")
Back to Journals » Cancer Management and Research » Volume 12
Identification of Core Genes Involved in the Metastasis of Clear Cell Renal Cell Carcinoma
Authors Peng R, Wang Y, Mao L, Fang F, Guan H
Received 12 August 2020
Accepted for publication 4 November 2020
Published 30 December 2020 Volume 2020:12 Pages 13437—13449
DOI https://doi.org/10.2147/CMAR.S276818
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Sanjeev K. Srivastava
Rui Peng,1,* Yahui Wang,2,* Likai Mao,3 Fang Fang,4 Han Guan1
1Department of Urology, First Affiliated Hospital of Bengbu Medical College, Bengbu, People’s Republic of China; 2Department of Urology, Sun Yat-Sen Memorial Hospital, Sun Yat-Sen University, Shenshan Central Hospital, Shanwei, People’s Republic of China; 3Department of Urology, Second Affiliated Hospital of Bengbu Medical College, Bengbu, People’s Republic of China; 4Department of Immunology, School of Laboratory Medicine, Anhui Provincial Key Laboratory of Infection and Immunity, Bengbu Medical College, Bengbu, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Han Guan
Department of Urology, First Affiliated Hospital of Bengbu Medical College, No. 87 Zhihuai Road, Longzhihu District, Bengbu, Anhui 233000, People’s Republic of China
Tel +86 138 5522 8689
Email [email protected]
Introduction: Renal cell carcinoma (RCC) is one of the most common malignancies globally, among which clear cell carcinoma (ccRCC) accounts for 85– 90% of all pathological types. This study aims to screen out potential genes in metastatic ccRCC so as to provide novel insights for ccRCC treatment.
Methods: GSE53757 and GSE84546 datasets in the Gene Expression Omnibus (GEO) were profiled to identify differentially expressed genes (DEGs) from ccRCC samples with or without metastasis. The Kyoto Encyclopedia of Genes and Genomes (KEGG) and the gene ontology (GO) analysis were performed to analyze pathway enrichment and functional annotation of DEGs. Protein–protein interaction (PPI) network was constructed, and survival analysis was conducted to evaluate the clinical values of the identified hub genes. In vitro loss-of-function assays were performed to explore the biological roles of these genes.
Results: The bioinformatic analysis indicated that 312 DEGs were identified, including 148 upregulated genes and 164 downregulated ones. Using PPI and Cytoscape, 10 hub genes were selected (C3, CXCR4, CCl4, ACKR3, KIF20A, CCNB2, CDCA8, CCL28, S1PR5, and CCL20) from DEGs which might be closely related with ccRCC metastasis. In Kaplan–Meier analysis, three potential prognostic biomarkers (KIF20A, CCNB2 and CDCA8) were identified. Finally, cell proliferative and invasive assays further verified that KIF20A, CCNB2 and CDCA8 were associated with the proliferation and invasion of ccRCC cells.
Conclusion: Our results demonstrated that metastatic ccRCC was partially attributed to the aberrant expression of KIF20A, CCNB2 and CDCA8, and more personalized therapeutic approaches should be explored targeting these hub genes.
Keywords: clear cell renal cell carcinoma, hub genes, biomarkers, metastasis, differentially expressed gene
Introduction
Renal cell carcinoma (RCC) is one of the most common malignant tumors in the world. It is estimated that approximately 74,000 new cases and 15,000 deaths of RCC may occur in the US in 2020, among which clear cell RCC (ccRCC) is the most common histological type of RCC, accounting for 85–90% of all RCC cases.1,2 The five-year survival rate of patients with distant metastasis drops to 12%, despite that the likelihood of survival in early and localized ccRCC patients is comparatively higher.3 The established studies in the past 10 years have been used in clinical diagnosis and treatment of renal tumors.4,5 For example, it has been demonstrated that VHL (Von Hippel–Lindau) deletion directly led to mRNA alteration and facilitated RCC development. Thus, a specific drug targeting pVHL-HIF-VEGF has been utilized in clinical practice and its efficacy was shown to be better than that of cytokine therapy.6 However, the clinical prognosis of advanced and metastatic ccRCC remained unsatisfactory due to chemotherapy and radiotherapy resistance. Therefore, it is of great necessity to develop specific markers that can better predict the prognosis of ccRCC, provide new insights for the development of related drugs, reduce the incidence of advanced ccRCC, and ultimately improve the survival rate.
In recent years, microarrays based on mathematical tools can be utilized to extract useful biological information from a mass of data, identifying and analyzing the association and molecular mechanisms between key genes and central signaling pathways in tumorigenesis. In addition, data mining from high-throughput platforms can be conducted to identify significant genetic or epigenetic changes in carcinogenesis, which in turn help to select promising biomarkers for cancer diagnosis and prognosis. Mounting studies were conducted to explore the molecular mechanisms involved in RCC development through mining public datasets,7–10 whereas few have been validated by experiments.
In this study, we downloaded gene expression profiles GSE53757 and GSE84546 from the Gene Expression Omnibus (GEO) containing paired advanced-stage ccRCC and normal samples to identify the differentially expressed genes (DEGs). In addition, gene ontology (GO) analysis was used to search for pathways, and a protein–protein interaction (PPI) network was constructed by the STRING database. The hub genes were identified by Cytoscape software. Finally, in vitro experiments were performed to verify the biological effects of typical hub genes. Collectively, the identified genes associated with the metastasis of ccRCC may shed light on novel therapeutic targets and pave the way for further exploration of the underlying mechanisms in ccRCC metastasis.
Methods
Download and Conversion of Microarray Data
Two gene expression profiles (GSE53757 and GSE84546)11,12 were downloaded from the GEO database (https://www.ncbi.nlm.nih.gov/geo/). GSE53757 dataset contained 15 metastatic ccRCC (mccRCC) samples and 15 paired normal samples and GSE84546 dataset contained three mccRCC samples and three paired normal samples (Table 1). GEO-2R online tools (http://www.ncbi.nlm.nih.gov/geo/geo-2r/) were performed to normalize and log 2 transform the matrix data of each GEO dataset and screen out DEGs in each microarray. The value of | log FC (fold change) | ≥2 with P<0.05 was considered of statistical significance. Hierarchical clustering analysis was performed to classify the data into two groups with similar expression patterns. The Venn diagram package of R software was used to make a Venn diagram and screen out DEGs for further study.
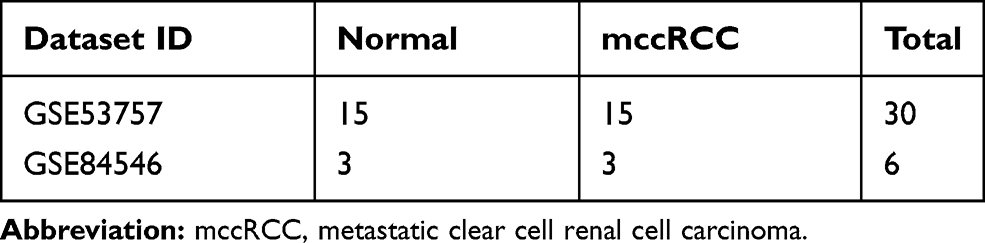 |
Table 1 Statistics of the Two Microarray Databases Derived from the GEO Database |
Enrichment Analysis of Functional Pathways
Cytoscape software was utilized to construct unified conceptual framework through integrating biomolecular interaction network, high-throughput expression data and other molecular states.13 In this study, GO term enrichment14 and the Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment pathway15 were conducted using the plug-in ClueGO16 in Cytoscape software to analyze DEGs. The enrichment analysis of GO terms includes biological process (BP), cell composition (CC) and molecular function (MC).
Protein–Protein Interaction (PPI) Network Construction and Hub Gene Analysis
STRING and Cytoscape software were used to construct PPI network and screen key genes of PPI network.17 In this study, STRING database (http://string-db.org) was used to conduct PPI network analysis of DEGs in ccRCC and normal renal samples. Key genes in PPI network were screened by CytoHubba and MCC (Maximum Clique Centrality) topology analysis algorithm was selected as the node sorting method.
Correlation Analysis of Hub Genes Expression and Prognosis in ccRCC
In order to verify the mRNA expression level of hub genes in ccRCC, we employed HPA database (http://www.proteinatlas.org/) to obtain IHC staining. The total survival analyses for hub genes were conducted through online cBioPortal platform (http://www.cbioportal.org). UALCAN database (http://ualcan.path.uab.edu/index.html) was used to identify survival rate for ccRCC patients. Genes significantly associated with overall survival were considered potential biomarkers of prognosis.
Cell Lines and Culture
The 786-O RCC cell line was purchased from the American Type Culture Collection (ATCC), and cultured in RPMI-1640 (Gibco, Thermo Fisher Scientific, USA) supplemented with 10% FBS and antibiotics. The cell line was maintained at 37°C in a humidified chamber supplemented with 5% CO2.
Oligonucleotide and Plasmid Transfection
Short interfering RNA (siRNA) against KIF20A, CCNB2 and CDCA8 (si-KIF20A, si-CCNB2 and si-CDCA8), and negative control siRNA with nonspecific sequences (si-NC) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). For cell transfection, an equal dose (40 pmol) of si-KIF20A, si-CCNB2, si-CDCA8, or si-NC was transiently transfected into 786-O cells, which were seeded in six-well plates. The cells were employed in in-vitro loss-of function assays 48 hafter transfection.
CCK-8 (Cell Counting Kit-8) Assay
Cells were seeded in 96-well plates with a density of 2×104 cells/well. Approximately 20 µl CCK-8 reagent was added to each well, incubated at 37°C for 10 min, and then placed on a shaker for five minutes. The OD value of each well was detected. Each assay was performed in triplicate and repeated three times.
Transwell Assay
After transfection by si-KIF20A, si-CCNB2, and si-CDCA8 for 48 h, 786-O cells were diluted to a final concentration of 5.0×106/mL for the invasion assay. The transfected cells were placed on the filter membrane of the upper chamber of transwell and incubated at 37°C for one hour. In the lower chamber, 600 µl of complete cell culture medium was added, and 100 µl of cell suspension was added in the upper chamber. Afterward, 786-O cells were fixed with methanol, stained with 0.2% crystal violet, counted, and photographed under a microscope.
RNA Extraction and Quantitative Real-Time PCR Analysis
Total cellular RNA isolation was carried out following the manufacturer’s protocol. The synthesis of cDNA was performed using the PrimeScript RT reagent (Takara, Kusatsu, Japan) after measuring the concentration and purity of the extracted RNA. The fluorescence quantitative PCR detection of mRNAs was conducted using SYBR Premix Ex Taq I. PCR amplification was carried out with reverse-transcribed cDNA as a template and U6 snRNA as an internal control. The experimental results were analyzed quantitatively by using the standard 2–ΔΔCt method.
Western Blot Analysis
Total protein was extracted from 786-O cells after lysis in RIPA buffer, and the concentration was detected by the BCA method. The buffer solution was added according to the protein concentration, and SDS-polyacrylamide gels were used in electrophoresis after adding samples. Next, the protein was transferred onto polyvinylidene fluoride (PVDF) membranes, which were then blocked using a 5% skimmed milk powder solution. The membranes were then placed in a shaker at 37°C for one hour, incubated with primary antibodies overnight at 4°C, washed by TBST buffer three times, and added with secondary antibody (1:2000) for one hour. ECL liquid was used for luminescence. X-ray film exposure was carried out in the darkroom. The primary antibodies used in this experiment were as follows: anti-KIF20A (ab70791, Abcam, USA), anti-CCNB2 (ab185622, Abcam, USA), and anti-CDCA8 antibody (ab67126, Abcam, USA). GAPDH was used as an internal control.
Statistical Analysis
SPSS version 24.0 software was used for all statistical analyses. The log-rank method was applied in Kaplan–Meier analysis to determine the survival difference of hub genes. Student’s-t test and one‐way ANOVA analysis were used to compare the significance of two groups. The qPCR and Western blot results were analyzed via Wilcoxon signed-rank tests. All experiments above were repeated three times. A P-value of <0.05 was considered statistically significant.
Results
Identification of DEGs
After the standardization of GSE53757 and GSE84546 profiles, Venn analysis was performed to obtain the DEG profiles intersection (2791 in GSE84546 and 923 in GSE53757). The result showed that a total of 312 genes were markedly differentially expressed in two groups. Limma R package was used to identify 312 DEGs, among which 148 genes were upregulated and 164 downregulated. The DEGs were analyzed by hierarchical cluster analysis (Figure 1).
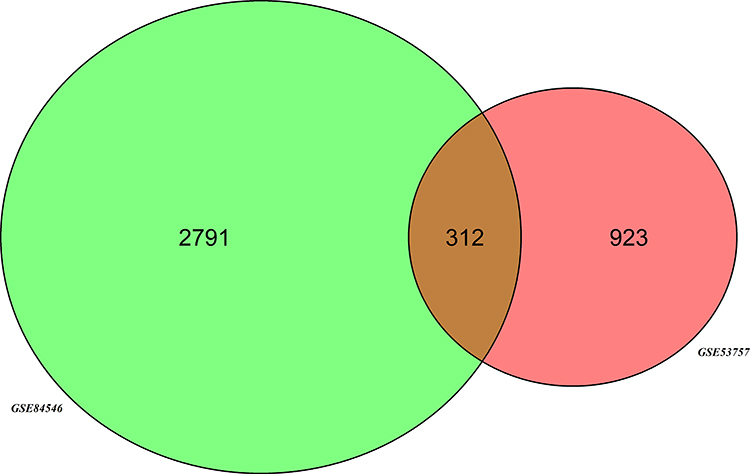 |
Figure 1 Identification of DEGs. Venn diagram of DEGs (2791 in GSE84546 and 923 in GSE53757) was used to obtain the DEG profiles intersection. Three hundred and twelve DEGs were differentially expressed in 2 groups. |
Enrichment Analysis
To better understand the targeted DEGs, GO function analysis was conducted by using Cytoscape plug-in ClueGo. The results presented that the DEGs were mostly enriched in the biological processes (BP) such as blood vessel development (25.0%), tube morphogenesis (23.33%) and regulation of cell adhesion (13.33%) (P<0.01) (Figure 2). Furthermore, KEGG pathway analysis demonstrated that DEGs were mostly aggregated in the intestinal immune network for IgA production (50.0%) and bile secretion (50.0%) (P<0.05) (Figure 3, Table 2).
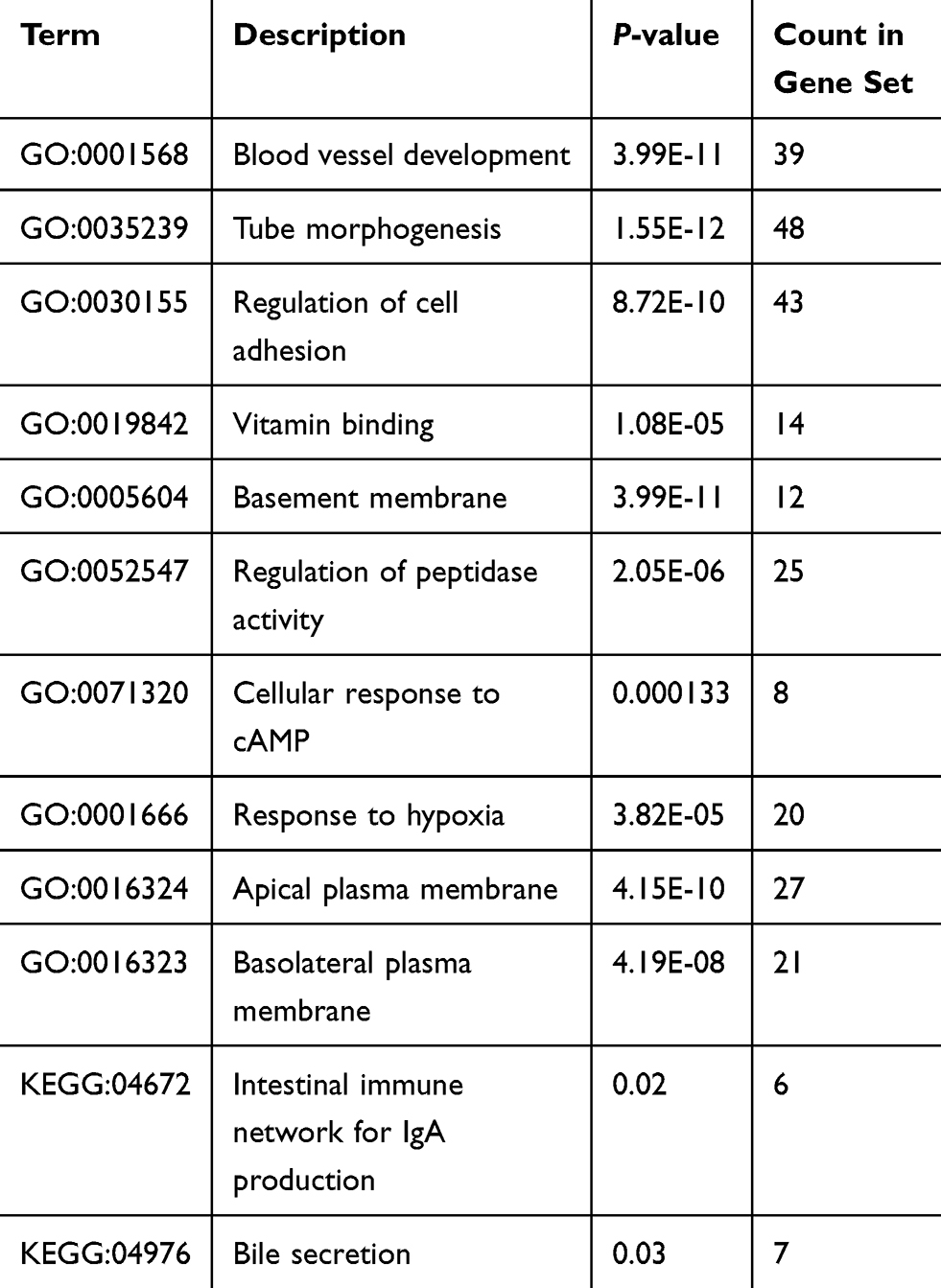 |
Table 2 Significantly Enriched GO Terms and KEGG Pathways of DEGs |
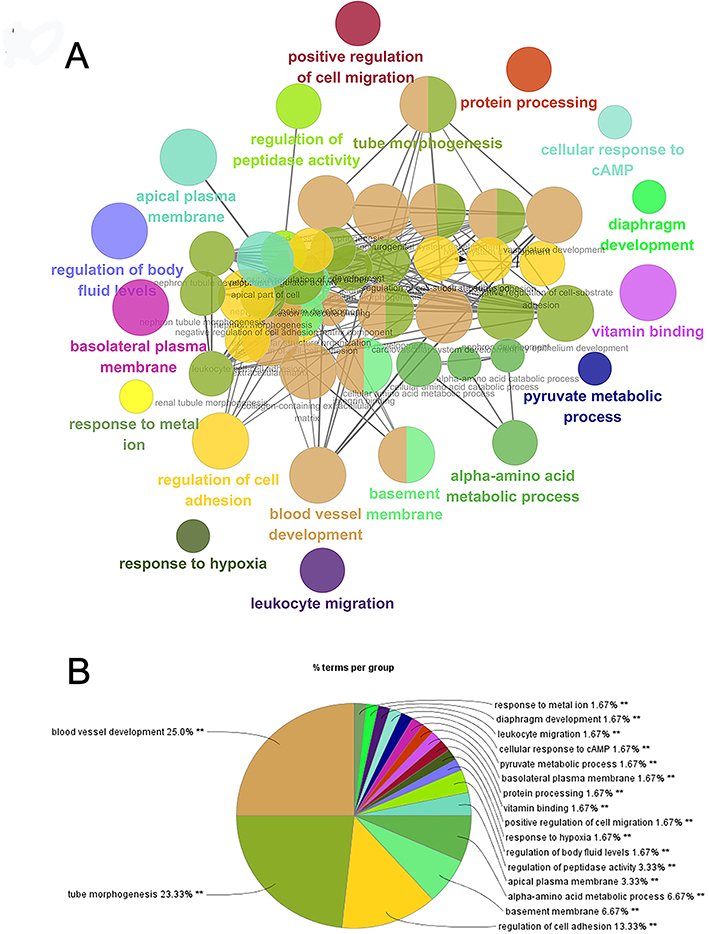 |
Figure 2 Enrichment analysis of functional pathways. (A) GO analysis of the DEGs of metastatic ccRCC. (B) Pie chart of different GO terms of the DEGs. |
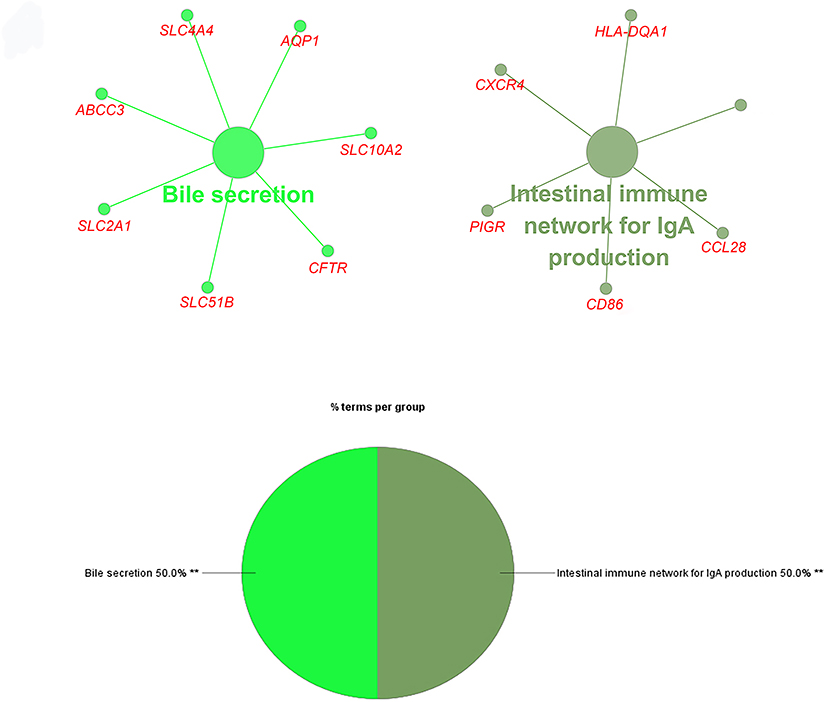 |
Figure 3 Enrichment analysis of functional pathways. Enriched key genes involved in KEGG analysis. |
PPI Network Analysis and Screening for Hub Genes
PPI analysis of DEGs was predicted by online STRING software. The processed data were imported into Cytoscape to generate PPI network (Interaction score setting >0.7, strong correlation). The results revealed 301 nodes and 215 edges among the DEGs, as shown in Figure 4A. MCC topology analysis algorithm of CytoHubba tool was conducted to screen the top 10 hub genes in PPI network, including downregulated C3, CXCR4, CCL4, ACKR3, KIF20A, CCNB2 and CDCA8. It upregulated CCL28, S1PR5 and CCL20, with the connectivity scores of the top 10 central genes listed in Table 3 (Figure 4B, Table 3).
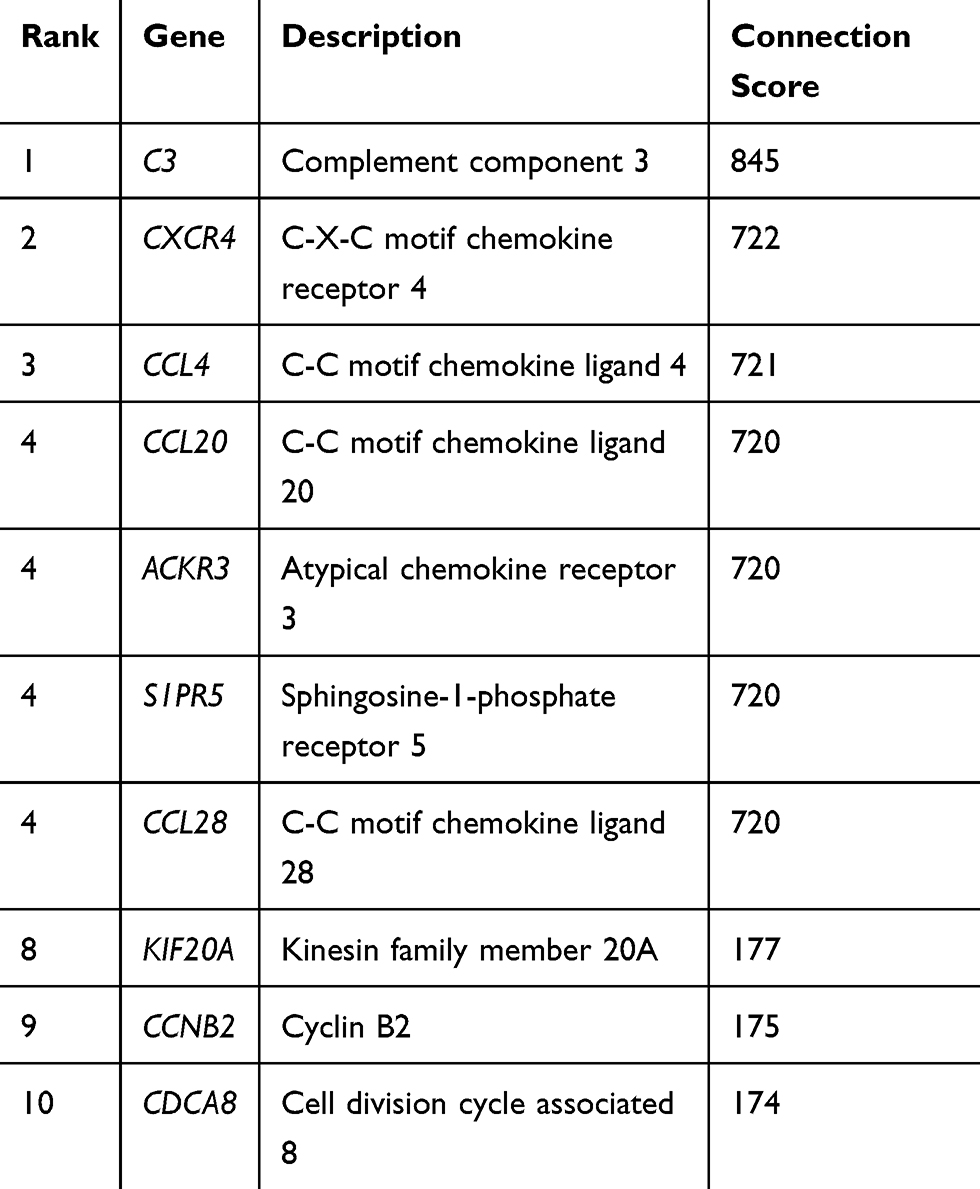 |
Table 3 The Top 10 Hub Genes in PPI Network with Highest Degree |
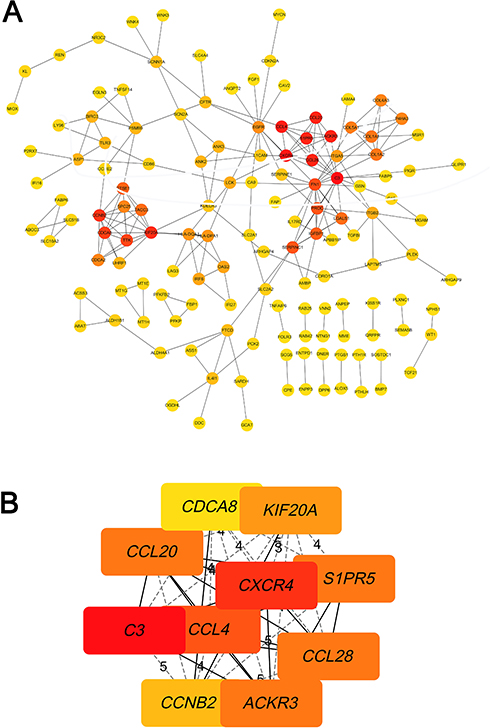 |
Figure 4 PPI network analysis and screening for hub genes. (A) PPI network of DEGs. (B) The PPI network of top 10 hub genes. |
Analysis of Hub Genes
The overall survival rate analysis of identified hub genes was conducted by using the Kaplan–Meier survival curve with a log-rank test. In the samples of the ccRCC patients at an advanced stage, CCL4, KIF20A, CCNB2, and CDCA8 were related to worse overall survival, whereas CCL20 and CCL28 were accompanied by a favorable prognosis (Figure 5). Next, UALCAN online tools were employed to validate the associations between these four genes and the poor prognosis of RCC patients. KIF20A, CCNB2, and CDCA8 were concordantly related to unfavorable survival rates. (Figure 6). Consistently, analysis of TCGA data from the UALCAN database illustrated that KIF20A, CCNB2, and CDCA8 were overexpressed in RCC tissues compared to normal ones, and the expressions of the screened hub genes were positively related to clinical stage and lymph node metastasis. This result suggested that KIF20A, CCNB2, and CDCA8 might play a crucial role in the development and progression of ccRCC (Figure 7). After analyzing the HPA database, it was also found that KIF20A, CCNB2, and CDCA8 were upregulated in RCC tissues (Figure 8). Afterward, the association between KIF20A, CCNB2, and CDCA8 was investigated. By using the LinkedOmics database, it was discovered that CDCA8 was positively correlated with CCNB2 and KIF20A, and CCNB2 was positively correlated with KIF20A in RCC (P<0.001) (Figure 9A–C).
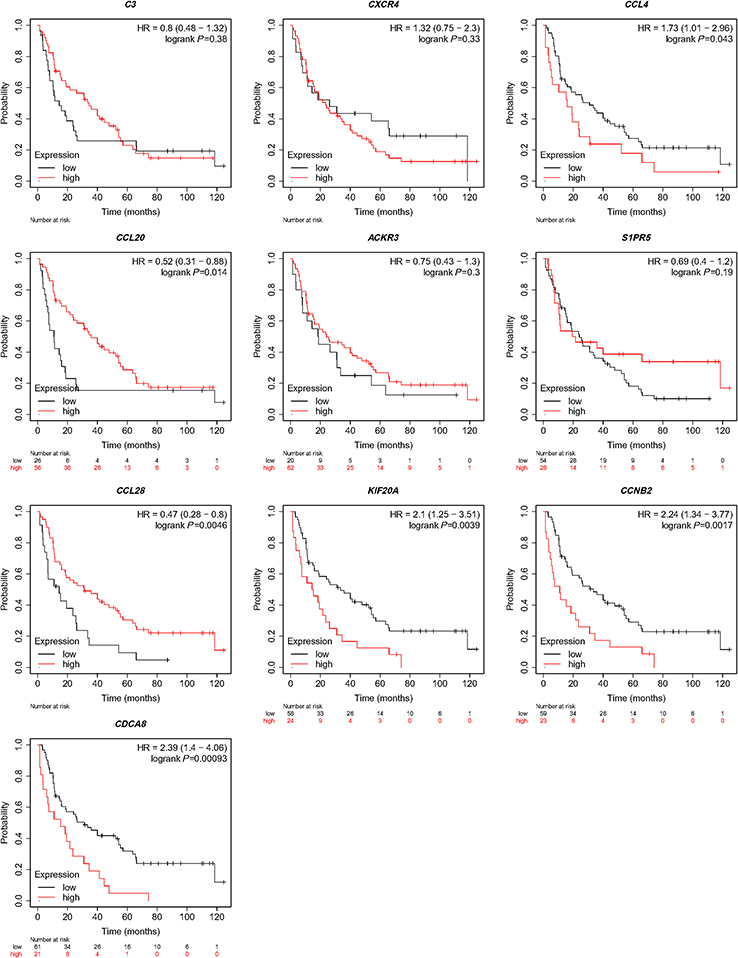 |
Figure 5 Analysis for hub genes. The overall survival rate of identified hub genes was analyzed using the Kaplan–Meier survival curve. |
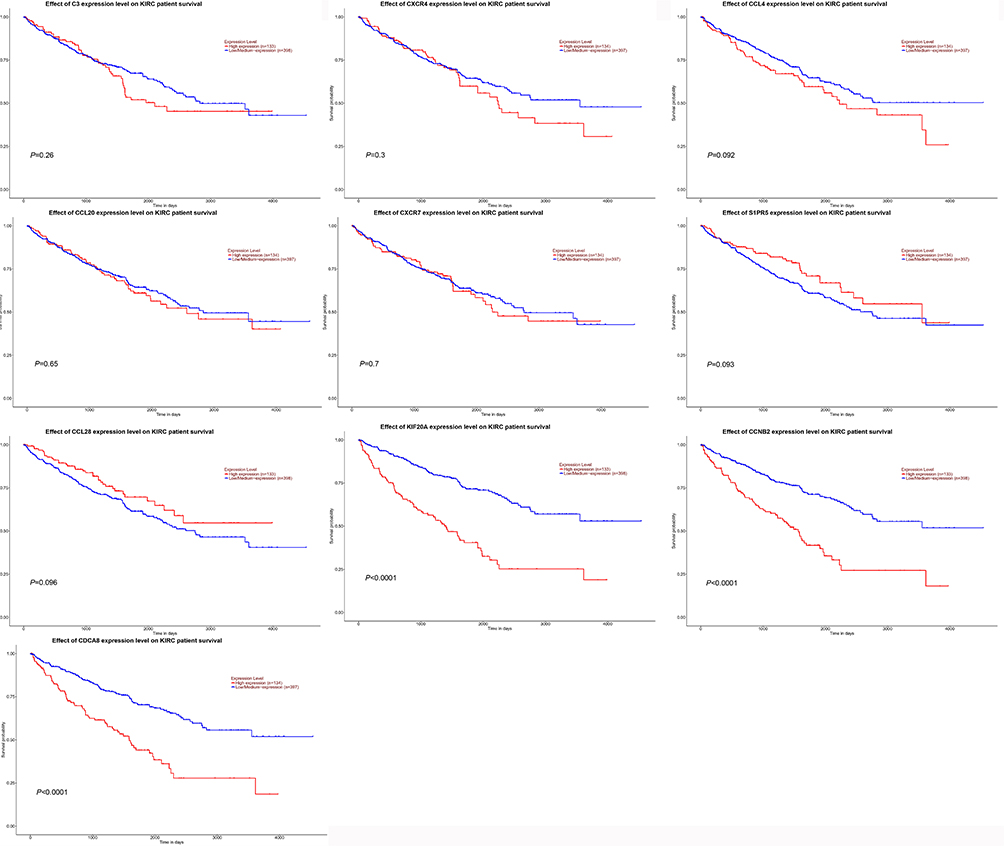 |
Figure 6 Analysis for hub genes. The overall survival analyses of hub genes were performed by cBioPortal online platform using data of 531 renal cell carcinoma patients from TCGA database. |
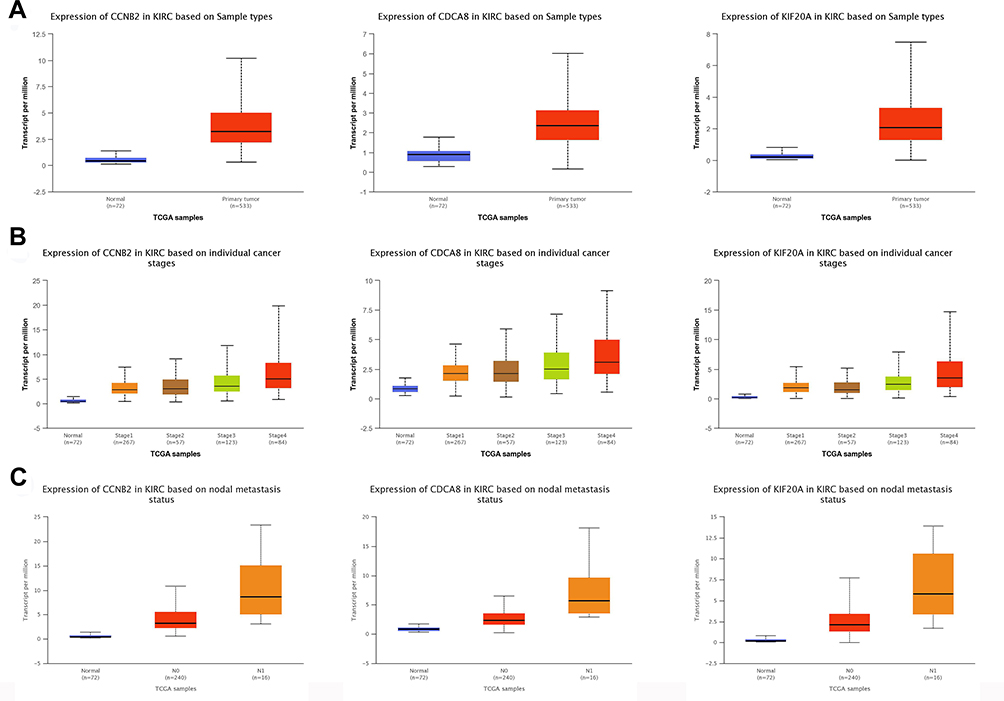 |
Figure 7 Analysis for hub genes. (A) Association of the expression of CCNB2, CDCA8, and KIF20A with normal and tumor tissues. (B) Association of the expression of CCNB2, CDCA8, and KIF20A with different clinical stages. (C) Association of the expression of CCNB2, CDCA8, and KIF20A with different nodal metastasis events. Abbreviations: N0, renal cell carcinoma without nodal metastasis; N1, renal cell carcinoma with nodal metastasis. |
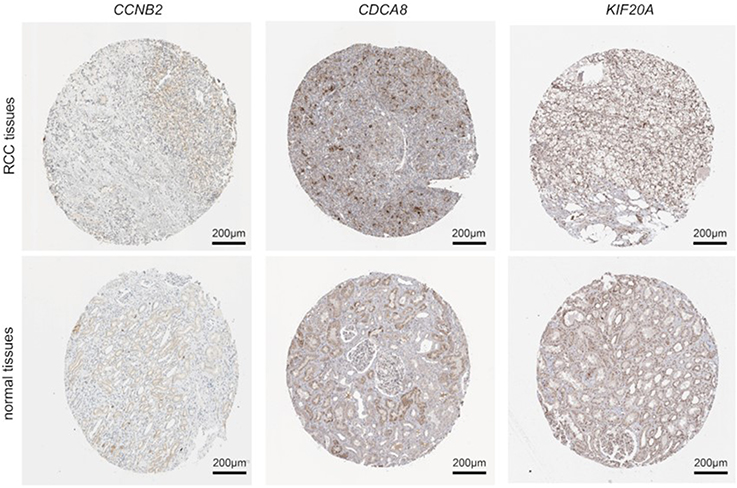 |
Figure 8 Analysis for hub genes. The protein expression levels of CCNB2, CDCA8 and KIF20A in normal and RCC tissues from HPA. |
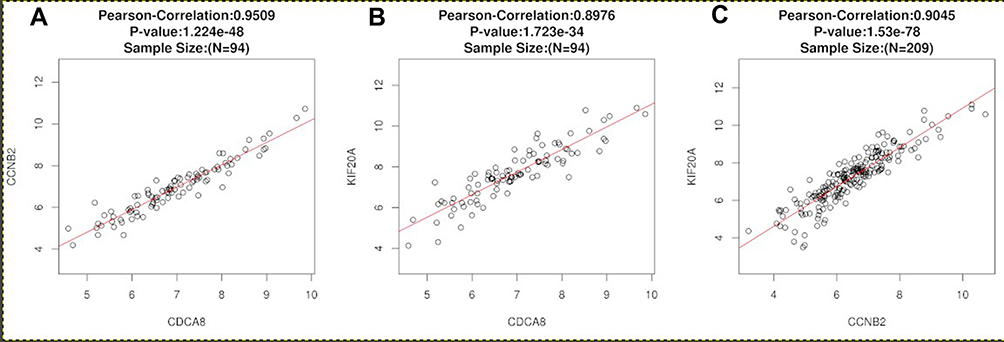 |
Figure 9 Analysis for hub genes. (A) The correction between CDCA8 and CCNB2 in ccRCC was analyzed by LinkedOmics. (B) The correction between CDCA8 and KIF20A in ccRCC was analyzed by LinkedOmics. (C) The correction between CCNB2 and KIF20A in ccRCC was analyzed by LinkedOmics. |
Verification of Hub Gene Expressions in RCC Tissues
Considering that KIF20A, CCNB2, and CDCA8 were closely associated with ccRCC metastasis, the effects of these genes on the biological activities of RCC cells were investigated. Firstly, we validated three hub genes in normal and RRCC cells were treated with the inhibitors of CCNB2, CDCA8, and KIF20A, respectively. The knockdown efficiency after siRNA transfection was detected via qPCR (Figure 10A, C and E). Western blot analysis was also conducted to measure the expression difference of hub genes after the knockdown (Figure 10B, D and F). The results demonstrated that the expression of these genes was markedly decreased at mRNA and protein levels. Expectedly, the knockdown of CCNB2, CDCA8, and KIF20A suppressed RCC cell growth and invasion ability compared with that of si-NC-transfected cells (Figure 11A–F).
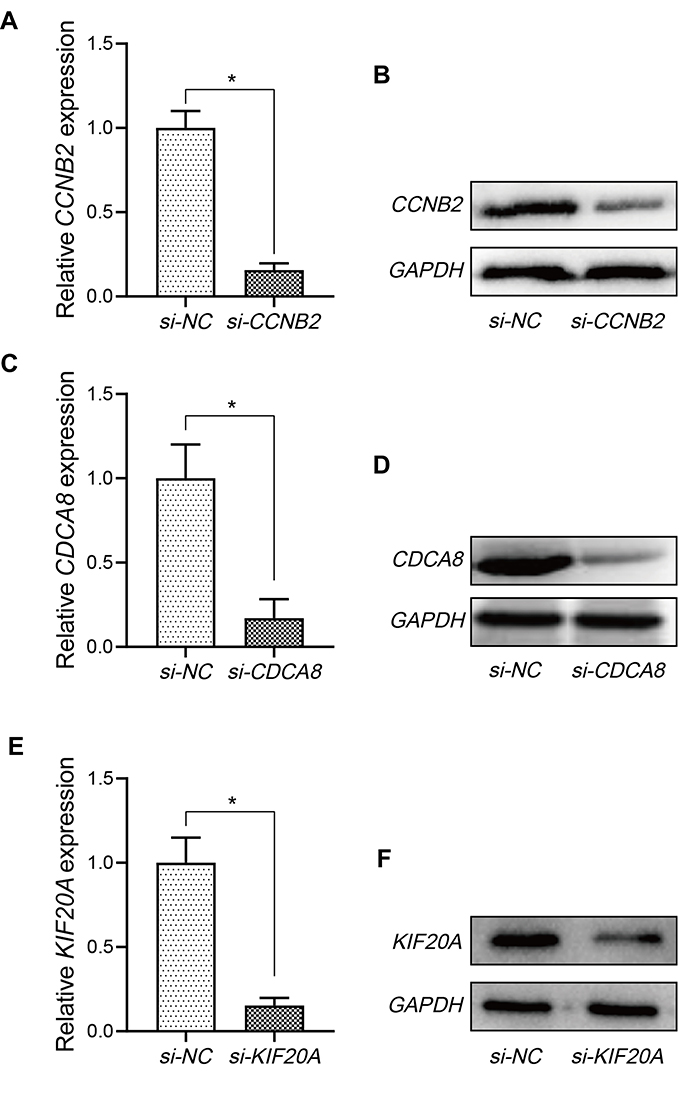 |
Figure 10 Analysis for hub genes. (A, C, and E) The knockdown efficiency after si-CCNB2, si-CDCA8, and si-KIF20A transfection was detected via qPCR. (B, D, and F) The knockdown efficiency after si-CCNB2, si-CDCA8, and si-KIF20A transfection was detected via Western blot. *P<0.05. |
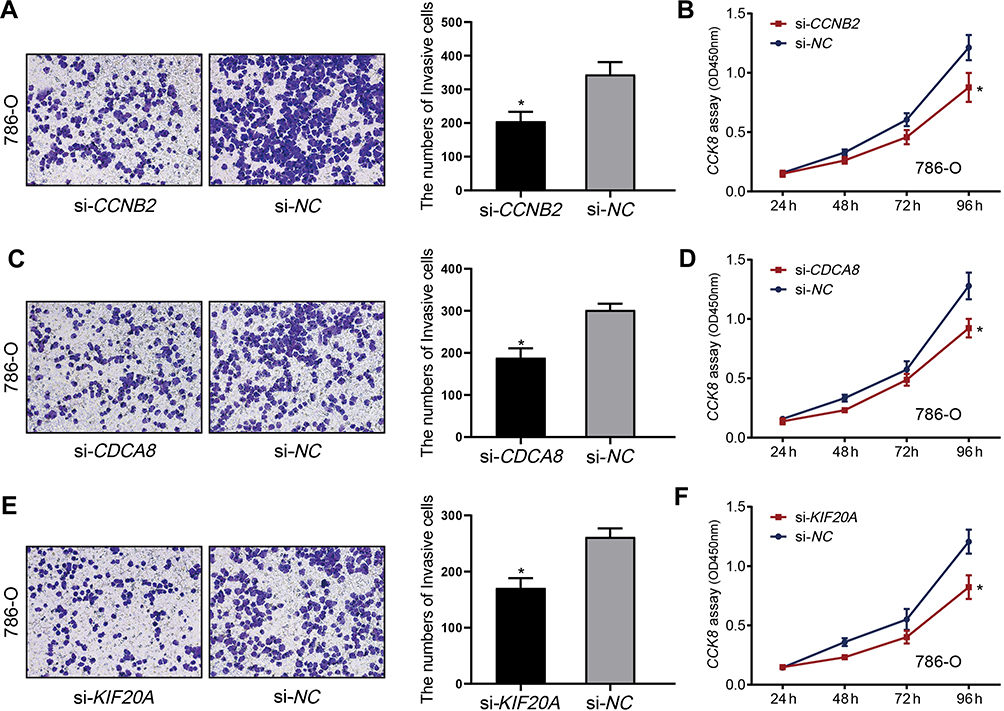 |
Figure 11 Verification of hub genes expression in RCC tissues. (A) CCK-8 assay was performed to determine cell viability of CCNB2 on 786-O cells. (B) Invasion assay was performed to detect invasion ability of CCNB2 on 786-O cells. (C) CCK-8 assay was performed to determine cell viability of CDCA8 on 786-O cells. (D) Invasion assay was performed to detect invasion ability of CDCA8 on 786-O cells. (E) CCK-8 assay was performed to determine cell viability of KIF20A on 786-O cells. (F) Invasion assay was performed to detect invasion ability of KIF20A on 786-O cells. Each bar represents the mean ±SD of three independent experiments. *P<0.05. |
Discussion
Despite that molecularly targeted therapies, representatively, drugs targeting vascular endothelial growth factor (VEGF) pathway and mammalian/mechanistic target of rapamycin (mTOR) pathway, have played a promising role in treating metastatic RCC, the median survival time is only 13 months, and the five-year survival rate is less than 10% due to the paucity of effective molecular targets. Even worse, the vast majority of the patients with metastatic RCC have eventually developed drug resistance, leading to unfavorable survival time. It is, therefore, important to further explore the underlying mechanisms or molecular markers for the diagnosis and treatment of ccRCC.
In this study, GO enrichment analysis demonstrated that DEGs are mainly enriched in the BPs such as blood vessel development, tube morphogenesis and regulation of cell adhesion. KEGG pathway analysis revealed that DEGs are mostly aggregated in intestinal immune network regulating IgA production and bile secretion. The top 10 hub genes were identified by PPI network analysis. By searching through HPA and UALCAN databases, we found that CCNB2, CDCA8, and KIF20A were significantly overexpressed in ccRCC tissues and closely associated with the poor prognosis of ccRCC patients. Survival analyses performed by the Kaplan–Meier method demonstrated that CCNB2, CDCA8, and KIF20A production were related to the survival rate of ccRCC patients. The correction between CCNB2, CDCA8 and KIF20A in RCC was also positively correlated with each other, illustrating that they might serve pivotal roles in the progression and metastasis of ccRCC.
CCNB2 (cyclin B2) is a member of cyclin family proteins, which are important components in the cell cycle regulation mechanism. It has been fully demonstrated to trigger G2/M transition by activating CDK1 kinase.18 Accumulating evidence proved that CCNB2 was overexpressed in various human cancers, such as lung cancer, colorectal adenocarcinoma, and ovarian carcinoma.19–21 Moreover, it has been found that advanced clinical stage and worse metastasis status were associated with the elevated expression of circulating CCNB2.22 As a biomarker, it has been reported that overexpression of CCNB2 could act as an independent predictor of poor prognosis in patients with lung adenocarcinoma and breast cancer. However, little is known about the prognostic role of CCNB2 in metastatic ccRCC. Integrating the results from survival analysis and the differential expression in different stages, we hypothesized that CCNB2 might act as a potential indicator and therapy target in mccRCC treatment.
CDCA8 is a member of the chromosomal passenger complex (CPC), which dominates cell division.23 It is commonly acknowledged that dysregulation of cell cycle and uncontrolled cell proliferation are hallmarks of cancer, and loss of CDCA8 leads to defective cell proliferation and early fetal death. Established studies have illustrated that CDCA8 overexpression is essential for tumor development and is associated with poor prognosis in patients with cutaneous melanoma and breast cancer.24,25 In this study, we observed that CDCA8 overexpression had the same effect in ccRCC patients, especially in stage IV patients with lymph node metastasis. Our study found that CDCA8 was upregulated in ccRCC and might act as a prognostic biomarker.
Previous studies have shown that KIF20A was essential for cell cycle mitosis, and its aberrant accumulation promoted cell proliferation.26,27 In addition, abnormal expression of KIF20A participates in the carcinogenesis of multiple malignancies, including cell differentiation, invasion, and distant metastasis, which ultimately lead to drug resistance28–30. It is worth noting that KIF20A can not only be used as a novel biomarker for predicting prognosis but also a new potential therapeutic target in various malignant tumors through different mechanisms. Representatively, Asahara et al conducted a single-center phase I/II clinical trial utilizing a cancer vaccine reagent KIF20A-66 to treat advanced pancreatic cancer, which significantly prolonged the overall survival rate of patients.31 Therefore, further study on the carcinogenic mechanism of KIF20A may contribute to the development of molecularly targeted drugs. Consistently, our results echoed with previous research showing that KIF20A might be a key molecule affecting the development of ccRCC and as a result impeding the prognosis.
ccRcc is the most common subtype of RCC, and its tumor progression and prognosis are associated with complex gene interactions. Therefore, exploring potential molecular mechanisms or markers of ccRCC is of great necessity for the diagnosis and treatment of ccRCC. However, there are a lack of molecular markers related to the pathological stage of ccRCC. Notably, few effective treatments for metastatic ccRCC have been developed, despite that the response rate to VEGFR inhibitors is relatively favorable (≈50%). Further exploration is required considering that there is a lack of available drugs to treat metastatic RCC. In this study, we identified DEGs in metastatic ccRCC samples by using bioinformatics analysis, and screened out three hub genes CCNB2, CDCA8, and KIF20A associated with mccRCC. These genes can be used as personalized therapeutic targets and diagnostic biomarkers for metastatic ccRCC. Our study may contribute to a better understanding of ccRCC and have important clinical significance in improving risk stratification, therapeutic strategy, and prognosis prediction of ccRCC patients.
Data Sharing Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Acknowledgments
This study was supported by grants from National Science Foundation of China [81801573], Science Foundation of Anhui Province [2008085QH358, KJ2018A0214, KJ2019A0355], First Affiliated Hospital of Bengbu Medical College Science Fund for Outstanding Young Scholars [2019BYYFYYQ09]. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Siegel R, Miller K, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. 2020;70(1):7–30. doi:10.3322/caac.21590
2. Vecchio S, Ellis R. Cabozantinib for the management of metastatic clear cell renal cell carcinoma. J Kidney Cancer. 2018;5(4):1–5. doi:10.15586/jkcvhl.2018.109
3. Atkins M, Tannir N. Current and emerging therapies for first-line treatment of metastatic clear cell renal cell carcinoma. Cancer Treat Rev. 2018;70:127–137. doi:10.1016/j.ctrv.2018.07.009
4. Thomas G, Tran C, Mellinghoff I, et al. Hypoxia-inducible factor determines sensitivity to inhibitors of mTOR in kidney cancer. Nat Med. 2006;12(1):122–127. doi:10.1038/nm1337
5. Vachhani P, George S. VEGF inhibitors in renal cell carcinoma. Clin Adv Hematol Oncol. 2016;14(12):1016–1028.
6. Kammerer-Jacquet S, Crouzet L, Brunot A, et al. Independent association of PD-L1 expression with noninactivated VHL clear cell renal cell carcinoma-A finding with therapeutic potential. Int J Cancer. 2017;140(1):142–148. doi:10.1002/ijc.30429
7. Yamamura S, Kawakami K, Hirata H, et al. Oncogenic functions of secreted Frizzled-related protein 2 in human renal cancer. Mol Cancer Ther. 2010;9(6):1680–1687. doi:10.1158/1535-7163.MCT-10-0012
8. Gu Y, Lu L, Wu L, et al. Identification of prognostic genes in kidney renal clear cell carcinoma by RNA‑seq data analysis. Mol Med Rep. 2017;15(4):1661–1667. doi:10.3892/mmr.2017.6194
9. Yuan L, Chen L, Qian K, et al. Co-expression network analysis identified six hub genes in association with progression and prognosis in human clear cell renal cell carcinoma (ccRCC). Genom Data. 2017;14:132–140. doi:10.1016/j.gdata.2017.10.006
10. Wei W, Lv Y, Gan Z, et al. Identification of key genes involved in the metastasis of clear cell renal cell carcinoma. Oncol Lett. 2019;17(5):4321–4328.
11. von Roemeling C, Radisky D, Marlow L, et al. Neuronal pentraxin 2 supports clear cell renal cell carcinoma by activating the AMPA-selective glutamate receptor-4. Cancer Res. 2014;74(17):4796–4810. doi:10.1158/0008-5472.CAN-14-0210
12. Khan M, Czarnecka A, Lewicki S, et al. Comparative gene expression profiling of primary and metastatic renal cell carcinoma stem cell-like cancer cells. PLoS One. 2016;11(11):e0165718. doi:10.1371/journal.pone.0165718
13. Smoot M, Ono K, Ruscheinski J, et al. Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics. 2011;27(3):431–432. doi:10.1093/bioinformatics/btq675
14. Ashburner M, Ball C, Blake J, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25(1):25–29. doi:10.1038/75556
15. Kanehisa M. The KEGG database. Novartis Found Symp. 2002;247:
16. Bindea G, Mlecnik B, Hackl H, et al. ClueGO: a Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics. 2009;25(8):1091–1093. doi:10.1093/bioinformatics/btp101
17. Lopes C, Franz M, Kazi F, et al. Cytoscape web: an interactive web-based network browser. Bioinformatics. 2010;26(18):2347–2348. doi:10.1093/bioinformatics/btq430
18. Li H, Tian X, Wang P, et al. MicroRNA-582-3p negatively regulates cell proliferation and cell cycle progression in acute myeloid leukemia by targeting cyclin B2. Cell Mol Biol Lett. 2019;24:66. doi:10.1186/s11658-019-0184-7
19. Fridley B, Dai J, Raghavan R, et al. Transcriptomic characterization of endometrioid, clear cell, and high-grade serous epithelial ovarian carcinoma. Cancer Epidemiol Biomarkers Prev. 2018;27(9):1101–1109. doi:10.1158/1055-9965.EPI-17-0728
20. Zhu X, Wang D, Lin Q, et al. Screening key lncRNAs for human rectal adenocarcinoma based on lncRNA-mRNA functional synergistic network. Cancer Med. 2019;8(8):3875–3891. doi:10.1002/cam4.2236
21. Niemira M, Collin F, Szalkowska A, et al. Molecular signature of subtypes of non-small-cell lung cancer by large-scale transcriptional profiling: identification of key modules and genes by weighted gene co-expression network analysis (WGCNA). Cancers. 2019;12(1):37. doi:10.3390/cancers12010037
22. Mo M-L, Chen Z, Li J, et al. Use of serum circulating CCNB2 in cancer surveillance. Int J Biol Markers. 2010;25(4):236–242. doi:10.5301/JBM.2010.6088
23. Hindriksen S, Meppelink A, Lens S. Functionality of the chromosomal passenger complex in cancer. Biochem Soc Trans. 2015;43(1):23–32. doi:10.1042/BST20140275
24. Ci C, Tang B, Lyu D, et al. Overexpression of CDCA8 promotes the malignant progression of cutaneous melanoma and leads to poor prognosis. Int J Mol Med. 2019;43(1):404–412.
25. Phan N, Wang C, Kl L, et al. Distinct expression of CDCA3, CDCA5, and CDCA8 leads to shorter relapse free survival in breast cancer patient. Oncotarget. 2018;9(6):6977–6992. doi:10.18632/oncotarget.24059
26. Yan G, Zou F, Dang B, et al. Genistein-induced mitotic arrest of gastric cancer cells by downregulating KIF20A, a proteomics study. Proteomics. 2012;12(14):2391–2399.
27. Qiu R, Runxiang Q, Geng A, et al. SEPT7 Interacts with KIF20A and regulates the proliferative state of neural progenitor cells during cortical development. Cereb Cortex. 2020;30(5):3030–3043. doi:10.1093/cercor/bhz292
28. Zhao X, Zhou L, Li X, et al. Overexpression of KIF20A confers malignant phenotype of lung adenocarcinoma by promoting cell proliferation and inhibiting apoptosis. Cancer Med. 2018;7(9):4678–4689. doi:10.1002/cam4.1710
29. Saito K, Ohta S, Kawakami Y, et al. Functional analysis of KIF20A, a potential immunotherapeutic target for glioma. J Neurooncol. 2017;132(1):63–74. doi:10.1007/s11060-016-2360-1
30. Khongkow P, Gomes A, Gong C, et al. Paclitaxel targets FOXM1 to regulate KIF20A in mitotic catastrophe and breast cancer paclitaxel resistance. Oncogene. 2016;35(8):990–1002. doi:10.1038/onc.2015.152
31. Asahara S, Takeda K, Yamao K, et al. Phase I/II clinical trial using HLA-A24-restricted peptide vaccine derived from KIF20A for patients with advanced pancreatic cancer. J Transl Med. 2013;11:291. doi:10.1186/1479-5876-11-291
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.