")
Back to Journals » The Application of Clinical Genetics » Volume 9
Identification of a novel pathogenic OTOF variant causative of nonsyndromic hearing loss with high frequency in the Ashkenazi Jewish population
Authors Fedick A, Jalas C, Swaroop A, Smouha E, Webb B
Received 29 May 2016
Accepted for publication 22 June 2016
Published 31 August 2016 Volume 2016:9 Pages 141—146
DOI https://doi.org/10.2147/TACG.S113828
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Martin Maurer
Anastasia M Fedick,1 Chaim Jalas,2 Ananya Swaroop,1 Eric E Smouha,3 Bryn D Webb1
1Department of Genetics and Genomic Sciences, Icahn School of Medicine at Mount Sinai, New York, NY, USA; 2Bonei Olam, Center for Rare Jewish Genetic Disorders, Brooklyn, NY, USA; 3Department of Otolaryngology, Icahn School of Medicine at Mount Sinai, New York, NY, USA
Abstract: Mutations in the OTOF gene have previously been shown to cause nonsyndromic prelingual deafness (DFNB9, OMIM 601071) as well as auditory neuropathy/dys-synchrony. In this study, the OTOF NM_194248.2 c.5332G>T, p.Val1778Phe variant was identified in a large Ashkenazi Jewish family as the causative variant in four siblings with hearing loss. Our analysis reveals a carrier frequency of the OTOF c.5332G>T, p.Val1778Phe variant of 1.27% in the Ashkenazi Jewish population, suggesting that this variant may be a significant contributor to nonsyndromic sensorineural hearing loss and should be considered for inclusion in targeted hearing loss panels for this population. Of note, the degree of hearing loss associated with this phenotype ranged from mild to moderately severe, with two of the four siblings not known to have hearing loss until they were genotyped and underwent pure tone audiometry and auditory brainstem response testing. The phenotypic variability along with the auditory neuropathy/dys-synchrony, which allows for the production of otoacoustic emissions, supports that nonsyndromic hearing loss caused by OTOF mutations may be much more common in the Ashkenazi Jewish population than currently appreciated due to a lack of diagnosis.
Keywords: hearing loss, OTOF, c.5332G>T, p.Val1778Phe, Ashkenazi Jewish, otoferlin
Introduction
Otoferlin is a six C2 domain transmembrane protein responsible for priming and fusing synaptic vesicles during auditory hair cell exocytosis to allow for sound encoding.1,2 In the inner hair cell ribbon synapse, otoferlin is the major calcium sensor and binds to membranes as well as syntaxin 1A/SNAP-25 heterodimers in a calcium-dependent manner suggesting a role in regulating SNARE activity.3,4 It also allows for calcium-dependent exocytosis in immature outer hair cells that act as sound signal amplifiers in the mature cochlea5 as well as in type 1 vestibular hair cells that play roles in sensing head acceleration and maintaining balance.6
Mutations in the OTOF gene cause nonsyndromic prelingual deafness (DFNB9, OMIM 601071) in humans7 and profound hearing loss in OTOF knockout mice.3 Specific phenotypes, including severe, profound, progressive, and temperature-sensitive hearing loss, have been consistently seen for different OTOF mutations, which have allowed for genotype–phenotype correlations to be established.8 OTOF mutations have also been associated with auditory neuropathy/dys-synchrony (also referred to as auditory neuropathy spectrum disorder [ANSD]), which is characterized by functioning outer hair cells, but damaged auditory nerve and/or inner hair cells leading to disturbed pure tone audiometry, the absence of auditory brainstem responses (ABRs), and the detection of otoacoustic emissions (OAEs).9–11
In this study, the OTOF NM_194248.2 c.5332G>T, p.Val1778Phe variant was identified in homozygous form in a nonconsanguineous Ashkenazi Jewish (AJ) family in siblings with symptoms of hearing loss presenting for genetic evaluation. We identified the carrier frequency of this variant to be 1.27% in the AJ population, suggesting that this variant may be a significant contributor to nonsyndromic sensorineural hearing loss (SNHL) and should be considered for inclusion in targeted hearing loss panels for this population.
Materials and methods
Ethics statement
All family members from the index case were enrolled in a Mount Sinai Institutional Review Board (IRB)-approved genetics research study. Written informed consent was obtained from all the family members, and investigations were conducted in accordance with the principles of the Declaration of Helsinki. For the carrier frequency study, all samples were obtained with written patient consent from self-identified Ashkenazi Jews to be used for research purposes. These individuals were a mix of male and female young adults, who were presumed to be healthy. Consent form information included that patient material would be used for clinical testing and that excess material would be de-identified and used for research purposes to characterize single gene disorders in the AJ population. IRB permission was not required for the samples used in the carrier frequency study because all sample identifiers were removed prior to receipt by our lab (45 CFR part 46.101(b)(4)).
Human subjects
DNA was isolated from peripheral blood samples obtained from the proband (II.8) as well as his siblings and parents (Figure 1). The subjects were all of Hungarian and Romanian AJ descent, and the parents denied consanguinity. Of the nine siblings, siblings II.6 and II.8 were reported to have hearing loss at the time of presentation. The proband (II.8) was a male, aged 5 years, who had mild-to-moderate progressive SNHL. The hearing loss was first noted at the age of 3 years due to articulation difficulties, although the patient had failed his hearing test at birth, and follow-up by OAEs indicated hearing within normal limits. He was treated with binaural Oticon Safari hearing aids and uses a frequency-modulated system in school. His hearing exam at the time of genetic testing indicated that he had high-frequency moderately severe SNHL bilaterally, but normal middle ear function, and distortion product otoacoustic emissions (DPOAEs) that were present and robust.12
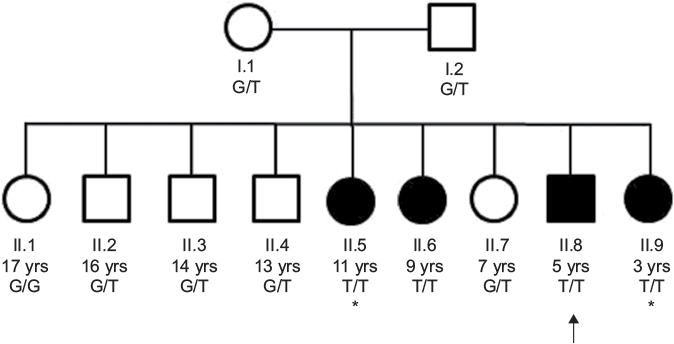 | Figure 1 Pedigree. Notes: This pedigree shows the autosomal recessive inheritance of the OTOF variant. Males are represented with squares and females are represented with circles. Affected individuals are shaded. Genotypes are shown for each family member, with affected individuals displaying homozygosity for the minor allele. The proband II.8 is indicated by the arrow. The asterisks indicate the two siblings (II.5 and II.9) that were not known to have hearing loss until they were genotyped and underwent hearing evaluations. Of note, the proband’s paternal great-grandmother had unilateral early-onset hearing loss of unknown etiology (not shown). Abbreviation: yrs, years. |
Sibling II.6 was a female, aged 9 years, who had bilateral mild SNHL. She had failed her newborn hearing screen, but follow-up OAE testing was within normal limits. At 4 years, her mother noticed that she was having difficulty hearing, which led to a full audiology evaluation. Sibling II.6 did not require hearing aids or assistive devices. Her hearing exam at the time of genetic testing revealed mild high-frequency hearing loss in the right ear, absence of contralateral acoustic reflexes in the left ear, and ANSD (patient had normal middle ear function, present and robust DPOAEs and present ipsilateral acoustic reflexes bilaterally) (Table 1).
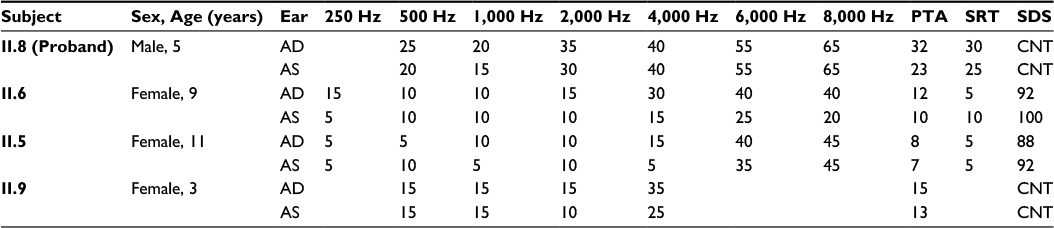 | Table 1 Audiometry findings Abbreviations: AD, right ear; AS, left ear; PTA, pure tone average; SRT, speech reception threshold; SDS, speech discrimination score; CNT, could not test. |
The Harvard OtoGenome testing panel, which sequences 71 genes involved in hereditary hearing loss, was ordered for the proband II.8. After completion of clinical testing for the proband, all the siblings and both the parents were enrolled into our research study for completion of segregation analysis with targeted genotyping. After genetic testing, two additional siblings were evaluated for hearing loss. Audiology testing of sibling II.5 revealed mild-to-moderate SNHL at 6–8 kHz bilaterally with normal hearing at ≤4 kHz, and robust DPOAEs with the absence of ABR waves 1–5 bilaterally, consistent with ANSD. Similarly, sibling II.9 was found to have normal hearing at 4 kHz and below bilaterally, slight high-frequency SNHL in the left ear and mild high-frequency SNHL in the right ear, and robust bilateral DPOAEs with the absence of ABR waves 1–5 with cochlear microphonic inversion bilaterally, consistent with ANSD (Table 1). In both children, speech and language development were normal and there were no communication impairments; the auditory deficits, which are specific for ANSD, would probably not have been suspected had the testing not been ordered.
Segregation analysis
PCR and Sanger sequencing were used to perform segregation analysis. Parents and all siblings were genotyped. Standard polymerase chain reaction (PCR) conditions were used at the following settings: hold at 95°C for 5 minutes, 35 cycles at 95°C for 30 seconds, 58°C for 30 seconds, and 72°C for 25 seconds, and a final hold at 72°C for 7 minutes along with forward (5′-GTCTCGTCCCAGGAGAACAT-3′) and reverse (5′-AGAGGAGGCAGAGGGAAGGT-3′) primers. PCR products were separated by agarose gel electrophoresis to ensure proper amplification, which was indicated by a single strong band at the expected amplicon size. The samples were then prepared for sequencing using BigDye and run on a 3730xl DNA Analyzer (Thermo Fisher Scientific, Waltham, MA, USA).
Carrier frequency study
File Builder software (Thermo Fisher Scientific) was used to design the forward (5′-GGCCAGCAGGAGGACAAG-3′) and reverse (5′-GTTGCCCTCGCCAGTGA-3′) primers as well as VIC (5′-AGGACACAGACGTCCACT-3′) and FAM (5′-AGGACACAGACTTCCACT-3′) labeled probes. The assay was validated on control samples obtained from the proband and his family, and then the carrier frequency study was performed on 1,021 samples. The samples were run on the GeneAmp® PCR System 9700 (Thermo Fisher Scientific) at the following PCR conditions: hold at 50°C for 2 minutes and 95°C for 10 minutes, then 40 cycles at 95°C for 15 seconds and 60°C for 1 minute. Allelic discrimination was then performed on the ABI PRISM® 7900 HT Sequence Detection System (Thermo Fisher Scientific), and data were analyzed using TaqMan Genotyper v1.2 software (Thermo Fisher Scientific).
Results
The OtoGenome testing panel, which screens 71 genes involved in hereditary hearing loss, revealed a homozygous, likely pathogenic missense variant NM_194248.2 c.5332G>T, p.Val1778Phe in the OTOF gene in the proband II.8. This missense change occurred at position chr2:26684765 (on assembly hg19/GRCh37) and is recorded in dbSNP as rs111033330. While OTOF mutations are known to cause autosomal recessive nonsyndromic hearing impairment associated with ANSD, the c.5332G>T, p.Val1778Phe variant has not been reported in any manuscripts previously, although this variant has been reported once in ClinVar. Computational analysis of this variant supported that the variant was pathogenic based on biochemical amino acid properties, conservation, Align-GVGD, PolyPhen2 (probably damaging by HumDiv, score 0.971; possibly damaging by HumVar, score 0.681), and SIFT (damaging, score 0.001). Additionally, there was a low allele frequency in ExAC in Europeans (0.0001799 [12/66720]), with no homozygotes reported, and the other ExAC populations reported no carriers (overall allele frequency =9.885e-05 [12/121390]). Additionally, the valine at amino acid 1778 of OTOF shows strong evolutionary conservation. This residue is conserved in rhesus monkey, mouse, dog, chicken, western clawed frog, zebrafish, and lamprey.
The family was then enrolled into an IRB-approved research study and segregation of the variant was assessed. Sanger sequencing confirmed that each parent was an obligate carrier of the variant and that sibling II.6 was homozygous for the variant (Figure 2). Interestingly, when the remaining seven siblings were genotyped, two additional siblings (siblings II.5 and II.9) were also found to be homozygous for the pathogenic variant, suggesting that they should also have hearing difficulties. Audiology exams, along with ABR and OAE testing, were subsequently performed for siblings II.5 and II.9. In accordance with her genotype, audiology testing on sibling II.5 revealed mild-to-moderate high-frequency SNHL bilaterally and ANSD. Similarly, sibling II.9 was found to have slight high-frequency SNHL in the left ear and mild high-frequency SNHL in the right ear, as well as a low conductive component and ANSD. In both the siblings, the audiometric findings were significant, specific for ANSD, and were not suspected clinically as the children had normal speech and language, and no outward signs of a hearing problem.
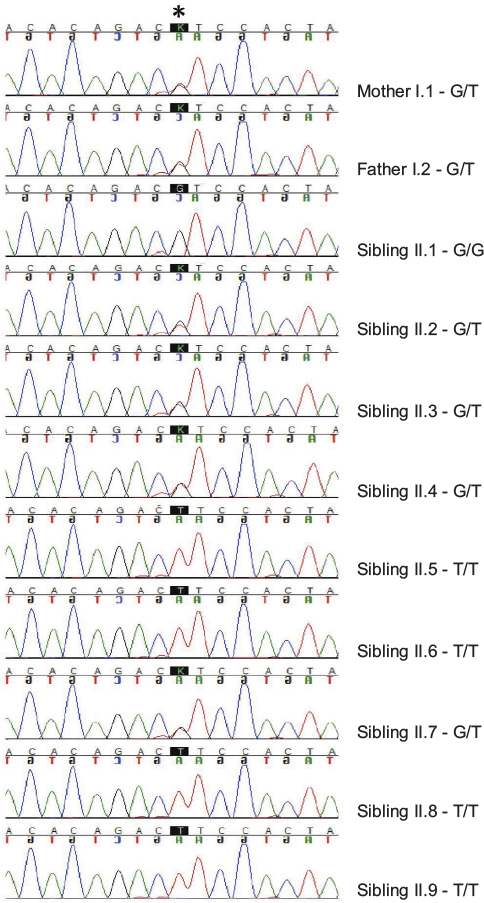 | Figure 2 Sanger sequencing results. Notes: The Sanger sequencing traces for the proband and his family are depicted in the figure. The asterisk denotes the location of the variant. The black trace indicates the reference G allele, while the red trace indicates the mutant T allele. |
As the proband was of AJ descent, we performed a carrier frequency study on 1,021 AJ samples, revealing a carrier frequency of 1.27% or ~1 in 79 individuals, based on the presence of 13 heterozygous carriers and 1,008 wild-type samples (Figure 3). All of the samples identified as being heterozygous carriers had their genotypes confirmed through Sanger sequencing, and all the results were concurrent.
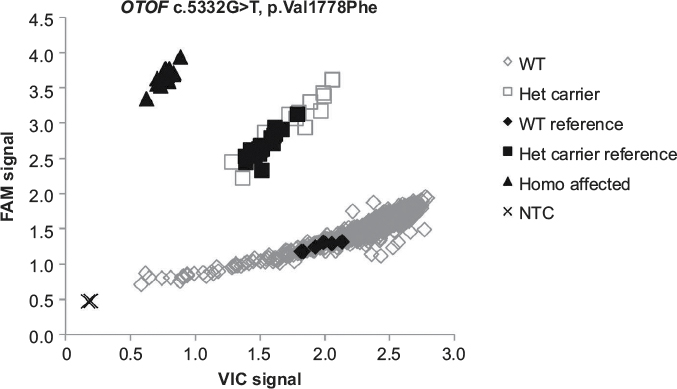 | Figure 3 TaqMan allelic discrimination carrier frequency plot. Notes: The VIC probe is represented by the x-axis and signifies the WT allele, and the FAM probe is represented by the y-axis and signifies the minor allele. Water was used as the NTC. Abbreviations: WT, wild type; NTC, no template control; Het, heterozygous; Homo, homozygous. |
Discussion
Most isolated childhood hearing loss cases have a genetic etiology. In the AJ population, two frameshift mutations in the gap junction beta 2 (GJB2) gene, c.167delT and c.30delG, are primarily responsible for congenital, nonsyndromic autosomal recessive SNHL with carrier frequencies of 4.03% and 0.73%, respectively.13 The OTOF c.5332G>T, p.Val1778Phe variant identified in this study may be another significant contributor to SNHL, and specifically ANSD, in the AJ population with a carrier frequency of 1.27%. This high frequency suggests that this variant is likely a founder mutation, and we recommend adding this variant to targeted hearing loss panels for this population.
Interestingly, a second case of the OTOF c.5332G>T, p.Val1778Phe variant has been reported in ClinVar (ClinVar submission # SCV000065265.3). This variant was identified in a pair of AJ siblings with ANSD, who were found to be compound heterozygous for the c.5332G>T, p.Val1778Phe variant and a known pathogenic OTOF variant (personal communication with Dr Heidi Rehm, Laboratory for Molecular Medicine, Harvard Medical School), further supporting the potential frequency of this variant in AJ patients with hearing loss. OTOF founder mutations have been recognized in other populations and have helped to increase the number of molecular diagnoses made for patients of these ethnicities. These variants include the Spanish c. 2485C>T, p.Q829X,14 Taiwanese c.5098G> C, p.E1700Q,15 Japanese c.5816G>A, p.R1939Q,16 and Argentinian c.2905_2923delinsCTCCGAGCGCA variants.9 It is important to now associate the c.5332G>T, p.Val1778Phe variant with the AJ population.
Of note, the degree of hearing loss associated with the homozygous OTOF c.5332G>T, p.Val1778Phe genotype ranged from mild to moderately severe, with two of the four siblings not reported to have hearing loss until they were genotyped and underwent pure tone audiometry, DPOAE, and ABR testing. The mild phenotype along with ANSD and the presence of OAEs support that nonsyndromic hearing loss caused by OTOF mutations may be much more common in the AJ population than currently appreciated and clinically unapparent. Additionally, newborn screening, which is based on OAEs, may fail to identify individuals with this mutation. It is therefore possible that affected individuals may not present for evaluation and/or may not be properly diagnosed and treated unless they receive specific testing.
Acknowledgments
The authors would like to thank the family for participating in the study. The authors would also like to acknowledge the Mount Sinai Audiology team who performed some of the hearing tests reported in this manuscript. Finally, the authors would like to thank Bonei Olam for providing funding for this project.
Disclosure
The authors report no conflicts of interest in this work.
References
Pangrsic T, Lasarow L, Reuter K, et al. Hearing requires otoferlin-dependent efficient replenishment of synaptic vesicles in hair cells. Nat Neurosci. 2010;13(7):869–876. | ||
Pangrsic T, Reisinger E, Moser T. Otoferlin: a multi-C2 domain protein essential for hearing. Trends Neurosci. 2012;35(11):671–680. | ||
Roux I, Safieddine S, Nouvian R, et al. Otoferlin, defective in a human deafness form, is essential for exocytosis at the auditory ribbon synapse. Cell. 2006;127(2):277–289. | ||
Johnson CP, Chapman ER. Otoferlin is a calcium sensor that directly regulates SNARE-mediated membrane fusion. J Cell Biol. 2010;191(1):187–197. | ||
Beurg M, Safieddine S, Roux I, Bouleau Y, Petit C, Dulon D. Calcium- and otoferlin-dependent exocytosis by immature outer hair cells. J Neurosci. 2008;28(8):1798–1803. | ||
Dulon D, Safieddine S, Jones SM, Petit C. Otoferlin is critical for a highly sensitive and linear calcium-dependent exocytosis at vestibular hair cell ribbon synapses. J Neurosci. 2009;29(34):10474–10487. | ||
Yasunaga S, Grati M, Cohen-Salmon M, et al. A mutation in OTOF, encoding otoferlin, a FER-1-like protein, causes DFNB9, a nonsyndromic form of deafness. Nat Genet. 1999;21(4):363–369. | ||
Yildirim-Baylan M, Bademci G, Duman D, Ozturkmen-Akay H, Tokgoz-Yilmaz S, Tekin M. Evidence for genotype-phenotype correlation for OTOF mutations. Int J Pediatr Otorhinolaryngol. 2014;78(6):950–953. | ||
Rodriguez-Ballesteros M, del Castillo FJ, Martin Y, et al. Auditory neuropathy in patients carrying mutations in the otoferlin gene (OTOF). Hum Mutat. 2003;22(6):451–456. | ||
Santarelli R, Del Castillo I, Rodriguez-Ballesteros M, et al. Abnormal cochlear potentials from deaf patients with mutations in the otoferlin gene. J Assoc Res Otolaryngol. 2009;10(4):545–556. | ||
Varga R, Kelley PM, Keats BJ, et al. Non-syndromic recessive auditory neuropathy is the result of mutations in the otoferlin (OTOF) gene. J Med Genet. 2003;40(1):45–50. | ||
Clark JG. Uses and abuses of hearing loss classification. ASHA 1981;23(7):493–500. | ||
Morell RJ, Kim HJ, Hood LJ, et al. Mutations in the connexin 26 gene (GJB2) among Ashkenazi Jews with nonsyndromic recessive deafness. New Engl J Med. 1998;339(21):1500–1505. | ||
Migliosi V, Modamio-Hoybjor S, Moreno-Pelayo MA, et al. Q829X, a novel mutation in the gene encoding otoferlin (OTOF), is frequently found in Spanish patients with prelingual non-syndromic hearing loss. J Med Genet. 2002;39(7):502–506. | ||
Chiu YH, Wu CC, Lu YC, et al. Mutations in the OTOF gene in Taiwanese patients with auditory neuropathy. Audiol Neurootol. 2010;15(6):364–374. | ||
Matsunaga T, Mutai H, Kunishima S, et al. A prevalent founder mutation and genotype-phenotype correlations of OTOF in Japanese patients with auditory neuropathy. Clin Genet. 2012;82(5):425–432. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.