")
Back to Journals » Research Reports in Clinical Cardiology » Volume 14
Identification of Diagnostic Signatures in Ischemic Cardiomyopathy Patients via Bioinformatics Analysis Integrated with Machine Learning
Authors Luo Y, Zhao J, Chen X , Huang R, Hou L , Su K, Lei Y, Li Y
Received 9 January 2023
Accepted for publication 13 April 2023
Published 18 April 2023 Volume 2023:14 Pages 13—20
DOI https://doi.org/10.2147/RRCC.S399277
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Richard Kones
Yinhua Luo,1,* Jinbo Zhao,2,* Xueru Chen,3,* Rui Huang,2 Ling Hou,2 Ke Su,2 Yuhua Lei,2 Yuanhong Li1,2
1Department of Central Hospital of Tujia and Miao Autonomous Prefecture, Hubei University of Medicine, Shiyan, Hubei Province, 442000, People’s Republic of China; 2Cardiovascular Disease Center, Central Hospital of Tujia and Miao Autonomous Prefecture, Hubei University of Medicine, Enshi Prefecture, Hubei Province, 445000, People’s Republic of China; 3Department of General Practice, Sun Yat-sen Memorial Hospital of Sun Yat-sen University, Guangzhou, Guangdong Province, 510120, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Yuanhong Li, Cardiovascular Disease Center, Central Hospital of Tujia and Miao Autonomous Prefecture, Hubei University of Medicine, Enshi Prefecture, People’s Republic of China, Email [email protected]
Background: Ischemic cardiomyopathy (ICM) with high mobility and mortality is closely linked to immunology, oxidative stress, inflammatory response and so on. Early diagnosis counts for the effective treatment of ICM. However, there are still no distinctive diagnostic signatures. This research aims to investigate effective signatures and build the diagnostic model for ICM.
Methods: The Gene Expression Omnibus was used to retrieve the microarray data of GSE9800 and GSE580, which were obtained from tissue biopsy samples. Differentially expressed genes (DEGs), GO, and KEGG analyses were then carried out on the microarray data. The PPI network was constructed via STRING database.Following that, CIBERSORT techniques in conjunction with the LM22 feature matrix were used to assess the immune infiltration of the samples.The expression of a few chosen genes served as the predictor variable, and the occurrence of ICM served as the responder variable, in the construction of the best subset stepwise regression model.
Results: A total of 28 DEGs were found. And according to the GO and KEGG studies, numerous biological processes were enriched. Patients with ICM and their normal counterparts had considerably distinct immune cell types infiltrating. For the construction of the PPI network, the top 20 most significant DEGs were selected and were used to build the original regression model. The optimal subset screened using stepwise regression analysis contained three pivotal genes (SCD, SNX25, WNT7B) and the area under the curve (AUC) values in this model was 0.891.
Conclusion: We identified several possible hub genes, including SCD, SNX25, and WNT7B, which may be strongly related to the development of ICM. Based on the three genes, the logistic regression model could be used to accurately diagnose ICM patients.
Keywords: ischemic cardiomyopathy, ICM, immunology, inflammatory responses, diagnosis, differently expressed genes, bioinformatic analysis, the optimal subset stepwise regression
Introduction
Ischemic cardiomyopathy (ICM), characterized by high mobility and high mortality, is so far one of the most prevalent etiologies of congestive heart failure.1 The development of left ventricular (LV) systolic dysfunction, which is typically brought on by prior acute myocardial infarction(s), or, alternatively, a sneaky process of gradual deterioration in systolic function without discernible episodes of acute coronary syndromes, is the mechanism by which ischemic heart disease results in heart failure (HF). Thus, the term ischemic cardiomyopathy describes the syndrome of HF due to chronic LV systolic dysfunction resulting from underlying coronary artery disease (CAD). It is estimated that about 125 million people worldwide have ICM, and that 720,000 people in the United States experience their first myocardial infarction each year, resulting in ICM.2–4 ICM ultimately has a great impact on people’s lives and even society as a whole.
The etiology of ICM is currently considered to be associated with immunology, oxidative stress, inflammatory response, living habits, and genetic susceptibility. ICM is diagnosed primarily through clinical and medical history examination, which is supported by histologic, cardiac ultrasonography and radiologic. It is characterized pathologically an enlarged heart with markedly reduced left ventricular systolic function and clinically by non-specific symptoms such as arrhythmias, heart failure and embolism, while the clinical symptoms are similar to those of dilated cardiomyopathy, with a low degree of specificity, leading to a high incidence of clinical misdiagnosis. As a result, research into precise diagnostic biomarkers for ICM is critical.
As a branch of information science, machine learning (ML) trains computers to complete tasks by identifying patterns in large datasets and then using those patterns to derive rules or algorithms that could maximize assignment production.5,6 Clinical uses of machine learning in ischemic heart disease patients have been studied.7–9 Logistic regression models, a typical IHD model, are used to forecast the outcomes of future instances rested on the individual’s predictive factors, hence identifying the underlying relationships between predictors and outcomes. Prior research frequently concentrated on identifying ICM risk variables by logistic regression models.7–9 The utility of optimal subset stepwise regression model to investigate biomarkers and maybe diagnose ICM is still not obvious, nevertheless.
In the current study, we coupled bioinformatic analysis with machine learning to identify the significant genes of ICM and found that many particular putative hub genes, including SCD, SNX25, and WNT7B, may be strongly connected with ICM. In this work, the stepwise regression model based on these three genes is capable of accurately forecasting the occurrence of ICM.
Materials and Methods
Data Source
The study was approved by the Ethics Committee of the Central Hospital of Enshi Autonomous Prefecture.The GEO was queried for two sets of expression profile data derived from tissue biopsies and associated clinical information ((http://www.ncbi.nlm.nih.gov/geo/). The GSE9800 dataset (quantified by Agilent-012097 Human 1A Microarray (V2) G4110B microarray platform) contained cardiac muscle samples from 2 ICM patients and 11 normal cardiac muscle samples. The GSE580 dataset (quantified by Affymetrix Human Genome U133 Plus 2.0 Array microarray platform) contained myocardial samples from 20 ICM patients. Above all, totally 22 ICM patients and 11 control group were included and integrated into one sample set, and then the COMBAT package was used to remove the batch effect between different datasets.
Identification of Differentially Expressed Genes (DEGs)
The bladderbatch R package was used to correct batch effects between GSE9800 and GSE570. Additionally, the samples from the GSE9800, GSE580 were subjected to a differential gene expression analysis using the limma R package [20]. After quantile normalization, the raw signals from microarrays were log2-transformed. The absolute value of fold change > 2 (|log2FC|> 1) and P value<0.05 (Student’s t-test) were used to filter the differentially expressed genes (DEG).
Functions and Pathways Enrichment Analysis
Using Gene Ontology (GO) enrichment analysis, the cellular components, biological processes, and molecular functions of DEGs were determined. Using Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis, the gene cluster’s pathway and associated functions were determined. The R cluster package was used to investigate GO enrichment and KEGG pathway analysis with an adjusted P<0.05 cut off.
PPI Analysis
Protein-protein interaction (PPI) networks were developed in our study using the Search Tool for the Retrieval of Interacting Genes/Proteins (STRING) online to identify functional relationships between DEGs. Cytoscape was used to visualize PPI networks. The Cytoscape plugin Cytohubba was used to determine the degree of each protein node in a co-expressed network. The top ten genes in our analysis were chosen as hub genes.
Statistical Analysis
The findings are shown as MeanSD. The scores of two different groups were compared using t-tests or non parametric tests. Statistical significance was defined as a two-tailed value of P<0.05.Use the optimal subset stepwise regression method (backward method) to screen out the strongest correlation with ICM from the pivotal genes and build a model. Statistics were done using R 4.0.5.
Results
Different Expressed Genes
We integrated the two datasets into one sample set, and then used the COMBAT package to remove the batch effect between different datasets (Figure 1). In the comparison between the two groups (ICM patients vs the normal counterpart), 92 DEGS, of which 28 genes were up-regulated and 3 genes were down-regulated in the ICM patients, were found (Figure 2A), as well as significant different in expression between the two groups (Figure 2B), suggesting that these genes were likely involved in the development of ICM.
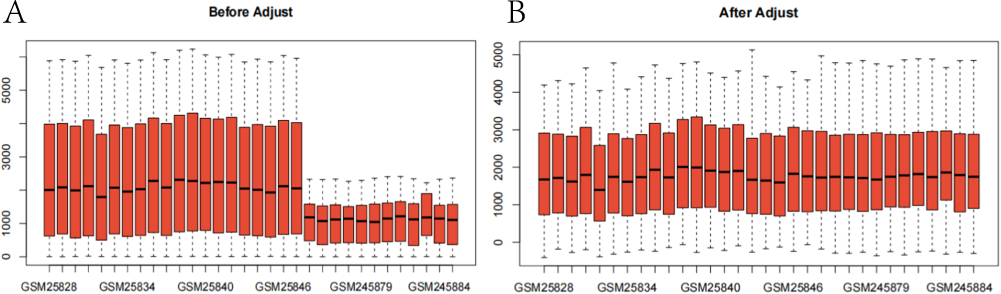 |
Figure 1 (A) depicts the box plot of gene expression of the datasets prior to correction; it is evident that the median gene expression of each sample is not linear. (B) In the box plot of gene expression of the datasets after debatching, the median gene expression of all samples is approximately a straight line, indicating that the debatching effect has been met. |
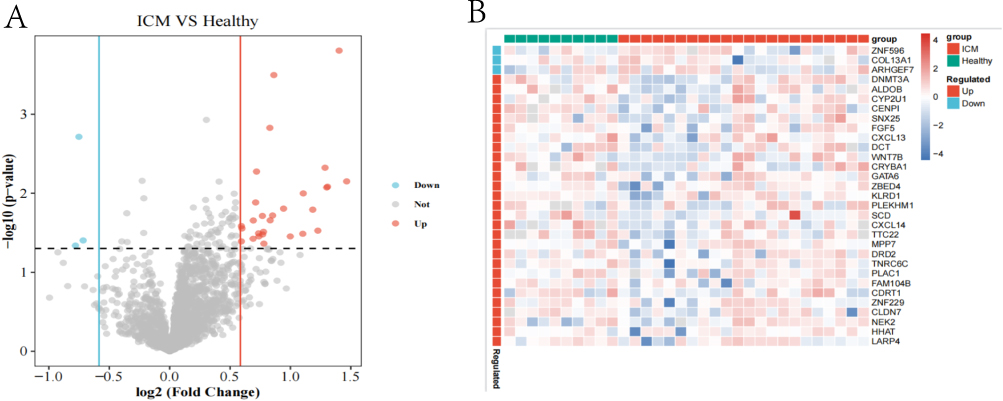 |
Figure 2 (A) The volcano diagram depicts genes with varying expression. X-axis: log2FC, Y-axis: log10 (FDR). Down-regulated genes are shown by blue dots; down-regulated genes are represented by blue dots. (B) The heat map exhibited differentially expressed genes. The samples are on the X-axis. Y-axis: various genes. |
Significantly Enriched GO Terms and KEGG Pathways
The clusterProfler package of R was used to perform Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis for the 1856 DEGs that were found. Based on the 1082 differentially expressed genes (Supplementary Table 1) and the top 32 significantly enriched GO items and top 13 most significantly enriched KEGG pathways (Supplementary Table 2), numerous GO terms and KEGG pathways were considerably enriched. KEGG pathways were shown in Figures 3. The GO and KEGG analyses showed that a number of key biological processes and signaling pathways, including the inhibition of protein kinase B signaling and cell junction assembly, were considerably enriched (Figure 3).
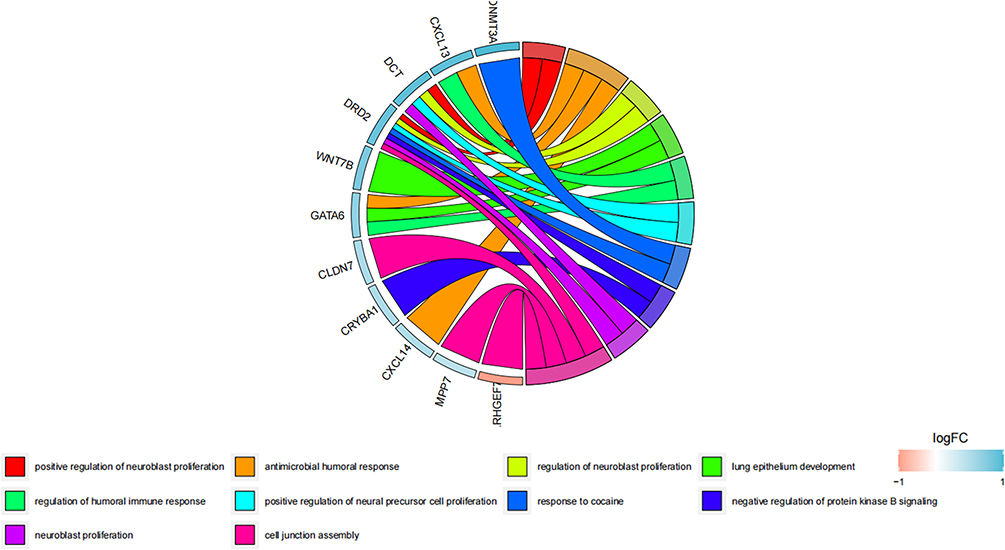 |
Figure 3 Analyses of functional enrichment. The top ten GO terms with the most substantial enrichment. The right semicircle represents these 10 Gene Ontology (GO) concepts, whereas the left semicircle indicates the genes that were enriched for these ten GO items. |
Immune Cell Infiltration
Our functional analysis, which was based on the 92 genes that displayed differential expression, was able to identify immune-related pathways as well. There was a significant difference between the ICM and healthy subjects in the composition of immune cells (Figure 4A). CD4 naive T cells, CD4 memory resting T cells, Dendritic cells activate, Dendritic cells resting, Macrophages M0, Macrophages M2, Mast cells resting and NK resting cells were significantly higher in the ICM subjects (Figure 4B). Otherwise, Eosinophils, Mast cells activated, Monocyte, Plasma cells, T cells CD4 memory resting, T cells CD4 naive and T cells regulatory were much lower in the ICM subjects (Figure 4B).
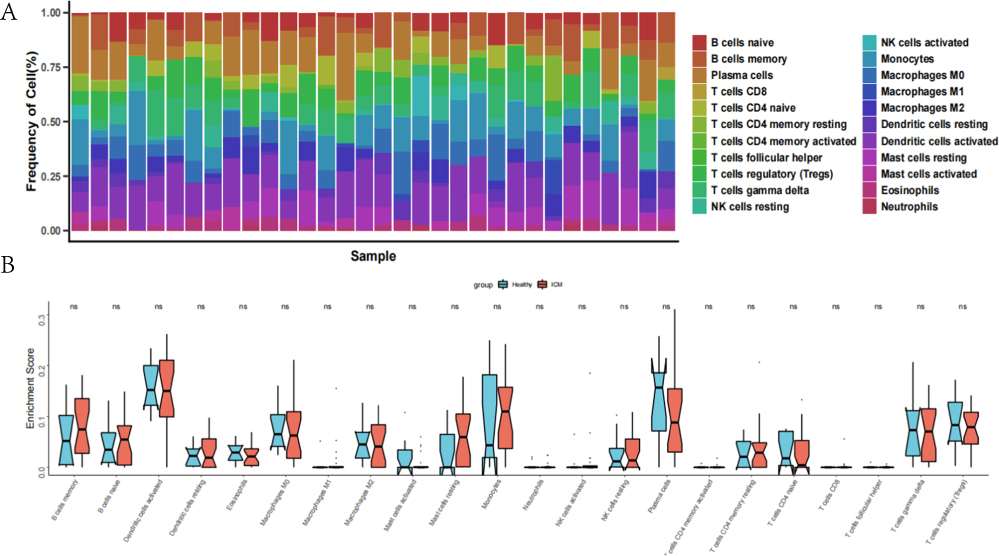 |
Figure 4 An examination of immunological infiltration. (A) Displays the relative proportion of immunological infiltration in each sample. (B) The immune cell violin chart illustrates the considerable difference between ICM patients and controls. X-axis: grouping of high and low risk; y-axis: enrichment Score. |
Construction of PPI Nets
To study the connection between the DEGs and ICM development, a protein-protein interaction (PPI) network was built using Cytoscape and the 20 DEGs from the STRING online database (https://string-db.org/cgi/input.pl) (Figure 5A). After applying the maximal clique centrality (MCC) method to the DEGs in the PPI network, the top five important genes were chosen, and a subnetwork made up of those 20 genes was also shown (Figure 5B).Many of these genes, including SCD and SNX25, which served as lipid metabolism10 and inflammatory responses,11 respectively, have been identified as being essential to ICM.
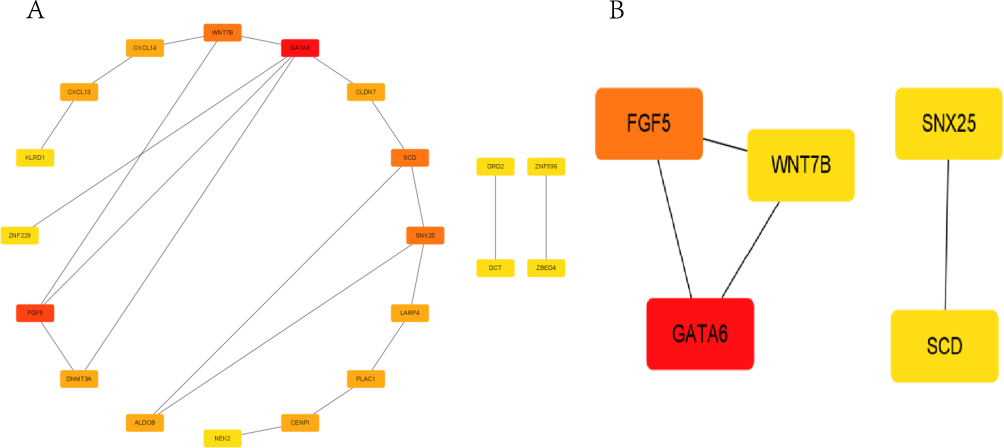 |
Figure 5 The development of the PPI network. (A) The protein–protein interaction (PPI) study of the twenty differentially expressed genes. (B) The subnetwork consisted of the five most significant genes with the highest degree inside the PPI network. The greater the degree, the darker the red color. |
The Development and Validation of a Model for Logistic Diagnosis
Then, using a stepwise regression method, 3 genes were chosen from the 20 identified genes in the PPI network, and it was discovered that 3 of these genes, including SCD, SNX25, and WNT8B, displayed P values 0.05, suggesting that these three genes are closely associated with the development of ICM. According to our data, this model fits well and no extreme point was found (Figure 6A). The AUC values of our model was 0.891 (Figure 6B). Together, it is found that the selected gene-based logistic model accurately predicts the occurrence of ICM.
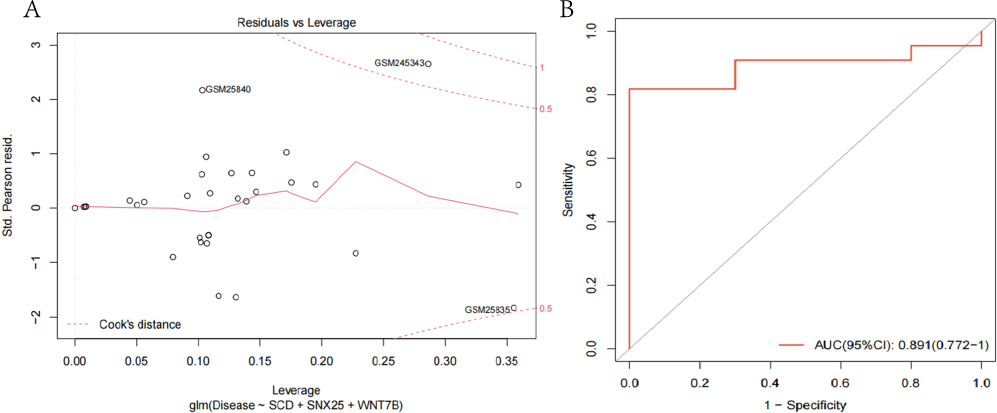 |
Figure 6 (A) construction of the model for logistic regression. Using logistic regression, a diagnostic model based on the three hub genes was created. The red dashed line demonstrates the COOK distance. (B) The ROC graphs. On the X-axis is the false positive rate (FPR), while the Y-axis is the genuine positive rate (TPR). The AUC value was used to measure the accuracy of the logistic regression model. |
Discussion
Heart failure has remained the leading cause of death globally for the last 20 years and its prevalence will continue to rise.12 ICM, with low five-year survival rate, is the main etiology of HF and characterized by high mortality and morbidity.12–14 The diagnosis of ICM15 based on patients’ medical history, clinical examination, which is supported by histologic, cardiac ultrasonography and radiologic. ICM has no specific symptoms in the early stage, and it is mostly in the middle and late stages when it is discovered.16 Thus, identifying specific diagnostic signatures in high-risk subjects counts for ICM, not only for preventing the occurrence but for early intervention of ICM. For the development of ICM, bioinformatics analysis gave information for the identification of many genes and proteins.
Although a few literature17–19 have revealed that several genes are associated with ICM, including SERPINA3, ASPN, HBB, MXRA5 and so on, limitations remain. Firstly, lack of literature revealed the identification of signature between ICM and healthy subjects. Secondly, the relationship between ICM and inflammatory responses is unclear and needs to be explore. In the present study, five co-DEGs (FGF5, GATA6, WNT7B, SNX25, SCD) were identified in two datasets (GSE9800, GSE580). GO enrichment and KEGG pathway analyses were conducted, and a PPI network was created to investigate other molecular pathways underlying ICM advancement. The PPI analysis results suggested that SCD, SNX25 and WNT8B were the most influential factors.
SCD (stearoyl-CoA desaturase) is the rate-limiting enzyme required by the body to produce monounsaturated fatty acids from saturated fatty acids. SCD deficiency alters cholesterol homeostasis in vivo and increases levels of the inflammatory cytokine IL-6,20–22 which in turn causes or exacerbates atherosclerosis. AS for SNX25 (Sorting nexin 25), it is an SNX family member, is reported to negatively regulate TGF-β signaling by enhancing TGF receptor degradation10 and inhibits the NF-κB signal and thereby regulates proinflammatory cytokine expression in macrophages.11 Obviously, SNX25 is closely related with inflammatory responses and may associated with atherosclerosis. Talking to the WNT7B, it is reported23 that the protective role of WNT7B in ox-LDL-induced cell apoptosis, inflammatory responses and EndMT, possibly via increasing miR-30c-5p and inactivating Wnt7b/β-catenin, and thus play pivotal roles in the pathogenesis and development of atherosclerosis.
Conclusion
In the present study, we utilized bioinformatics analysis integrated with machine learning to build a diagnostic model with the hope that it can serve to early identify high risk subjects and intervene the disease in the early stage. The ICM prediction model exhibited adequate prognostic value (AIC 0.891).
Nevertheless, several limitations remained in this study. This is a paper on bioinformatics mining of purely public database data, and further basic experimental validation is needed, but unfortunately further validation is not possible for the time being due to the limitations of our hospital experimental conditions.
Data Sharing Statement
The datasets for this study can be found in the Gene Expression Omnibus (GEO) (http://www.ncbi.nlm.nih.gov/geo/).
Author Contributions
Yinhua Luo and Jinbo Zhao should be regarded as co-first authors. All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was funded by the National Natural Science Foundation of China (No.82160072) and the Science andTechnology Support Project of Enshi Prefecture Science and Technology Bureau (D20210024).
Disclosure
The authors declare that they have no conflict of interests.
References
1. Panza JA, Chrzanowski L, Bonow RO. Myocardial viability assessment before surgical revascularization in ischemic Cardiomyopathy: JACC review topic of the week. J Am Coll Cardiol. 2021;78(10):1068–1077. doi:10.1016/j.jacc.2021.07.004
2. Bakaeen FG, Gaudino M, Whitman G, et al. 2021: the American association for thoracic surgery expert consensus document: coronary artery bypass grafting in patients with ischemic cardiomyopathy and heart failure. J Thorac Cardiovasc Surg. 2021;162(3):829–850.e821. doi:10.1016/j.jtcvs.2021.04.052
3. Khan MA, Hashim MJ, Mustafa H, et al. Global epidemiology of ischemic heart disease: results from the global burden of disease study. Cureus. 2020;12(7):e9349. doi:10.7759/cureus.9349
4. Virani SS, Alonso A, Aparicio HJ, et al. Heart disease and stroke statistics-2021 Update: a report from the American heart association. Circulation. 2021;143(8):e254–e743. doi:10.1161/CIR.0000000000000950
5. Lu J, Wang Z, Maimaiti M, et al. Identification of diagnostic signatures in ulcerative colitis patients via bioinformatic analysis integrated with machine learning. Hum Cell. 2022;35(1):179–188. doi:10.1007/s13577-021-00641-w
6. Luo Y, Tan N, Zhao J, et al. A nomogram for predicting in-stent restenosis risk in patients undergoing percutaneous coronary intervention: a population-based analysis. Int J Gen Med. 2022;15:2451–2461. doi:10.2147/IJGM.S357250
7. Alimadadi A, Manandhar I, Aryal S, et al. Machine learning-based classification and diagnosis of clinical cardiomyopathies. Physiol Genomics. 2020;52(9):391–400. doi:10.1152/physiolgenomics.00063.2020
8. Aronis KN, Prakosa A, Bergamaschi T, et al. Characterization of the electrophysiologic remodeling of patients with ischemic cardiomyopathy by clinical measurements and computer simulations coupled with machine learning. Front Physiol. 2021;12:684149. doi:10.3389/fphys.2021.684149
9. Rogers AJ, Selvalingam A, Alhusseini MI, et al. Machine learned cellular phenotypes in cardiomyopathy predict sudden death. Circ Res. 2021;128(2):172–184. doi:10.1161/CIRCRESAHA.120.317345
10. Hao X, Wang Y, Ren F, et al. SNX25 regulates TGF-β signaling by enhancing the receptor degradation. Cell Signal. 2011;23(5):935–946. doi:10.1016/j.cellsig.2011.01.022
11. Nishimura K, Tanaka T, Takemura S, et al. SNX25 regulates proinflammatory cytokine expression via the NF-κB signal in macrophages. PLoS One. 2021;16(3):e0247840. doi:10.1371/journal.pone.0247840
12. Mosterd A, Hoes AW. Clinical epidemiology of heart failure. Heart. 2007;93(9):1137–1146. doi:10.1136/hrt.2003.025270
13. McMurray JJ, Pfeffer MA. Heart failure. Lancet. 2005;365(9474):1877–1889. doi:10.1016/S0140-6736(05)66621-4
14. Tanai E, Frantz S. Pathophysiology of Heart Failure. Compr Physiol. 2015;6(1):187–214.
15. Sekulic M, Zacharias M, Medalion B. Ischemic cardiomyopathy and heart failure. Circ Heart Fail. 2019;12(6):e006006. doi:10.1161/CIRCHEARTFAILURE.119.006006
16. Schinkel AF, Poldermans D, Rizzello V, et al. Why do patients with ischemic cardiomyopathy and a substantial amount of viable myocardium not always recover in function after revascularization? J Thorac Cardiovasc Surg. 2004;127(2):385–390. doi:10.1016/j.jtcvs.2003.08.005
17. Cao J, Liu Z, Liu J, et al. Bioinformatics analysis and identification of genes and pathways in ischemic cardiomyopathy. Int J Gen Med. 2021;14:5927–5937. doi:10.2147/IJGM.S329980
18. Li GM, Zhang CL, Rui RP, et al. Bioinformatics analysis of common differential genes of coronary artery disease and ischemic cardiomyopathy. Eur Rev Med Pharmacol Sci. 2018;22(11):3553–3569. doi:10.26355/eurrev_201806_15182
19. Chen C, Tian J, He Z, et al. Identified three interferon induced proteins as novel biomarkers of human ischemic cardiomyopathy. Int J Mol Sci. 2021;22(23):13116. doi:10.3390/ijms222313116
20. Yang ZH, Pryor M, Noguchi A, et al. Dietary palmitoleic acid attenuates atherosclerosis progression and hyperlipidemia in low-density lipoprotein receptor-deficient mice. Mol Nutr Food Res. 2019;63(12):e1900120. doi:10.1002/mnfr.201900120
21. MacDonald ML, van Eck M, Hildebrand RB, et al. Despite antiatherogenic metabolic characteristics, SCD1-deficient mice have increased inflammation and atherosclerosis. Arterioscler Thromb Vasc Biol. 2009;29(3):341–347. doi:10.1161/ATVBAHA.108.181099
22. Brown JM, Chung S, Sawyer JK, et al. Combined therapy of dietary fish oil and stearoyl-CoA desaturase 1 inhibition prevents the metabolic syndrome and atherosclerosis. Arterioscler Thromb Vasc Biol. 2010;30(1):24–30. doi:10.1161/ATVBAHA.109.198036
23. Wu H, Liu T, Hou H. Knockdown of LINC00657 inhibits ox-LDL-induced endothelial cell injury by regulating miR-30c-5p/Wnt7b/β-catenin. Mol Cell Biochem. 2020;472(1–2):145–155. doi:10.1007/s11010-020-03793-9
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.