")
Back to Journals » International Journal of General Medicine » Volume 14
Identification of Hub Genes Associated with the Pathogenesis of Intracranial Aneurysm via Integrated Bioinformatics Analysis
Authors Zhong A , Ding N, Zhou Y, Yang G, Peng Z , Zhang H, Chai X
Received 18 May 2021
Accepted for publication 9 July 2021
Published 30 July 2021 Volume 2021:14 Pages 4039—4050
DOI https://doi.org/10.2147/IJGM.S320396
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Scott Fraser
Aifang Zhong,1,2 Ning Ding,1,2 Yang Zhou,1,2 Guifang Yang,1,2 Zhenyu Peng,1,3 Hongliang Zhang,1,3 Xiangping Chai1,2
1Department of Emergency Medicine, The Second Xiangya Hospital, Central South University, Changsha, Hunan, People’s Republic of China; 2Trauma center, The Second Xiangya Hospital, Central South University, Changsha, Hunan, People’s Republic of China; 3Emergency Medicine and Difficult Disease Institute, The Second Xiangya Hospital, Central South University, Changsha, Hunan, People’s Republic of China
Correspondence: Xiangping Chai
Central South University, 139 Renmin Middle Road, Changsha City, Hunan Province, People’s Republic of China
Email [email protected]
Background: At present, the pathogenesis of intracranial aneurysms (IA) remains unclear, which significantly hinders the development of novel strategies for the clinical treatment. In this study, bioinformatics methods were used to identify the potential hub genes and pathways associated with the pathogenesis of IA.
Methods: The gene expression datasets of patients with intracranial aneurysm were downloaded from the Gene Expression Database (GEO), and the different data sets were integrated by the robust rank aggregation (RRA) method to identify the differentially expressed genes between patients with intracranial aneurysm and the controls. The functional enrichment analyses of the significant differentially expressed genes (DEGs) were performed and the protein–protein interaction (PPI) network was constructed; thereafter, the hub genes were screened by cytoHubba plug-in of Cytoscape, and finally sequencing dataset GSE122897 was used to verify the hub genes.
Results: The GSE15629, GSE75436, GSE26969, and GSE6551 expression profiles have been included in this study, including 34 intracranial aneurysm samples and 26 control samples. The four datasets obtained 136 significant DEGs (45 up-regulated, 91 down-regulated). Enrichment analysis showed that the extracellular matrix structural constituent and the ECM-receptor interaction were closely related to the occurrence of IA. It was finally determined that eight hub genes associated with the development of IA, including VCAN, COL1A1, COL11A1, COL5A1, COL5A2, POSTN, THBS2, and CDH2.
Conclusion: The discovery of potential hub genes and pathways could enhance the understanding of the molecular mechanisms associated with the development of IA. These hub genes may be potential therapeutic targets for the management and new biomarker for the diagnosis of IA.
Keywords: intracranial aneurysm, hub genes, robust rank aggregation
Introduction
Intracranial aneurysm (IA) is a cerebrovascular disease that can be caused by several factors, such as inflammation, hemodynamic changes, genetics, smoking history, etc.1–4 The global prevalence of IA is about 3.2%, and the main risk associated with it is that of rupture and bleeding.5,6 The available treatment modalities for IA include surgical clipping and various endovascular therapy options (such as endovascular coiling, shunts, and intravascular devices).6–9 However, there are still controversies about the choice of treatment modalities and the start time of treatment for IA under different conditions (ruptured or unruptured). In addition, the existing main treatment methods are all invasive procedures, which may cause various complications.10 IA and the subarachnoid hemorrhage (SAH) caused by its rupture have caused a significant burden on the global health.5,11 Therefore, effective management of unruptured IA (UIA) and treatment of IA still remains a significant clinical challenge.10 Therefore, detailed studies on the pathogenesis of IA may potentially help to improve the treatment strategies for this devastating disease.
However, so far, no studies have clearly explained the specific mechanisms of aneurysm formation.12–15 In recent years, with the advancement of genetic technology, several studies have performed gene expression profiling on the vascular wall tissue of IA patients in order to discover the expression of various key genes and changes in pathways involved in the formation of aneurysm. The differentially expressed genes between aneurysm as well as the normal samples were identified in a study by Yu et al13 and mainly involved genes regulating immune and inflammatory responses, cell growth, proliferation, differentiation and migration, and intercellular signal transduction. The validation analysis by Wang et al showed that VCAM1, MAGI2, PPP2R2, PPP2R3A genes may be related to the occurrence and development of IA, and these genes were mainly involved in encoding cell adhesion molecules (CAMs) and extracellular regulated protein kinase (ERK)/c-JunN-terminal kinase (JHK) signaling pathways.16 The team of Wei and coworkers found that CD40, CD40LG, NOS1, STRN, MCM4, MYH11 and miR-125b may play an important role in the pathogenesis of IA.14 In fact, due to the difficulty in obtaining materials from IA tissues, different data processing methods and platform conditions, the results of screening differential genes with microarray and sequencing data might vary significantly.
In order to overcome the shortcomings of limited or inconsistent results due to applications of the different technology platforms and small sample sizes, robust rank aggregation (RRA) method was used in this study to merge the different gene data sets. This method has been used in many other diseases (such as preeclampsia, acute myocardial infarction, thyroid cancer) to screen for the various differentially expressed genes (DEGs),17–19 but it is currently used less in the studies related to IA. In this study, the gene expression datasets were downloaded from the Gene Expression Omnibus (GEO) database, and the RRA method was used to screen the significant genes between the IA tissue and the control tissue, and perform a functional enrichment analysis for the significant genes. The cytoHubba plug-in of Cytoscape software was used to identify the various hub genes. The expression of different hub genes was further verified in the sequencing dataset.
Materials and Methods
Microarray Datasets of IA
The gene expression microarray datasets of IA analyzed in this study were acquired from the Gene Expression Omnibus GEO (http://www.ncbi. nlm.nih.gov/geo). We downloaded raw data or series matrix files from GEO, including GSE15629,20 GSE7543621 and GSE26969,22 GSE6551.23
Identification of DEGs in IA
We used the corresponding annotation package in R software (version 4.0.3; 64-bit; https://www.R-project.org/) or annotation document to map the microarray probes to gene symbols, and to calculate the mean value when multiple probes mapped to the same gene symbol. Next, the package of “limma” in R software was used to identify the DEGs. The |log2 fold change (FC)|>1 and P-value < 0.05 were treated as the criteria to screen for the DEGs, and the DEGs were visualized with volcano plots.
RRA Analysis
The RRA method assumes that genes are randomly arranged in each dataset. When a gene ranks high in all datasets, the P value will decrease, and it is more likely to be a significant gene. The differential genes obtained in each dataset were sorted into a list of up-regulated and down-regulated genes. The “Robust Rank Aggregation” package in R software was used to rank different up-regulated and down-regulated genes and to identify the significance of the different genes. The conditions for the screening of the significant genes were set to the fold change >1 and P-value < 0.05.
Functional and Pathway Enrichment Analyses of DEGs
The “clusterProfiler” package in R was used to perform Gene Ontology (GO) functional enrichment and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analyses of the significant genes screened out by RRA analysis.
Protein–Protein Interaction (PPI) Network Analysis
The STRING database (http://string-db.org/) was used to establish a PPI network with a confidence of 0.4 to predict the various protein–protein interactions. Thereafter, the results were imported into Cytoscape 3.8.1 to build a network model. Using cytoHubba plug-in of Cytoscape, the top 10 DEGs were selected with a high connectivity in the gene expression network as the hub genes according to the degree algorithm.
Hub Genes Expression in RNA-Seq Dataset
We further verified the hub genes by using data from an RNA-seq dataset (GSE122897,24 including 44 IA, 16 controls). The RNA-seq reading count data was analyzed using DESeq2 function to identify the various DEGs. The genes with P-value <0.05 were considered to be significant.
Results
Information on Included Datasets
The microarray data GSE15629 contains 14 IA samples and 5 control samples. GSE75436 contains 15 IA samples and 15 control samples. GSE26969 has 3 IA samples and 3 control samples. GSE6551 consists of 2 IA samples and 3 control samples. The detailed information of these datasets has been shown in Table 1.
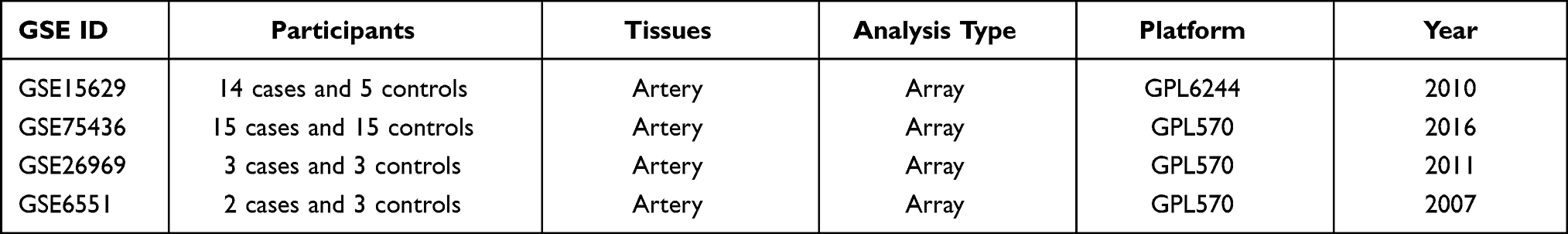 |
Table 1 Characteristics of the Included Microarray Datasets |
Identification of DEGs
In order to ensure homogeneity, the four data sets were evaluated for quality, and the results have been shown in Supplementary Figure 1. According to the set threshold, the “limma” package in R was used to screen the DEGs. The results of the DEGs in each dataset have been represented by volcano plots in Figure 1.
 |
Figure 1 Volcano plots of the four chip datasets. Red points: upregulated genes; Blue points: downregulated genes; Gray points: genes with no significant difference. (A) GSE15629, (B) GSE75436, (C) GSE26969, (D) GSE6551. |
Results of RRA Integrated Analysis
According to the threshold conditions that were set, a total of 136 significant genes were identified by RRA integrated analysis, which included 45 up-regulated genes and 91 down-regulated genes and described in Supplementary Table 1. Then, the top ten significant genes among the up-regulated and down-regulated genes were selected to draw a heatmap, as shown in Figure 2.
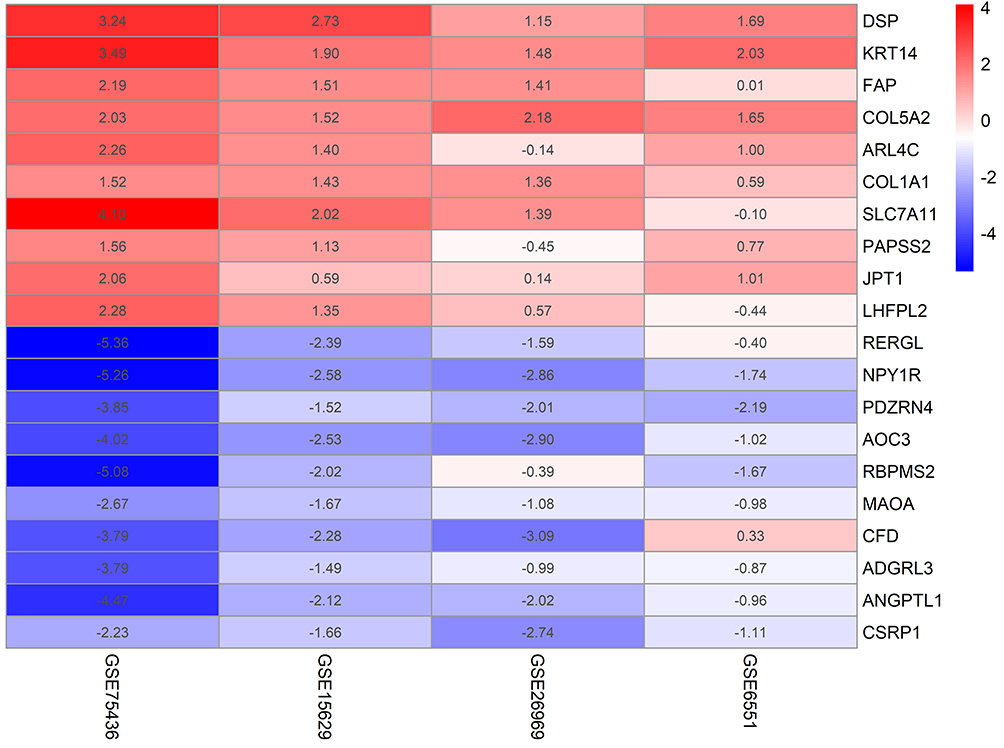 |
Figure 2 Heatmap of the top ten up-regulated and down-regulated genes in the RRA analysis. Different colors represent the level of gene expression. Red: high expression. Blue: low expression. |
Functional Enrichment Analysis of Significantly Different Genes
We performed gene enrichment analysis on the significant genes. The results showed that the degree of bio-enrichment of extracellular matrix (ECM) structural constituent (GO: 0005201, P-value = 9.02E-14) was the most significant, and the all enrichment pathways of GO are listed in Table 2. In the KEGG enrichment analysis, it was found that ECM-receptor interaction (hsa04512; P-value =0.0008) and protein digestion and absorption were significantly enriched (Table 3). Figure 3 depicts the proportion of the significant genes and the degree of enrichment in the GO and KEGG enriched pathways.
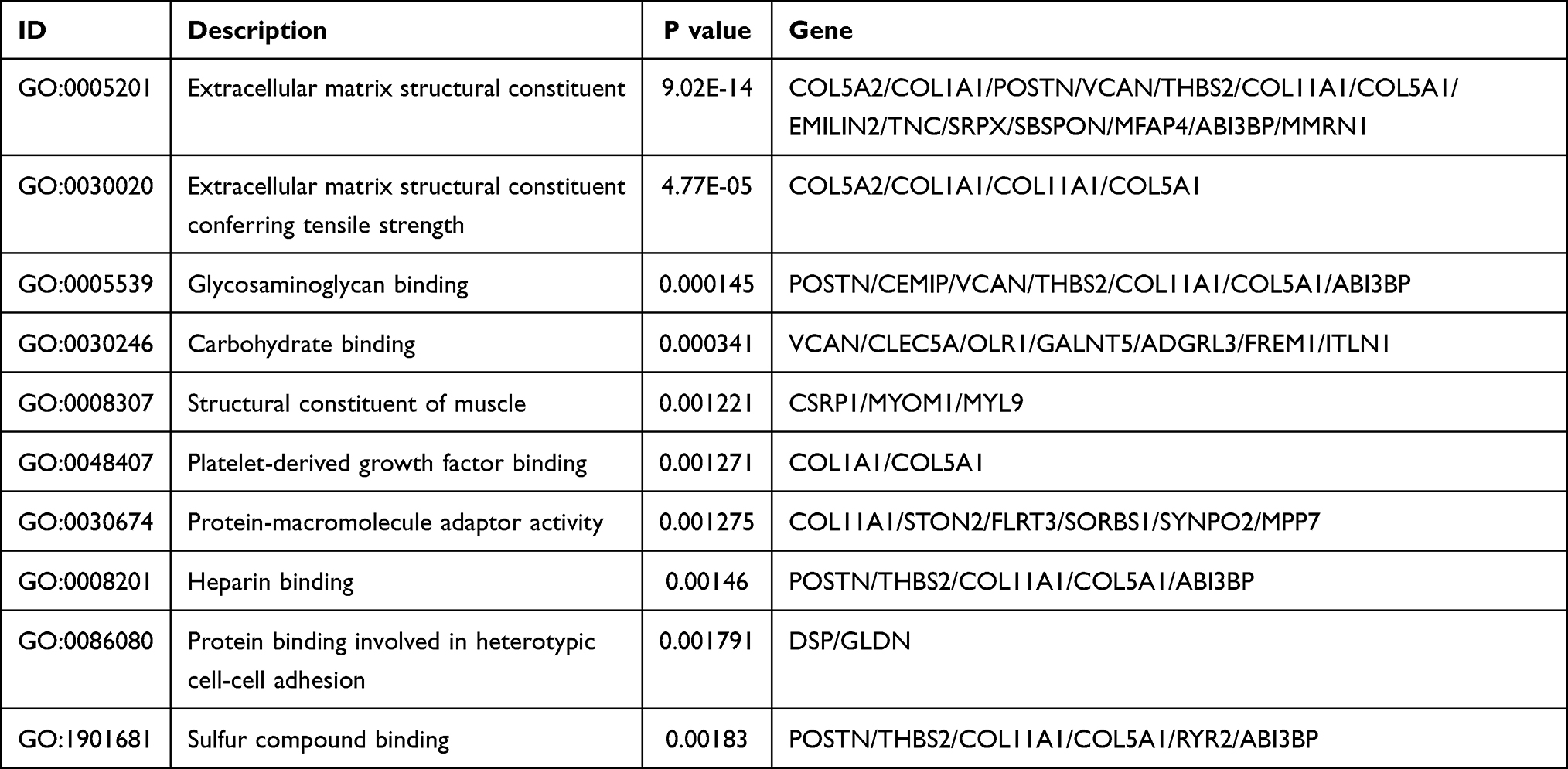 |
Table 2 GO Analysis of Integrated DEGs |
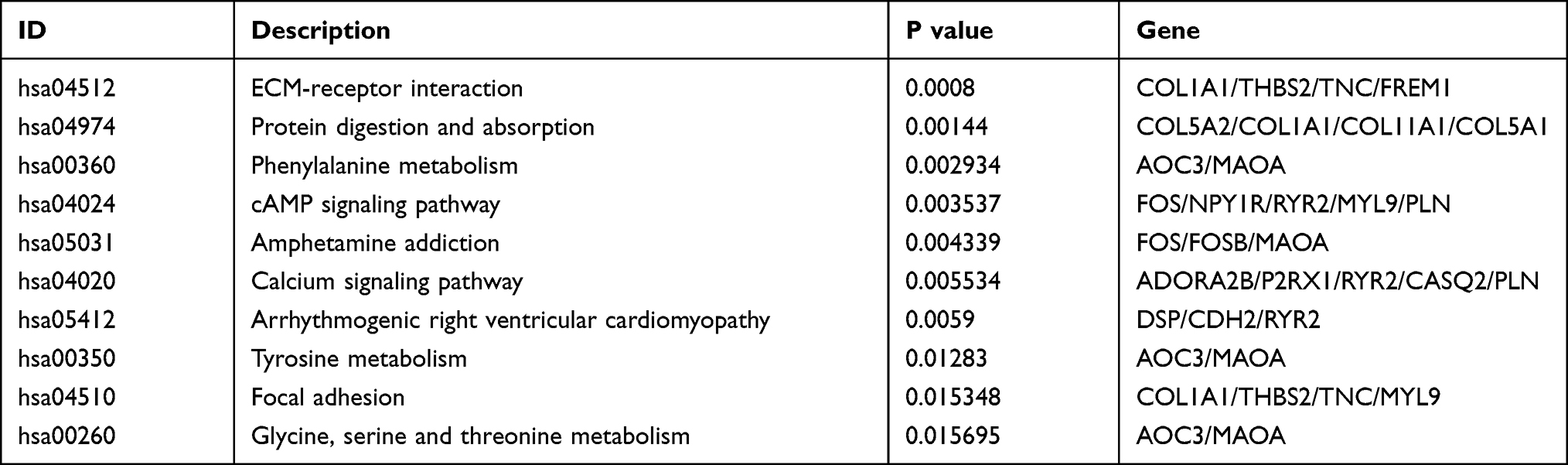 |
Table 3 KEGG Analysis of Integrated DEGs |
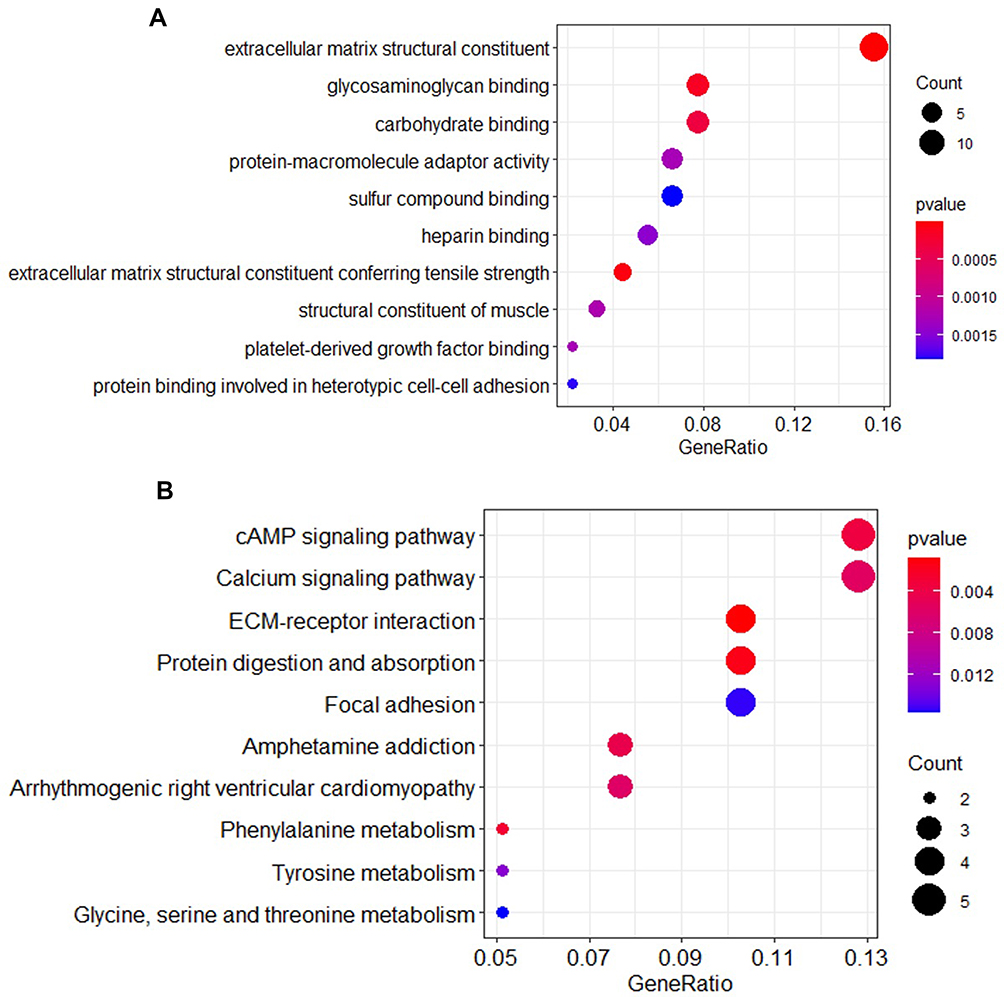 |
Figure 3 Bubble chart of GO and KEGG enrichment pathway. (A) GO enrichment pathway; (B) KEGG enrichment pathway. |
PPI Network Analysis and Identification of Hub Genes
In order to understand the possible interactions between the proteins encoded by the significant genes in IA, a PPI network was constructed using the STRING online database, and then the Cytoscape was used to visualize the network and identify the different hub genes. Sixty-seven of 136 significantly different genes were used for the construction of the PPI network. The results of network analysis performed by Cytoscape software is shown in Figure 4A. The top ten hub genes screened by cytoHubba were VCAN, COL1A1, COL11A1, COL5A1, COL5A2, TNC, POSTN, THBS2, CHRDL1 and CDH2. The connection network between them is shown in Figure 4B.
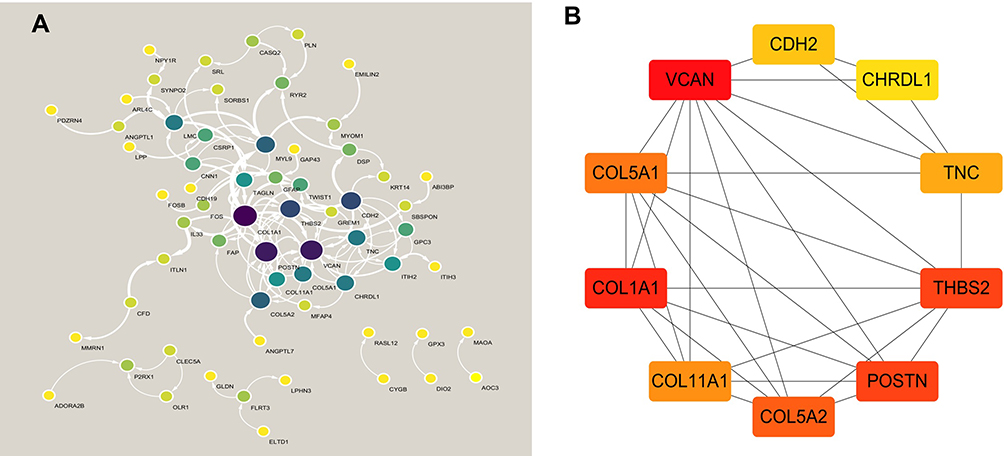 |
Figure 4 PPI network and hub genes identified by cytoHubba plug-in of Cytoscape. (A) PPI network. (B) hub genes. |
Analysis of the Expression Level of the Different Hub Genes
We validated the expression level of different hub genes using sequencing dataset GSE122897, and found that eight out of the 10 hub genes had significant differences in the verification dataset. Only TNC and CHRDL1 genes did not show significant differences in the validation dataset (P-value > 0.05). The difference in the expression level of hub genes between IA and the control group is shown in Figure 5.
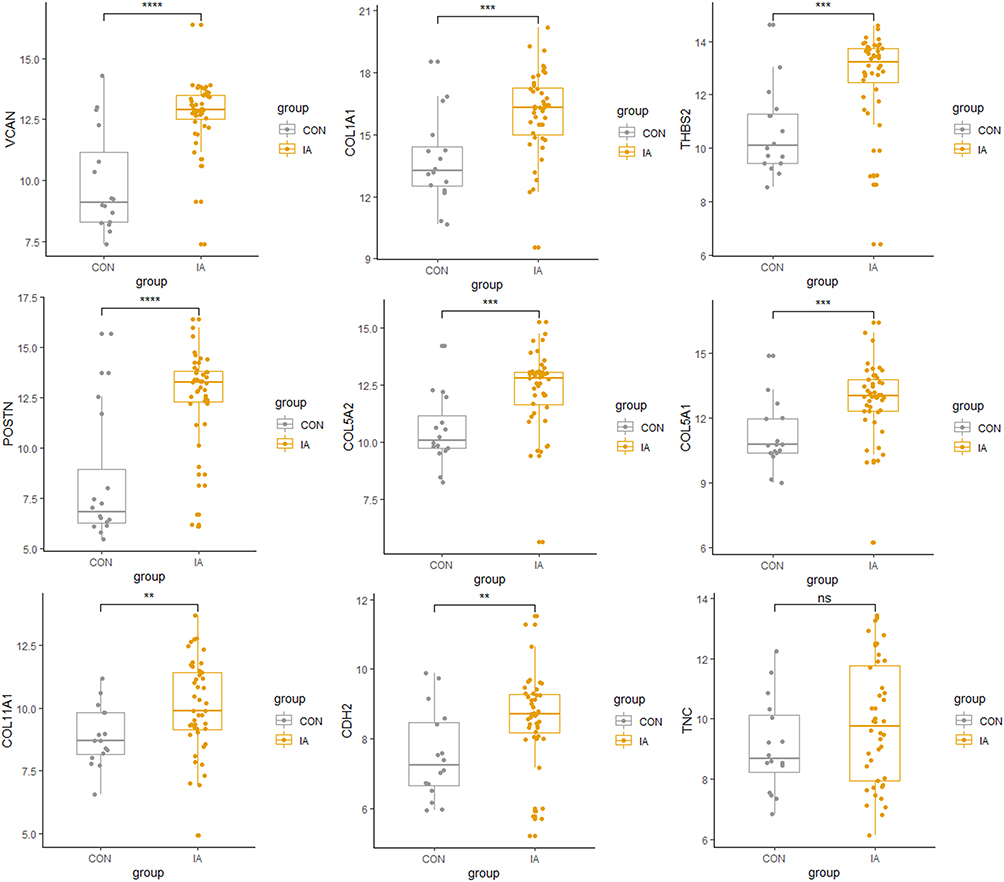 |
Figure 5 The validation of the expression of the hub genes in dataset GSE122897. **P-value < 0.01; ***P-value < 0.001; ****P-value < 0.0001; ns indicates non-significant. |
Discussion
Four datasets from GEO were found to be downloaded in this study, and RRA was used to perform integration analysis on them to screen for the presence of the significant genes. A total of 136 significant DEGs were screened from the four datasets, including 45 up-regulated genes and 91 down-regulated genes. The top ten hub genes screened by cytoHubba were VCAN, COL1A1, COL11A1, COL5A1, COL5A2, TNC, POSTN, THBS2, CHRDL1 and CDH2. In the sequencing dataset, 8 hub genes were verified to have differences, including VCAN, COL1A1, COL11A1, COL5A1, COL5A2, POSTN, THBS2 and CDH2.
Apoptosis and migration of the vascular smooth muscles, extracellular matrix remodeling of intracranial blood vessels, and inflammatory reactions have been widely accepted to actively participate in IA formation.1,2,25,26
Moreover, enrichment analysis showed that extracellular matrix structural constituent and ECM-receptor interaction may be involved in the formation of IA. At the same time, it was found that the various genes encoding multiple types of collagen were involved in the structural composition of ECM. Collagen is the most important fibrin outside the cell, and it is also the skeleton of the ECM. Normal extracellular matrix components can maintain the elasticity of the arterial wall. A number of studies have found that when the synthesis of type III collagen decreases and the proportion of type I collagen increases, the blood vessel wall can undergo changes, thereby causing hemangiomas or rupture.27 Yu et al found that the type I collagen gene (COL1A1) was involved in the destruction of the cellular matrix, and COL11A1 was the most significant up-regulated gene among all genes analyzed.13 A recent study showed that COL1A1 and COL5A2 were significantly up-regulated in the aneurysm group and could effectively regulate phosphoinositide 3-kinase (PI3K)/Akt signaling pathway.28 Similar to the results of previous studies, we found that the five genes involved in encoding collagen (COL5A2, COL1A1, COL11A1, COL5A1) were all up-regulated in the intracranial aneurysm group, and may play an important role in maintaining normal extracellular matrix components.
The genes affecting ECM structural constituent also include VCAN, POSTN and THBS2. VCAN is a chondroitin sulfate proteoglycan, which has five different subtypes (V0, V1, V2, V3, V4).29–31 VCAN can exist in a variety of the tissues and is an important component of the ECM.29,31,32 It plays a pivotal part in regulating cell proliferation, differentiation, adhesion and migration, and can promote angiogenesis and inflammatory tumor microenvironment formation.30,33 It has been confirmed that VCAN is over-expressed in the tumor stroma and the cancer cells of a variety of malignancies, such as breast cancer,34 endometrial cancer,35 testicular germ cell tumor,36 bladder carcinoma,37 gastric cancer.38 The study by Shen et al38 found that a high expression of VCAN in gastric cancer tissue was associated with a larger depth of tumor invasion and a poor prognosis. Zhang et al37 also confirmed similar observations. They analyzed the lesion tissues of 417 bladder cancer patients and found that the overexpression of VCAN was related to the number of tumors, depth of invasion, lymph node metastasis, distant metastasis, and histological grade. When VCAN was silenced, it was found to effectively inhibit the growth of tumors in situ, and when combined with endostatin, it was more effective to prolong the survival time of mice.39 The above studies have clearly established that an abnormal expression of VCAN in the tumors can significantly promote the tumor growth and metastasis, which is usually related to the poor prognosis of tumors. VCAN is not only related to the development and prognosis of malignant tumor diseases but also can contribute to the development of vascular diseases. VCAN affects the composition of the ECM and controls the formation of elastic fiber fibrils, which is of great significance for the remodeling of the ECM in the vascular diseases, and also plays a key role in atherosclerosis and restenosis.40,41 ECM remodeling plays an important role in the formation of aneurysms, and the reduction of ECM is a significant feature found in IA. A previous study42 reported that a similar phenotype displayed by the V2 subtype of VCAN gene may help to inhibit the growth of smooth muscle and other vascular entities, thus leading to IA. Thus, VCAN gene might be an important candidate gene involved in the pathogenesis of IA. Additionally, similar to the results of previous studies, we also found that VCAN was significantly up-regulated in diseased IA and was involved in the composition of ECM.
The POSTN gene is located on the long arm of human chromosome 13 and can encode POSTN.43 POSTN is a protein secreted by ECM, which is involved in the regulation of cell adhesion, differentiation, blood vessel formation and calcification, tissue fibrosis as well as other pathophysiological processes.44–47 Some studies have found that POSTN is highly expressed in the tissues or the serum in a variety of diseases (such as pulmonary fibrosis, cartilage-like tumors, head and neck cancer), and has been reported to be associated with poor prognosis.48–50 Therefore, POSTN may serve as a potential prognostic biomarker and treatment target for various diseases. The findings of Luo et al also established this point of view.51 They found that the serum POSTN concentration in patients with SAH increased, and it was related to the clinical severity and poor prognosis. At present, the expression of POSTN in various diseases is not fully understood, and the mechanisms involved in regulating its expression in the different diseases might also be different. Studies have found that POSTN can participate in the regulation of cell growth, tumorigenesis and drug response through activating the PI3K/Akt pathway.50,52 Researchers found that when POSTN expression was reduced, it could significantly inhibit the formation of colorectal tumors in mice.47 They proposed that POSTN can activate integrin-focal adhesion kinase (FAK)-Src kinase by binding to integrin, thereby activating YAK/TA2 to cause the release of interleukin-6 (IL-6), which ultimately leads to an inflammatory response. A previous study has confirmed that inflammation can serve as an important mechanism for the formation of hemangioma, and proteomics analysis has found that the deposition of POSTN in the extracellular matrix of abdominal aortic aneurysm tissue was increased.53 The results reported by Yamashita et al also showed that POSTN was highly expressed in the cell infiltration and elastin degradation area of human abdominal aortic aneurysm tissue and was upregulated in the stage of abdominal aortic aneurysm expansion caused by continuous inflammation in the mice.54 Therefore, it could be speculated that POSTN is also related to the occurrence and development of aneurysms. In our analysis, it was found that the expression of POSTN in IA tissue was significantly higher than that in the normal control group, and it was involved in the composition of ECM, which indicated that POSTN may be involved in the formation and development of IA.
THBS2 is a multifunctional stromal cell protein encoded by the THBS2 gene and belongs to the thrombin-sensitive protein family.55 It has been reported to be involved in the regulation of angiogenesis, proliferation, apoptosis, tumor migration, autophagy and transforming growth factor (TGF) β activation.56–59 In arteries, THBS2 is mainly secreted by the smooth muscle cells and partly by endothelial cells. A large number of studies have shown that THBS2 may be involved in the formation and development of aneurysms. For instance, a previous study has suggested that THBS2 gene polymorphism is related to thoracic aortic aneurysms, but the specific mechanism is not clear.60 Recent studies have found that the expression of THBS2 in the aortic samples of aortic dissection (AD) patients and the concentration of THBS2 in the circulation were significantly higher than those in the control group, and the levels of THBS2 in the circulation of AD patients could positively correlate with the levels of various inflammatory factors.56 Similarly, the circulating concentration of THBS2 in patients with abdominal aortic aneurysm was also increased, which was independently related to the risk of cardiovascular death in patients.61 Another study also confirmed that THBS2 expression was increased in abdominal aortic aneurysm tissue, and further found that THBS2 could act as a potential target of miR-195-5p, which may be involved in the formation of abdominal aortic aneurysm.55 We found that THBS2 was highly expressed in the tissues of IA. This is similar to the results of the various previous studies, which prove that THBS2 may be involved in the formation of aneurysms, but the specific mechanism needs to be further studied.
CDH2 is a transmembrane protein that plays an important role in the cell adhesion and is encoded by the CDH2 gene.62 Apoptosis of the vascular smooth muscle cells can function as an important regulator of aneurysm formation. Studies have found that cadherin can regulate the behavior of the vascular smooth muscle cells through the mediation of matrix metalloproteinases.63 It has been reported that the protein level of full-length nitro-cadherin in the human abdominal aortic aneurysm samples was significantly lower, but more nitro-cadherin fragments were observed.63 Interestingly, they found that the combination of matrix metalloproteinase-7 deletion and overexpression of EC4-Fc (soluble N-cadherin and a smaller soluble N-cadherin truncation) significantly increased the severity of abdominal aortic aneurysms. In contrary, other study has reported that the expression of CDH2 in patients with thoracic aortic aneurysms was significantly increased.64 After testing the cerebrospinal fluid of SAH patients with the ruptured anterior circulation aneurysm, Takase et al found that the level of soluble vascular endothelial cadherin in the cerebrospinal fluid was significantly increased, and the clinical severity of this index could positively correlate with the level of cerebrospinal fluid tumor necrosis factor-α.65 We found that CDH2 expression was increased in the tissue samples from IA patients, which indicates that CDH2 may be involved in the formation of IA, but its involvement in specific pathways requires further analysis.
In summary, our results found that various genes differentially expressed in IA were mainly involved in the ECM structural constituent and ECM-receptor interactions. The hub genes involved in the various physiological and pathological processes, such as inflammation, angiogenesis, cell growth, proliferation, etc., are closely related to aneurysm diseases and may serve as a potential therapeutic target and new biomarker for IA. This study has certain limitations, the sample size was still small, and more experiments need to be performed to verify the expression changes of the genes and the specific mechanisms involved in the development of IA.
Data Statement and Linking
All datasets used and analyzed in the current research are available from the Gene Expression Omnibus GEO (http://www.ncbi.nlm.nih.gov/geo).
Acknowledgments
We thank all the participants.
Funding
The present study was supported by the Key Research and Development Program of Hunan Province (2020SK1014-2).
Disclosure
The authors declare that they have no competing interests.
References
1. Signorelli F, Turjman F, Gory B, Labeyrie P, Pelissou-Guyotat I, Riva R. Hemodynamics, inflammation, vascular remodeling, and the development and rupture of intracranial aneurysms: a review. Neuroimmunol Neuroinflammation. 2015;2(2):59. doi:10.4103/2347-8659.154885
2. Signorelli F, Sela S, Gesualdo L, et al. Hemodynamic stress, inflammation, and intracranial aneurysm development and rupture: a systematic review. World Neurosurg. 2018;115:234–244. doi:10.1016/j.wneu.2018.04.143
3. Zhou S, Dion PA, Rouleau GA. Genetics of intracranial aneurysms. Stroke. 2018;49(3):780–787. doi:10.1161/STROKEAHA.117.018152
4. Si H, Yu N, Li Y, Hao Z, Liu Z, Li M-H. NianZu Yu YL, Zheng Hao, Zheng Liu, Li M. A Meta-Analysis of Risk Factors for the Formation of de novo Intracranial Aneurysms. Neurosurgery. 2019;85(4):454–465. doi:10.1093/neuros/nyy332
5. Vlak MH, Algra A, Brandenburg R, Rinkel GJ. Prevalence of unruptured intracranial aneurysms, with emphasis on sex, age, comorbidity, country, and time period: a systematic review and meta-analysis. Lancet Neurol. 2011;10(7):626–636. doi:10.1016/S1474-4422(11)70109-0
6. Thompson BG, Brown RD, Amin-Hanjani S, et al. Guidelines for the management of patients with unruptured intracranial aneurysms. Stroke. 2015;46(8):2368–2400. doi:10.1161/STR.0000000000000070
7. Toccaceli G, Diana F, Cagnazzo F, et al. Microsurgical clipping compared with new and most advanced endovascular techniques in the treatment of unruptured middle cerebral artery aneurysms: a Meta-Analysis in the modern era. World Neurosurg. 2020;137:451–464.e1. doi:10.1016/j.wneu.2019.12.118
8. Signorelli F, Pop R, Ganau M, et al. Endovascular versus surgical treatment for improvement of oculomotor nerve palsy caused by unruptured posterior communicating artery aneurysms. J Neurointerv Surg. 2020;12(10):964–967. doi:10.1136/neurintsurg-2020-015802
9. Connolly ES, Rabinstein AA, Carhuapoma JR, et al. Guidelines for the management of aneurysmal subarachnoid hemorrhage. Stroke. 2012;43(6):1711–1737. doi:10.1161/STR.0b013e3182587839
10. Brown RDP, Broderick JPP. Unruptured intracranial aneurysms: epidemiology, natural history, management options, and familial screening. Lancet Neurol. 2014;13(4):393–404. doi:10.1016/S1474-4422(14)70015-8
11. Tacconi L, Spinelli R, Signorelli F. Subarachnoid hemorrhage in the eighties: to treat or not to treat. J Neurosurg Sci. 2019. doi:10.23736/S0390-5616.19.04743-X
12. Bo L, Wei B, Wang Z, Li C, Gao Z, Miao Z. Bioinformatic analysis of gene expression profiling of intracranial aneurysm. Mol Med Rep. 2018;17(3):3473–3480. doi:10.3892/mmr.2017.8367
13. Yu L, Fan J, Wang S, et al. Gene expression profiles in intracranial aneurysms. Neurosci Bull. 2014;30(1):99–106. doi:10.1007/s12264-013-1398-8
14. Wei L, Wang Q, Zhang Y, et al. Identification of key genes, transcription factors and microRNAs involved in intracranial aneurysm. Mol Med Rep. 2017;17(1):891–897. doi:10.3892/mmr.2017.7940
15. Liu Y, Song Y, Liu P, et al. Comparative bioinformatics analysis between proteomes of rabbit aneurysm model and human intracranial aneurysm with label‐free quantitative proteomics. CNS Neurosci Ther. 2021;27(1):101–112. doi:10.1111/cns.13570
16. Wang J, Yu L, Zhang D, Wang S, Zhao J. Analysis of gene expression in intracranial aneurysms. Chinese Neurosurgical Journal. 2017;3(1). doi:10.1186/s41016-017-0098-z
17. Shen Y, Dong S, Liu J, et al. Identification of potential biomarkers for thyroid cancer using bioinformatics strategy: a study based on GEO datasets. Biomed Res Int. 2020;2020:1–21. doi:10.1155/2020/9710421
18. Chen D, Kong X, Shen X, et al. Identification of differentially expressed genes and signaling pathways in acute myocardial infarction based on integrated bioinformatics analysis. Cardiovasc Ther. 2019;2019:1–13. doi:10.1155/2019/8490707
19. Kang Q, Li W, Xiao J, et al. Integrated analysis of multiple microarray studies to identify novel gene signatures in preeclampsia. Placenta. 2021;105:104–118. doi:10.1016/jplacenta.2021.01.023
20. Pera J, Korostynski M, Krzyszkowski T, et al. Gene expression profiles in human ruptured and unruptured intracranial aneurysms. Stroke. 2010;41(2):224–231. doi:10.1161/STROKEAHA.109.562009
21. Wang W, Li H, Yu L, et al. Aberrant expression of lncRNAs and mRNAs in patients with intracranial aneurysm. Oncotarget. 2017;8(2):2477–2484. doi:10.18632/oncotarget.13908
22. Li L, Yang X, Jiang F, Dusting GJ, Wu Z. Transcriptome-wide characterization of gene expression associated with unruptured intracranial aneurysms. Eur Neurol. 2009;62(6):330–337. doi:10.1159/000236911
23. Weinsheimer S, Lenk GM, van der Voet M, et al. Integration of expression profiles and genetic mapping data to identify candidate genes in intracranial aneurysm. Physiol Genomics. 2007;32(1):45–57. doi:10.1152/physiolgenomics.00015.2007
24. Kleinloog R, Verweij BH, van der Vlies P, et al. RNA sequencing analysis of intracranial aneurysm walls reveals involvement of lysosomes and immunoglobulins in rupture. Stroke. 2016;47(5):1286–1293. doi:10.1161/STROKEAHA.116.012541
25. Penn DL, Witte SR, Komotar RJ, Sander connolly E. The role of vascular remodeling and inflammation in the pathogenesis of intracranial aneurysms. J Clin Neurosci. 2014;21(1):28–32. doi:10.1016/j.jocn.2013.07.004
26. Gareev I, Beylerli O, Aliev G, et al. The role of long Non-Coding RNAs in intracranial aneurysms and subarachnoid hemorrhage. Life. 2020;10(9):155. doi:10.3390/life10090155
27. Schwarze U, Schievink WI, Petty E, et al. Haploinsufficiency for one COL3A1 allele of type III procollagen results in a phenotype similar to the vascular form of Ehlers-Danlos syndrome, Ehlers-Danlos syndrome type IV. Am J Hum Genet. 2001;69(5):989–1001. doi:10.1086/324123
28. Pan Y, Lu J, Yang B, Lenahan C, Zhang J, Shao A. Construction of competitive endogenous RNA network reveals regulatory role of long non-coding RNAs in intracranial aneurysm. BMC Neurosci. 2021;22(1):15. doi:10.1186/s12868-021-00622-7
29. Islam S, Watanabe H. Versican: a dynamic regulator of the extracellular matrix. J Histochem Cytochem. 2020;68(11):763–775. doi:10.1369/0022155420953922
30. Wight TN, Kang I, Evanko SP, et al. Versican—a critical extracellular matrix regulator of immunity and inflammation. Front Immunol. 2020;11:512. doi:10.3389/fimmu.2020.00512
31. Du WW, Yang W, Yee AJ. Roles of versican in cancer biology–tumorigenesis, progression and metastasis. Histol Histopathol. 2013;28(6):701. doi:10.14670/HH-28.701
32. Keire PA, Bressler SL, Mulvihill ER, Starcher BC, Kang I, Wight TN. Inhibition of versican expression by siRNA facilitates tropoelastin synthesis and elastic fiber formation by human SK-LMS-1 leiomyosarcoma smooth muscle cells in vitro and in vivo. Matrix Biol. 2016;50:67–81. doi:10.1016/j.matbio.2015.12.010
33. Ricciardelli C, Sakko AJ, Ween MP, Russell DL, Horsfall DJ. The biological role and regulation of versican levels in cancer. Cancer Metast Rev. 2009;28(1–2):233–245. doi:10.1007/s10555-009-9182-y
34. Dos Reis DC, Damasceno KA, de Campos CB, et al. Versican and Tumor-Associated macrophages promotes tumor progression and metastasis in canine and murine models of breast carcinoma. Front Oncol. 2019;9:577. doi:10.3389/fonc.2019.00577
35. Kodama J, Kusumoto T. Prognostic significance of stromal versican expression in human endometrial cancer. Ann Oncol. 2007;18(2):269–274. doi:10.1093/annonc/mdl370
36. Labropoulou VT, Theocharis AD, Ravazoula P, et al. Versican but not decorin accumulation is related to metastatic potential and neovascularization in testicular germ cell tumours. Histopathology. 2006;49(6):582–593. doi:10.1111/j.1365-2559.2006.02558.x
37. Zhang Q, Wu J, Chen X, Zhao M, Zhang D, Gao F. Upregulation of versican associated with tumor progression, metastasis, and poor prognosis in bladder carcinoma. Biomed Res Int. 2021;2021:1–11. doi:10.1155/2021/6949864
38. Shen X, Lin W, Xu M, et al. Prognostic significance of Versican expression in gastric adenocarcinoma. Oncogenesis. 2015;4(11):e178–e178. doi:10.1038/oncsis.2015.36
39. Wang ZI, Li Z, Wang Y, et al. Versican silencing improves the antitumor efficacy of endostatin by alleviating its induced inflammatory and immunosuppressive changes in the tumor microenvironment. Oncol Rep. 2015;33(6):2981–2991. doi:10.3892/or.2015.3903
40. Rahmani M, Wong BW, Ang L, et al. Versican: signaling to transcriptional control pathways. Can J Physiol Pharm. 2006;84(1):77–92. doi:10.1139/y05-154
41. Wight TN, Merrilees MJ. Proteoglycans in atherosclerosis and restenosis. Circ Res. 2004;94(9):1158–1167. doi:10.1161/01.RES.0000126921.29919.51
42. Sathyan S, Koshy LV, Balan S, et al. Association of Versican (VCAN) gene polymorphisms rs251124 and rs2287926 (G428D), with intracranial aneurysm. Meta Gene. 2014;2:651–660. doi:10.1016/j.mgene.2014.07.001
43. Conway SJ, Izuhara K, Kudo Y, et al. The role of periostin in tissue remodeling across health and disease. Cell Mol Life Sci. 2014;71(7):1279–1288. doi:10.1007/s00018-013-1494-y
44. Kudo A, Kii I. Periostin function in communication with extracellular matrices. J Cell Commun Signal. 2018;12(1):301–308. doi:10.1007/s12079-017-0422-6
45. Xiao H, Chen J, Duan L, Li S. Role of emerging vitamin K-dependent proteins: growth arrest-specific protein 6, Gla-rich protein and periostin (Review). Int J Mol Med. 2021;47(3):1. doi:10.3892/ijmm.2020.4835
46. Idolazzi L, Ridolo E, Fassio A, et al. Periostin: the bone and beyond. Eur J Intern Med. 2017;38:12–16. doi:10.1016/j.ejim.2016.11.015
47. Ma H, Wang J, Zhao X, et al. Periostin promotes colorectal tumorigenesis through Integrin-FAK-Src Pathway-Mediated YAP/TAZ activation. Cell Rep. 2020;30(3):793–806.e6. doi:10.1016/j.celrep.2019.12.075
48. Kim D, Jeong JY, Han M, et al. Periostin is a novel histological biomarker for the diagnosis of chondroid tumor. Transl Cancer Res. 2021;10(1):434–444. doi:10.21037/tcr-20-2499
49. Dwyer O, Moore BB. The role of periostin in lung fibrosis and airway remodeling. Cell Mol Life Sci. 2017;74(23):4305–4314. doi:10.1007/s00018-017-2649-z
50. Liu C, Feng X, Wang B, et al. Bone marrow mesenchymal stem cells promote head and neck cancer progression through Periostin-mediated phosphoinositide 3-kinase/Akt/mammalian target of rapamycin. Cancer Sci. 2018;109(3):688–698. doi:10.1111/cas.13479
51. Luo W, Wang H, Hu J. Increased concentration of serum periostin is associated with poor outcome of patients with aneurysmal subarachnoid hemorrhage. J Clin Lab Anal. 2018;32(5):e22389. doi:10.1002/jcla.22389
52. Chu L, Wang F, Zhang W, Li H, Xu J, Tong X. Periostin secreted by Carcinoma-Associated fibroblasts promotes ovarian cancer cell platinum resistance through the PI3K/Akt signaling pathway. Technol Cancer Res T. 2020;19:153303382097753. doi:10.1177/1533033820977535
53. Didangelos A, Yin X, Mandal K, et al. Extracellular matrix composition and remodeling in human abdominal aortic aneurysms: a proteomics approach. Mol Cell Proteomics. 2011;10(8):
54. Yamashita O, Yoshimura K, Nagasawa A, et al. Periostin links mechanical strain to inflammation in abdominal aortic aneurysm. PLoS One. 2013;8(11):e79753. doi:10.1371/journal.pone.0079753
55. Plana E, Gálvez L, Medina P, et al. Identification of novel microRNA profiles dysregulated in plasma and tissue of abdominal aortic aneurysm patients. Int J Mol Sci. 2020;21(13):4600. doi:10.3390/ijms21134600
56. Qi L, Wu K, Shi S, Ji Q, Miao H, Bin Q. Thrombospondin-2 is upregulated in patients with aortic dissection and enhances angiotensin II-induced smooth muscle cell apoptosis. Exp Ther Med. 2020;20(6):150. doi:10.3892/etm.2020.9279
57. Rusnati M, Borsotti P, Moroni E, et al. The calcium-binding type III repeats domain of thrombospondin-2 binds to fibroblast growth factor 2 (FGF2). Angiogenesis. 2019;22(1):133–144. doi:10.1007/s10456-018-9644-3
58. Morris AH, Stamer DK, Kunkemoeller B, Chang J, Xing H, Kyriakides TR. Decellularized materials derived from TSP2-KO mice promote enhanced neovascularization and integration in diabetic wounds. Biomaterials. 2018;169:61–71. doi:10.1016/j.biomaterials.2018.03.049
59. Helkin A, Maier KG, Gahtan V. Thrombospondin-1, −2 and −5 have differential effects on vascular smooth muscle cell physiology. Biochem Biophys Res Commun. 2015;464(4):1022–1027. doi:10.1016/j.bbrc.2015.07.044
60. Kato K, Oguri M, Kato N, et al. Assessment of genetic risk factors for thoracic aortic aneurysm in hypertensive patients. Am J Hypertens. 2008;21(9):1023–1027. doi:10.1038/ajh.2008.229
61. Golledge J, Clancy P, Hankey GJ, Norman PE. Relation between serum thrombospondin-2 and cardiovascular mortality in older men screened for abdominal aortic aneurysm. Am J Cardiol. 2013;111(12):1800–1804. doi:10.1016/j.amjcard.2013.02.038
62. Mayosi BM, Fish M, Shaboodien G, et al. Identification of Cadherin 2 (CDH2) Mutations in Arrhythmogenic Right Ventricular Cardiomyopathy. Circ Cardiovasc Genet. 2017;10(2). doi:10.1161/CIRCGENETICS.116.001605
63. Lyon CA, Williams H, Bianco R, et al. Aneurysm severity is increased by combined mmp-7 deletion and n-cadherin mimetic (EC4-Fc) Over-Expression. Sci Rep-Uk. 2017;7(1). doi:10.1038/s41598-017-17700-8
64. Sulkava M, Raitoharju E, Mennander A, et al. Differentially expressed genes and canonical pathways in the ascending thoracic aortic aneurysm–the Tampere Vascular Study. Sci Rep-Uk. 2017;7(1). doi:10.1038/s41598-017-12421-4
65. Takase H, Chou SH, Hamanaka G, et al. Soluble vascular endothelial-cadherin in CSF after subarachnoid hemorrhage. Neurology. 2020;94(12):e1281–e1293. doi:10.1212/WNL.0000000000008868
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.