")
Back to Journals » Cancer Management and Research » Volume 11
Identification of a transcription factor-microRNA network in esophageal adenocarcinoma through bioinformatics analysis and validation through qRT-PCR
Authors Chen D, Lu T , Tan J, Zhao K, Li Y, Zhao W, Li H, Wang Q, Wang Y , Wei L
Received 11 January 2019
Accepted for publication 25 February 2019
Published 18 April 2019 Volume 2019:11 Pages 3315—3326
DOI https://doi.org/10.2147/CMAR.S201274
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Chien-Feng Li
Di Chen,1 Tong Lu,2 Junying Tan,1 Kun Zhao,1 Yuli Li,1 Wenjie Zhao,1 Hao Li,1 Qiuyue Wang,1 Yuanyong Wang,2 Liangzhou Wei1
1Department of Gastroenterology, Affiliated Hospital of Qingdao University, Qingdao 266003, People’s Republic of China; 2Department of Thoracic Surgery, Affiliated Hospital of Qingdao University, Qingdao 266003, People’s Republic of China
Purpose: The rapidly rising incidence of esophageal adenocarcinoma (EAC), which is usually diagnosed late with a poor prognosis, has become a growing problem. This study investigated the potential transcription factor (TF)-related molecular mechanisms of EAC by using bioinformatics analysis and qRT-PCR validation.
Methods: Expression profile datasets for mRNAs (GSE92396, GSE13898, GSE26886 and GSE1420) and miRNAs (GSE16456) were downloaded from the GEO database. Overlapping differentially expressed genes (DEGs) and differentially expressed miRNAs (DEMs) were identified through integrative analysis. Then, a TF-miRNA-mRNA network was constructed based on bioinformatics data from the TRRUST, TRED and miRTarBase database. Furthermore, overall survival analysis for the mRNAs and miRNAs in the TF-miRNA-mRNA network was performed with data from TCGA, and qRT-PCR was used to validate the results.
Results: A total of 294 overlapping DEGs were identified in EAC tissues compared to normal tissues, including 181 downregulated and 113 upregulated genes. Then, 16 TFs that could target the DEGs and were related to cancer were predicted based on public databases, and 41 DEGs that could be targeted were identified as key genes. Additionally, 12 DEMs were predicted through miRTarBase to be associated with the key genes, and TP53-(miR-125b)-ID2 and JUN-(miR-30a)-IL1A from the TF-miRNA-mRNA network were identified to potentially play significant roles in EAC. Furthermore, CCL20, IL1A, ABCC3, hsa-miR-23b, and hsa-miR-191, which are involved in the TF-miRNA-mRNA network, were found to be significantly associated with patient survival in EAC. Finally, the expression of a miRNA-mRNA pair (hsa-miR-30a-5p and IL1A) was revealed to be correlated with prognosis.
Conclusion: In this study, a TF-miRNA-mRNA network was constructed to analyze the potential molecular mechanisms of EAC. Key genes and miRNAs associated with patient survival were identified, which may reveal promising approaches for EAC diagnosis and therapy.
Keywords: esophageal adenocarcinoma, prognosis, differentially expressed genes, microRNA, transcription factor
Introduction
Esophageal carcinoma has a five-year survival rate of 12–20% in Western populations1,2 and induces the deaths of more than 400,000 people worldwide annually.3 The incidence of esophageal adenocarcinoma (EAC), one major histologic subtype of esophageal carcinoma, has increased by approximately 6-fold in the past 40 years.4 EAC is a form of cancer with a rapidly increasing incidence in Western countries.5,6 In the last few decades, the prognosis of patients with EAC has mildly improved, but it remains worse than that of most other types of cancer.7–9 However, the molecular mechanisms giving rise to EAC remain unclear. Therefore, a thorough exploration of EAC pathogenesis is urgently needed to improve EAC diagnosis, treatment and prognosis.
Transcription factors (TFs) are DNA-binding proteins that can function as tumor suppressors or oncogenes.10 TFs play significant roles in the regulation of gene expression and can induce avoidance of apoptosis and uncontrolled growth.11 microRNAs (miRNAs) are short, noncoding RNAs consisting of 18–25 nucleotides that regulate the translation of mRNAs.12 Mature miRNAs can recognize and bind to the 3ʹ untranslated region of mRNAs and regulate translation at the posttranscriptional level through repression or degradation of the targeted mRNAs.13 Numerous studies have revealed that TFs regulate gene expression by interacting with miRNAs. For instance, Chandra et al found that two TFs, ZEB1 and ZEB2, could contribute to epithelial-mesenchymal transition (EMT) through interaction with miR-101–1.14 In addition, Hua et al demonstrated that ETS1 could inhibit cell proliferation, invasion and migration by regulating miR-139-5p in hepatocellular carcinoma (HCC).15 In addition to miRNAs, lncRNAs and circRNAs were also proven to interact with TFs. Sun et al reported that NRG-1-ICD could be regulated by circACTA2 via the NRG-1-ICD/circACTA2/miR-548f-5p axis in vascular smooth muscle cells (VSMCs).16 Additionally, Guo et al found that KIF25-AS1, LINC01355 and AC092171.2 were regulated by POLR2A in ovarian cancer tissues.17
Recently, bioinformatics analysis has gradually been utilized to explore the mechanisms of cancer initiation and progression to identify important cancer-related genes, noncoding RNAs and TFs for further experimental verification. In this study, dysregulated genes and miRNAs in EAC were identified from the Gene Expression Omnibus (GEO) database. Subsequently, to understand the underlying molecular mechanisms in EAC, we used a public database to find the TFs that regulate the dysregulated genes. miRNAs were further predicted according to the TFs that targeted the DEGs, and interactions between miRNAs and TFs were predicted with miRTarBase. Based on our findings, TF-miRNA-mRNA networks were constructed to reveal a transcriptional regulation model and identify key genes and miRNAs associated with EAC. Functional enrichment analysis and protein-protein interaction (PPI) network analysis were also performed, and we used the Cancer Genome Atlas (TCGA) database to identify correlations between the expression of key genes and miRNAs in the TF-miRNA-mRNA network and overall survival. Finally, quantitative real-time PCR (qRT-PCR) was utilized to validate the results.
Methods
Collection of datasets
A flowchart of the study design is shown in Figure 1. Both mRNA and miRNA expression profiles were downloaded from the GEO database (
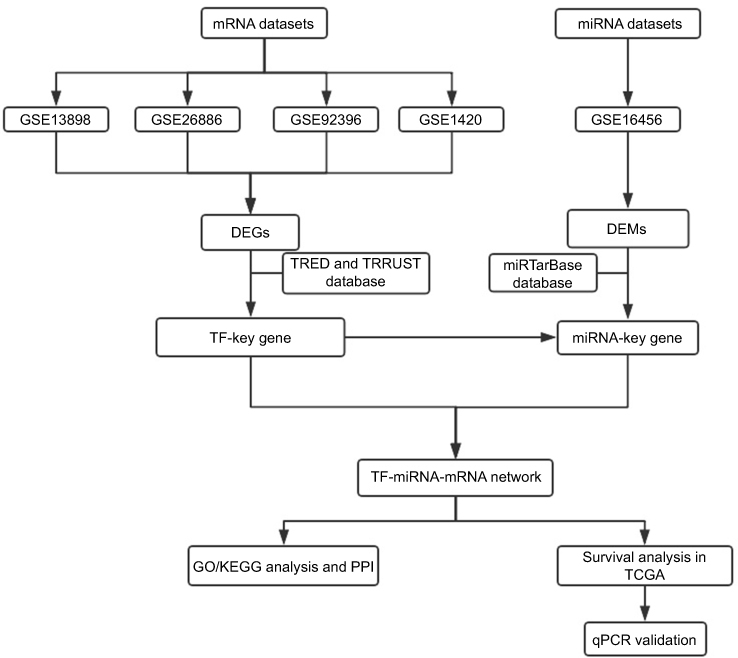 | Figure 1 Flowchart of the bioinformatics analysis. Abbreviations: DEGs, differentially expressed genes; DEMs, differentially expressed miRNAs; miRNA, microRNA; PPI, protein-protein interaction; TCGA, the Cancer Genome Atlas; TF, transcription factor; TRED, Transcriptional Regulatory Element Database; TRRUST, Transcriptional Regulatory Relationships Unraveled by Sentence-based Text mining. |
Aberrantly expressed miRNA and DEG analysis
The limma algorithm package18 in R software was utilized to identify DEGs and aberrantly expressed miRNAs in EAC tissues compared with NE tissues. DEGs were identified based on the following criteria: (1) they had a |log2FC|≥1 and a P-value <0.05, (2) they were shared among the four datasets, and (3) they showed consistent trends in all four datasets. The Benjamini and Hochberg false discovery rate (FDR) and adjusted P-values (adj.p) were applied to provide a balance between the probability of obtaining false positives and the probability of discovering statistically significant genes. The cutoff criteria for aberrantly expressed miRNAs were a |log2FC|≥1 and a P-value <0.05. Finally, an UpSet plot was constructed to visualize the DEGs in the different mRNA datasets.
Prediction of TFs and miRNAs and construction of a TF-miRNA-mRNA network
The Transcriptional Regulatory Relationships Unraveled (TRRUST,
Functional enrichment and pathway analysis
Gene ontology (GO) analysis and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis were conducted to analyze the functions of the key genes. Then, the Database for Annotation Visualization and Integrated Discovery (DAVID:
PPI network construction
To identify relevant PPIs, the Search Tool for the Retrieval of Interacting Genes/Proteins (STRING;
Survival analysis validation
TCGA, which collates information from the National Human Genome Research Institute (NHGRI) and the National Cancer Institute (NCI), has generated multidimensional and comprehensive maps for multiple types of cancers. To verify the results obtained thus far, data for 89 EAC tissue samples and the clinical data of the patients were downloaded from TCGA, and the prognostic value of the key genes and miRNAs was identified by Kaplan-Meier analysis. The survival package in R software was used for survival analysis. A P-value <0.05 was set as the cutoff criterion.
Total RNA extraction and qRT-PCR validation
A total of 20 EAC tissues and adjacent noncancerous tissues were collected from Qingdao University Hospital. This study was approved by the Ethical Committee of Qingdao University Hospital and was conducted in accordance with the Declaration of Helsinki, and each patient offered written informed consent. All tissues were immediately frozen in liquid nitrogen after resection and stored at −80 °C.
Tissue sample RNA was extracted using TRIzol (Invitrogen, CA, USA) according to the manufacturer’s protocol. Total RNA including miRNAs was reverse transcribed into cDNA with a miRNA First Strand cDNA Synthesis Kit (Sangon, China) and a 1st strand cDNA Synthesis Kit (Takara, China). miRNA and mRNA expression was then examined by qRT-PCR with a MicroRNAs qPCR Kit (Sangon, China), a Biosystems 7500 Fast Real-Time PCR System, and TransStart® Top Green qPCR SuperMix (TransGen Biotech, China). miRNA and mRNA expression was normalized to U6/GAPDH expression. The 2−∆∆Ct method was used to calculate the levels of miRNA and mRNA expression. The primers were designed using Primer 5.0 software, and the primer sequences are shown in Table 1. GraphPad Prism version 7.0 was used to analyze the qRT-PCR data, and comparisons between two groups were performed using Student’s t-test, with a P-value <0.05 considered significant.
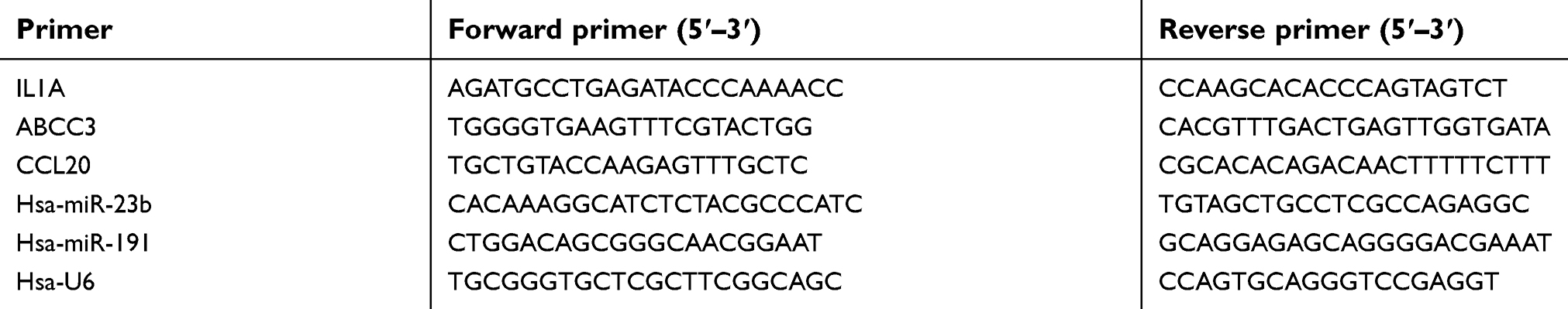 | Table 1 Primer sequences of qRT-PCR |
Results
Microarray datasets from the GEO
miRNA profiles were obtained from the GSE16456 dataset, while mRNA expression profiles were obtained from the GSE13898, GSE26886, GSE92396 and GSE1420 datasets. Twenty Barrett’s esophagus, 19 normal and 21 EAC tissues were included in the GSE26886 dataset, while 15 Barrett’s esophagus, 28 normal and 64 EAC tissues were included in the GSE13898 dataset. The GSE92396 dataset included a total of 9 normal tissues and 12 EAC tissues, while the GSE1420 dataset included 8 Barrett’s esophagus, 8 normal and 8 EAC tissues. The miRNA dataset GSE16456 included 10 Barrett’s esophagus, 16 normal and 6 EAC tissues. All relevant details are shown in Table 2.
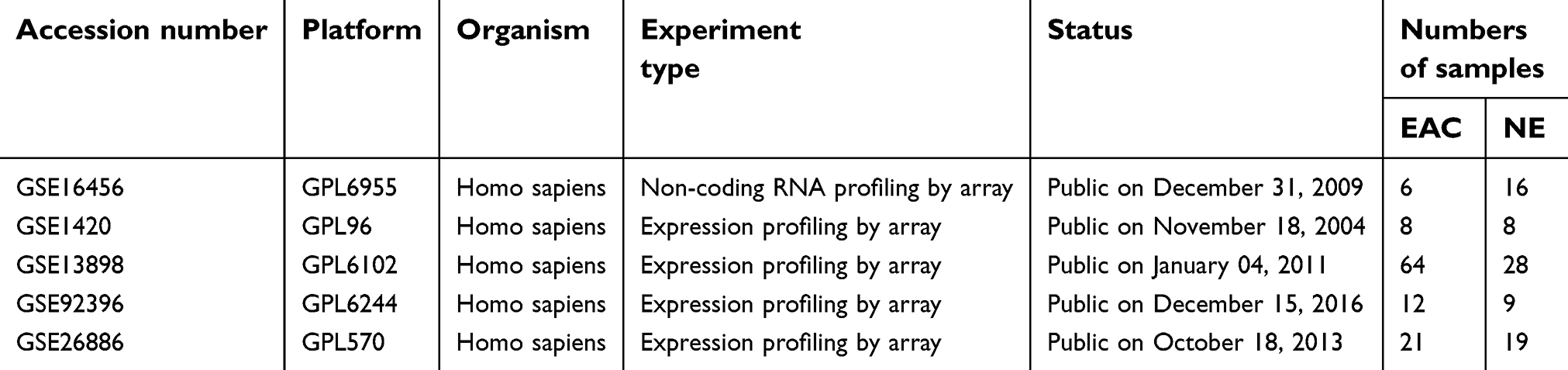 | Table 2 Information of expression profiling microarray datasets |
Analysis of DEGs and differentially expressed miRNAS
To identify DEGs, the GEO mRNA expression profile datasets were analyzed. A total of 294 DEGs were obtained through examining genes that showed the same expression trends among the mRNA datasets (Figure 2). Among the DEGs, 113 genes were upregulated in EAC, while the remaining 181 genes were downregulated (|log2FC|≥1, adjusted P-value <0.05)
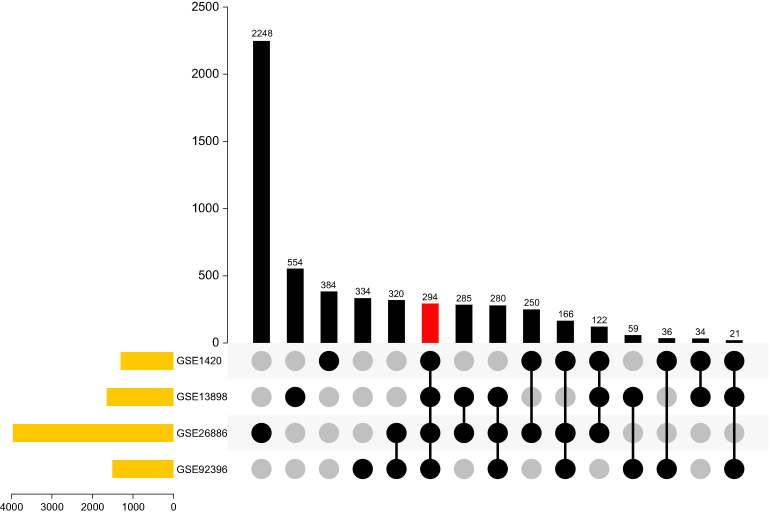 | Figure 2 Identification of overlapping DEGs in the mRNA expression profile datasets GSE1420, GSE26886, GSE13898, and GSE92396.Abbreviation: DEG, differentially expressed gene. |
A total of 61 DEMs were identified between the EAC and NE tissues in the GSE16456 dataset. Among them, 37 miRNAs were significantly downregulated in EAC, while the other 24 miRNAs were upregulated (|log2FC|≥1, adjusted P-value <0.05).
Construction of the TF-miRNA-mRNA network
Upon analysis of 294 DEGs for potential TFs in the TRRUST database, which is a database of transcriptional regulatory networks, 46 TFs and 70 TF-targeted genes were identified. Then, we cross-referenced the above results with the 36 cancer-related TF families and TF-target gene regulatory networks in the TRED database and ultimately identified 16 TFs and 41 key genes in EAC. Subsequently, miRTarBase was used to identify miRNAs predicted to be linked to the 41 key genes and to predict interactions between the TFs and miRNAs. Furthermore, 12 overlapping miRNAs were identified from among the DEMs in the GSE16456 dataset and the miRNAs predicted from miRTarBase. A TF-miRNA-mRNA interaction network was constructed based on the TRED, TRRUST and miRTarBase analyses. Finally, TP53-(miR-125b)-ID2 and JUN-(miR-30a)-IL1A were identified as possibly playing significant roles in EAC. Cytoscape software was used to visualize the TF-miRNA-mRNA network in EAC (Figure 3 and Table 3).
 | Table 3 Regulatory relationships among TFs, key genes, and miRNAs |
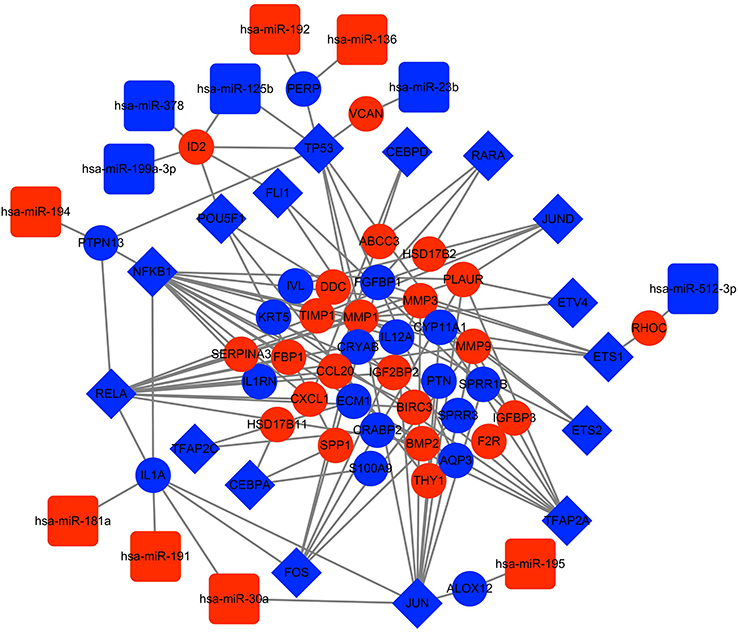 | Figure 3 TF-miRNA-mRNA network. The blue rhombuses indicate TFs. The blue circles indicate downregulated genes, and the red circles indicate upregulated genes. The blue squares represent downregulated miRNAs, and the red squares represent upregulated miRNAs. Abbreviations: DEG, differentially expressed gene; TF, transcription factor; miRNA, microRNA. |
Functional and pathway analysis of key genes
To further study the functional roles of key genes in the pathogenesis of EAC, GO and KEGG pathway enrichment analyses were performed. In the GO biological process (BP) group, the key genes were mainly enriched for terms related to the inflammatory response, extracellular matrix disassembly, positive regulation of I-kappaB kinase/NF-kappaB signaling, and epidermis development, among others. In the cellular component (CC) group, the key genes were mainly enriched for the extracellular space term, while in the molecular function (MF) group, the key genes were mainly enriched for the cytokine activity term (Figure 4 and Table 4).
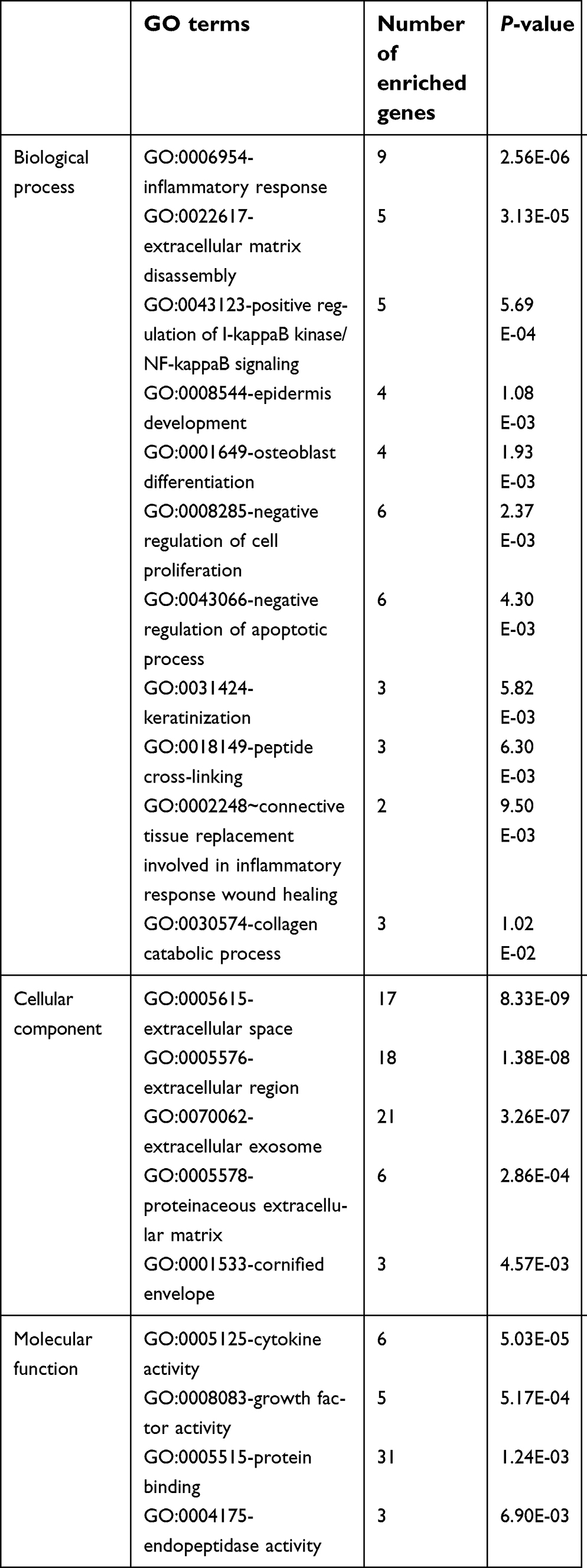 | Table 4 GO terms of key genes |
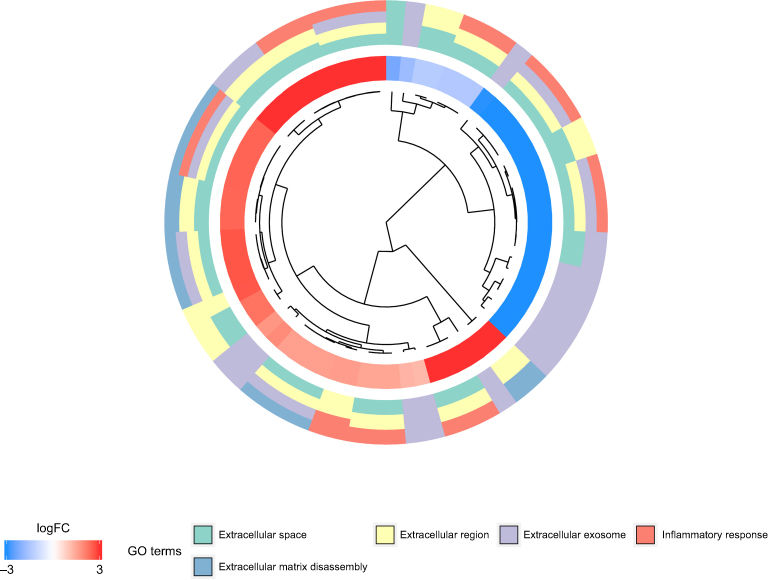 | Figure 4 GO analysis results for the key genes. Abbreviation: GO, Gene ontology. |
The results of KEGG pathway analysis suggested that the key genes were significantly enriched in the TNF signaling pathway, rheumatoid arthritis, cytokine-cytokine receptor interaction, transcriptional misregulation in cancer and pathways in cancer (Figure 5 and Table 5). In particular, the KEGG results further verified that the regulation of the key genes was associated with TFs.
 | Table 5 KEGG pathways of key genes |
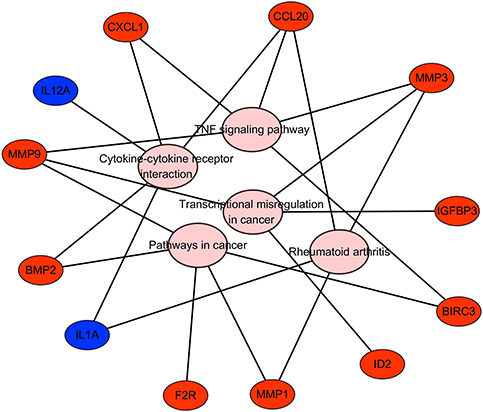 | Figure 5 KEGG pathway analysis results for the key genes. Abbreviation: KEGG, Kyoto Encyclopedia of Genes and Genomes. |
PPI analysis
PPI networks, which are used to analyze the interaction of proteins, were constructed in the STRING database with a combined score >0.4 as the cutoff criterion. Then, we used the MCODE plug-in to identify functional modules. One module consisting of 10 nodes and 31 edges was found and included IGFBP3, MMP3, SPP1, MMP1, PLAUR, VCAN, IL1A, MMP9, ECM1, and CXCL1 (Figure 6).
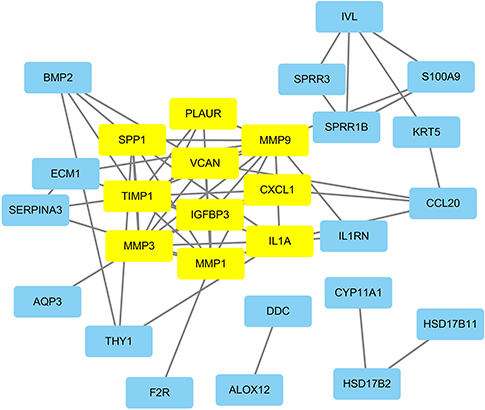 | Figure 6 PPI network of the key genes. The yellow rectangles represent the genes in the functional modules of the PPI network. Abbreviation: PPI, protein-protein interaction. |
Survival analysis validation in TCGA
The clinical data and RNA expression data of patients from TCGA were used to validate the key genes and miRNAs. The survival R package was used to analyze patient survival, and three key genes (CCL20, IL1A, and ABCC3) and two miRNAs (hsa-miR-23b and hsa-miR-191) were identified as being significantly associated with prognosis in EAC (Figure 7). More importantly, one miRNA-gene pair (hsa-miR-30a-5p and IL1A) was identified as being related to prognosis in EAC (Figure 8).
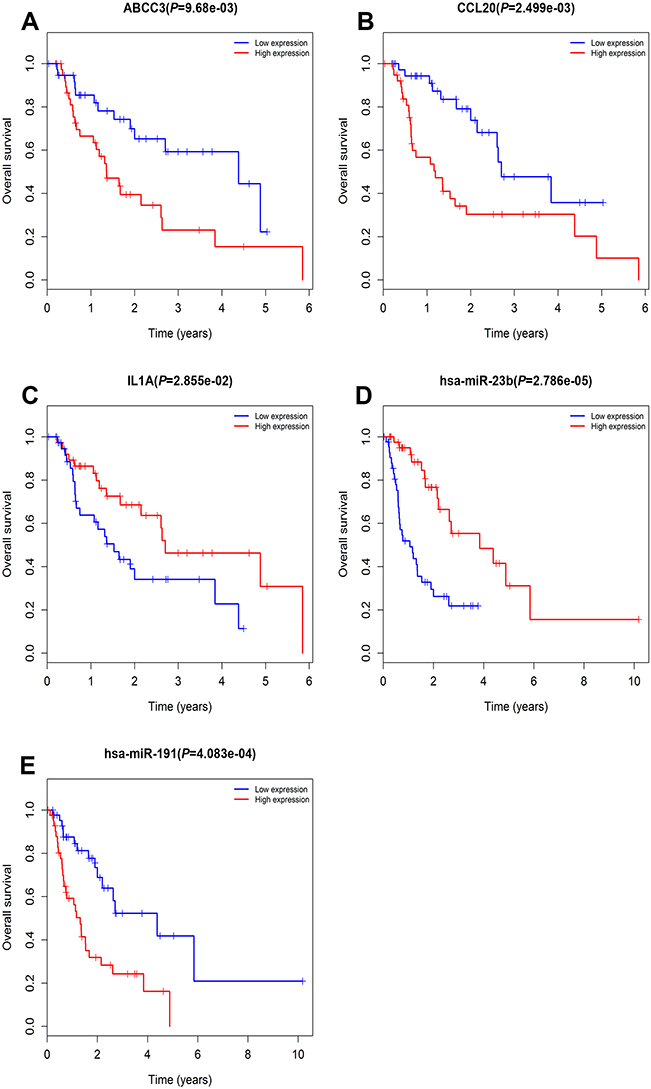 | Figure 7 Prognostic value of ABCC3, CCL20, IL1A, hsa-miR-23b, and hsa-miR-191 in EAC patients from TCGA datasets (A–E). Abbreviations: EAC, esophageal adenocarcinoma; TCGA, the Cancer Genome Atlas. |
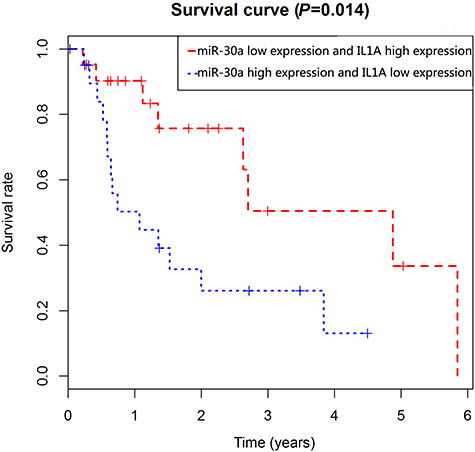 | Figure 8 Prognostic value of hsa-miR-30a-5p and IL1A in EAC patients from TCGA datasets. Abbreviations: EAC, esophageal adenocarcinoma; TCGA, the Cancer Genome Atlas. |
Validation of miRNAs and key genes by qRT-PCR
qRT-PCR was performed to validate the expression of the key genes and miRNAs. We found that three key genes (CCL20, IL1A, and ABCC3) and two miRNAs (hsa-miR-23b and hsa-miR-191) were significantly dysregulated in EAC. Of these genes and miRNAs, IL1A and miRNA-23b were significantly downregulated, while CCL20, ABCC3 and hsa-miR-191 were significantly upregulated in EAC (P<0.001). The outcome revealed that the expression of the miRNAs and key genes was consistent with the results of our survival analysis, indicating that the results were reproducible and reliable (Figure 9).
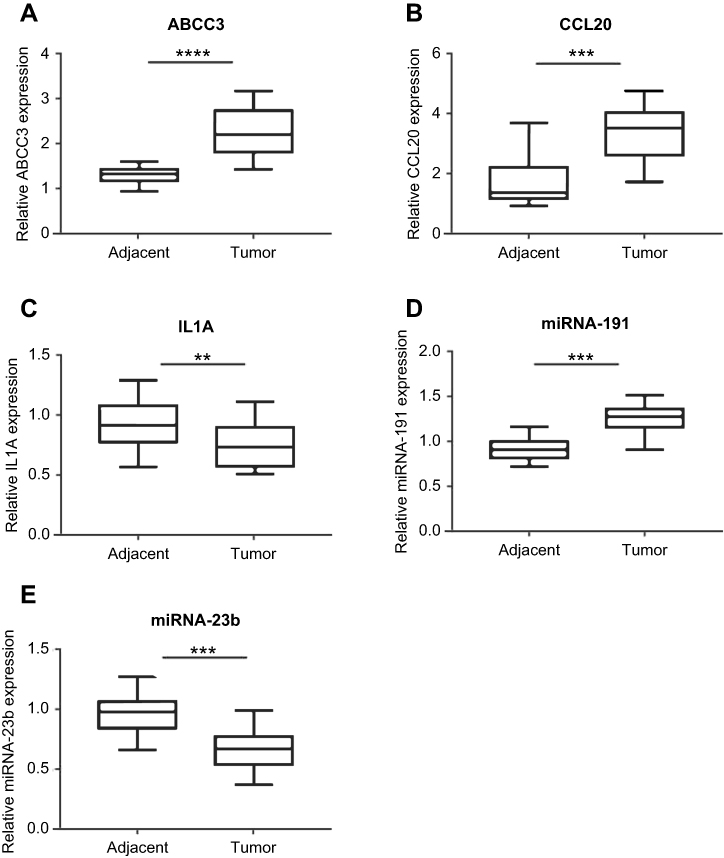 | Figure 9 qRT-PCR validation of the expression of ABCC3, CCL20, IL1A, hsa-miR-191 and hsa-miR-23b in EAC patients (A–E). Notes: **P<0.01, ***P<0.001, ****P<0.0001. Abbreviations: EAC, esophageal adenocarcinoma; qRT-PCR, quantitative real-time PCR. |
Discussion
EAC is one of the most common types of esophageal cancer, and the five-year survival rate is 12–20% in Western populations.1,2 However, the precise pathological mechanism of EAC remains unclear. Therefore, exploring the mechanisms of EAC progression is essential to improve survival and prevent progression. In recent years, microarray technology has been widely used to investigate gene expression in multiple tumor types, providing a novel approach for identifying important genes and leading to further study on tumor initiation and progression.
In this study, 294 DEGs were identified in four mRNA datasets, including 181 downregulated and 113 upregulated genes. Then, public databases were used to predict the TFs that targeted the DEGs. Generally, not all DEGs correspond to TFs. Through a series of comprehensive analyses, 16 TFs (TP53, NFKB1, JUND, RELA, POU5F1, CEBPA, ETS1, JUN, FOS, ETS2, TFAP2A, FLI1, ETV4, TFAP2C, CEBPD and RARA) and 41 key genes were identified to be related to cancer progression. In recent years, many studies have stated that TFs regulate gene expression by interacting with miRNAs. Thus, we further studied the correlations between the 41 key genes and miRNAs and the interactions between the TFs and miRNAs with the miRTarBase database. Subsequently, a TF-miRNA-target gene network was constructed to study the potential molecular mechanisms, and we found that TP53-(miR-125b)-ID2 and JUN-(miR-30a)-IL1A might play significant roles in EAC. Once the network was constructed, enrichment analysis and PPI analysis were performed to understand the underlying functions of these key genes. Intriguingly, the key genes were enriched in the TNF signaling pathway, transcriptional misregulation in cancer and pathways in cancer, which further verified that the key genes were associated with misregulation of TFs leading to tumorigenesis. Furthermore, clinical data and RNA-Seq data from TCGA were used to analyze the correlations of the key genes and miRNAs with patient survival. The analysis predicted ABCC3, IL1A, and CCL20 as independent factors associated with EAC prognosis. ABCC3 is a member of the ATP-binding cassette (ABC) family that is associated with the prognosis of several cancers.25 Zhao et al revealed that overexpression of ABCC3 is related to TNM stage and poor prognosis in non-small cell lung cancer.26 Liu et al showed that increased expression of ABCC3 is correlated with worse overall survival in urinary bladder cancer patients.27 CCL20 belongs to the chemotactic cytokine family, which is correlated with the occurrence, invasion, and metastasis of cancer.28–30 A recent study indicated that the expression of CCL20 is increased in esophageal squamous cell carcinoma and can help classify patients into low-risk, medium-risk and high-risk groups.31 In addition, the prognostic value of CCL20 in multiple tumors has been evaluated. For example, it has been reported that elevated expression of CCL20 is inversely associated with prognosis in smoker patients with lung cancer, providing a potential therapeutic target for this deadly disease.32 IL1A is a member of the interleukin-1 family of cytokines that have multiple functions in cancer and inflammation; when expressed on the cytomembrane, IL1A heightens the immunity of antitumor cells, as indicated by tumor regression.33 However, IL1A has received less attention as a potential tumor suppressor gene than ABCC3 and CCL20. Although the prognostic value of ABCC3, CCL20 and IL1A in EAC has not been reported in previous studies, the importance of these three genes should not be underestimated. In addition, we discovered that hsa-miR-23b and hsa-miR-191 were significantly associated with patient survival in EAC. Many previous studies have reported miR-23b as a tumor suppressor that combats the occurrence and progression of different cancers. The expression of miR-23b is reduced in clear cell renal cell carcinoma, and decreased miR-23b expression predicts poor survival and increased risk of disease progression.34 Downregulation of miR-23b is significantly correlated with poor prognosis in epithelial ovarian cancer (EOC) patients and may be a potential prognostic marker for EOC.35 However, the prognostic value of miR-23b and miR-191 in EAC has not been validated in previous studies. miR-191 has been reported to participate in the initiation and progression of multiple cancers, including pancreatic cancer, colon cancer, lung cancer, and gastric cancer. Overexpression of miR-191 has been discovered in four gastric cancer cell lines and in gastric tumor tissues, indicating miR-191 that may be a novel biomarker for gastric cancer.36 Additionally, inhibition of miR-191 decreases HCC cell proliferation and tumor growth,37 and miR-191 has been found to be closely correlated with TNM stage, metastasis and poor prognosis in pancreatic cancer.38 In our study, a miRNA-mRNA pair (hsa-miR-30a and IL1A) was found to be a prognostic marker for EAC. However, the prognostic value of hsa-miR-23b, hsa-miR-191, and the hsa-miR-30/IL1A pair in EAC has not previously been evaluated. Thus, qRT-PCR was performed to verify the results of bioinformatics analysis and revealed that the expression of the miRNAs and key genes was consistent with our analysis.
Conclusion
In this study, we constructed a TF-miRNA-mRNA network to analyze the potential molecular mechanisms of EAC and identified 16 TFs, 41 key genes, 12 miRNAs and the TF-miRNA-mRNA sets TP53-(miR-125b)-ID2 and JUN-(miR-30a)-IL1A. Among the identified molecules, CCL20, IL1A, ABCC3, miR-23b and miR-191 were determined to be potential independent prognostic factors via survival analysis. qRT-PCR was used to validate the results. In addition, a miRNA-mRNA pair (hsa-miR-30a-5p and IL1A) was determined to be significantly associated with patient survival through integrative survival analysis; this finding might provide a novel direction for further experiments, and the pair could be a potential target for precision treatment. The dysregulated genes identified in this study may be promising biomarkers for EAC diagnosis, treatment and prognosis and can provide a basis for follow-up experiments.
Disclosure
The authors report no conflicts of interest in this work.
References
1. De Angelis R, Sant M, Coleman MP, et al. Cancer survival in Europe 1999–2007 by country and age: results of EUROCARE–5-a population-based study. Lancet Oncol. 2014;15(1):23–34. doi:10.1016/S1470-2045(13)70546-1
2. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66(1):7–30. doi:10.3322/caac.21332
3. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. doi:10.3322/caac.21492
4. Coleman HG, Xie SH, Lagergren J. The epidemiology of esophageal adenocarcinoma. Gastroenterology. 2018;154(2):390–405. doi:10.1053/j.gastro.2017.07.046
5. Edgren G, Adami HO, Weiderpass E, Nyren O. A global assessment of the oesophageal adenocarcinoma epidemic. Gut. 2013;62(10):1406–1414. doi:10.1136/gutjnl-2012-302412
6. Lagergren J, Lagergren P. Recent developments in esophageal adenocarcinoma. CA Cancer J Clin. 2013;63(4):232–248. doi:10.3322/caac.21185
7. Gavin AT, Francisci S, Foschi R, et al. Oesophageal cancer survival in Europe: a EUROCARE-4 study. Cancer Epidemiol. 2012;36(6):505–512. doi:10.1016/j.canep.2012.07.009
8. Njei B, McCarty TR, Birk JW. Trends in esophageal cancer survival in United States adults from 1973 to 2009: a SEER database analysis. J Gastroenterol Hepatol. 2016;31(6):1141–1146. doi:10.1111/jgh.13289
9. Launoy G, Bossard N, Castro C, Manfredi S;
10. Hughes TR. Introduction to “a handbook of transcription factors”. Subcell Biochem. 2011;52:1–6. doi:10.1007/978-90-481-9069-0_1
11. Bhagwat AS, Vakoc CR. Targeting transcription factors in cancer. Trends Cancer. 2015;1(1):53–65. doi:10.1016/j.trecan.2015.07.001
12. Doench JG, Sharp PA. Specificity of microRNA target selection in translational repression. Genes Dev. 2004;18(5):504–511. doi:10.1101/gad.1184404
13. Ebert MS, Sharp PA. Roles for microRNAs in conferring robustness to biological processes. Cell. 2012;149(3):515–524. doi:10.1016/j.cell.2012.04.005
14. Mangalhara KC, Manvati S, Saini SK, et al. ERK2-ZEB1-miR-101-1 axis contributes to epithelial–mesenchymal transition and cell migration in cancer. Cancer Lett. 2017;391:59–73. doi:10.1016/j.canlet.2017.01.016
15. Hua S, Lei L, Deng L, et al. miR-139-5p inhibits aerobic glycolysis, cell proliferation, migration, and invasion in hepatocellular carcinoma via a reciprocal regulatory interaction with ETS1. Oncogene. 2018;285(3):1060–1061.
16. Sun Y, Yang Z, Zheng B, et al. A novel regulatory mechanism of smooth muscle alpha-actin expression by NRG-1/circACTA2/miR-548f-5p axis. Circ Res. 2017;121(6):628–635. doi:10.1161/CIRCRESAHA.117.311441
17. Guo Q, He Y, Sun L, et al. Identification of potential prognostic TF-associated lncRNAs for predicting survival in ovarian cancer. J Cell Mol Med. 2019;23(3):1840–1851. doi:10.1111/jcmm.14084
18. Ritchie ME, Phipson B, Wu D, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43(7):e47. doi:10.1093/nar/gkv007
19. Han H, Cho JW, Lee S, et al. TRRUST v2: an expanded reference database of human and mouse transcriptional regulatory interactions. Nucleic Acids Res. 2018;46(D1):D380–D386. doi:10.1093/nar/gkx1013
20. Jiang C, Xuan Z, Zhao F, Zhang MQ. TRED: a transcriptional regulatory element database, new entries and other development. Nucleic Acids Res. 2007;35(Database):D137–D140. doi:10.1093/nar/gkl1041
21. Chou CH, Shrestha S, Yang CD, et al. miRTarBase update 2018: a resource for experimentally validated microRNA-target interactions. Nucleic Acids Res. 2018;46(D1):D296–D302. doi:10.1093/nar/gkx1067
22. Huang Da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4(1):44–57. doi:10.1038/nprot.2008.211
23. Szklarczyk D, Franceschini A, Wyder S, et al. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015;43(Database issue):D447–D452. doi:10.1093/nar/gku1003
24. Bader GD, Hogue CW. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinformatics. 2003;4:2.
25. Sodani K, Patel A, Kathawala RJ, Chen ZS. Multidrug resistance associated proteins in multidrug resistance. Chin J Cancer. 2012;31(2):58–72. doi:10.5732/cjc.011.10329
26. Zhao Y, Lu H, Yan A, et al. ABCC3 as a marker for multidrug resistance in non-small cell lung cancer. Sci Rep. 2013;3:3120. doi:10.1038/srep03120
27. Liu X, Yao D, Liu C, et al. Overexpression of ABCC3 promotes cell proliferation, drug resistance, and aerobic glycolysis and is associated with poor prognosis in urinary bladder cancer patients. Tumour Biol. 2016;37(6):8367–8374. doi:10.1007/s13277-015-4703-5
28. Ding X, Wang K, Wang H, et al. High expression of CCL20 is associated with poor prognosis in patients with hepatocellular carcinoma after curative resection. J gastrointestinal surg. 2012;16(4):828–836. doi:10.1007/s11605-011-1775-4
29. Sutherland A, Mirjolet JF, Maho A, Parmentier M. Expression of the chemokine receptor CCR6 in the Lewis lung carcinoma (LLC) cell line reduces its metastatic potential in vivo. Cancer Gene Ther. 2007;14(10):847–857. doi:10.1038/sj.cgt.7701074
30. Giuliani N, Lisignoli G, Colla S, et al. CC-chemokine ligand 20/macrophage inflammatory protein-3alpha and CC-chemokine receptor 6 are overexpressed in myeloma microenvironment related to osteolytic bone lesions. Cancer Res. 2008;68(16):6840–6850. doi:10.1158/0008-5472.CAN-08-0402
31. Liu JY, Li F, Wang LP, et al. CTL- vs Treg lymphocyte-attracting chemokines, CCL4 and CCL20, are strong reciprocal predictive markers for survival of patients with oesophageal squamous cell carcinoma. Br J Cancer. 2015;113(5):747–755. doi:10.1038/bjc.2015.290
32. Wang GZ, Cheng X, Li XC, et al. Tobacco smoke induces production of chemokine CCL20 to promote lung cancer. Cancer Lett. 2015;363(1):60–70. doi:10.1016/j.canlet.2015.04.005
33. Voronov E, Dotan S, Krelin Y, et al. Unique versus redundant functions of IL-1alpha and IL-1beta in the tumor microenvironment. Front Immunol. 2013;4:177. doi:10.3389/fimmu.2013.00177
34. Ishihara T, Seki N, Inoguchi S, et al. Expression of the tumor suppressive miRNA-23b/27b cluster is a good prognostic marker in clear cell renal cell carcinoma. J Urol. 2014;192(6):1822–1830. doi:10.1016/j.juro.2014.07.001
35. Li W, Liu Z, Chen L, Zhou L, Yao Y. MicroRNA-23b is an independent prognostic marker and suppresses ovarian cancer progression by targeting runt-related transcription factor-2. FEBS Lett. 2014;588(9):1608–1615. doi:10.1016/j.febslet.2014.02.055
36. Peng WZ, Ma R, Wang F, Yu J, Liu ZB. Role of miR-191/425 cluster in tumorigenesis and diagnosis of gastric cancer. Int J Mol Sci. 2014;15(3):4031–4048. doi:10.3390/ijms15034031
37. Elyakim E, Sitbon E, Faerman A, et al. hsa-miR-191 is a candidate oncogene target for hepatocellular carcinoma therapy. Cancer Res. 2010;70(20):8077–8087. doi:10.1158/0008-5472.CAN-10-1313
38. Liu H, Xu XF, Zhao Y, et al. MicroRNA-191 promotes pancreatic cancer progression by targeting USP10. Tumour Biol. 2014;35(12):12157–12163. doi:10.1007/s13277-014-2521-9
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.