androgen receptor-mediated signaling in prostate cancer cell lines. Oncogene. 2004;23(20):3541–3549. | ||
Stegh AH. Targeting the p53 signaling pathway in cancer therapy. Expert Opin Ther Targets. 2012;16(1):67–83. | ||
Massie CE, Mills IG, Lynch AG. The importance of DNA methylation in prostate cancer development. J Steroid Biochem Mol Biol. 2016;166:1–15. | ||
Tan H, Sood A, Rahimi HA. Rb loss is characteristic of prostatic small cell neuroendocrine carcinoma Rb loss is characteristic of prostatic small cell. Clin Cancer Res. 2014;20(4):890–903. | ||
Kinoshita H, Shi Y, Sandefur C, et al. Methylation of the androgen receptor minimal promoter silences transcription in human prostate cancer. Cancer Res. 2000;60(13):3623–3630. | ||
Gravina GL, Marampon F, Di Staso M, et al. 5-azacitidine restores and amplifies the bicalutamide response on preclinical models of androgen receptor expressing or deficient Prostate tumors. Prostate. 2010;1178:1166–1178. | ||
Gravina GL, Festuccia C, Millimaggi D, et al. Chronic azacitidine treatment results in differentiating effects, sensitizes against bicalutamide in androgen-independent prostate cancer cells. Prostate. 2008;68(7):793–801. | ||
Zorn CS, Wojno KJ, Mccabe MT, Kuefer R, Gschwend JE, Day ML. Cancer therapy: preclinical 5-Aza-2′-deoxycytidine delays androgen-independent disease and improves survival in the transgenic adenocarcinoma of the mouse prostate mouse model of prostate cancer. Clin Cancer Res. 2007;13(7):2136–2144. | ||
Andreeva AV, Kutuzov MA. Cadherin 13 in cancer. Genes Chromosomes Cancer. 2010;49(9):775–790. | ||
Martignano F, Gurioli G, Salvi S, et al. GSTP1 methylation and protein expression in prostate cancer: diagnostic implications. Dis Markers. 2016;2016:1–6. | ||
Liu L, Yoon J, Dammann R, Pfeifer GP. Frequent hypermethylation of the RASSF1A gene in prostate cancer. Oncogene. 2002;21:6835–6840. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.