")
Back to Journals » International Journal of General Medicine » Volume 14
Identification of an Innate Immune-Related Prognostic Signature in Early-Stage Lung Squamous Cell Carcinoma
Authors Li L, Yu X, Ma G, Ji Z, Bao S, He X, Song L, Yu Y, Shi M, Liu X
Received 4 October 2021
Accepted for publication 15 November 2021
Published 30 November 2021 Volume 2021:14 Pages 9007—9022
DOI https://doi.org/10.2147/IJGM.S341175
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
Liang Li,1 Xue Yu,2 Guanqiang Ma,1 Zhiqi Ji,1 Shihao Bao,1 Xiaopeng He,1,3 Liang Song,1,3 Yang Yu,1,3 Mo Shi,1,3 Xiangyan Liu1,3
1Department of Thoracic Surgery, Shandong Provincial Hospital, Cheeloo College of Medicine, Shandong University, Jinan, Shandong, 250021, People’s Republic of China; 2Department of Pediatrics, Wuhan Children’s Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, Hubei, 420100, People’s Republic of China; 3Department of Thoracic Surgery, Shandong Provincial Hospital Affiliated to Shandong First Medical University, Jinan, Shandong, 250021, People’s Republic of China
Correspondence: Mo Shi; Xiangyan Liu
Department of Thoracic Surgery, Shandong Provincial Hospital, Cheeloo College of Medicine, Shandong University, Jinan, Shandong, 250021, People’s Republic of China
Email [email protected]; [email protected]
Background: Early-stage lung squamous cell carcinoma (LUSC) progression is accompanied by changes in immune microenvironments and the expression of immune-related genes (IRGs). Identifying innate IRGs associated with prognosis may improve treatment and reveal new immunotherapeutic targets.
Methods: Gene expression profiles and clinical data of early-stage LUSC patients were obtained from the Gene Expression Omnibus and The Cancer Genome Atlas databases and IRGs from the InnateDB database. Univariate and multivariate Cox regression and LASSO regression analyses were performed to identify an innate IRG signature model prognostic in patients with early-stage LUSC. The predictive ability of this model was assessed by time-dependent receiver operator characteristic curve analysis, with the independence of the model-determined risk score assessed by univariate and multivariate Cox regression analyses. Overall survival (OS) in early-stage LUSC patients was assessed using a nomogram and decision curve analysis (DCA). Functional and biological pathways were determined by gene set enrichment analysis, and differences in biological functions and immune microenvironments between the high- and low-risk groups were assessed by ESTIMATE and the CIBERSORT algorithm.
Results: A signature involving six IRGs (SREBF2, GP2, BMX, NR1H4, DDX41, and GOPC) was prognostic of OS. Samples were divided into high- and low-risk groups based on median risk scores. OS was significantly shorter in the high-risk than in the low-risk group in the training (P < 0.001), GEO validation (P = 0.00021) and TCGA validation (P = 0.034) cohorts. Multivariate Cox regression analysis showed that risk score was an independent risk factor for OS, with the combination of risk score and T stage being optimally predictive of clinical benefit. GSEA, ESTIMATE, and the CIBERSORT algorithm showed that immune cell infiltration was higher and immune-related pathways were more strongly expressed in the low-risk group.
Conclusion: A signature that includes these six innate IRGs may predict prognosis in patients with early-stage LUSC.
Keywords: early-stage lung squamous cell carcinoma, prognosis, risk score, gene signature, innate immune-related genes, immune cell infiltration
Introduction
Lung cancer is the second most commonly diagnosed cancer (11.4%) and the leading cause of cancer-related deaths (18%) worldwide.1 Lung cancer can be classified generally into two types: non-small cell lung cancer (NSCLC), which accounts for approximately 85% of tumors, and small cell lung cancer (SCLC), which accounts for approximately 15%.2 About 30% of patients with NSCLC are diagnosed with lung squamous-cell carcinoma (LUSC),3 with these patients having a median overall survival (OS) approximately 30% shorter than in patients with other NSCLC subtypes.4 Poor prognosis in patients with LUSC may be attributed to older age, more advanced disease at diagnosis and a more central location predisposing to the invasion of larger blood vessels.5,6 Additionally, many novel small-molecule inhibitors shown to provide clinical benefit to patients with lung adenocarcinoma are ineffective in patients with LUSC.7 Early-stage diagnosis and more effective treatment of LUSC can therefore enhance patient outcomes.
The innate immune system is the first line of defense against invading microbes. Pathogen recognition receptors on immune system cells activate innate immune signaling pathways, leading to the production of various proinflammatory cytokines and chemokines, subsequently inducing adaptive immune responses.8,9 Innate immune responses also indirectly influence the tumor microenvironment (TME) by controlling T cell fate and critically sculpting the TME.10 Distinct signal regulation modes in the TME can alter the biological functions of certain innate immune system cells, which may lead to tumor proliferation and evasion of immune surveillance. Additionally, specific immunophenotypes of partial innate immune cell compositions depend on the type, pathological type, and even stage of malignancy. Therefore, understanding the characteristics of the innate immune landscape in specific tumor types and stages may provide a theoretical basis for subsequent monitoring and treatment.
Immune-checkpoint inhibitors (ICIs) have shown promise in the treatment of NSCLC, with some of these agents approved for first-line treatment. These include pembrolizumab monotherapy for advanced NSCLC patients with PD-L1 TPS ≥ 1% and without EGFR/ALK gene alterations, atezolizumab monotherapy for NSCLC patients with high PD-L1 expression and without EGFR/ALK genetic alterations, and combinations of pembrolizumab plus chemotherapy and nivolumab plus ipilimumab.11–14 Some patients, however, do not benefit clinically from ICI treatment. For example, ICI treatment has not been shown to prolong survival in patients with early-stage LUSC, and some patients develop resistance to ICIs after an initial or develop immune-related adverse events. Owing to the complexity of the tumor immune microenvironment (TIME), no single indicator, including immunohistochemical expression of PD-L1, tumor mutational burden, number of CD8+ tumor-infiltrating lymphocytes or IFN-γ-related gene signature, is optimal for screening responders.15,16 These drawbacks may be overcome by elucidating the detailed mechanisms of action of ICIs. Although all currently available ICIs target the adaptive immune system to activate T cells,17 immune sensing and escape in the TIME depend on interactions between the innate and adaptive immune systems. Metabolic changes in innate immune system cells can alter immune activity and antitumor effects.18 Alternatively, newly developed ICIs targeting the innate immune system could overcome resistance to currently approved ICIs.17 In addition, screening for specific gene signatures related to innate immunity may also enhance the effects of immunotherapy. Several signature models related to innate immunity, as determined by RNA-sequencing and microarray assays, have successfully predicted OS in patients with hepatoblastoma,19 head and neck squamous cell carcinoma,20 ovarian cancer21 and colorectal cancer.22 Less is known, however, about the association between LUSC and innate immunity.
The present study systematically investigated the relationships between innate immune-related genes (IRGs) and prognosis in patients with early-stage LUSC. Our findings showed that innate IRGs can be used to classify patients with early-stage LUSC based on their clinical and molecular features. Furthermore, this study assessed differences in the immune microenvironment in high- and low-risk patients, providing a potential theoretical basis for future immunotherapy.
Materials and Methods
Mining from GEO and TCGA Databases
Cohorts I (GSE157009) and II (GSE157010) contain large numbers of specimens and datasets from multiple institutions, based on assays by independent laboratories and locked test parameters, thus ensuring objective quality standards.23 Microarray Suite version 5.0 (MAS 5.0), containing data on normalized mRNA expression and relevant clinical information (including age, gender, T stage, OS and disease-free survival [DFS]), were acquired from the Gene Expression Omnibus (GEOhttps://www.ncbi.nlm.nih.gov/geo/). Samples were included if (1) gene expression data were available; (2) clinical information, including age, gender and T stage, were available; and (3) follow-up time was greater than 30 days. The training cohort consisted of 245 patients in GSE157009, and the validation cohort consisted of 232 patients in GSE157010. Transcriptome profiles in FPKM format were derived from The Cancer Genome Atlas (TCGA) database. Based on the above-described criteria, 438 patients with early stage (ranging from IA-IIIA) LUSC were selected as a second validation cohort.
Establishment of a Prognostic Signature of Innate IRGs
Univariate Cox regression analysis was performed to identify possible prognostic genes associated with OS in the training cohort, with a P-value < 0.001 considered the filtering criterion. A comparison of these genes with innate IRGs obtained from InnateDB was used to select overlapping genes, which were considered candidate genes for the construction of a prognostic signature. The least absolute shrinkage and selection operator (LASSO) method was then applied using the glmnet package (https://CRAN.R-project.org/package=glmnet) in R24 for feature selection to narrow the number of IRGs with non-zero coefficients. Genes with non-zero coefficients were included in multivariate Cox regression analysis, and genes with P-values < 0.05 included in a prognostic signature model. The prognostic risk score of each sample was calculated by the “predict” function in “stats” R package using the formula:
Risk score=h0 (t)*exp (β1X1+β2X2+β3X3+β4X4+β5X5+β6X6),
where h0(t) represents the baseline risk value (unknown constants), β represents the regression coefficient calculated by multivariate Cox regression analysis, and X represents the gene expression level.
Evaluation and Validation of Innate IRG Signatures for Predicting Survival
Patients in the training cohort were divided into low- and high-risk groups based on the median risk score. Kaplan–Meier (K-M) survival curves and time-dependent receptor operating characteristic (time-ROC) curves were used to evaluate the accuracy of this signature. Moreover, the above-described formula and statistical methods were applied to the independent validation cohorts to validate the prognostic capacity of the gene signature.
Construction and Evaluation of a Predictive Nomogram
Risk scores were dichotomized into high- and low-risk score groups based on median risk score and treated as an independent clinicopathological parameter. Univariate and multivariate Cox regression analyses were performed to determine the clinical characteristics (age, gender, T stage and risk score) associated with OS. Factors differing significantly were visualized using a nomogram and their predictive accuracy and clinical efficacy determined. Decision curve analysis (DCA) was performed to estimate their possible clinical applications,25 and ROC curves were used to measure the accuracy of the model.
Functional Enrichment Analysis
Log fold-changes (logFCs) in the levels of gene expression in the high- and low-risk groups were calculated using the “limma” package in R, with these genes sorted from the highest to lowest logFCs. Gene set enrichment analysis (GSEA) was performed using the “clusterProfiler” package in R,26 with results considered significant if |NES| was >1, NOM p-value was <0.05, and FDR q-value was <0.25. Function and pathway enrichment were visualized using Ridgeplot.
Immune Profile Analysis
TIME differences between the high- and low-risk groups in the training and validation cohorts were evaluated using the “estimate” package in R. The relative proportions of 22 tumor-infiltrating immune cells (TICs) in the training cohort were calculated using the CIBERSORT algorithm.27
Statistical Analysis
Univariate and multivariate Cox regression analyses were performed using the “survminer” package in R, and LASSO analysis was performed using the “glmnet” package in R. Kaplan–Meier analyses were performed using the “survival” and “survminer” packages in R, and ROC analysis was performed using the “timeROC” package in R. The “rms”, “ggplot2”, “ggforest”, “cowplot”, “gseaplot2”, “pheatmap” and “VennDiagram” packages in R (version 4.0.3) were used for visualization. A p-value <0.05 was considered statistically significant.
Results
Cohort Characteristics
The flow chart of the present study is shown in Figure 1A. After screening for eligibility criteria, the training cohort consisted of 245 patients in GSE157009, and the independent validation cohort consisted of 232 patients in GSE157010, with a second validation cohort from TCGA consisting of 438 patients. The 245 patients in the training cohort included 88 women and 157 men, of median age 70 years (range: 43–92 years). The GEO validation cohort of 232 patients included 81 women and 151 men, of median age 68 years (range: 46–89 years), and the TCGA validation cohort of 438 patients included 116 women and 322 men, of median age 68 years (range: 39–90 years).
 |
Figure 1 Process used to construct the prognostic signature model based on innate immune-related genes. (A) Work flow. (B) Overlap of 147 genes associated with patient prognosis and 1376 genes associated with innate immunity, followed by selection of 29 genes by LASSO Cox regression analysis. (C and D) Construction of a LASSO Cox regression model from the 29 innate immune-related prognostic genes, followed by calculation of the tuning parameter (λ) based on partial likelihood deviance with 10-fold cross-validation. The vertical black line indicates the optimal log λ value. |
Establishment of Innate IRG Signatures Model in Training Cohort
Univariate Cox regression analyses identified 147 significantly prognostic genes (P < 0.001) in the training cohort. Of the 1376 innate IRGs downloaded from the innateDB database, 29 overlapped with genes in the training cohort (Figure 1B). LASSO-penalized Cox analysis identified 18 genes based on the optimal value of λ (Figure 1C and D). Finally, multivariate Cox regression analysis identified six genes (SREBF2, GP2, BMX, NR1H4, DDX41, GOPC) that were prognostically significant (P < 0.05). These genes were used to build a prognostic signature and calculate corresponding regression coefficients. Evaluation of risk scores in each sample yielded the formula:

The median risk score served as the boundary that separated patients in the training cohort into high- and low-risk groups (Figure 2A), with a scatter diagram showing poorer survival outcomes in the high-risk group (Figure 2B). Kaplan–Meier analysis confirmed that OS was significantly shorter in the high- than in the low-risk group (Figure 2C, P < 0.0001). A heatmap showed model genes differentially expressed in the high- and low-risk groups of the training cohort (Figure 2D). Evaluation of the predictive performance of the model using time-dependent ROC curves showed that the areas under the curve (AUCs) at 1, 3, and 5 years were 0.713, 0.722, and 0.708, respectively (Figure 2E).
 |
Figure 2 Risk score distribution, Kaplan-Meier survival analysis and time-dependent ROC curves in the training cohort (GSE157009). (A and B) Distribution of risk scores and survival status. (C) Kaplan-Meier analysis, showing that overall survival (OS) rates were significantly higher in the low- than in the high-risk group (P <0.0001 by Log rank test). (D) Distribution of risk scores as a function of gene expression characteristics. (E) AUC of time-dependent ROC curves measuring the ability of the model to predict OS. |
Validation of the Prognostic Model in an Independent Cohort
Two independent validation cohorts were utilized to further investigate the prognostic value of the innate IRGs signatures model. The 232 samples in the GEO cohort GSE157010 were categorized into high- and low-risk groups based on the median risk score, calculated as described above. The risk curve (Figure 3A) and the scatter diagram for survival (Figure 3B) were similar to those observed in the training cohort. Kaplan–Meier analysis showed that OS was significantly shorter in the high-risk group (P = 0.00021; Figure 3C). A heatmap showed the expression of the six genes in the high- and low-risk groups (Figure 3D), and time-dependent ROC curves showed that the average AUCs at 1, 3, and 5 years were 0.661, 0.643, and 0.640, respectively (Figure 3E).
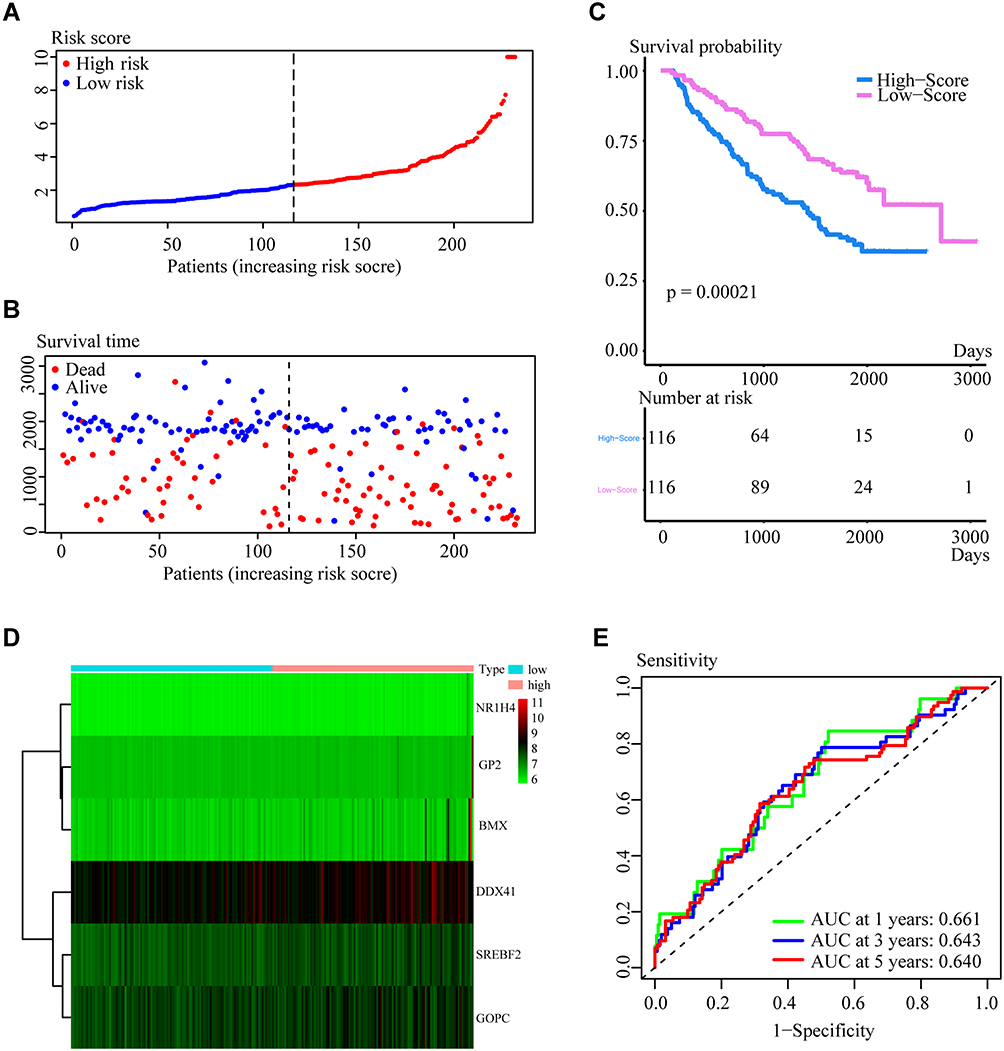 |
Figure 3 Risk score distribution, Kaplan-Meier survival analysis and time-dependent ROC curves in the validation cohort (GSE157010). (A and B) Distribution of risk scores and survival status. (C) Kaplan-Meier analysis, showing that overall survival (OS) rates were significantly higher in the low- than in the high-risk group (P= 0.00021 by Log rank test). (D) Distribution of risk scores as a function of gene expression characteristics. (E) AUC of time-dependent ROC curves measuring the ability of the model to predict OS. |
The same classification methods were applied to the TCGA cohort. Kaplan–Meier analysis showed that OS was markedly shorter in the high-risk group (Supplementary Figure 1A), and time-dependent ROC analysis showed that the average AUCs in 3, 5, and 7 years, were 0.482, 0.580 and 0.634, respectively (Supplementary Figure 1B).
Independent Prognostic Value of the IRG Signature Model
The independence and integration effects of the IRG signature model among multiple clinical parameters were assessed by drawing Forest plots and a nomogram, followed by univariate and multivariate Cox regression analysis. Univariate Cox regression analyses showed that age (hazard ratio [HR]=1.480, p = 0.019), T-stage (HR = 1.740, P < 0.001), and risk score (HR = 2.250, p < 0.001) were significantly associated with OS in the training cohort. Subsequent multivariate Cox regression analysis showed that age (HR = 1.560, p = 0.008), T-stage (HR = 1.600, P < 0.001) and risk score (HR = 2.060, p < 0.001) remained significantly associated with OS (Figure 4A and B). In the validation cohort, univariate Cox regression analysis showed that T-stage (HR = 1.450, p = 0.011) and risk score (HR = 2.010, p < 0.001), but not age, were significantly associated with OS. Multivariate Cox regression analysis also showed that T-stage (HR = 1.440, p = 0.014) and risk score (HR = 1.930, p < 0.001) were significantly associated with OS (Figure 4C and D). Taken together, these findings showed that both risk score and T-stage were independently predictive of OS in the training cohort. These factors were incorporated into a nomogram to quantify the predicted individual survival probability at 1, 3 and 5 years (Figure 5A). The possible application of this model to future clinical decision-making was further estimated by DCA, which quantifies the clinical value of a nomogram by analyzing the clinical results at different periods. This analysis showed that the combined model was more predictive of outcomes than any single model at 1, 3 and 5 years (Figure 5B), and ROC analyses showed that the average AUCs at 1, 3, and 5 years were 0.752, 0.727, and 0.715, respectively (Figure 5C).
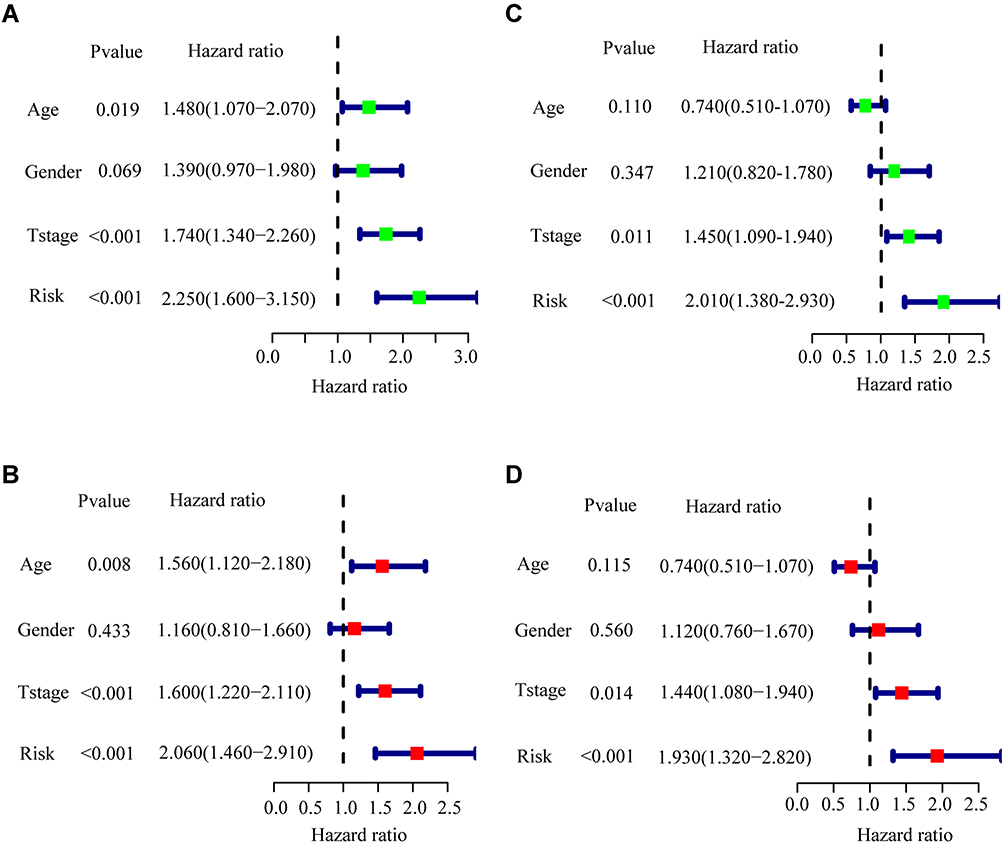 |
Figure 4 Univariate and multivariate Cox regression analysis of the risk scores and other clinical characteristics. (A) Univariate Cox regression analysis in the training cohort. (B) Multivariate Cox regression analysis in the training cohort. (C) Univariate Cox regression analysis in the validation cohort. (D) Multivariate Cox regression analysis in the validation cohort. |
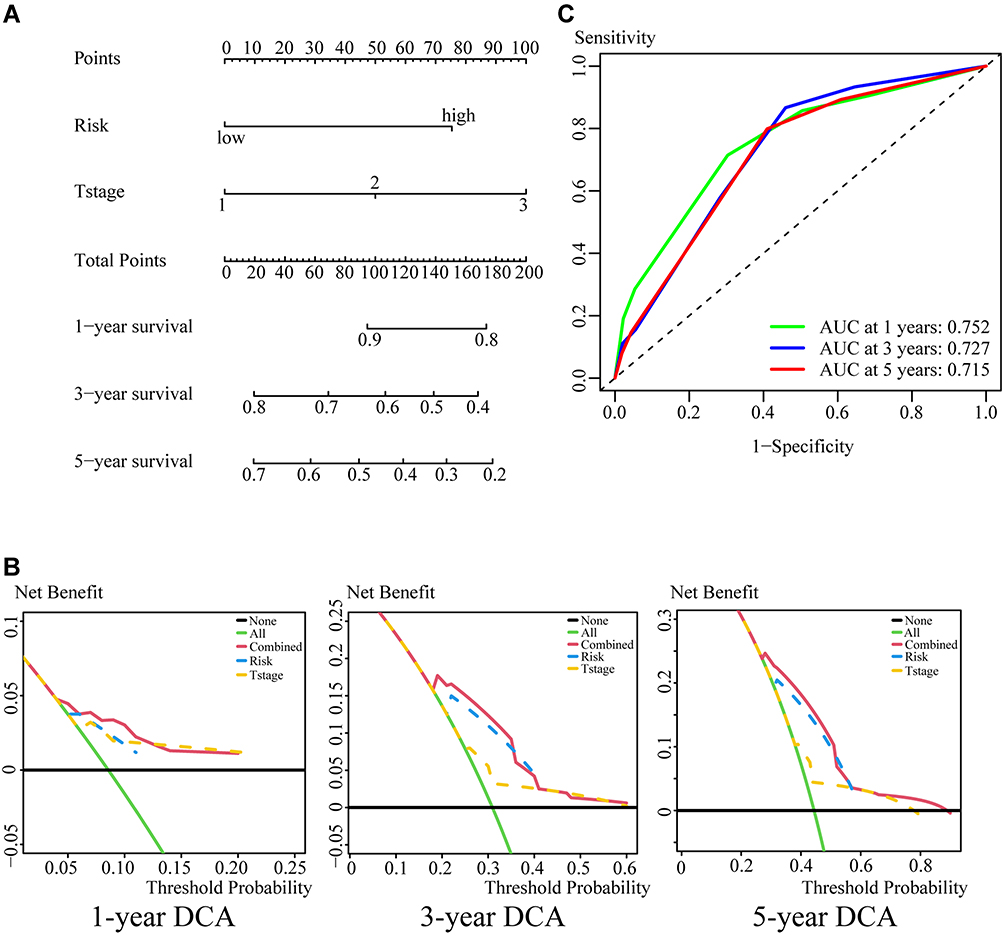 |
Figure 5 Construction and validation of a predictive nomogram. (A) Nomogram predicting the 1-, 3-, and 5-year OS of patients with early stage LUSC. (B) DCA curves showing that the nomogram is superior to a single model in providing optimal clinical decision-making benefits. (C) Time–ROC curves evaluating the predictive efficiency of the combined model at 1-, 3-, and 5-year. |
Functional Analysis in the Training and Validation Cohorts
To identify the biological functions and pathways associated with risk scores, samples in the high- and low-risk groups were subjected to differential analyses, and total logFC values were sorted from highest to lowest and included in GSEA. The top 20 biological functions and pathways enriched in both cohorts were visualized by Ridgeplot (adjusted P < 0.05, Figure 6A–D). After integrating, we found that carcinoma-related pathways, such as cell cycle, cellular senescence and microRNAs, were up-regulated in cancers in the high-risk group of the training cohort, while immune and cell death-related pathways, such as cytokine–cytokine receptor interactions, chemokine signaling pathways and phagosome pathways, were down-regulated (Figure 6E). Similar results were observed in the validation cohort, with cell cycle, thermogenesis and chemical carcinogenesis-reactive oxygen species pathways up-regulated in the high-risk group, while apoptosis, cytokine–cytokine receptor interactions and chemokine and NOD-like receptor signaling pathways being down-regulated (Figure 6F).
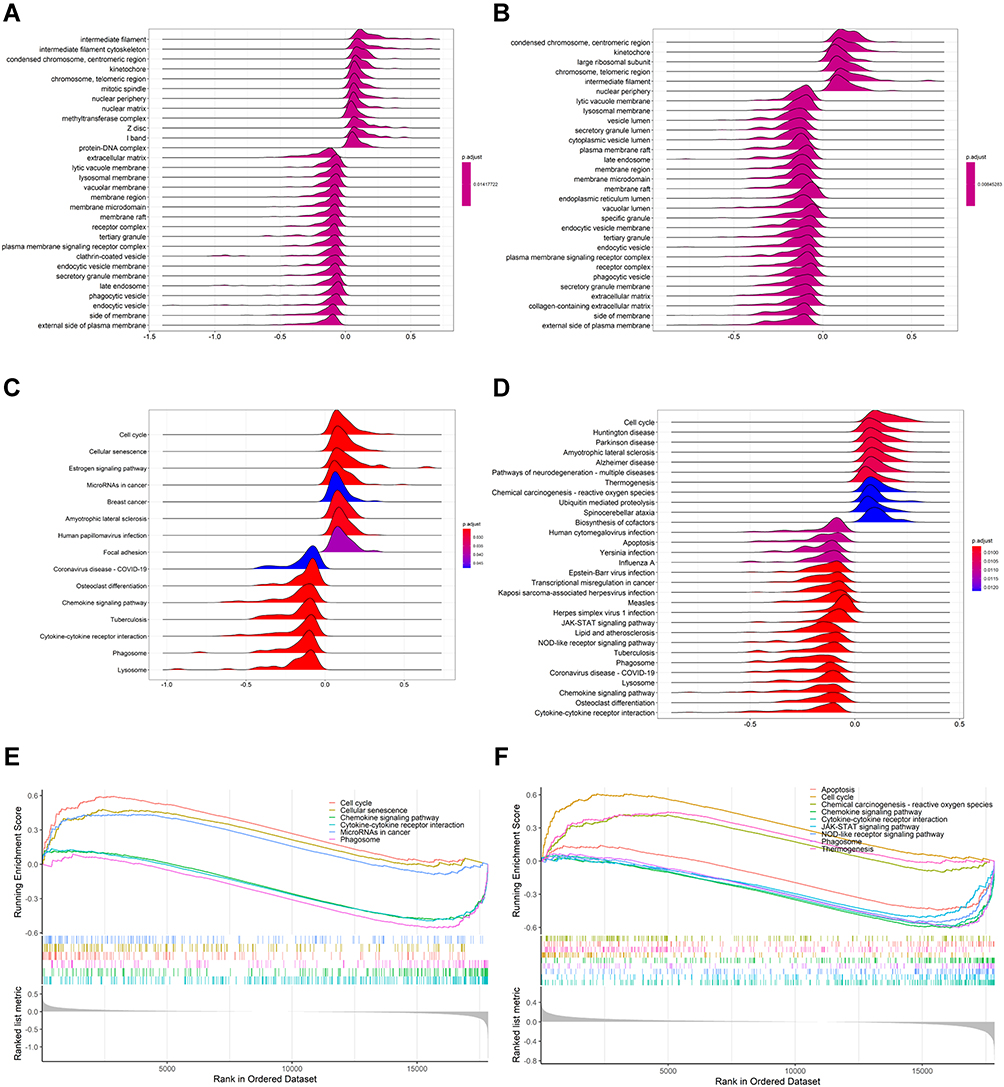 |
Figure 6 Enrichment analyses of biological functions and pathways in the high- and low-risk group. (A and B) Ridgeplots showing biological function enrichment in the training and validation cohorts, with the horizontal coordinate corresponding to the peak being the enrichment fraction. (C and D) Pathway enrichment in the training and validation cohorts. (E and F) Gene sets with |NES| > 1, NOM p-value <0.05, and FDR q-value < 0.25 were considered significantly enriched. |
TIME Score and Enrichment Analysis of 22 TICs
GSEA results showed that the differential functions and pathways between the high- and low-risk groups mainly included cytokine–cytokine receptor interactions and the chemokine signaling and carcinoma-related pathways. Evaluation of the contents of the immune-stromal components in TME of the training cohort showed that the high-risk group had a higher TumorPurity score but lower StromaScore and ImmuneScore than the low-risk group (Figure 7A), with similar results in the validation cohort (Figure 7B). Evaluation of the proportions of tumor-infiltrating immune subpopulations using the CIBERSORT algorithm showed the relative distributions of 22 TICs in the training cohort (Figure 7C), as well as correlations among these 22 TICs (Figure 7D). A violin plot (Figure 7E) showed that the numbers of naïve B cells, memory activated CD 4 T cells, and gamma delta T cells were markedly higher in the low- than in the high-risk group, whereas the numbers of memory B cells, resting NK cells and activated mast cells were significantly lower in the low-risk than in the high-risk group.
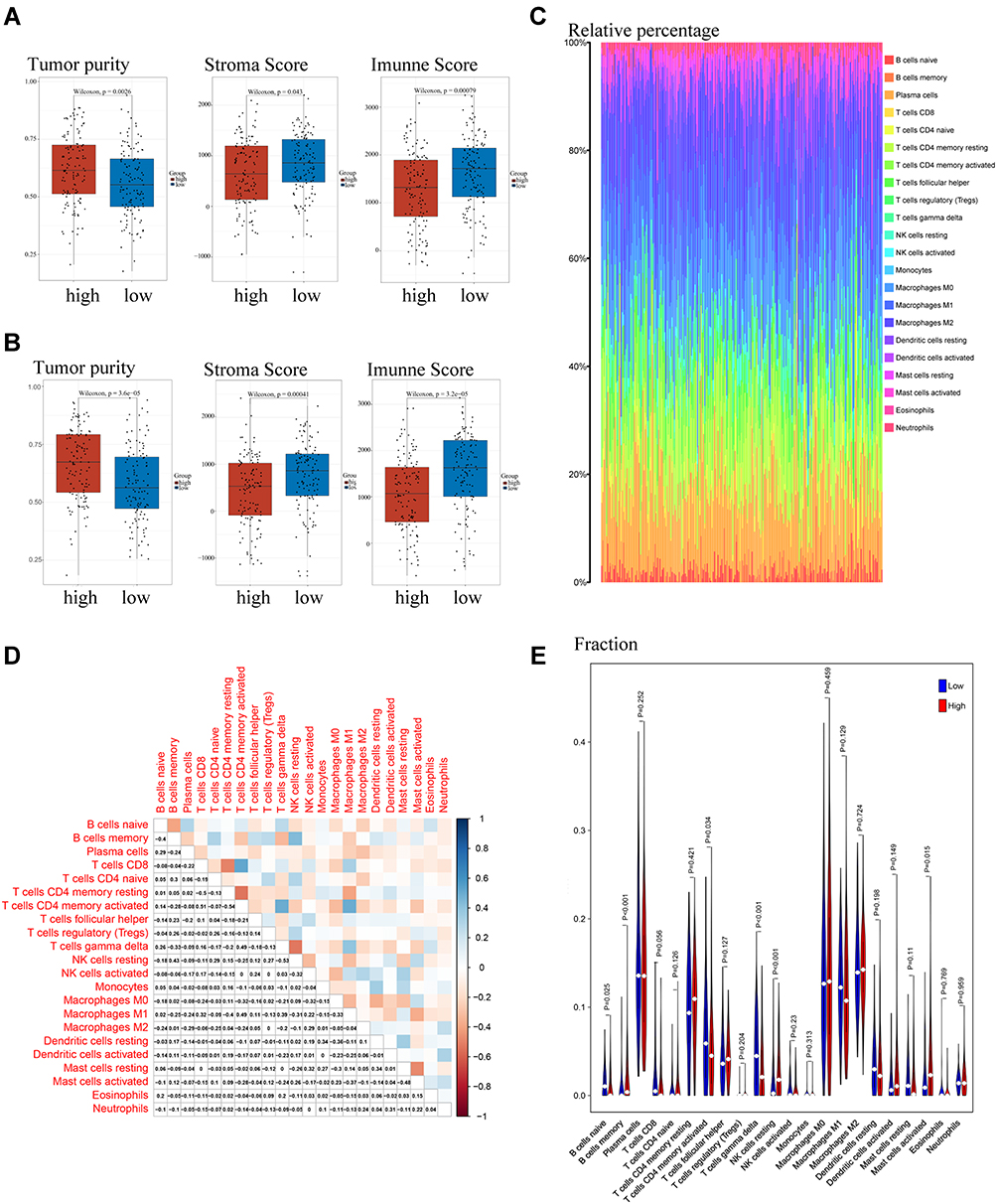 |
Figure 7 Component analysis of the tumor immune microenvironment. (A and B) Evaluation of tumor purity, Stroma score and Immune score by the “estimate” package in R and visualization by boxplots in the (A) training and (B) validation cohorts. (C) Relative content distribution of 22 TICs. (D) Analysis of correlations among the 22 TICs. (E) Violin plot showing the ratio of each of the 22 TICs between the high- and low-risk groups. (p < 0.05 was significant difference by Wilcoxon rank sum tests). |
Validation of the Six-Gene Signature by Evaluating Expression and Mutations
The expression patterns of the six gene signature in LUSC and normal lung tissue were further confirmed using the Human Protein Atlas (HPA; https://www.proteinatlas.org/). Immunohistochemical results from the HPA database showed that levels of expression of SREBF2, DDX41, GOPC and GP2 were higher to varying degrees in LUSC than in normal lung tissue samples, whereas the expression of BMX did not differ significantly (Figure 8A) and NR1H4 was not included in the HPA. The levels of mRNAs encoded by these six signature genes were downloaded from the Gene Expression Profiling Interactive Analysis (GEPIA) database. Except for BMX mRNA, the level of which was markedly lower in LUSC samples, there were no significant differences in expression of the other five genes (Figure 8B). Evaluation of mutations in these six signature genes, including missense, splice and truncating mutations, in 1176 samples from three independent TCGA cohorts using the cBioPortal instrument (http://www.cbioportal.org/) showed that GP2 and NR1H4 were mutated in 5% of these samples (Figure 8C).
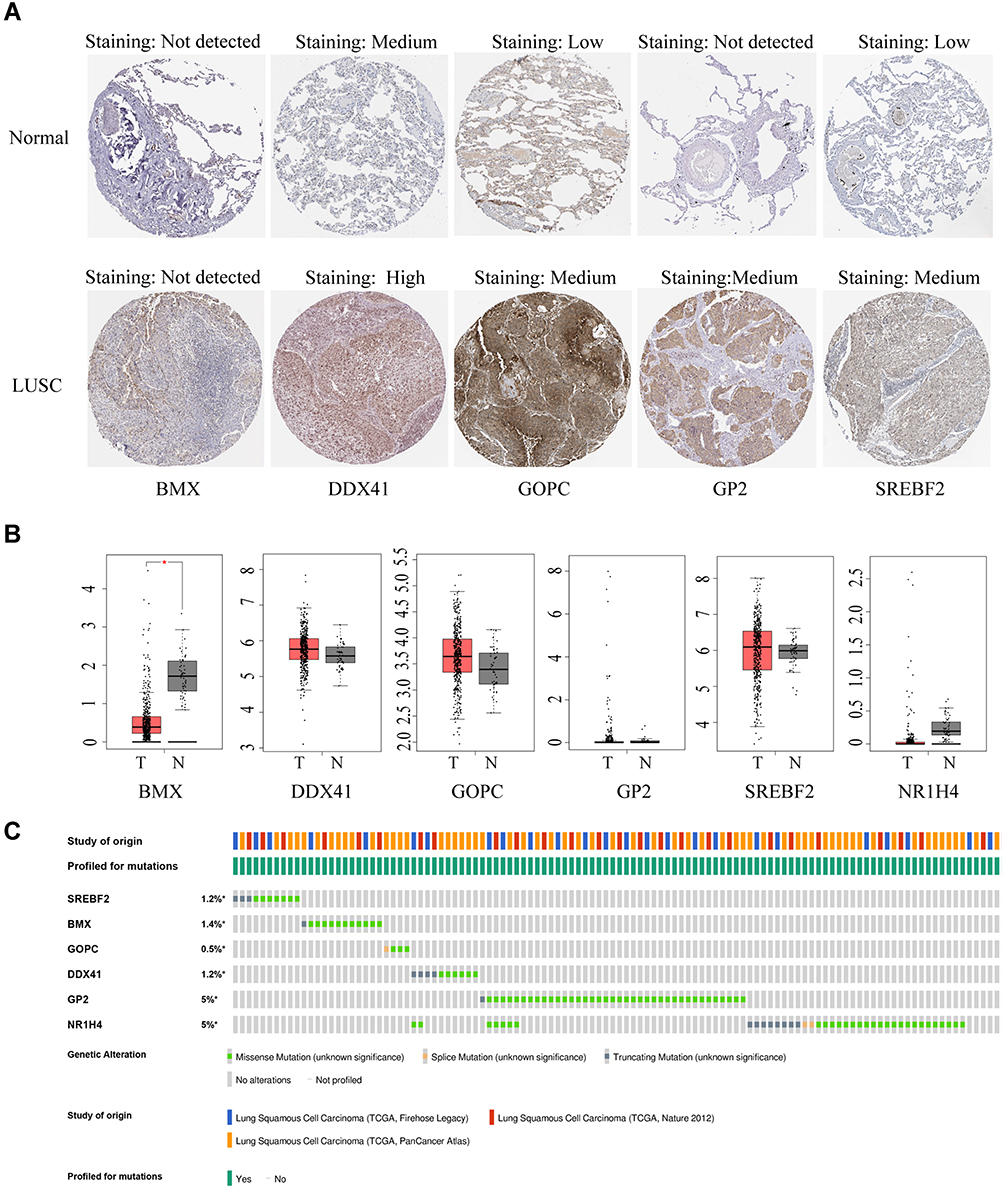 |
Figure 8 Differential expression of the six-genes in LUSC and normal lung tissue and gene mutations in LUSC. (A) Representative immunohistochemical results of LUSC and normal tissue samples from the Human Protein Atlas (HPA) assessing levels of expression of signature genes. (B) Levels of expression of mRNAs encoded by the six signature genes from the GEPIA database, with expression from the TCGA-LUSC and standardized by log2 (TPM+1). (C) Type and frequency of mutations of signature genes in three TCGA cohorts from the cBioPortal website. |
Discussion
The present study identified the prognostic innate immune-related signature associated with OS in early-stage LUSC using one cohort (GSE157009) and validated this signature in a second cohort (GSE157010). LASSO and subsequent multivariate Cox regression analyses identified a signature composed of six innate IRGs (SREBF2, GP2, BMX, NR1H4, DDX41, GOPC) as being significantly associated with OS in early-stage LUSC. SREBF2,28,29 BMX,30,31 GP2,32 DDX4133–35 and NR1H436–39 have been reported to correlate positively and GOPC negatively with tumor-related phenotypes and adverse events in several types of cancer.40 Bone marrow X kinase (BMX) was associated with chemoresistance in SCLC through its modulation of autophagy.30 Moreover, BMX signaling was found to mediate radiation resistance in the vascular endothelium of lung tumors in mice.31 A mutation in the gene encoding nuclear receptor subfamily 1 group H member 4 (NR1H4) was found to correlate with distant metastasis of LUSC.36 An SNP, rs12296850 at 12q23.1, may be associated with the expression of NR1H4 and high risk in LUSC.37 Indeed, this study found that the NR1H4 mutation frequency in LUSCs was 5%. NR1H4 has been reported to participate in colorectal cancer tumorigenesis, cell proliferation and cell survival via Wnt/β-catenin and the regulation of c-MYC stability.38,39 Additional studies are needed to assess the potential role of NR1H4 in LUSC development.
After integrating these six innate IRG genes into a prognostic signature, we calculated the risk score of each sample. Univariate and multivariate Cox regression analyses showed that both risk score and T stage were independently prognostic of OS. DCA and time-ROC curve showed that risk alone had a better capacity than T-stage alone in estimating 3- and 5-year OS in early-stage LUSC.
GSEA of both the training and validation cohorts showed that the high-risk group was enriched in genes associated with tumor-related pathways, such as the cell cycle, microRNAs in cancer and thermogenesis, in agreement with the evaluation of tumor purity in the TME. Moreover, genes associated with cytokine–cytokine receptor interactions and chemokine signal pathways were significantly negatively correlated with risk scores in both the training and validation cohorts. Specific cytokines, such as IFN, IL-2, IL-4, IL13, and IL15,41 can modulate both innate and adaptive immunity by interacting with their receptors on various immune cells, with chemokines playing crucial roles in inflammation and immunity. Owing to their expression in both tumor cells and leukocytes in the TME, chemokines represent an ideal target for immunotherapy.42 These functional analysis results were in accordance with immune microenvironment scores, with the low-risk group having a higher proportion of immune cells (quantified as ImmuneScore) and stromal cells (quantified as StromaScore). The TIME has been confirmed important during tumor development and progression. LUSCs have the highest leukocyte fractions among all solid tumors analyzed,16,43 making further exploration of the TIME immunity.
Although the adaptive immune system is primarily responsible for cytotoxicity and tumor suppression, various cytokines secreted by innate immune components within the TME can activate or suppress the immune system. For example, IL-4 in the TME initiates STAT6 signaling, polarizing macrophages to immune suppressive M2 macrophages;10 Neutrophils promote angiogenesis, tumor progression, and invasion by secreting neutrophil elastase, oncostatin-M and PGE244,45 factors that can modulate the curative effects of ICIs. Innate immune components and immunophenotypes in the TIME can change dynamically during tumor development, therefore, suggesting that efforts to develop therapeutic approaches should consider the specific conditions of innate immune components in different types and stages of cancer.
Genes representative of specific immune-cell types can predict TME components. The evolutionary trajectory of the immune response, from precancerous lesions to pre-invasive stages of LUSC46 has been found to include increases in the numbers of activated T cells, total neutrophils, M1 macrophages, memory B cells and the myeloid signature, accompanied by decreases in the abundance of naive B cells. In addition, resting mast cells were found to be more abundant in early than in later developmental stages, whereas activated mast cells increased during development, with both resting and activated NK cells remaining at low levels. The present study found that some innate immune components differed significantly in the high- and low-risk groups, as estimated by the CIBERSORT algorithm-based TIC analysis. Specifically, the numbers of γδ T cells were higher, while the numbers of resting NK cells and activated mast cells were lower. γδ T cells are considered a rapid lymphoid stress-surveillance system, rapidly responding to stress-induced tissue perturbation, and a bridge between innate and adaptive immunity.47,48 Moreover, increased numbers of γδ T cells have been found to correlated with prolonged OS in patients with LUSC.11 NK cells have potent antitumor and antimetastatic activity. Although NK cell infiltration did not correlate with clinical outcomes in NSCLC,49 activated NK cells in the TIME were found to correlate with OS in patients with LUSC.11 These findings may be due to NK cell dysfunction cells in the TIME,50 a hypothesis based on the higher ratio of resting NK cells in high-risk patients. Mast cells in the TME have both pro- and anti-tumorigenic activities, depending on tumor type and developmental stage.51 Mast cells infiltrating into tumor islets enhance the expression of TNFα, prolonging survival time in patients with NSCLC.52 In contrast, our results demonstrated that activated mast cells were more abundant in the high-risk group, suggesting a need for further studies to determine whether activated mast cells have pro- or anti-tumorigenic activities in early-stage LUSC. Despite the innate IRG signature identified in this study, components of adaptive immunity also differed in the high- and low-risk groups. For example, levels of activated memory CD4+ T cells and naïve B cells were higher, while levels of memory B cells were lower. These phenomena may be due to crosstalk between innate and adaptive immunity, which could determine the immune states in the TME and affect patient prognosis.
The present study had several limitations. First, clinical information about patients in these two cohorts in the GEO database was not complete. Although all samples were from patients with stage IA-IIIA LUSC (AJCC 8th),23 accurate clinical staging for each patient was unavailable. Thus, T stage was included for independent prognostic analysis. Second, further studies in larger numbers of samples are needed to validate the present results. Third, the functions of IRGs in early-stage LUSC have not been fully determined.
Conclusion
Based on bioinformatics analysis, a prognostic model that included six innate immune-related genes was constructed to predict OS in patients LUSC. The efficacy of the model was further validated in two independent cohorts. Risk scores, as determined by this model, were negatively associated with immune cell infiltration and functional performance. These findings may improve understanding of the roles of innate immune-related genes in early stages of LUSC and to further explore dynamic changes in the TIME.
Abbreviations
LUSC, lung squamous cell carcinoma; ICIs, immune-checkpoint inhibitors; IRGs, immune-related genes; TIME, tumor immune microenvironment; AUC, area under the curve; Time-ROC, time-dependent receptor operating characteristics; TICs, tumor-infiltrating immune cells; SREBF2, sterol regulatory element-binding transcription factor 2; BMX, BMX non-receptor tyrosine kinase; GOPC, Golgi-associated PDZ and coiled-coil motif containing; GP2, glycoprotein 2; DDX41, DEAD-box helicase 41; NR1H4, nuclear receptor subfamily 1 group H member 4.
Ethics and Consent to Participate
Because all data sources were from public databases, the Biomedical Ethics Committee of Shandong Provincial Hospital waived the requirement for an ethics statement (Supplementary Figure 2).
Acknowledgments
The authors thank the GEO, HPA, GEPIA and TCGA databases for making their data available.
Funding
This work was supported by grants from the National Natural Science Foundation of China (81902418) and the Natural Science Foundation of Shandong Province (ZR2020MH244).
Disclosure
The authors report no conflicts of interest.
References
1. Sung H, Ferlay J, Siegel RL, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71(3):209–249. doi:10.3322/caac.21660
2. Zappa C, Mousa SA. Non-small cell lung cancer: current treatment and future advances. Transl Lung Cancer Res. 2016;5(3):288–300. doi:10.21037/tlcr.2016.06.07
3. Yuan H, Liu J, Zhang J. The current landscape of immune checkpoint blockade in metastatic lung squamous cell carcinoma. Molecules. 2021;26(5):1392. doi:10.3390/molecules26051392
4. Socinski MA, Obasaju C, Gandara D, et al. Current and emergent therapy options for advanced squamous cell lung cancer. J Thorac Oncol. 2018;13(2):165–183. doi:10.1016/j.jtho.2017.11.111
5. Hirsch FR, Spreafico A, Novello S, Wood MD, Simms L, Papotti M. The prognostic and predictive role of histology in advanced non-small cell lung cancer: a literature review. J Thorac Oncol. 2008;3(12):1468–1481. doi:10.1097/JTO.0b013e318189f551
6. Nichols L, Saunders R, Knollmann FD. Causes of death of patients with lung cancer. Arch Pathol Lab Med. 2012;136(12):1552–1557. doi:10.5858/arpa.2011-0521-OA
7. Lazzari C, Karachaliou N, Gregorc V, et al. Second-line therapy of squamous non-small cell lung cancer: an evolving landscape. Expert Rev Respir Med. 2017;11(6):469–479. doi:10.1080/17476348.2017.1326822
8. Cui J, Chen Y, Wang HY, Wang RF. Mechanisms and pathways of innate immune activation and regulation in health and cancer. Hum Vaccin Immunother. 2014;10(11):3270–3285. doi:10.4161/21645515.2014.979640
9. Takeda K, Akira S. Toll-like receptors in innate immunity. Int Immunol. 2005;17(1):1–14. doi:10.1093/intimm/dxh186
10. Hinshaw DC, Shevde LA. The tumor microenvironment innately modulates cancer progression. Cancer Res. 2019;79(18):4557–4566. doi:10.1158/0008-5472.CAN-18-3962
11. Zhou F, Qiao M, Zhou C. The cutting-edge progress of immune-checkpoint blockade in lung cancer. Cell Mol Immunol. 2021;18(2):279–293. doi:10.1038/s41423-020-00577-5
12. Reck M, Rodríguez-Abreu D, Robinson AG, et al. Pembrolizumab versus chemotherapy for PD-L1-positive non-small-cell lung cancer. N Engl J Med. 2016;375(19):1823–1833. doi:10.1056/NEJMoa1606774
13. Herbst RS, Giaccone G, de Marinis F, et al. Atezolizumab for first-line treatment of PD-L1-selected patients with NSCLC. N Engl J Med. 2020;383(14):1328–1339. doi:10.1056/NEJMoa1917346
14. Herbst RS, Baas P, Kim DW, et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEY-NOTE-010): a randomised controlled trial. Lancet. 2016;387(10027):1540–1550. doi:10.1016/S0140-6736(15)01281-7
15. Doroshow DB, Sanmamed MF, Hastings K, et al. Immunotherapy in non-small cell lung cancer: facts and hopes. Clin Cancer Res. 2019;25(15):4592–4602. doi:10.1158/1078-0432.CCR-18-1538
16. Chi A, He X, Hou L, et al. Classification of non-small cell lung cancer’s tumor immune micro-environment and strategies to augment its response to immune checkpoint blockade. Cancers (Basel). 2021;13(12):2924. doi:10.3390/cancers13122924
17. Lentz RW, Colton MD, Mitra SS, Messersmith WA. Innate immune checkpoint inhibitors: the next breakthrough in medical oncology? Mol Cancer Ther. 2021;20(6):961–974. doi:10.1158/1535-7163.MCT-21-0041
18. Talty R, Olino K. Metabolism of innate immune cells in cancer. Cancers (Basel). 2021;13(4):904. doi:10.3390/cancers13040904
19. Sun R, Li S, Zhao K, Diao M, Li L. Identification of ten core hub genes as potential biomarkers and treatment target for hepatoblastoma. Front Oncol. 2021;11:591507. doi:10.3389/fonc.2021.591507
20. Zhang F, Liu Y, Yang Y, Yang K. Development and validation of a fourteen- innate immunity-related gene pairs signature for predicting prognosis head and neck squamous cell carcinoma. BMC Cancer. 2020;20(1):1015. doi:10.1186/s12885-020-07489-7
21. Zhang B, Nie X, Miao X, Wang S, Li J, Wang S. Development and verification of an immune-related gene pairs prognostic signature in ovarian cancer. J Cell Mol Med. 2021;25(6):2918–2930. doi:10.1111/jcmm.16327
22. Ma XB, Xu YY, Zhu MX, Wang L. Prognostic signatures based on thirteen immune-related genes in colorectal cancer. Front Oncol. 2021;10:591739. doi:10.3389/fonc.2020.591739
23. Bueno R, Richards WG, Harpole DH, et al. Multi-institutional prospective validation of prognostic mRNA signatures in early stage squamous lung cancer (Alliance). J Thorac Oncol. 2020;15(11):1748–1757. doi:10.1016/j.jtho.2020.07.005
24. Friedman J, Hastie T, Tibshirani R. Regularization paths for generalized linear models via coordinate descent. J Stat Softw. 2010;33(1):1–22. doi:10.18637/jss.v033.i01
25. Vickers AJ, Cronin AM, Elkin EB, Gonen M. Extensions to decision curve analysis, a novel method for evaluating diagnostic tests, prediction models and molecular markers. BMC Med Inform Decis Mak. 2008;8:53. doi:10.1186/1472-6947-8-53
26. Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16(5):284–287. doi:10.1089/omi.2011.0118
27. Newman AM, Steen CB, Liu CL, et al. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat Biotechnol. 2019;37(7):773–782. doi:10.1038/s41587-019-0114-2
28. Li X, Wu JB, Li Q, Shigemura K, Chung LW, Huang WC. SREBP-2 promotes stem cell-like properties and metastasis by transcriptional activation of c-Myc in prostate cancer. Oncotarget. 2016;7(11):12869–12884. doi:10.18632/oncotarget.7331
29. Zhong C, Fan L, Li Z, Yao F, Zhao H. SREBP2 is upregulated in esophageal squamous cell carcinoma and co-operates with c-Myc to regulate HMGCR expression. Mol Med Rep. 2019;20(4):3003–3010. doi:10.3892/mmr.2019.10577
30. Wang Q, Zeng F, Sun Y, et al. Etk interaction with PFKFB4 modulates chemoresistance of small-cell lung cancer by regulating autophagy. Clin Cancer Res. 2018;24(4):950–962. doi:10.1158/1078-0432.CCR-17-1475
31. Tu T, Thotala D, Geng L, Hallahan DE, Willey CD. Bone marrow X kinase-mediated signal transduction in irradiated vascular endothelium. Cancer Res. 2008;68(8):2861–2869. Erratum in: Cancer Res. 2008;68(19):8189. doi:10.1158/0008-5472.CAN-07-5743
32. Yun JW, Lee S, Ryu D, et al. Biomarkers associated with tumor heterogeneity in prostate cancer. Transl Oncol. 2019;12(1):43–48. doi:10.1016/j.tranon.2018.09.003
33. Sébert M, Passet M, Raimbault A, et al. Germline DDX41 mutations define a significant entity within adult MDS/AML patients. Blood. 2019;134(17):1441–1444. doi:10.1182/blood.2019000909
34. Polprasert C, Schulze I, Sekeres MA, et al. Inherited and somatic defects in DDX41 in myeloid neoplasms. Cancer Cell. 2015;27(5):658–670. doi:10.1016/j.ccell.2015.03.017
35. Qin K, Jian D, Xue Y, et al. DDX41 regulates the expression and alternative splicing of genes involved in tumorigenesis and immune response. Oncol Rep. 2021;45(3):1213–1225. doi:10.3892/or.2021.7951
36. Wu Y, Ni H, Yang D, et al. Driver and novel genes correlated with metastasis of non-small cell lung cancer: a comprehensive analysis. Pathol Res Pract. 2021;224:153551. doi:10.1016/j.prp.2021.153551
37. Dong J, Jin G, Wu C, et al. Genome-wide association study identifies a novel susceptibility locus at 12q23.1 for lung squamous cell carcinoma in Han Chinese. PLoS Genet. 2013;9(1):e1003190. doi:10.1371/journal.pgen.1003190
38. Yu J, Li S, Guo J, Xu Z, Zheng J, Sun X. Farnesoid X receptor antagonizes Wnt/β-catenin signaling in colorectal tumorigenesis. Cell Death Dis. 2020;11(8):640. doi:10.1038/s41419-020-02819-w
39. Lee YJ, Lee EY, Choi BH, Jang H, Myung JK, You HJ. The role of nuclear receptor subfamily 1 group H member 4 (NR1H4) in colon cancer cell survival through the regulation of c-Myc stability. Mol Cells. 2020;43(5):459–468. doi:10.14348/molcells.2020.0041
40. Ohara N, Haraguchi N, Koseki J, et al. Low expression of the GOPC is a poor prognostic marker in colorectal cancer. Oncol Lett. 2017;14(4):4483–4490. doi:10.3892/ol.2017.6817
41. Spangler JB, Moraga I, Mendoza JL, Garcia KC. Insights into cytokine-receptor interactions from cytokine engineering. Annu Rev Immunol. 2015;33:139–167. doi:10.1146/annurev-immunol-032713-120211
42. Mollica Poeta V, Massara M, Capucetti A, Bonecchi R. Chemokines and chemokine receptors: new targets for cancer immunotherapy. Front Immunol. 2019;10:379. doi:10.3389/fimmu.2019.00379
43. Thorsson V, Gibbs DL, Brown SD, et al. The immune landscape of cancer. Immunity. 2018;48(4):812–830. Erratum in: Immunity. 2019;51(2):411–412. doi:10.1016/j.immuni.2018.03.023
44. Demers M, Wong SL, Martinod K, et al. Priming of neutrophils toward NETosis promotes tumor growth. Oncoimmunology. 2016;5(5):e1134073. doi:10.1080/2162402X.2015.1134073
45. He M, Peng A, Huang XZ, et al. Peritumoral stromal neutrophils are essential for c-Met-elicited metastasis in human hepatocellular carcinoma. Oncoimmunology. 2016;5(10):e1219828. doi:10.1080/2162402X.2016.1219828
46. Mascaux C, Angelova M, Vasaturo A, et al. Immune evasion before tumour invasion in early lung squamous carcinogenesis. Nature. 2019;571(7766):570–575. doi:10.1038/s41586-019-1330-0
47. Chen H, Zou M, Teng D, Hu Y, Zhang J, He W. Profiling the pattern of the human T-cell receptor γδ complementary determinant region 3 repertoire in patients with lung carcinoma via high-throughput sequencing analysis. Cell Mol Immunol. 2019;16(3):250–259.
48. Kakimi K, Matsushita H, Murakawa T, Nakajima J. γδ T cell therapy for the treatment of non-small cell lung cancer. Transl Lung Cancer Res. 2014;3(1):23–33. doi:10.3978/j.issn.2218-6751.2013.11.01
49. Platonova S, Cherfils-Vicini J, Damotte D, et al. Profound coordinated alterations of intratumoral NK cell phenotype and function in lung carcinoma. Cancer Res. 2011;71(16):5412–5422. doi:10.1158/0008-5472.CAN-10-4179
50. Cong J, Wei H. Natural killer cells in the lungs. Front Immunol. 2019;10:1416. doi:10.3389/fimmu.2019.01416
51. Varricchi G, Galdiero MR, Loffredo S, et al. Are mast cells MASTers in cancer? Front Immunol. 2017;8:424. doi:10.3389/fimmu.2017.00424
52. Shikotra A, Ohri CM, Green RH, Waller DA, Bradding P. Mast cell phenotype, TNFα expression and degranulation status in non-small cell lung cancer. Sci Rep. 2016;6:38352. doi:10.1038/srep38352
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.