")
Back to Journals » Neuropsychiatric Disease and Treatment » Volume 14
Identification of a novel mutation in APP gene in a Thai subject with early-onset Alzheimer's disease
Authors Giau VV , Senanarong V, Bagyinszky E, Limwongse C, An SSA , Kim SY
Received 13 July 2018
Accepted for publication 24 September 2018
Published 8 November 2018 Volume 2018:14 Pages 3015—3023
DOI https://doi.org/10.2147/NDT.S180174
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Taro Kishi
Vo Van Giau,1,* Vorapun Senanarong,2,* Eva Bagyinszky,1 Chanin Limwongse,2 Seong Soo A An,1 SangYun Kim3
1Department of Bionano Technology, Gachon Bionano Research Institute, Gachon University, Seongnam, South Korea; 2Faculty of Medicine Siriraj Hospital, Mahidol University, Bangkok, Thailand; 3Department of Neurology, Seoul National University College of Medicine & Neurocognitive Behavior Center, Seoul National University Bundang Hospital, Seongnam, South Korea
*These authors contributed equally to this work
Introduction: Early-onset Alzheimer’s disease (AD) accounts for than less 1% of all AD cases, with large variation in the reported genetic contributions of known dementia genes. Mutations in the amyloid precursor protein (APP) gene were the first to be recognized as the cause of AD.
Methods: Here, a male patient with probable early-onset AD at the age of 55 years from Thailand was investigated by next-generation sequencing.
Results: A novel mutation in exon 14 of APP (c.1810C>T, p.V604M) was found. He initially illustrated the clinical manifestations of progressive nonfluent aphasia in 2011. However, he was finally diagnosed with AD presenting logopenic aphasia in 2013. The follow-up magnetic resonance imaging scan showed progression of hippocampal trophy compared with the initial image. A 3D protein structure model revealed that V604M exchange could result in significant changes in the APP protein due to the increased hydrophobicity of methionine in the helix, which could result in altering of the APP functions.
Conclusion: Additional studies to characterize APP p.V604M are necessary to further understand the effects of this mutation.
Keywords: APP, Alzheimer’s disease, Thailand, V604M
Introduction
Alzheimer’s disease (AD) is the most common form of dementia in the elderly. AD can be categorized into two forms with age division: early-onset AD (EOAD) for before onset before 65 years of age and late-onset AD for onset at or after 65 years of age. Familial AD is associated with mutations in presenilin-1 (PSEN1) (MIM #104311), presenilin-2 (PSEN2) (MIM #600759), and the amyloid precursor protein (APP) (MIM #104760), which would sequentially be cleaved by the enzymes β- and γ-secretase to yield the amyloid β (Aβ) peptide.1 With at least 32 mutations in APP reported as pathogenic in the Alzforum database (www.alzforum.org/mutations), it has become the second most commonly involved gene after PSEN1. APP encodes the Aβ precursor protein, which would be processed by the β- and γ-secretase complex, leading to the production of the Aβ peptide, a key event in AD pathogeny. APP mutations, located within the Aβ1–43 region of the protein or adjacent to the N- or C-termini of the Aβ sequence, could contribute to a cascade of events of overproduction of Aβ and may lead to AD. Typically, the mutations adjacent to the N-terminus of the Aβ sequence led to an increased production of all Aβ species, while the ones near the carboxyl terminus led to an increase in the Aβ42/Aβ40 ratio.2,3 However, the discovery of APP mutations in EOAD launched many functional investigations of APP.4–6
No additional causative gene(s) were reported for AD except the above three genes, but additional possible risk factor genes were also discovered, such as ABCA7, SORL1, or CLU.7,8 In addition, the overlapping clinical and neuropathologic features between AD and other neurodegenerative dementias: frontotemporal dementia (FTD), corticobasal degeneration, progressive supranuclear palsy, and Creutzfeldt–Jakob disease, led to a misdiagnosis in 17%–30% of AD cases.7–10 This raised the question of whether the genetic risk factors in dementia could play a role in terms of their pathologies, clinical phenotypes and affected brain regions. Even though the standard sequencing was used widely, it may not be enough for extensive genetic analysis of patients with neurodegenerative dementias, due to the time and cost, especially with availability of the next-generation sequencing (NGS) technologies for the diagnosis.10
In this manuscript, the clinical and neuropathological phenotypes associated with a novel mutation (g.21,27284152, c.1810C>T; p.V604M) in the APP gene is presented. This mutation was found in a Thai male patient with EOAD at the age of 55 years with family history. Structural characterization from the 3D protein prediction and gene ontology studies with other missense variants were also performed to understand the consequences and the characteristics of this variant.
Materials and methods
Subjects
The study subject provided written informed consent allowing genetic and clinical data to be used for research purposes. A diagnosis of probable AD according to the criteria of the National Institute of Neurological and Communicative Disorders and Stroke Alzheimer’ Disease and Related Disorders Association.11 This study was conducted with approval from the Institutional Review Board of Seoul National University College of Medicine & Neurocognitive Behavior Center, Seoul National University Bundang Hospital, Korea (B-1302/192-006) and the Faculty of Medicine Siriraj Hospital, Mahidol University, Bangkok, Thailand. The study subject provided written informed consent allowing genetic and clinical data to be used for research purposes, and for the details to be published.
Imaging scan
In 2011 (17/06/2011), brain magnetic resonance imaging (MRI) analyses were performed with following coronal T2-weighted turbo spin echo (T2W/TSE), and T1-weighted (T1W) 3D Sagittal for the comparison study. In 2013, MRI analyses with Axial T1W, T2W/TSE, T2W fluid-attenuated inversion recovery, diffusion-weighted imaging (DWI), and gradient echo techniques were performed, and the results were compared, including the data from 2011.
Family history
The proband’s family history was complex and included several family members affected by dementia (Figure 1). This case could be a familial case of mutation, since other family members (I-1; I-3, and II-1) presented similar symptoms. His father (I-3) had memory problems in late 50s and died at the age of 70 years. Remarkably, his uncle (I-1) also had memory problem after retirement at 60 and died at 80 years of age. The patient was one of the 4 siblings, comprising 3 brothers and 1 sister. At the time of this study, the first brother (II-1) also had delusions and memory problems in late 50s and had been treated by psychiatrist, married with 2 children, and displayed normal cognitive function. All living family members and relatives refused genetic testing, and declined to provide any additional information regarding their health.
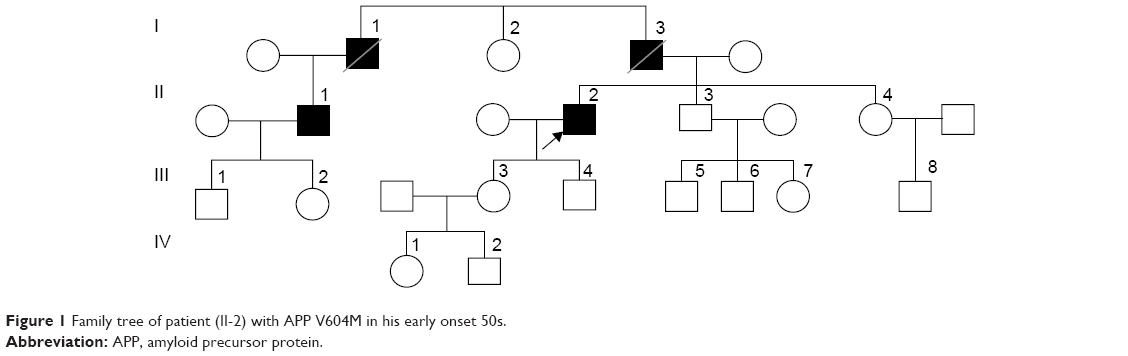 | Figure 1 Family tree of patient (II-2) with APP V604M in his early onset 50s. |
Biomarkers
Cerebrospinal fluid (CSF) was collected from the proband patient in the morning into polypropylene tubes by following the standard operating procedures. The sample was aliquoted in 1 mL each and frozen within 8 hours from the collection and stored at –80°C CSF until use. Total tau, phosphorylated tau, and Aβ42 were measured using ELISAs (Fujirebio Inc., Tokyo, Japan) by following the manufacturer’s instructions. Levels of protein and glucose in CSF were measured by following the standard operating procedures in the laboratory medicine. Red and white blood cells were counted.
Genetic testing
Complex genetic screening investigation by NGS was performed on the proband patient. Targeted NGS approaches were performed with a panel of 50 causative and risk factor genes in neurodegenerative diseases by Theragen Etex Bio Institute on IonProton device.10 The standard Sanger sequencing verified the NGS data by the BioNeer Inc. (http://eng.bioneer.com/home.aspx, Bioneer Inc., Dajeon, Korea).12
In silico analysis, structure, and predictions
Variants were checked in the Korean Genome Reference Database (http://152.99.75.168/KRGDB/menuPages/firstInfo.jsp) for their novelty, which provided the full genome sequences of 622 asymptomatic individuals by whole genome sequencing. In addition, variants were also monitored in larger genome reference databases, including the 1000 Genomes (http://www.internationalgenome.org/) and Exome Aggregation Consortium (ExAC; http://exac.broadinstitute.org) databases.
Variants were screened by PolyPhen-2 (bulkiness, polarity or hydrophobicity (Kyte and Doolittle) index, http://genetics.bwh.harvard.edu/pph2/), sorting intolerant from tolerant algorithm (SIFT; http://sift.jcvi.org/), and PROVEAN (http://provean.jcvi.org/index.php) softwares, which were simple and easy-to-use online tools. They provided the variant predictions on the possibly damaging properties of missense mutations.8 In addition, prediction of mutational change on the APP protein properties was also performed by using PyMOL programs at ExPASy server (https://www.expasy.org/) based on bulkiness parameter. A 3D protein structure prediction model was determined by an online Raptor X software (http://raptorx.uchicago.edu/) and the APP variant was compared with the normal X-ray structure. Superimposed images of variant and normal proteins were processed by Discovery Studio 3.5 Visualizer software, designed by Accerlrys.13
Results
Clinical findings
The proband (II-2, Figure 1) was a 55-year-old male patient clinically diagnosed with EOAD, presenting with progressive forgetfulness. He frequently forgot things, appointment, and the name of his friends. Language problems, which were non-fluent and naming difficulty. His fluency had decreased, and he could not name objects. Since his retirement at the age of 60 years, he became anxious and started to complain of insomnia. He occasionally needed lorazepam prescriptions from his doctor. He was on rivastigmine from the memory clinic. Upon his neurologic examination, he had impairments in objects naming, auditory and visual memory, and verbal fluency and executive functions. Unremarkable general physical and neurological examinations, especially no evidence of primitive releasing signs.
An MRI scan of the proband’s brain showed diffuse marked brain atrophy, especially in the frontoparietal regions (Figure 2). In addition, MRI revealed slightly increased atrophy in his right hippocampus with increased atrophic changes from moderate to severe, whereas diffuse cortical brain atrophy seemed to be at the moderate degree. Abnormal signal intensity of brain parenchyma was absent. Microbleeds or white matter changes were not detected. The size of pituitary glands was normal. The ventricular systems were not dilated. No midline shifting was detected. Posterior fossa, the mastoid, peripheral nervous system, and orbits were unremarkable.
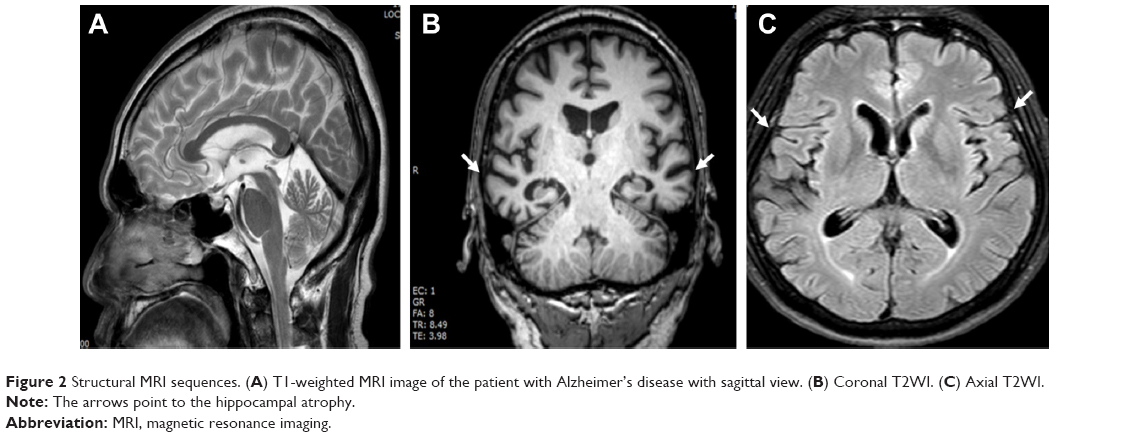 | Figure 2 Structural MRI sequences. (A) T1-weighted MRI image of the patient with Alzheimer’s disease with sagittal view. (B) Coronal T2WI. (C) Axial T2WI. |
Biomarkers
The Multimer Detection System is an enzyme-linked immunosorbent assay (ELISA) that is used for measuring Aβ oligomer in plasma as previously described.14 Levels of Aβ42, T-tau, and P-tau in CSF were 555, 285, and 37 pg/mL, respectively. These values did not support the AD diagnosis clearly with the CSF biomarkers, since the cutoff values of Aβ42, T-tau, and P-tau were 550, 350, and 60 pg/mL, respectively. Interestingly, the levels of Aβ42 in normal healthy controls and patients with mild cognitive impairment were 800 and 600 pg/mL, respectively, the severe atrophy could not reveal the relatively high Aβ42 in this patient.
Protein level in CSF was at 65 mg/dL, slightly higher than normal range between 15 and 60 mg/dL. Glucose level in CSF was at 67 mg/dL within normal range of 50–80 mg/dL. RBC counts were 7.4 million/Cumm, slightly higher than the normal range of 4.2–6.5 million/Cumm. WBC counts were 10,000/Cumm, which fell into the normal range of 4,000–11,000/Cumm.
Mutation analysis
A heterozygous C>T substitution (chromosome 21, g.27284152, c.1810C>T) was identified and confirmed to occur in the APP coding region using both NGS and standard sequencing. This mutation caused the change from valine to methionine (c.1810C>T; p.Val604Met) substitution, located at exon 14 of APP gene, and in topological domain-II (TD-II) of the APP protein (Figure 3). The APOE genotype of the patient was ε3/ε3. APP Val604Met is likely a novel mutation, as it was not included in either AD genetic databases (AD and FTD mutation database http://www.molgen.ua.ac.be/admutations/; AlzGene database http://www.alzforum.org/mutations), neither in the KRGDB, the ExAC, or 1000 Genomes databases.
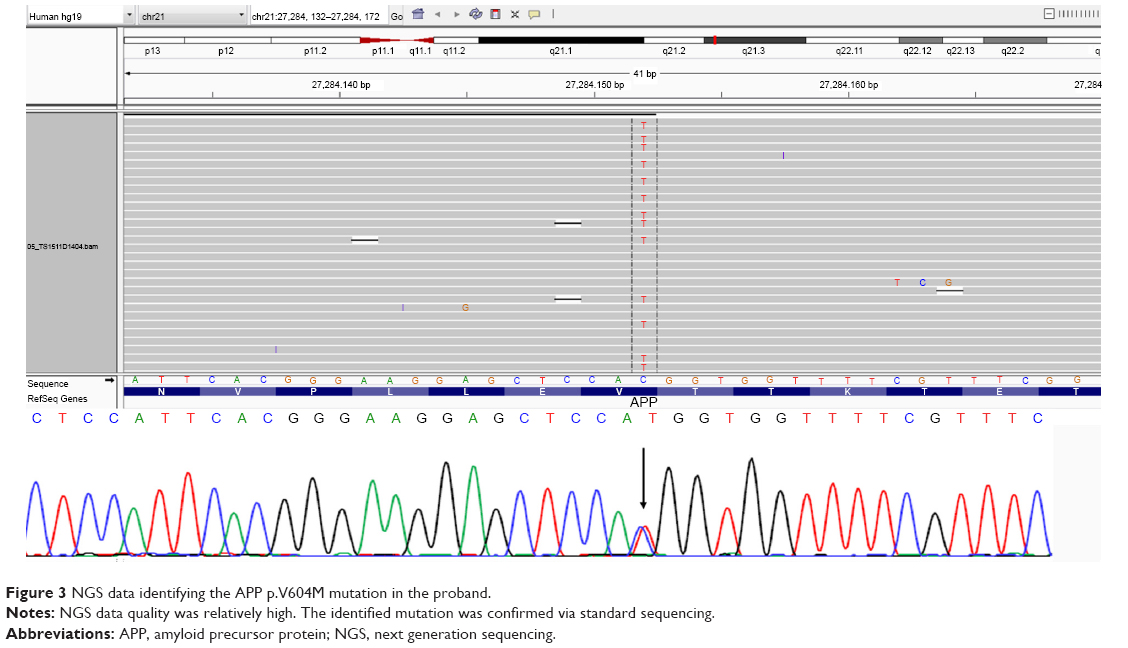 | Figure 3 NGS data identifying the APP p.V604M mutation in the proband. |
No additional pathogenic mutations were found in other dominant causative genes of AD, FTD, or other neurodegenerative diseases, such as PSEN1, PSEN2, PGRN, or MAPT. However, additional 22 missense variants from other AD risk factor genes, CR1, CTNNA3, SORL1, and ABCA7 genes, as well as in other dementia (such as PINK1, CSF1R, PRNP, and NOTCH), were found, as indicated in the Table S1.
In silico predictions
Scores from PolyPhen2 analysis suggested this mutation as possibly damaging with HumVar score. SIFT also revealed V604M as tolerant variant with the score of 0.095. PROVEAN also revealed V604M as neutral, with the score of −0.72. ExPASy tools revealed that mutation could affect significantly the APP structure through bulkiness parameter. Scores of bulkiness dropped significantly due to V604M (17.008) compared with normal APP (17.599), and several amino acids nearby were also affected (Figure 4). The results revealed significant structural changes in the mutation spot, this also affected the neighboring residues from residue 600 to 608. Hence, these analyses suggest that the APP Val604Met mutation could possibly be a strong damaging variant.
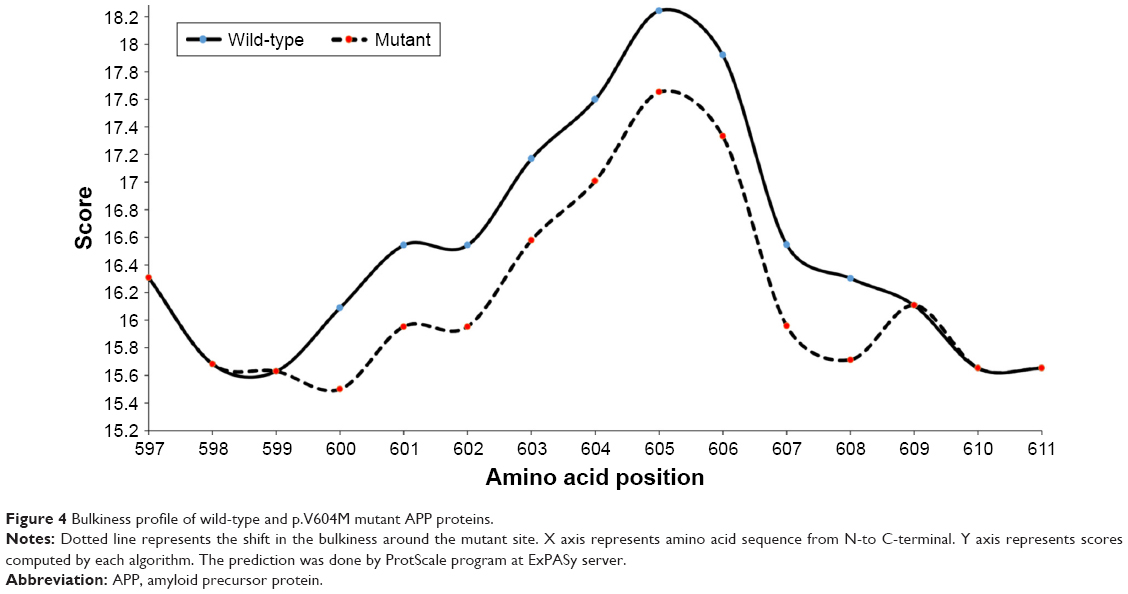 | Figure 4 Bulkiness profile of wild-type and p.V604M mutant APP proteins. |
Comparing the mutant and normal proteins by structure predictions revealed significant changes in the TD helix, as well as amino acid interactions with neighboring residues. Differences between the normal and mutant protein were highlighted where valine was labeled with yellow whereas methionine with purple. Significant changes could be seen in the structure of mutant APP due to the charge differences between valine and methionine (Figure 5). Furthermore, the side-chains of Val and met 604 residues reveal noticeable changes of extending toward opposite direction. By this sound, a V604M mutation would probably result in disturbances of the helical structure by counter electrostatic interaction.
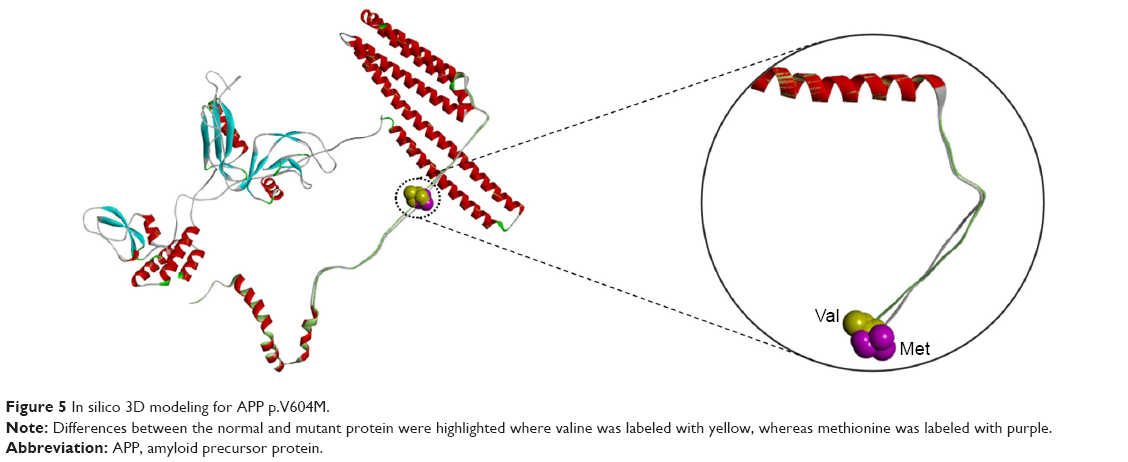 | Figure 5 In silico 3D modeling for APP p.V604M. |
Discussion
In the current study, a novel mutation of APP Val604Met was discovered in a Thai EOAD patient with probable autosomal dominant inheritance pattern. No other pathogenic mutation has been reported at APP codon 604. PolyPhen-2, SIFT, PROVEAN, and ExPASY software analysis, predicted that V604M may not be a damaging mutation. However, the 3D structure revealed that valine and methionine are nonpolar and hydrophobic, while methionine is larger in size. The overall structures were similar. Perhaps, because the mutation is located in the TD-II structure, this exchange could result in extra stress inside the protein (Figure 5). This hydrophobic nature of the substituted amino acids was suggested to cause dysfunction, which can be further modulated by features, such as propensity of peptides to aggregate as oligomers and fibrils.6 All these findings confirmed the “extra stress” properties of methionine. Since the mutation V604M was not tested for levels of APP expressions, this variant could not be fully characterized as the genetic determinism of probable EOAD.
Approximately, 25 APP mutations were reported as pathogenic variants among 32 coding mutations in APP gene, and several of them presented in autosomal dominant pattern of EOAD.6,15 Overproductions of total Aβ or changes in the Aβ1–40:Aβ1–42 ratio toward formation of more toxic Aβ1–42 peptide were the results from the variants at or near the β- and γ-proteolytic sites.16 On the other hand, the variants within the region of Aβ could result in the formation of Aβ oligomers with increased propensity for aggregation.16 Not only mutations located near the cleavage sites of β, and γ-secretase enzymes might be involved in the onset of AD, but also mutations near the cleavage site of γ-secretase (Glu693Lys, Glu693Gly, Glu693del, and Asp694Asn) might change the processing of APP, in enhancing the proteolytic resistance of Aβ peptide.17,18 In addition, mutations located in critical regions of APP, including the region that generates Aβ, cause familial susceptibility to AD,19–21 whereas several mutations outside the Aβ region, including the substituted amino acids at 595 and 596 also associated with familial Alzheimer’s have been found to dramatically increase production of Aβ.22
Recent studies screened the entire sequence of APP, and several variants were found outside of amyloid processing area (exon 16–17).23–26 APP K496Q was found in a Caucasian British patient diagnosed with AD <65 years of age. However, no detailed clinical description was available on this patient. Mutation was missing among controls, and predicted as damaging in silico.25 Y538H mutation was also found in a 69-year-old AD patient, without any family history of disease.26 APP E599K appeared in AD and Parkinson’s disease (PD) patients.23,24,26 Schulte et al found this variant in 1 PD and 2 PD+ dementia cases, and its frequency seemed to be higher among patients than among controls. Neuropathology revealed Lewy bodies pathology with cerebral amyloid angiopathy.23APP P620A was discovered in a French EOAD patient, but was missing in controls. No detailed description is available on this patient.24 P620L also appeared in an EOAD patient, who developed disease at the age of 65 years. In silico prediction predicted it as non-damaging.26 None of these findings defined the exact association between neurodegeneration and APP mutations, outside of amyloid progression. However, several of these rare variants were missing in controls, but their role in AD may not be ignored. It may be possible that mutations, outside of exon 16–17 could act somehow in disease progression, for example, through altered APP expression, metabolism. It is unknown, whether APP Val604Met could be a damaging mutation. MRI of the proband patient (Figure 1) revealed hippocampal atrophy, especially in the frontoparietal regions. These results suggested that the p.Val604Met mutation may contribute to disease by a mechanism other than enhancing amyloid fibril formation, but a lot of further study is needed.
Among the variants (Table S1), reported in our patient, APP Val604Met could be the most dominant mutation, which could alternate the amyloid production, resulting in disease phenotype. The main limitation of this study was that no cell study was done for the mutation yet, which could definitely verify or refute its role in AD progression. In addition, segregation of mutation could not be verified, since all living relatives of patient refused to undergo genetic testing. Functional studies are definitely needed to verify, how APP p.Val604Met could be related and contribute to AD progression.
Conclusion
We suggest that APP p.Val604Met could be a novel probable pathogenic mutation, involved in EOAD. The patient was diagnosed with early-onset dementia at the age of 55 years. Since no possible pathogenic mutations were found in the known AD-associated genes, APP p.Val604Met may be considered as a causative mutation in the proband. APP Val604Met mutation was a novel mutation, since it was missing in 622 asymptomatic Korean individuals from KRGDB database, and also in large international genetic variation databases, such as ExAC and 1000 Genomes. This case of mutation could be associated with a familial case of AD/dementia, since several additional family members, affected with similar symptoms were described. Even though we could not perform in vitro functional studies, our in silico models revealed that mutation may affect the flexibility of APP TD-II region, which may be associated with the EOAD phenotype. Future cell studies will be performed to investigate the role of mutation in disease progression, or whether it could affect the splicing of APP transcript.
Acknowledgment
This research was supported by National Research Foundation of Korea grants awarded by the Korean government (Ministry of Education, Science & Technology [MEST]; MEST numbers 2017R1A2B4012636 and 2017R1C1B5017807).
Disclosure
The authors report no conflicts of interest in this work.
References
Weggen S, Beher D. Molecular consequences of amyloid precursor protein and presenilin mutations causing autosomal-dominant Alzheimer’s disease. Alzheimers Res Ther. 2012;4(2):9. | ||
Bertram L, Tanzi RE. Thirty years of Alzheimer’s disease genetics: the implications of systematic meta-analyses. Nat Rev Neurosci. 2008;9(10):768–778. | ||
Benilova I, Karran E, De Strooper B. The toxic Aβ oligomer and Alzheimer’s disease: an emperor in need of clothes. Nat Neurosci. 2012;15(3):349–357. | ||
Rovelet-Lecrux A, Hannequin D, Raux G, et al. APP locus duplication causes autosomal dominant early-onset Alzheimer disease with cerebral amyloid angiopathy. Nat Genet. 2006;38(1):24–26. | ||
Sleegers K, Brouwers N, Gijselinck I, et al. APP duplication is sufficient to cause early onset Alzheimer’s dementia with cerebral amyloid angiopathy. Brain. 2006;129(Pt 11):2977–2983. | ||
Hunter S, Brayne C. Understanding the roles of mutations in the amyloid precursor protein in Alzheimer disease. Mol Psychiatry. 2018;23(1):81–93. | ||
Beach TG, Monsell SE, Phillips LE, Kukull W. Accuracy of the clinical diagnosis of Alzheimer disease at National Institute on Aging Alzheimer Disease Centers, 2005–2010. J Neuropathol Exp Neurol. 2012;71(4):266–273. | ||
Giau VV, Bagyinszky E, An SSA, Kim S. Clinical genetic strategies for early onset neurodegenerative diseases. Mol Cell Toxicol. 2018;14(2):123–142. | ||
Kovacs GG. Molecular pathological classification of neurodegenerative diseases: turning towards precision medicine. Int J Mol Sci. 2016;17(2):E189. | ||
van Giau V, An SSA, Bagyinszky E, Kim S. Gene panels and primers for next generation sequencing studies on neurodegenerative disorders. Mol Cell Toxicol. 2015;11(2):89–143. | ||
McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology. 1984;34(7):939–944. | ||
Giau VV, Wang MJ, Bagyinszky E, Youn YC, An SSA, Kim S. Novel PSEN1 p.Gly417Ala mutation in a Korean patient with early-onset Alzheimer’s disease with parkinsonism. Neurobiol Aging. Epub 2018; Aug 9. | ||
Källberg M, Wang H, Wang S, et al. Template-based protein structure modeling using the RaptorX web server. Nat Protoc. 2012;7(8):1511–1522. | ||
Yang Y, Giau VV, An SSA, Kim S. Plasma oligomeric beta amyloid in Alzheimer’s disease with history of agent orange exposure. Dement Neurocogn Disord. 2018;17(2):41–49. | ||
Giau VV, Lee H, Shim KH, Bagyinszky E, An SSA. Genome-editing applications of CRISPR-Cas9 to promote in vitro studies of Alzheimer’s disease. Clin Interv Aging. 2018;13:221–233. | ||
Brouwers N, Sleegers K, Van Broeckhoven C. Molecular genetics of Alzheimer’s disease. Ann Med. 2008;40(8):562–583. | ||
Tomiyama T, Nagata T, Shimada H, et al. A new amyloid beta variant favoring oligomerization in Alzheimer’s-type dementia. Ann Neurol. 2008;63(3):377–387. | ||
Roks G, van Harskamp F, de Koning I, et al. Presentation of amyloidosis in carriers of the codon 692 mutation in the amyloid precursor protein gene (APP692). Brain. 2000;123(Pt 10):2130–2140. | ||
Goate A, Chartier-Harlin MC, Mullan M, et al. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature. 1991;349(6311):704–706. | ||
Murrell J, Farlow M, Ghetti B, Benson MD. A mutation in the amyloid precursor protein associated with hereditary Alzheimer’s disease. Science. 1991;254(5028):97–99. | ||
Chartier-Harlin MC, Crawford F, Houlden H, et al. Early-onset Alzheimer’s disease caused by mutations at codon 717 of the beta-amyloid precursor protein gene. Nature. 1991;353(6347):844–846. | ||
Citron M, Oltersdorf T, Haass C, et al. Mutation of the beta-amyloid precursor protein in familial Alzheimer’s disease increases beta-protein production. Nature. 1992;360(6405):672–674. | ||
Schulte EC, Fukumori A, Mollenhauer B, et al. Rare variants in β-Amyloid precursor protein (APP) and Parkinson’s disease. Eur J Hum Genet. 2015;23(10):1328–1333. | ||
Nicolas G, Wallon D, Charbonnier C, et al. Screening of dementia genes by whole-exome sequencing in early-onset Alzheimer disease: input and lessons. Eur J Hum Genet. 2016;24(5):710–716. | ||
Sassi C, Guerreiro R, Gibbs R, et al. Exome sequencing identifies 2 novel presenilin 1 mutations (p.L166V and p.S230R) in British early-onset Alzheimer’s disease. Neurobiol Aging. 2014;35(10):2422.e13–e16. | ||
Sassi C, Guerreiro R, Gibbs R, et al. Investigating the role of rare coding variability in Mendelian dementia genes (APP, PSEN1, PSEN2, GRN, MAPT, and PRNP) in late-onset Alzheimer’s disease. Neurobiol Aging. 2014;35(12):2881.e1–2881.e6. |
Supplementary material
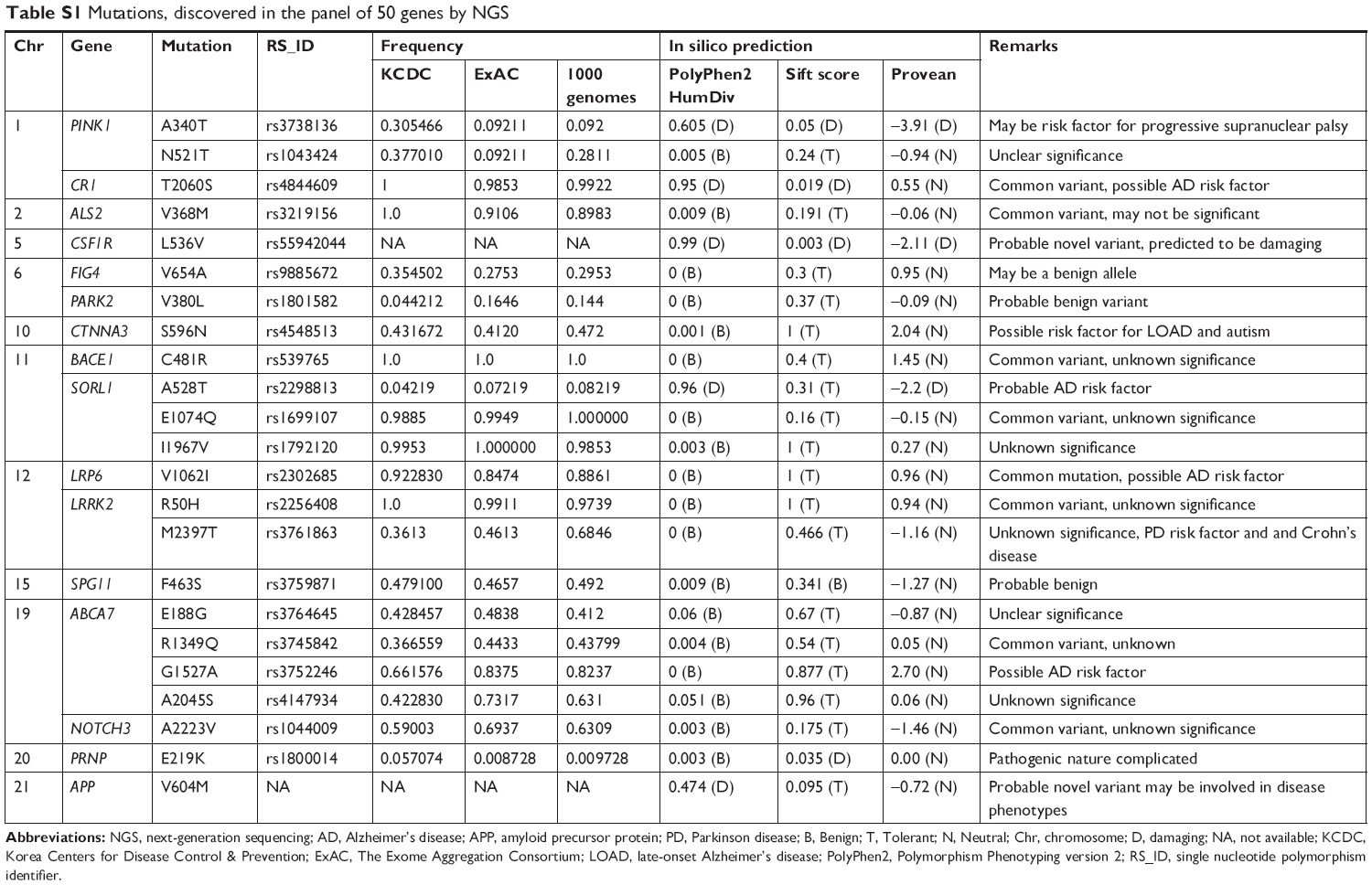 | Table S1 Mutations, discovered in the panel of 50 genes by NGS |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.