")
Back to Journals » Therapeutics and Clinical Risk Management » Volume 17
Identification of Hub Gene TIMP1 and Relative ceRNAs Regulatory Network in Colorectal Cancer
Authors Qin YF, Li GM, Wang G, Kong DJ, Wang HD, Zhao YM, Hao JP, Qin H, Sun DQ, Wang H
Received 20 May 2021
Accepted for publication 16 August 2021
Published 27 August 2021 Volume 2021:17 Pages 889—901
DOI https://doi.org/10.2147/TCRM.S321101
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Deyun Wang
Ya-Fei Qin,1,2 Guang-Ming Li,1,2 Grace Wang,3 De-Jun Kong,1,2 Hong-Da Wang,1,2 Yi-Ming Zhao,1,2 Jing-Peng Hao,4 Hong Qin,1,2 Da-Qing Sun,5 Hao Wang1,2
1Department of General Surgery, Tianjin Medical University General Hospital, Tianjin, People’s Republic of China; 2Tianjin General Surgery Institute, Tianjin Medical University General Hospital, Tianjin, People’s Republic of China; 3Faculty of Medicine, University of Toronto, Toronto, Ontario, Canada; 4Department of Anorectal Surgery, The Second Hospital of Tianjin Medical University, Tianjin, People’s Republic of China; 5Department of Pediatric Surgery, Tianjin Medical University, Tianjin, People’s Republic of China
Correspondence: Da-Qing Sun; Hao Wang Email [email protected]; [email protected]; [email protected]
Objective: This study aimed to discover the ceRNAs network in the pathophysiological development of human colorectal cancer (CRC) and to screen biomarkers for target therapy and prognosis by using integrated bioinformatics analysis.
Methods: Data on gene expressions of mRNAs, miRNAs, and circRNAs and clinical information were downloaded from The Cancer Genome Atlas and Gene Expression Omnibus databases, respectively. Differentially expressed mRNAs (DEmRNAs) were identified by using the DESeq2 package of R software. Functional enrichment analysis was conducted using the ClusterProfiler package of R software. The protein–protein interaction (PPI) network was shown by the STRING website. Survival analysis of hub genes was performed using the survival package in R software. Interactions among hub genes, differentially expressed miRNAs (DEmiRNAs), and differentially expressed circRNAs (DEcircRNAs) were used to construct the ceRNAs network.
Results: A total of 412 DEmRNAs including 82 upregulated and 330 downregulated genes were screened out between 473 CRC and 41 normal samples. Two hundred and sixty DEcircRNAs including 253 upregulated and 7 downregulated genes were altered between 23 CRC and 23 normal samples. One hundred and ninety DEmiRNAs including 82 upregulated and 108 downregulated genes were obtained between 450 CRC and 8 normal samples. A ceRNAs and PPI network were successfully constructed, and TIMP1 associated with prognosis was employed.
Conclusion: The present study identified a novel circRNAs-miRNAs-mRNA ceRNAs network, which implied that TIMP1 and related miRNAs, circRNAs were potential biomarkers underlying the development of CRC, providing new insights for survival predictions and therapeutic targets.
Keywords: circRNAs, colorectal cancer, ceRNAs, TIMP1, bioinformatic analysis
Introduction
Colorectal cancer (CRC) has become the predominant cancer worldwide with more than 1.8 million new cases diagnosed annually.1,2 Furthermore, the five-year survival rate for patients with advanced-stage metastatic cancer is approximately 10%.1 Like other cancers, CRC is considered to be a heterogeneous disease in which gene aberrations, cellular context, and environmental influences concur with tumor initiation, progression, and metastasis.3 Despite advances in laparoscopic and robotic surgery, more aggressive resection of metastatic disease, radiotherapy, as well as neoadjuvant and palliative chemotherapies, the new treatments had an insignificant effect on long-term survival.4 Thus, it is critical to make a thorough inquiry into the underlying biological mechanism of the occurrences and metastases of cancers associated with prognosis so as to discover novel biomarkers for target therapies and prognosis predictions. Although accumulating evidence has demonstrated that multiple genes and cellular pathways participate in the occurrence and development of CRC,5,6 a paucity of knowledge regarding the potential precise molecules and potential mechanisms underlying CRC progression currently limits the ability to treat this disease.
Bioinformatics analyses, including the analysis of gene interaction networks, microarray expression profiles, and gene annotations are being utilized as powerful tools for identifying potential diagnostic and treatment-relevant biomarkers of cancers.7,8 For example, by analyzing data from the Gene Expression Omnibus (GEO) database, Cao et al9 identified five genes as potential biomarkers and therapeutic targets for gastric cancer. In addition, by analyzing data from GEO and The Cancer Genome Atlas (TCGA), Zhu et al found that high expression of cyclin-dependent kinase 1 (CDK1) is a prognostic factor for hepatocellular carcinoma (HCC), making it a potential therapeutic target and biomarker for the diagnosis of HCC.10 In particular, the method of integrated bioinformatics analysis can be used to overcome inaccuracies in sequencing arising from small sample sizes. Circular RNAs (circRNAs) are a novel class of endogenous non-coding RNAs that form a covalently closed-continuous loop by back-splicing events via exon or intron circularization.11 Due to the development of high-throughput sequencing, researchers have discovered that thousands of circRNAs are involved in the progression of oncogenesis, invasion, and metastasis by playing the role of “sponges” to microRNAs (miRNAs).12 For instance, Wang et al13 verified that circDLGAP4 regulated lung cancer cell biological processes by sponging miRNA-143 to regulate CDK1 expression and circDLGAP4 may serve as a potential biomarker for the diagnosis and treatment of lung cancer. However, at present, most studies involving circRNAs have been limited to the sequencing of a few samples or exploring the biological function of single circRNAs. To the best of our knowledge, currently, no researchers have used integrated analysis to investigate CRC-related circRNAs.
In this study, differentially expressed mRNAs (DEmRNAs) between human CRC tissues and adjacent non-tumor tissues were identified via analysis of public TCGA datasets. Next, to explore the roles of these DEmRNAs, functional enrichment analyses and pathway enrichment analyses were performed. Then, protein–protein interaction (PPI) networks were successfully constructed. The key genes and significant modules in the networks were identified. Kaplan–Meier analysis was performed to evaluate the prognostic value of these hub genes. Furthermore, three additional circRNA expression profiles were analyzed to identify differentially expressed circRNAs (DEcircRNAs) and differentially expressed miRNAs (DEmiRNAs) between CRC and adjacent non-tumor tissues. Finally, circRNAs–miRNAs–mRNA ceRNAs network was constructed. The research is expected to help to further elucidate the ceRNAs interactions in CRC and generate insight into the potential biomarkers and targets for the diagnosis, prognosis, and therapy of CRC.
Materials and Methods
Data Collection and Preprocessing
CRC gene expression profile data were downloaded from TCGA (https://www.cancer.gov/about-nci/organization/ccg/research/structural-genomics/tcga) and standardized, including 41 normal samples and 473 tumor samples and their clinical information. Previous studies have demonstrated that without adjustment, TCGA-COAD READ data set could be generated by merging samples from TCGA-COAD data set and TCGA-READ data set, since principal components analyses and unsupervised hierarchical clustering showed no significant differences.14,15 CRC miRNA expression profiles from Illumina HiSeqmiRNASeq platforms, including 8 normal samples and 450 tumor samples, were downloaded from TCGA and standardized. In addition, 4 circRNA expression profiles (GSE121895, GSE126094, GSE138589, GSE142837) from Illumina HiSeqRNASeq platforms, including 23 tumor samples and 23 normal samples, were downloaded from GEO (http://www.ncbi.nlm.nih.gov/geo) by searching for the term “CRC” (July 2020), and batch effects were removed using the combat function in the R “sva” package.16
Identification of Differentially Expressed RNAs
DEcircRNAs, DEmRNAs, and DEmiRNAs were identified using an R package “DESeq2” (http://www.bioconductor.org/packages/release/bioc/html/DESeq2.html). |log2FC| >2 and FDR <0.05 were set as the cutoff criteria (FC, fold change; FDR, false discovery rate) based on the Benjamini‐Hochberg method for DEmRNAs and DEmiRNAs.17 DEcircRNAs were screened by |log2FC| >1 and FDR<0.05. R was used to visualize differential genes. For DEcircRNAs, we used Surrogate Variable Analysis to handle multiple GSE profiles as described above. Volcano maps were plotted based on the volcano map of R.
Functional Enrichment Analysis
To identify the biological function of the ceRNAs network, Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) analyses are widely used for gene annotation terms and pathway enrichment analysis. GO is a widely used tool for annotating genes with functions, especially molecular function (MF), biological pathways (BP), and cellular components (CC). KEGG Enrichment Analysis is a practical resource for the analytical study of gene functions and associated high-level genome functional information. ClusterProfiler package of R was performed to analyze and visualize functional profiles. A P-value < 0.05 was the threshold for significance for GO and KEGG terms.
Construction of PPI
The PPI network was conducted to analyze the functional interactions between proteins, providing insights into the mechanisms for the development of CRC. The minimum required interaction score is 0.5. The STRING website (https://string-db.org/) was employed to construct the PPI network.
Prognostic Survival Assessment of DEmRNAs
Based on the median expression level of each DEmRNAs, the CRC patients were divided into high and low-expression groups. Kaplan‐Meier analysis and the Log rank test were utilized to paint the survival curves to find the DEmRNAs that were significantly associated with the survival of CRC patients. A P-value < 0.05 was set as the threshold.
Construction of the ceRNAs Network
DEcircRNAs and DEmRNA matched by DEmiRNAs were retrieved using starBase database.18 Moreover, the prediction results of TargetScan, miRTarBase, and miRDB were integrated by starBase.19–21 The candidates searched in three databases were associated with the most important DEmiRNAs. Finally, the circRNAs–miRNAs–mRNA ceRNAs network was constructed and visualized using R.
Quantitative Real-Time PCR (qRT-PCR) of DEmiRNAs and TIMP1 in Colon Cancer and Normal Tissue
Total RNA was prepared from colonic tissue using an RNA extraction kit (TIANGEN BIOTECH, Beijing, China), according to the manufacturer’s instructions. The extracted RNA was synthesized to form cDNA using a FastKing one-step kit (TIANGEN BIOTECH, Beijing, China). qRT-PCR was performed using a RealUniversal Color PreMix (SYBR Green) kit (TIANGEN BIOTECH, Beijing, China) to assess the expression of target genes. U6 was used as an internal control for DEmiRNAs. GAPDH was used as internal control for TIMP1. In addition, the relative expression of RNAs was quantified by using the 2−ΔΔCt method.
Results
Identification of DEcircRNAs, DEmiRNAs, and DEmRNAs in Human CRC
Table 1 shows the clinicopathological data of 473 patients with CRC. According to the cutoff threshold, a total of 412 DEmRNAs (including 82 upregulated and 330 downregulated) were screened out between 473 CRC and 41 normal samples with the standard of logFC> 2 and an adjusted P value (adj.P.Val) <0.05 (Figure 1A). Two hundred and sixty DEcircRNAs (including 253 upregulated and 7 downregulated) were altered significantly between 23 CRC and 23 normal samples by log2FC > 1 and an adj.P.Val < 0.05 (Figure 1B). To further establish an circRNAs–miRNAs–mRNAs ceRNAs network, we also matched DEmiRNA expression profiles in the 450 CRC and 8 normal samples. As a result, 190 DEmiRNAs reached the inclusion criteria including 82 upregulated and 108 downregulated miRNAs (Figure 1C). The top 10 DEcircRNAs, DEmiRNAs, and DEmRNAs are presented in Table 2.
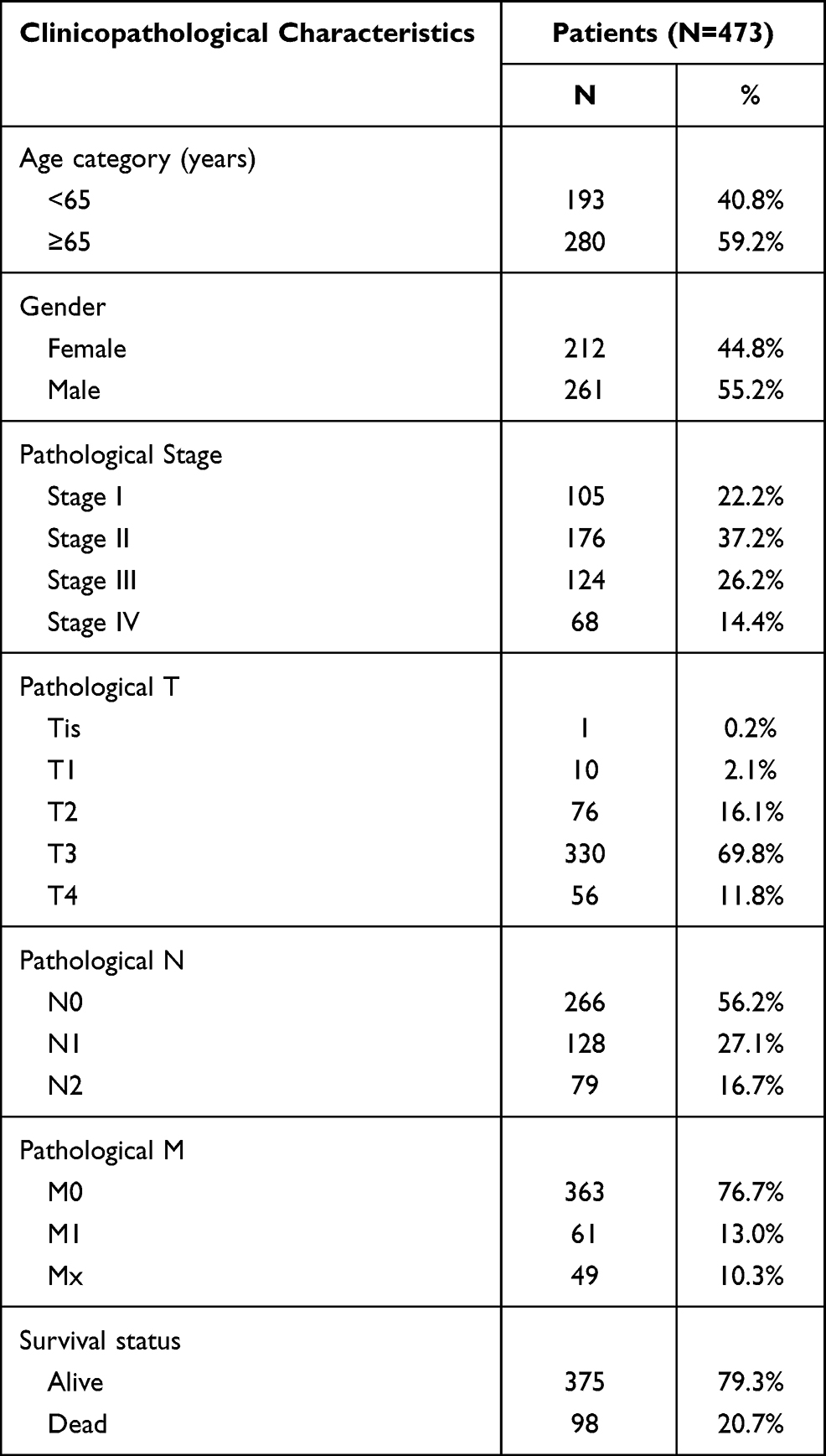 |
Table 1 Clinicopathological Characteristics of 473 CRC Patients |
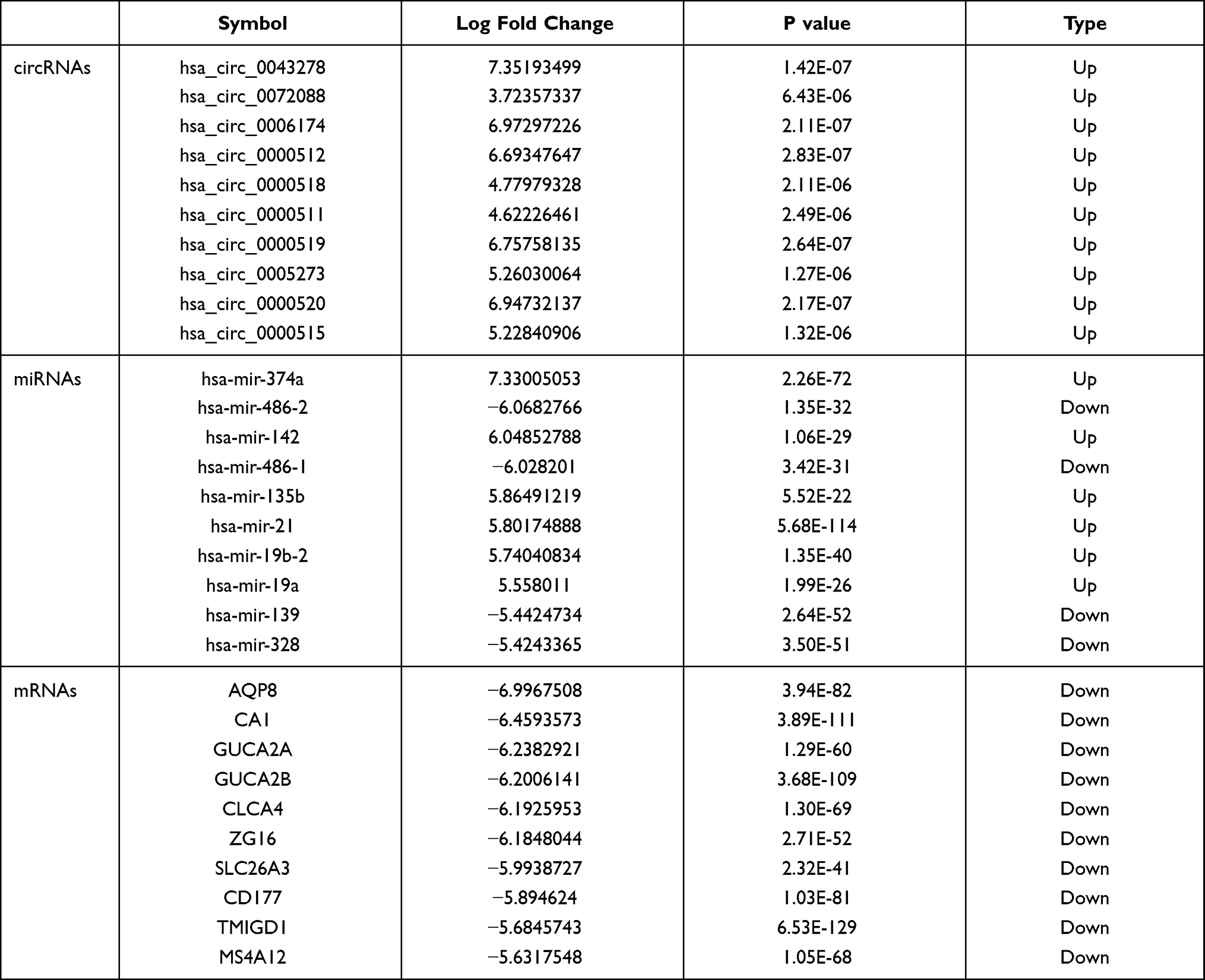 |
Table 2 Top 10 DEcircRNAs, DEmiRNAs and DEGs in Human CRC |
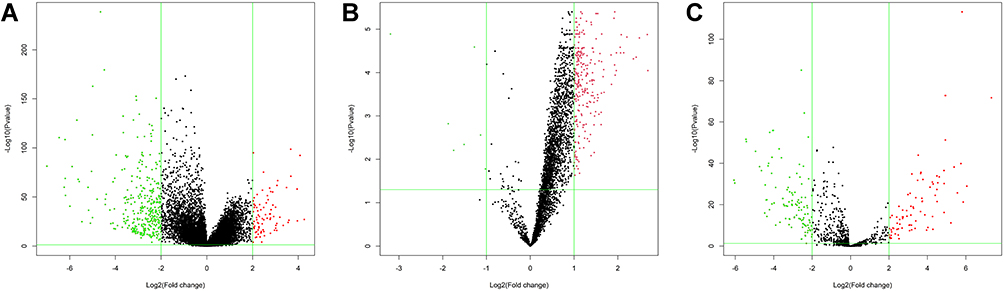 |
Figure 1 The volcano maps of DEGs between CRC samples and normal samples. (A) A total of 412 DEmRNAs including 82 upregulated and 330 downregulated genes. (B) A total of 260 DEcircRNAs including 253 upregulated and 7 downregulated genes. (C) A total of 190 DEmiRNAs including 82 upregulated and 108 downregulated genes. Red represents upregulated genes and green represents downregulated genes. |
Functional Enrichment Analysis of GO and KEGG Analysis of DEmRNAs
To further analyze the functional characteristics of DEmRNAs in CRC, GO and KEGG pathway analyses were performed using ClusterProfiler package of R. DEmRNAs were functionally classified into the biological process (BP), cellular component (CC), and molecular function (MF categories). In the BP category, four of the nine most enriched terms were “regulation of protein processing”, “protein activation cascade”, “regulation of acute inflammatory response” and “complement activation”. In the CC category, the four most enriched terms were “extracellular matrix”, “collagen-containing extracellular matrix”, “blood microparticle” and “apical part of cell”. In the MF categories, the three most enriched terms were “antigen binding”, “receptor regulator activity” and “receptor ligand activity” (Figure 2A). In addition, almost 16 KEGG pathways were significantly enriched in our analysis. The three most enriched terms were “cytokine-cytokine receptor interaction”, “kineral absorption” and “steroid hormone biosynthesis” (Figure 2B).
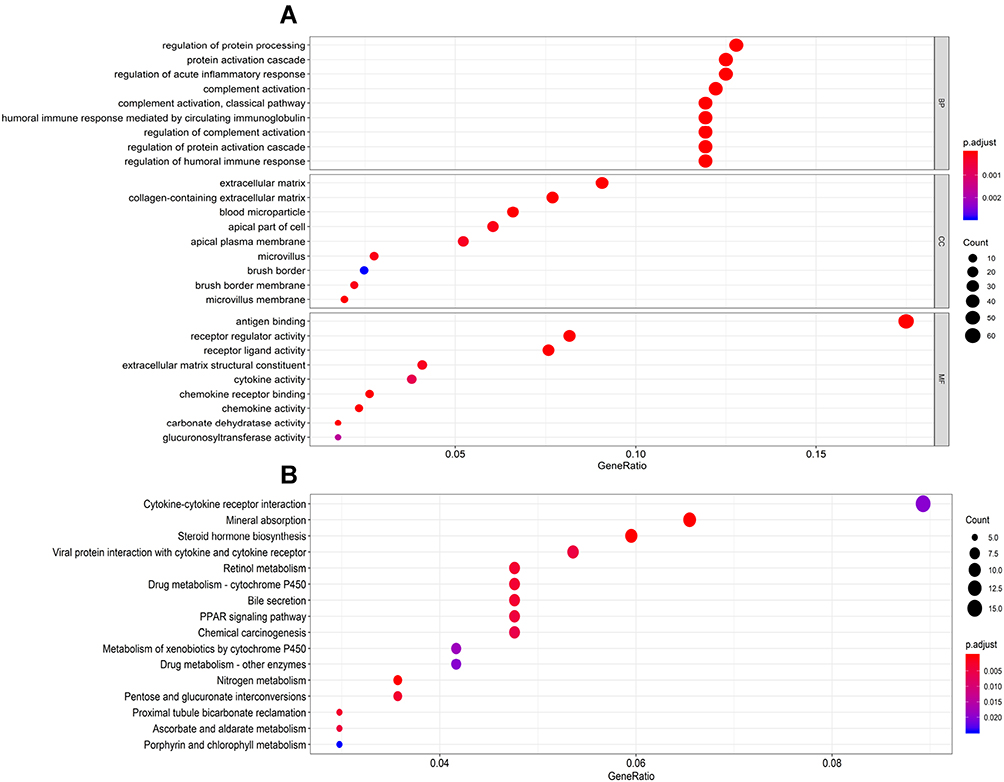 |
Figure 2 Functional enrichment analysis of DEmRNAs. (A) The top 9 enrichment scores in GO enrichment analysis of the DEmRNAs including biological process enrichment analysis, cellular components enrichment analysis, molecular function enrichment analysis. (B) The top 16 enrichment scores in KEGG enrichment analysis of the DEmRNAs. |
PPI Network Construction and Survival Analysis
A total of 412 DEmRNAs (82 upregulated and 330 downregulated) were used to construct the PPI networks, which included 226 nodes and 478 edges. The combined minimum required interaction score >0.5 was considered statistically significant (Figure 3). In addition, the degree distribution of each gene in the PPI network was analyzed, the top five genes [C-X-C chemokine receptor type 8 (CXCL8), TIMP1 (tissue inhibitor of metalloproteinase 1), CXCL1, secreted phosphoprotein 1 (SPP1) and CXCL12] with high connectivity were confirmed as hub genes and next were underwent survival analysis.
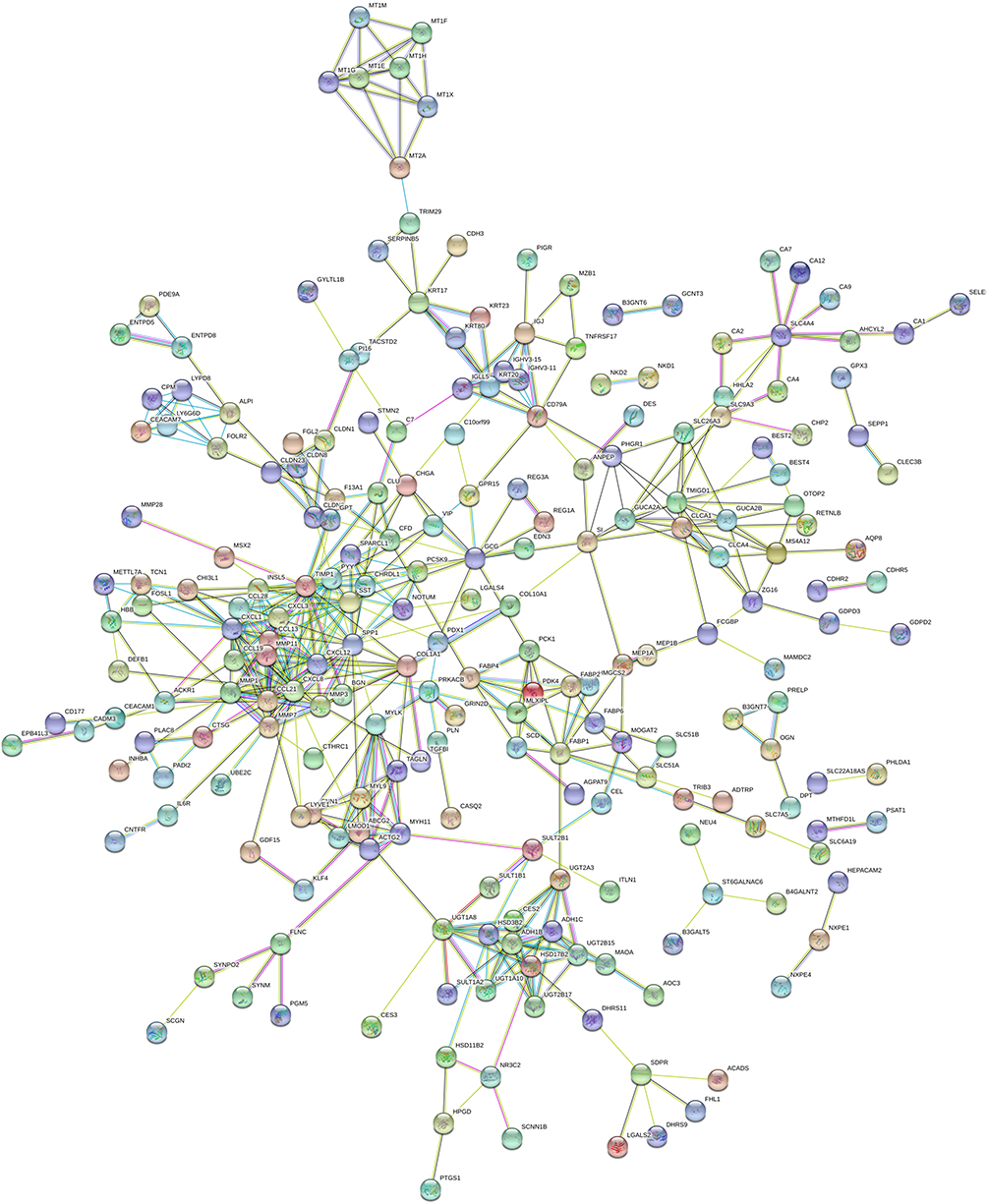 |
Figure 3 The plot of the PPI network of DEmRNAs including 226 nodes and 478 edges by the online database STRING. The combined minimum required interaction score>0.5 was considered statistically significant. |
Survival Analysis of Hub Genes
The prognostic values of the five hub genes were assessed in CRC patients using Kaplan‐Meier analysis and Log rank test. The results indicated that CRC patients with high expression of TIMP1 showed worse overall survival (P=0.004). In contrast, the other four hub genes (CXCL8, CXCL1, SPP1, and CXCL12) were not related to the overall survival of CRC patients (P > 0.05) (Figure 4).
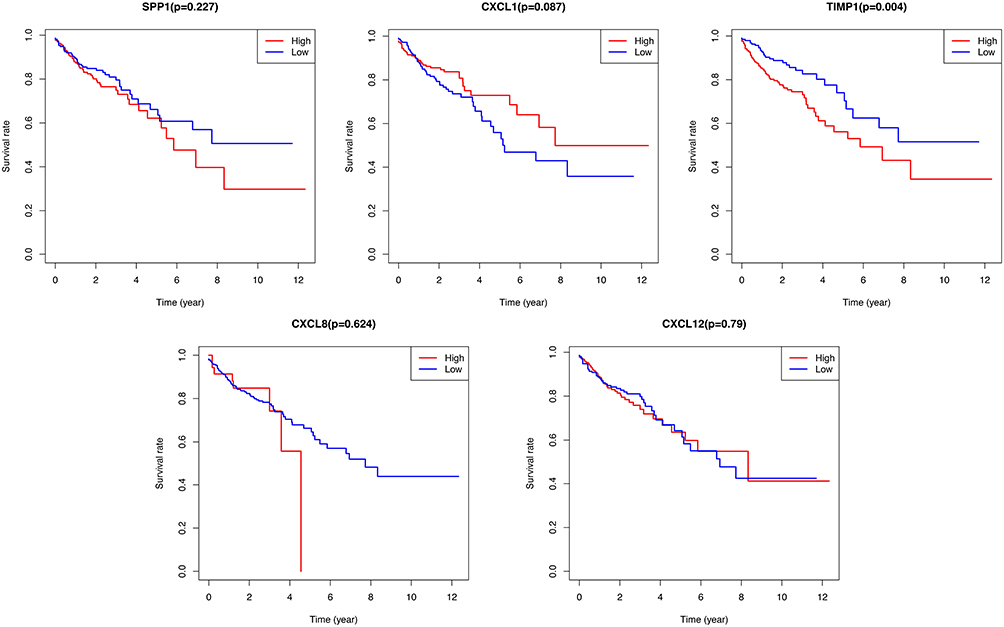 |
Figure 4 Kaplan-Meier survival curves for the top five hub genes including SPP1, CXCL1, TIMP1, CXCL8, and CXCL12. TIMP1 was significantly associated with survival rate of CRC patients. |
ceRNAs Regulatory Network Construction
miRNAs-mRNA interactivity was taken into account, in addition to the circRNAs-miRNAs, to construct an integrated ceRNAs network. Based on the starBase database, which masters the function of transcriptome-wide mircoRNA targeting prediction, we matched 61 DEcircRNAs and 3 DEmiRNAs. To clearly show the interaction in ceRNAs, the regulatory network contained some well-described biomarkers, including, hsa-miR-671-5p, hsa-miR-17-3p, hsa-miR-328-3p and TIMP1. This ceRNAs network is particularly informative in locating potential biomarkers for CRC. For instance, hsa-miR-671-5p interacted with TIMP1 and was mediated by hsa-circ-0002191. hsa-miR-17-3p interacted with TIMP1 and was mediated by has-circ-0023397 (Figure 5).
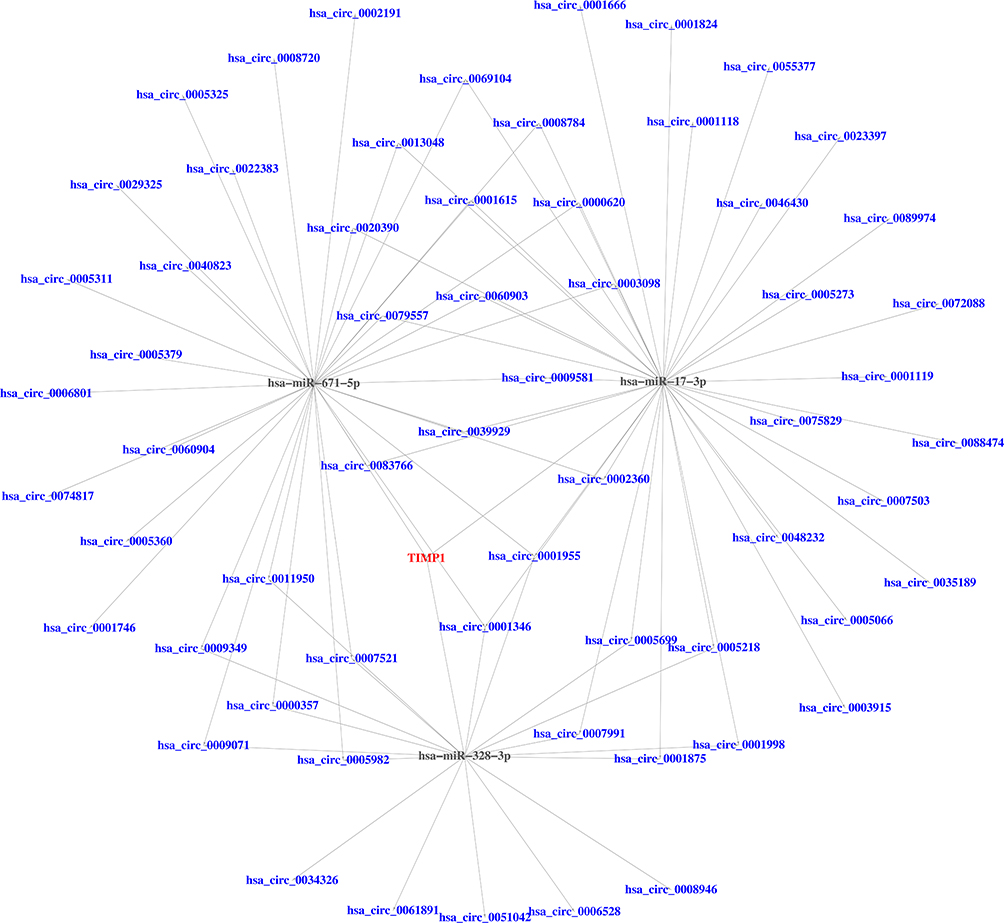 |
Figure 5 The ceRNAs network of circRNAs-miRNAs-mRNA in CRC. Blue represents DEcircRNAs; Black represents DEmiRNAs; Red represents DEmRNA. |
Expression Levels of DEmiRNAs and DEmRNAs in Human Colon Cancer Tissue
To identify the authenticity and feasibility of the ceRNAs regulatory network, some vital DEmiRNAs and DEmRNAs are evaluated in colon cancer tissue and normal tissues. We found that TIMP1 is highly expressed in colon cancer tissue compared to normal tissue (P < 0.001). In contrast, the expression levels of hsa-miR-671-5p, hsa-miR-17-3p, and hsa-miR-328-3p were significantly decreased in colon cancer tissue (Figure 6).
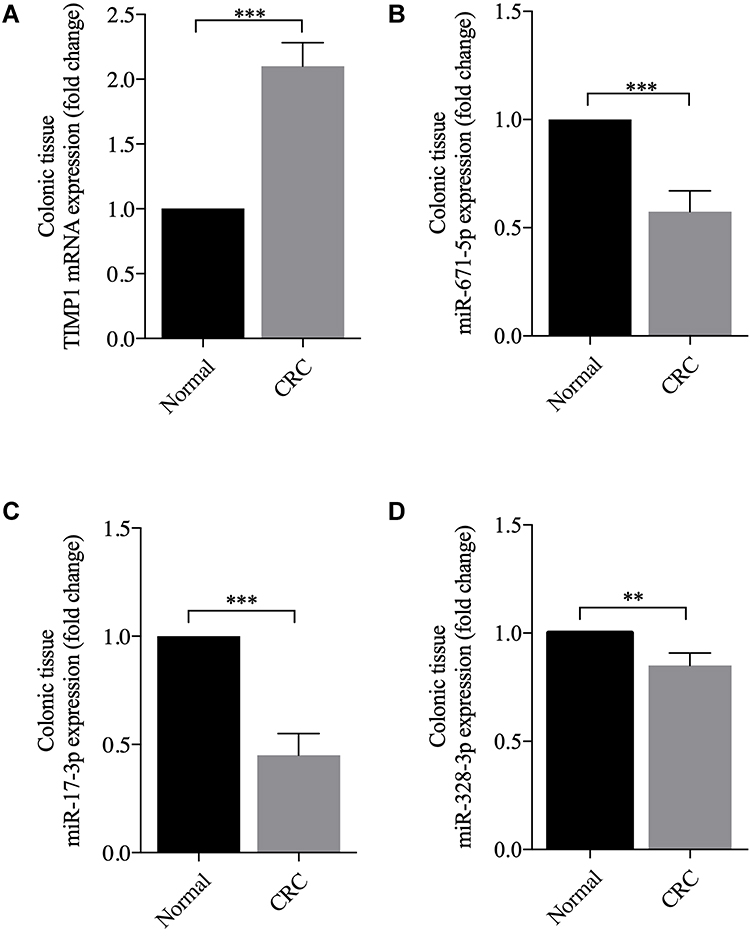 |
Figure 6 The expression levels of DEmRNA and DEmiRNAs in colon cancer patients compared with those of normal samples. (A) The TIMP1 is highly expressed in colon cancer tissue. (B–D) The miR-671-5p, miR-17-3p and miR-328-3p is low expression in colon cancer tissue, **P <0.01, and ***P <0.001. |
Discussion
CRC, the third most commonly diagnosed malignancy and the second leading cause of cancer-related deaths with notably aggressive biological behavior and poor survival rates, has always drawn close attention from researchers.2 It is crucial to identify reliable therapeutic targets and biomarkers in order to improve the clinical outcome for CRC patients. “The ceRNAs hypothesis” presents a new pattern of gene expression regulation that cicrRNAs could regulate mRNAs by competing with the corresponding miRNAs.22 Subsequently, benefits from developments in sequencing technology and the applications of bioinformatics confirm the increasingly important biological role in the initiation, progression, and metastasis of tumors.19–21 CircRNAs differ from other long non-coding RNAs in the structure, which is characterized by covalently linked 5ʹ- and 3ʹ-ends. CircRNAs functionally act as miRNAs sponges, RNA-binding protein sponges, and gene expression regulators. Therefore, circRNAs regulate their target genes expression and proteins network in both transcriptional and post-transcriptional patterns.23 Increasingly, clinicians consider that circRNAs-miRNAs-mRNAs ceRNAs networks could provide an integrated view of regulatory crosstalk between these CRC-specific RNA transcripts.24,25
In this study, DEmRNAs were identified between tumor samples and normal control tissues. Then, GO and KEGG analyses were performed to further understand the role of DEmRNAs. The results of GO analyses showed that the DEmRNAs were enriched in regulation of protein processing, protein activation cascade, and acute inflammatory which is confirmed by the knowledge that protein-induced pathology and inflammatory networks underlying CRC are the main cause for tumor development and progression.26–28 Furthermore, KEGG analyses showed that cytokine–cytokine receptor interaction is a substantial factor in the occurrence of CRC. Cytokines such as TNF-α and IL-6 are classically regarded as central players in CRC by driving activation of the NF-κB and STAT329. Cytokines including IL-11, IL-17A, and IL-22 have gained attention as facilitators of CRC.29
The top degree hub genes (CXCL8, TIMP1, CXCL1, SPP1 and CXCL12) were presented in the PPI network with DEmRNAs. SPP1, also named Osteopontin, has been proven to be overexpressed in various malignant neoplasms including breast cancer, lung cancer, and gastric cancer.30–32 Although Seo et al33 have evaluated the expression of SPP1 in 174 stage II and III CRC specimens and found SPP1 is significantly associated with cell invasion and adherence in CRC, the underlying mechanism was not revealed. Wang et al34 has shown that SPP1 functions as an enhancer of cell growth in hepatocellular carcinoma (HCC) targeted by miR-181c. Further studies have shown that SPP1 promotes the metastasis of CRC by activating epithelial-mesenchymal transition (EMT).35 CXCL8, as a prototypical chemokine, is responsible for the recruitment and activation of neutrophils and granulocytes to the site of inflammation which demonstrated that CXCL8 played a crucial role in facilitating tumor growth and progression in breast cancer, prostate cancer, lung cancer, colorectal carcinoma, and melanoma.36 Phosphorylation of Src-kinases and focal adhesion kinase (FAK) in cancer cells were increased in CXCL8 signaling, which contributed to cell proliferation and chemoresistance.37,38 The level of CXCL1 are elevated in CRC and increased level of CXCL1 are associated with tumor size, advancing stage, and patient survival.39,40 It was reported that CXCL1 could promote tumor growth by inducing angiogenesis and the recruitment of neutrophils into the tumor-associated microenvironment.41,42 CXCL1, the most abundant secreted chemokine by tumor-associated macrophages has been implicated in the promotion of breast cancer growth and metastasis via activating NF-κB/SOX4 signaling.43 The similar phenomenon has been observed in human bladder cancer.44 Some researchers have indicated that CXCL1 could increase oncogenes expression in colon cancer, including forkhead box O1 (FOXO1) and transcription factor 4 (TCF4) in CXCL1-treated SW620 cells according to transcriptome analyses.45 CXCL1 is also vital for pre-metastatic niche formation and metastasis in CRC.46 CXCL12 also known as SDF-1 is widely distributed in human tissues and more than 23 different types of cancers.47 Importantly, it has been found that CXCL4 and its ligand CXCL12 are implicated in cell proliferation, angiogenesis, migration, EMT, and tumor metastasis.48 TIMP1 belongs to the tissue inhibitor of the metalloproteinases family which includes TIMP1, TIMP2, TIMP3, and TIMP4.49 In the present study, TIMP1 has been reported to indicate poor prognosis in CRC (P=0.004), which is consistent with the research of Song et al.50 Song et al considers that the expression of TIMP1 was clearly associated with the regional lymph node and distant metastasis. In addition, research by Song et al indicated that TIMP1 was an independent prognostic indicator for the progression and metastasis of colon cancer through FAK-PI3K/AKT and MAPK pathway.50 Moreover, TIMP1 could promote receptor tyrosine kinase c-Kit signaling in CRC, while c-Kit is an important oncogene in CRC and plays a role in cell proliferation and migration.51 For other cancers, TIMP1 inhibited the chemosensitivity of breast cancer cells through the PI3K/AKT/NF-kB pathway.52 TIMP1 is in favor of cell survival in melanoma by activating the 3-phosphoinositide dependent kinase-1 signaling pathway.53 TIMP1/CD63/ERK signaling axis induces the formation of neutrophil extracellular traps and facilitates the development of pancreatic cancer.54 Clinical studies have demonstrated that the elevated level of TIMP1 was associated with poor prognosis in various tumors, such as breast cancer,55 cutaneous melanoma,56 and gastric cancer.57 The elevated plasma level of TIMP1 predicted a reduced response to second-line hormone therapy and low survival in women with metastatic breast cancer.58 Therefore, TIMP1 may be a potential biomarker to predict the prognosis of cancer and play a critical role as a therapeutic target. The TIMP-miRNAs axis has been believed to be a potential therapeutic target against aggressive or drug-resistant variants of human cancers.59–61 For instance, angiogenesis and tumor growth were increased when TIMP1 banded to CD63 and stimulated miR-210 accumulation by activating PI3K–AKT–HIF1α signaling in the lung adenocarcinoma.60 As the hub elements of the ceRNAs network, miRNAs exhibited key roles among different RNA transcripts. In fact, hsa-miR-671-5p have been proven to interact with TIMP1 directly by cross-linking immunoprecipitation.62 However, the interaction between hsa-miR-671-5p and TIMP1 still needs to be verified in the occurrence and progress of CRC. The miR-671-5p had a protective role in gastric cancer by targeting upregulator of cell proliferation.63 Meanwhile, miR-671-5p inhibits EMT by directly downregulating FOXM1 in breast cancer.64 Interestingly, the levels of miR-671-5p are not only increased in colon cancer tissue but also increased cell proliferation, migration, and invasion by targeting tripartite motif containing 67.65 This finding runs against our findings. The same miRNAs can regulate multiple mRNAs molecules and produce different physiological effects. MiR-328-3p was identified in bladder cancer and suppressed cell proliferation, migration, and invasion by targeting integrin subunit alpha 5 as well as by inhibiting EMT and inactivated PI3K/AKT pathway.66 Similar tumor suppression effects were observed in colon cancer.67
The present study identifies a novel ceRNAs network, which implies that TIMP1 is a potential biomarker underlying the development of CRC, providing new insights into survival predictions and therapeutic targets. However, the limitation of the study still needs investigation. Too many circRNAs were chosen in this study, a more advanced approach to narrow the scope of research is needed. The results of the present CRC-related ceRNAs regulatory network are required to be verified by clinical trials and molecular experiments.
Conclusion
The present study identified a novel circRNAs-miRNAs-mRNA ceRNAs network and provided candidate prognostic biomarkers for predicting the outcome of patients with CRC. Especially, TIMP1 is a potential indicator underlying the development of CRC. This study provided new insights for the survival predictions and therapeutic targets of CRC.
Ethics Approval and Consent to Participate
All the experiments were approved by the Ethics Committee of Tianjin Medical University General Hospital (Tianjin, China).
Author Contributions
All authors contributed to data analysis, drafting or revising the article, gave final approval for the version to be published, agreed to the submitted journal, and agreed to be accountable for all aspects of the work. Ya-Fei Qin, Guang-Ming Li and Grace Wang are co-first authors of this paper.
Funding
This work was supported by grants to Hao Wang from the National Natural Science Foundation of China (No. 82071802), Tianjin Application Basis and Cutting-Edge Technology Research Grant (No. 14JCZDJC35700), Li Jieshou Intestinal Barrier Research Special Fund (No. LJS_201412), Natural Science Foundation of Tianjin (No. 18JCZDJC35800), and Tianjin Medical University Talent Fund; by a grant to Dejun Kong from Tianjin Research Innovation Project for postgraduate students (No. 2019YJSS184).
Disclosure
The authors declare that they have no competing interests.
References
1. Kuipers EJ, Grady WM, Lieberman D, et al. Colorectal cancer. Nat Rev Dis Primers. 2015;1(1):15065. doi:10.1038/nrdp.2015.65
2. Keum N, Giovannucci E. Global burden of colorectal cancer: emerging trends, risk factors and prevention strategies. Nat Rev Gastroenterol Hepatol. 2019;16(12):713–732.
3. Murphy N, Ward HA, Jenab M, et al. Heterogeneity of colorectal cancer risk factors by anatomical subsite in 10 European countries: a multinational cohort study. Clin Gastroenterol Hepatol. 2019;17(7):1323–1331.e1326.
4. Wahab SMR, Islam F, Gopalan V, Lam AK. The identifications and clinical implications of cancer stem cells in colorectal cancer. Clin Colorectal Cancer. 2017;16(2):93–102.
5. Lech G, Słotwiński R, Słodkowski M, Krasnodębski IW. Colorectal cancer tumour markers and biomarkers: recent therapeutic advances. World J Gastroenterol. 2016;22(5):1745–1755.
6. Liang B, Li C, Zhao J. Identification of key pathways and genes in colorectal cancer using bioinformatics analysis. Med Oncol. 2016;33(10):111.
7. Lu DY, Qu RX, Lu TR, Wu HY. Cancer bioinformatics for updating anticancer drug developments and personalized therapeutics. Rev Recent Clin Trials. 2017;12(2):101–110.
8. Tao Z, Shi A, Li R, Wang Y, Wang X, Zhao J. Microarray bioinformatics in cancer- a review. J BUON. 2017;22(4):838–843.
9. Cao L, Chen Y, Zhang M, et al. Identification of hub genes and potential molecular mechanisms in gastric cancer by integrated bioinformatics analysis. Peer J. 2018;6:e5180.
10. Zhu Q, Sun Y, Zhou Q, He Q, Qian H. Identification of key genes and pathways by bioinformatics analysis with TCGA RNA sequencing data in hepatocellular carcinoma. Mol Clin Oncol. 2018;9(6):597–606.
11. Ebbesen KK, Kjems J, Hansen TB. Circular RNAs: identification, biogenesis and function. Biochim Biophys Acta. 2016;1859(1):163–168.
12. Hsiao KY, Sun HS, Tsai SJ. Circular RNA - new member of noncoding RNA with novel functions. Exp Biol Med. 2017;242(11):1136–1141.
13. Wang B, Hua P, Zhao B, Li J, Zhang Y. Circular RNA circDLGAP4 is involved in lung cancer development through modulating microRNA-143/CDK1 axis. Cell Cycle. 2020;1–11:2007–2017.
14. Wang X, Wang D, Liu J, Feng M, Wu X. A novel CpG-methylation-based nomogram predicts survival in colorectal cancer. Epigenetics. 2020;15(11):1213–1227.
15. Wang X, Wang D, Zhang H, Feng M, Wu X. Genome-wide analysis of DNA methylation identifies two CpG sites for the early screening of colorectal cancer. Epigenomics. 2020;12(1):37–52.
16. Leek JT, Storey JD. Capturing heterogeneity in gene expression studies by surrogate variable analysis. PLoS Genet. 2007;3(9):1724–1735.
17. Madar V, Batista S. FastLSU a more practical approach for the Benjamini-Hochberg FDR controlling procedure for huge-scale testing problems. Bioinformatics. 2016;32(11):1716–1723.
18. Li JH, Liu S, Zhou H, Qu LH, Yang JH. starBase v2.0: decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res. 2014;42(Database issue):D92–97.
19. Agarwal V, Bell GW, Nam JW, Bartel DP. Predicting effective microRNA target sites in mammalian mRNAs. eLife. 2015;4:e05005.
20. Chou CH, Chang NW, Shrestha S, et al. miRTarBase 2016: updates to the experimentally validated miRNA-target interactions database. Nucleic Acids Res. 2016;44(D1):D239–247.
21. Wong N, Wang X. miRDB: an online resource for microRNA target prediction and functional annotations. Nucleic Acids Res. 2015;43(Databaseissue):D146–152.
22. Heery R, Finn SP, Cuffe S, Gray SG. Long non-coding RNAs: key regulators of epithelial-mesenchymal transition, tumour drug resistance and cancer stem cells. Cancers. 2017;9:4.
23. Jiang C, Li X, Zhao H, Liu H. Long non-coding RNAs: potential new biomarkers for predicting tumor invasion and metastasis. Mol Cancer. 2016;15(1):62.
24. Hsiao KY, Lin YC, Gupta SK, et al. Noncoding effects of circular RNA CCDC66 promote colon cancer growth and metastasis. Cancer Res. 2017;77(9):2339–2350.
25. Zeng K, Chen X, Xu M, et al. CircHIPK3 promotes colorectal cancer growth and metastasis by sponging miR-7. Cell Death Dis. 2018;9(4):417.
26. Ko PJ, Dixon SJ. Protein palmitoylation and cancer. EMBO Rep. 2018;19:10.
27. Saini J, Sharma PK. Clinical, prognostic and therapeutic significance of heat shock proteins in cancer. Curr Drug Targets. 2018;19(13):1478–1490.
28. Murata M. Inflammation and cancer. Environ Health Prev Med. 2018;23(1):50.
29. West NR, McCuaig S, Franchini F, Powrie F. Emerging cytokine networks in colorectal cancer. Nat Rev Immunol. 2015;15(10):615–629.
30. Giopanou I, Lilis I, Papaleonidopoulos V, et al. Tumor-derived osteopontin isoforms cooperate with TRP53 and CCL2 to promote lung metastasis. Oncoimmunology. 2017;6(1):e1256528.
31. Raja UM, Gopal G, Shirley S, Ramakrishnan AS, Rajkumar T. Immunohistochemical expression and localization of cytokines/chemokines/growth factors in gastric cancer. Cytokine. 2017;89:82–90.
32. Sangaletti S, Tripodo C, Sandri S, et al. Osteopontin shapes immunosuppression in the metastatic niche. Cancer Res. 2014;74(17):4706–4719.
33. Seo KJ, Kim M, Kim J. Prognostic implications of adhesion molecule expression in colorectal cancer. Int J Clin Exp Pathol. 2015;8(4):4148–4157.
34. Wang J, Hao F, Fei X, Chen Y. SPP1 functions as an enhancer of cell growth in hepatocellular carcinoma targeted by miR-181c. Am J Transl Res. 2019;11(11):6924–6937.
35. Xu C, Sun L, Jiang C, et al. SPP1, analyzed by bioinformatics methods, promotes the metastasis in colorectal cancer by activating EMT pathway. Biomed Pharmacother/Biomedecine & Pharmacotherapie. 2017;91:1167–1177.
36. Liu Q, Li A, Tian Y, et al. The CXCL8-CXCR1/2 pathways in cancer. Cytokine Growth Factor Rev. 2016;31:61–71.
37. Kopetz S, Shah AN, Gallick GE. Src continues aging: current and future clinical directions. Clin Cancer Res. 2007;13(24):7232–7236.
38. Siesser PM, Hanks SK. The signaling and biological implications of FAK overexpression in cancer. Clin Cancer Res. 2006;12(11 Pt 1):3233–3237.
39. Ogata H, Sekikawa A, Yamagishi H, et al. GROα promotes invasion of colorectal cancer cells. Oncol Rep. 2010;24(6):1479–1486.
40. Wang D, Dubois RN, Richmond A. The role of chemokines in intestinal inflammation and cancer. Curr Opin Pharmacol. 2009;9(6):688–696.
41. Yuan M, Zhu H, Xu J, Zheng Y, Cao X, Liu Q. Tumor-derived CXCL1 promotes lung cancer growth via recruitment of tumor-associated neutrophils. J Immunol Res. 2016;2016:6530410.
42. Yu PF, Huang Y, Han YY, et al. TNFα-activated mesenchymal stromal cells promote breast cancer metastasis by recruiting CXCR2(+) neutrophils. Oncogene. 2017;36(4):482–490.
43. Wang N, Liu W, Zheng Y, et al. CXCL1 derived from tumor-associated macrophages promotes breast cancer metastasis via activating NF-κB/SOX4 signaling. Cell Death Dis. 2018;9(9):880.
44. Miyake M, Hori S, Morizawa Y, et al. CXCL1-mediated interaction of cancer cells with tumor-associated macrophages and cancer-associated fibroblasts promotes tumor progression in human bladder cancer. Neoplasia. 2016;18(10):636–646.
45. Hsu YL, Chen YJ, Chang WA, et al. Interaction between tumor-associated dendritic cells and colon cancer cells contributes to tumor progression via CXCL1. Int J Mol Sci. 2018;19:8.
46. Wang D, Sun H, Wei J, Cen B, DuBois RN. CXCL1 is critical for premetastatic niche formation and metastasis in colorectal cancer. Cancer Res. 2017;77(13):3655–3665.
47. Bertolini G, D’Amico L, Moro M, et al. Microenvironment-modulated metastatic CD133+/CXCR4+/EpCAM- lung cancer-initiating cells sustain tumor dissemination and correlate with poor prognosis. Cancer Res. 2015;75(17):3636–3649.
48. Onoue T, Uchida D, Begum NM, Tomizuka Y, Yoshida H, Sato M. Epithelial-mesenchymal transition induced by the stromal cell-derived factor-1/CXCR4 system in oral squamous cell carcinoma cells. Int J Oncol. 2006;29(5):1133–1138.
49. Batra J, Robinson J, Soares AS, Fields AP, Radisky DC, Radisky ES. Matrix metalloproteinase-10 (MMP-10) interaction with tissue inhibitors of metalloproteinases TIMP-1 and TIMP-2: binding studies and crystal structure. J Biol Chem. 2012;287(19):15935–15946.
50. Song G, Xu S, Zhang H, et al. TIMP1 is a prognostic marker for the progression and metastasis of colon cancer through FAK-PI3K/AKT and MAPK pathway. J Exp Clin Cancer Res. 2016;35(1):148.
51. Nordgaard C, Doll S, Matos A, et al. Metallopeptidase inhibitor 1 (TIMP-1) promotes receptor tyrosine kinase c-Kit signaling in colorectal cancer. Mol Oncol. 2019;13(12):2646–2662.
52. Fu ZY, Lv JH, Ma CY, Yang DP, Wang T. Tissue inhibitor of metalloproteinase-1 decreased chemosensitivity of MDA-435 breast cancer cells to chemotherapeutic drugs through the PI3K/AKT/NF-small ka, CyrillicB pathway. Biomed Pharmacother. 2011;65(3):163–167.
53. Toricelli M, Melo FHM, Hunger A, Zanatta D, Strauss BE, Jasiulionis MG. Timp1 promotes cell survival by activating the PDK1 signaling pathway in melanoma. Cancers. 2017;9:4.
54. Schoeps B, Eckfeld C, Prokopchuk O, et al. TIMP1 triggers neutrophil extracellular trap formation in pancreatic cancer. Cancer Res. 2021;81(13):3568–3579.
55. Wurtz SO, Schrohl AS, Mouridsen H, Brunner N. TIMP-1 as a tumor marker in breast cancer–an update. Acta Oncol. 2008;47(4):580–590.
56. Zurac S, Neagu M, Constantin C, et al. Variations in the expression of TIMP1, TIMP2 and TIMP3 in cutaneous melanoma with regression and their possible function as prognostic predictors. Oncol Lett. 2016;11(5):3354–3360.
57. Wang YYLL, Zhao ZS, Wang HJ. Clinical utility of measuring expression levels of KAP1, TIMP1 and STC2 in peripheral blood of patients with gastric cancer. World J Surg Oncol. 2013;11:81.
58. Lipton A, Ali SM, Leitzel K, et al. Elevated plasma tissue inhibitor of metalloproteinase-1 level predicts decreased response and survival in metastatic breast cancer. Cancer. 2007;109(10):1933–1939.
59. Ivanovic RF, Viana NI, Morais DR, et al. miR-618: possible control over TIMP-1 and its expression in localized prostate cancer. BMC Cancer. 2018;18(1):992.
60. Cui H, Seubert B, Stahl E, et al. Tissue inhibitor of metalloproteinases-1 induces a pro-tumourigenic increase of miR-210 in lung adenocarcinoma cells and their exosomes. Oncogene. 2015;34(28):3640–3650.
61. Huang Y, Chang JT, Liao CT, et al. OncomiR-196 promotes an invasive phenotype in oral cancer through the NME4-JNK-TIMP1-MMP signaling pathway. Mol Cancer. 2014;13:218.
62. Balakrishnan I, Yang X, Brown J, et al. Genome-wide analysis of miRNA-mRNA interactions in marrow stromal cells. Stem Cells (Dayton, Ohio). 2014;32(3):662–673.
63. Qiu T, Wang K, Li X, Jin J. miR-671-5p inhibits gastric cancer cell proliferation and promotes cell apoptosis by targeting URGCP. Exp Ther Med. 2018;16(6):4753–4758.
64. Tan X, Fu Y, Chen L, et al. miR-671-5p inhibits epithelial-to-mesenchymal transition by downregulating FOXM1 expression in breast cancer. Oncotarget. 2016;7(1):293–307.
65. Jin W, Shi J, Liu M. Overexpression of miR-671-5p indicates a poor prognosis in colon cancer and accelerates proliferation, migration, and invasion of colon cancer cells. Onco Targets Ther. 2019;12:6865–6873.
66. Yan T, Ye XX. MicroRNA-328-3p inhibits the tumorigenesis of bladder cancer through targeting ITGA5 and inactivating PI3K/AKT pathway. Eur Rev Med Pharmacol Sci. 2019;23(12):5139–5148.
67. Pan S, Ren F, Li L, et al. MiR-328-3p inhibits cell proliferation and metastasis in colorectal cancer by targeting Girdin and inhibiting the PI3K/Akt signaling pathway. Exp Cell Res. 2020;390(1):111939.
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.