")
Back to Journals » OncoTargets and Therapy » Volume 11
Icotinib inhibits the proliferation of hepatocellular carcinoma cells in vitro and in vivo dependently on EGFR activation and PDL1 expression
Authors Sun J, Jiang W, Tian D, Guo Q, Shen Z
Received 11 July 2018
Accepted for publication 18 October 2018
Published 21 November 2018 Volume 2018:11 Pages 8227—8237
DOI https://doi.org/10.2147/OTT.S179844
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Leo Jen-Liang Su
Jisan Sun, Wentao Jiang, Dazhi Tian, Qingjun Guo, Zhongyang Shen
Department of Liver Transplantation, Tianjin First Center Hospital, First Clinical Institute of Tianjin Medical University, Nan Kai District, Tianjin 300192, People’s Republic of China
Purpose: Hepatocellular carcinoma (HCC) accounts for one of the most prevalent tumor types in the world and still lacks an effective treatment regimen. The EGFR tyrosine-kinase inhibitor icotinib is capable of inhibiting proliferation of several kinds of cancer cells, but its anticancer effect in HCC is still not verified.
Methods: In the current study, ten HCC cell lines were selected to test their original EGFR-activation status and PDL1 protein level, and in vitro antiproliferation assays were also conducted to analyze the IC50 and further investigate the correlation between IC50 and protein level of phosphorylated EGFR and PDL1. A in vivo nude mouse xenograft animal model was used as well to analyze its anticancer effect.
Results: Icotinib showed significant inhibitory effects only on HCC cell lines that had both higher p-EGFR and PDL1 protein level. This specific HCC cell line was subcutaneously injected to establish the in vivo xenograft tumor model, and icotinib reduced tumor weight remarkably and growth dose dependently. Molecular mechanism study revealed that icotinib inhibited the phosphorylation of EGFR and PDL1 expression in cancer cells and activated apoptosis. Knocking down PDL1 significantly reduced the inhibitory effect of icotinib on HCC, and knocking in PDL1 increased the sensitivity of icotinib in HCC.
Conclusion: The current research suggests that icotinib has an inhibitory effect on a subgroup of HCC cells that have both higher p-EGFR and PDL1. This hints at the potential clinical usage of icotinib in HCC based on PDL1-biomarker examination.
Keywords: hepatocellular carcinoma, epidermal growth factor receptor, icotinib, PDL1 protein
Introduction
Liver cancer is one of the most common cancers worldwide. Hepatocellular carcinoma (HCC) is the most common form of liver cancer in adults and is characterized by high malignancy and poor prognosis.1,2 The International Agency for Research on Cancer estimated that there were about 782,000 new cases of liver cancer in 2012 globally, of which 83% occurred in developing countries, with China accounted for 50%.3 Statistics from a cancer center in China have shown that the incidence of liver cancer in different regions and sexes are decreasing, after adjusting for age.4 However, the large population base and increasing elderly population aggravate the burden of prevention and treatment of liver cancer, and the burden of liver cancer is still very serious in China.
Hepatectomy is an effective method for the treatment of liver cancer. Many patients already have metastases outside the liver, and up to 40% of cancer relapses 1 year after hepatectomy.5 Current nonsurgical therapies for liver cancer include local ablation, biotherapy (targeted therapy, immunotherapy), hormone therapy, liver transplantation, and chemotherapy. Although basic and clinical studies are progressing rapidly in liver cancer, less effective medicine still plagues its prognosis.
The EGFR pathway plays an important role in the carcinogenesis of HCC,6 and several EGFR tyrosine-kinase inhibitors (TKIs), such as gefitinib, have also been demonstrated to be effective for HCC,7,8 although TKIs are mostly used for lung cancer chemotherapy. Several studies have indicated that EGFR is frequently overexpressed and positively correlated with early tumor recurrence in HCC, so anti-EGFR may be a promising therapeutic strategy in HCC.8–10 However, although anti-EGFR therapy has achieved huge success in both lung cancer and colorectal cancer, it has failed in HCC in clinical trials, and the mechanisms behind this remain unclear.11,12 Possible reasons include lack of a predictive biomarker of response and irrational clinical trial design, which might limit EGFR-inhibitor usage against HCC. Therefore, searching for which HCC subgroup is sensitive to EGFR TKIs and further investigating possible TKI-sensitive biomarkers is very important to guide EGFR-TKI utilization against HCC. Studies on lung cancer chemotherapy have indicated that EGFR TKIs can decrease PDL1 expression in lung cancer cells, as well as decrease the phosphorylation of EGFR,13–15 which suggests that EGFR TKIs may be more sensitive to cancer cells with higher expression of PDL1 and provide some clues for clinical usage of EGFR TKIs against liver cancer.
Icotinib was developed by a Chinese pharmaceutical enterprise for use as a new type of small-molecule anticancer therapeutic. Preclinical studies have revealed that icotinib is a specific and potent EGFR TKI. Further, an in vitro study also indicated that icotinib inhibits EGFR-TK activity effectively at a concentration of 5 nM, including L858R, (99%), L861Q (96%), EGFR (91%), T790M (61%), and T790M and L858R (61%).16 In vitro and in vivo experimental studies both demonstrate that icotinib was effective in inhibiting lung cancer proliferation, not dependent on EGFR mutation. However, there has been no report about its usage in liver cancer. In the current study, ten HCC cell lines were selected to test icotinib inhibitory effect. Based on their differential sensitivity patterns to icotinib, cellular EGFR and PDL1 status were examined, and we further investigated whether PDL1-expression levels correlated with icotinib inhibitory capability.
Methods
Cell culture and chemical agents
The HCC cancer cell lines BEL7402, HepG2, SMMC7721, Huh7, SNU423, SNU449, SNU475, SNU182, SNU387, and SNU398 were obtained from the American Type Culture Collection. All cells were cultured in RPMI 1640 (Thermo Fisher Scientific, Waltham, MA, USA) containing 10% FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin in a 37°C incubator containing 5% CO2. When growth had reached the logarithmic phase, cells were seeded on 96-well plates for further study.
Protein levels of EGFR and PDL1 by Western blot
HCC cells were lysed for protein extraction. Proteins were extracted with RIPA buffer with 1 mM phenylmethylsulfonyl fluoride (Sigma-Aldrich Co., St Louis, MO, USA). Concentrations were determined with a bicinchoninic acid assay kit (Thermo Fisher Scientific). The same amount of protein (10 μg/lane) was subjected to SDS-PAGE with a constant voltage of 80 V for 120 minutes. Proteins were transferred into polyvinylidene fluoride membrane (Millipore, Bedford, MA, USA). The membrane was blocked with 5% nonfat milk in Tris-buffered saline (TBS) for 1 hour at room temperature. After the blockage, the membrane was incubated with the primary antibodies, including anti-p-EGFR (Cell Signaling Technology, Danvers, MA, USA), anti-PDL1 (Abcam, Cambridge, UK), and anti-β-actin (Abcam) antibodies overnight at 4°C. The next morning, the membrane was washed with TBS containing 0.05% Tween 20 (TBST) and probed with the horseradish peroxidase-linked secondary antibodies at 37°C for 1 hour. After being washed with TBST, membranes were developed with an enhanced chemiluminescence kit and visualized with an LAS 4000 imaging system (GE Healthcare, Little Chalfont, UK). Bands were quantified with ImageJ software by measuring target-gene band intensity normalized to internal control β-actin. All reactions were done in duplicate, and experiments were repeated to ensure reproducible results. Protein levels of cleaved caspase 3 (Cell Signaling Technology) and BCL2 (Cell Signaling Technology) in icotinib-treated HCC cells were also tested by Western blot following the same procedure.
MTT proliferation assay
Cells in the logarithmic growth period were digested with 0.25% trypsin and diluted to 5×104 cell/mL suspension by medium containing 10% FBS. Cells were inoculated in 96-well plate with 100 μL per well. A total of 100 μL icotinib with serial concentrations of 0, 0.1, 0.5, 1, 2, 10, 15, 25, and 30 μmol/L was added after culture for 24 hours, five parallel wells were prepared, and medium culture served as blank control. After being cultured for another 72 hours, the supernatant was abandoned and 100 μL MTT added and cultured for another 4 hours. Dimethyl sulfoxide (Sigma-Aldrich) was added, OD490 measured with WellScan MK3 ELISA (Labsystems, Vantaa, Finland), and IC50 was calculated by GraphPad prism 6.0 (GraphPad Software, La Jolla, CA, USA):
|

PDL1 RNAi lentivirus generation, infection, and cell sorting
The human HCC cell lines SMMC7721 and Huh7 were used in the PDL1 RNAi knockdown study. shRNA against human PDL1 (NM_014143.2; GenBank) cloned into lentiviral gene-transfer vector encoding GFP was obtained from GeneChem (Shanghai, People’s Republic of China). The targeting sequence of PDL1 shRNA was 5′-GACCTATATGTGGTAGAGTAT-3′. The recombinant PDL1-targeting lentivirus (LV-PDL1-siRNA virus) and untargeted control mock lentivirus (LV-NC virus) were prepared and transfected to SMMC7721 and Huh7 cell lines according to the manufacturer’s instructions. Infected cells were designated LV-PDL1-siRNA and LV-NC. Finally, infected cells were analyzed by flow cytometry (Canto II; BD, Franklin Lakes, NJ, USA) and positive cell sorting done using GFP tags with FL1 channel in a flow sorter (Aria II; BD).
Construction of PDL1 overexpression in BEL7402 and HepG2 cells
Full-length PDL1 was synthesized by Sangon Biotech (Shanghai, People’s Republic of China) and amplified by PCR. Primers were designed: forward 5′-TAGAATTCATGAGGATATTTGCTGTCTT-3′; reverse 5′-TAGGATCCTTACGTCTCCTCCAAATGTG-3′; The PCR fragment was subcloned into the EcoRI and BamHI sites in the pLV-IRES-ZsGreen-T2A-puro vector (Promega, Fitchburg, WI, USA). Then, BEL7402 and HepG2 cells were transfected with the recombinant lentivirus and sorted using a GFP tag. The positive sorted cells were then named PDL1-over-BEL7402 and HepG2 and LV-vector-control (BEL7402 and HepG2 cells overexpressing vector control).
Real-time reverse-transcription (RT- PCR)
mRNA expression of PDL1 in HCC cells after siRNA transfection was quantified by real-time reverse-transcription polymerase chain reaction (RT-PCR). Total RNA from both cell lines was extracted using Trizol (Thermo Fisher Scientific), and PCR reactions were attained using an ABI 7600 system (Thermo Fisher Scientific) according to the manufacturer’s instructions. Primer sequences for detection of the reference gene GAPDH and the target gene PDL1 were synthesized: human GAPDH forward primer 5′-TGACTTCAACAGCGACACCCA-3′, human GAPDH reverse primer 5′-CACCCTGTTGCTGTAGCCAAA-3′, human PDL1 forward primer 5′-ACTGGCATTTGCTGAACG-3′, and PDL1 reverse primer 5′-TCCTCCATTTCCCAATAGAC-3′. Data were analyzed by the 2−ΔΔCt method and expressed as fold change compared to GAPDH expression. RT-PCR products for GAPDH and PDL1 were also confirmed by electrophoresis on 1.8% agarose gel containing 0.1% ethidium bromide.
Xenografts in nude mice
Huh7 cell suspension (0.2 mL, 5×105/mL) was injected subcutaneously into the armpit of BALB/c male nude mice, with eight mice in each group. Sodium carboxymethylcellulose 0.5% was used for the solvent control group, and icotinib was administered at dosages of 50 mg/kg and 100 mg/kg. Icotinib was administered orally for 3 weeks once tumor sizes had reached 100 mm3, once every 2 days. Tumor volumes were measured with a Vernier caliper every 3 days. After treatment, mice were killed and tumors stripped and weighed. The length of the longest axis tumor (mm) was recorded, and the narrowest axis length was recorded as the width of the tumor (mm). Tumor volume (mm3) and tumor-growth-inhibition rate were calculated:
|
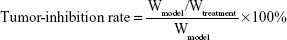
where W means the weight of the tumor. In this study, an inhibition rate ≥60% was defined as effective. All animal experiments and the study protocol were approved by the Ethics Committee of Laboratory Animals of Tianjin Medical University in Tianjin, People’s Republic of China (005412, April 2017). All experimental procedures were conducted in accordance with our institutional guidelines and international standards for manipulation and care of laboratory animals (Guide for the Care and Use of Laboratory Animals).29
Cell apoptosis by TUNEL
A TUNEL assay was performed using an in situ cell-death-detection kit, fluorescein (11684795910; Hoffman-La Roche, Basel, Switzerland) following the manufacturer’s instructions. SMMC7721 and Huh7 cells were pretreated with or without icotinib (0.5 and 5 μM) for 48 hours. Cells in the six-well plate were fixed in 4% paraformaldehyde for 20 minutes at room temperature, washed with PBS, and permeabilized with freshly prepared 0.1% Triton X-100 and 0.1% sodium citrate for 2 minutes on ice. After being washed with PBS, cells were overlaid with TUNEL reaction mixture (100 μL) according to the manufacturer’s instructions and incubated for 1 hour at 37°C. Finally, cells were washed in PBS and coverslips mounted onto slides, Cells were imaged using fluorescence microscopy (AX70; Olympus). Excitation wavelength in the range of 450–500 nm and detection wavelength range of 515–565 nm was used for TUNEL fluorescence. The magnification of the photographs was 20×, and cells that were positively labeled were quantified in ten fields per slide by Image-Pro Plus 6.0 software (Media Cybernetics, Rockville, MD, USA).
Data analysis
Continuous variable data are presented as means with SD. Statistical analyses were performed using SPSS 21.0 software (IBM Corporation, Armonk, NY, USA). Differences between groups were compared with Student’s t-tests or Mann–Whitney nonparametric tests. Differences among multiple groups were compared with ANOVA. P<0.05 was considered statistically significant.
Results
EGFR and PDL1 expression
All protein levels of p-EGFR and PDL1 from HCC cells were examined by Western blot. As shown in Figure 1, we found that BEL7402, HepG2, and SNU475 had relatively lower PDL1 expression, while SMMC7721 and Huh7 had relatively higher PDL1 expression. For p-EGFR protein levels, HepG2 and Huh7 had highest expression, while BEL7402 had lower p-EGFR expression. Relative protein quantification for each cell line is shown in Table 1.
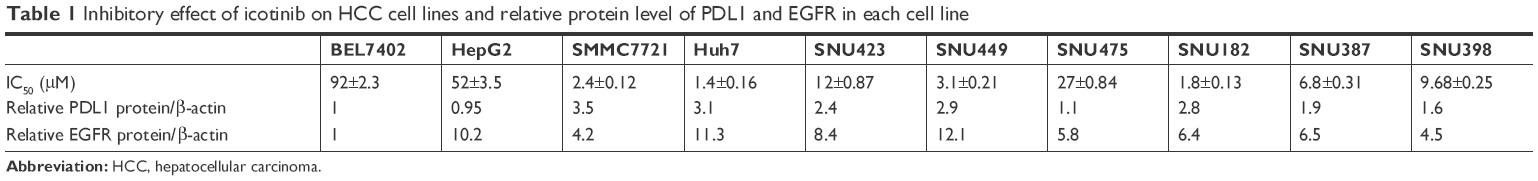 | Table 1 Inhibitory effect of icotinib on HCC cell lines and relative protein level of PDL1 and EGFR in each cell line |
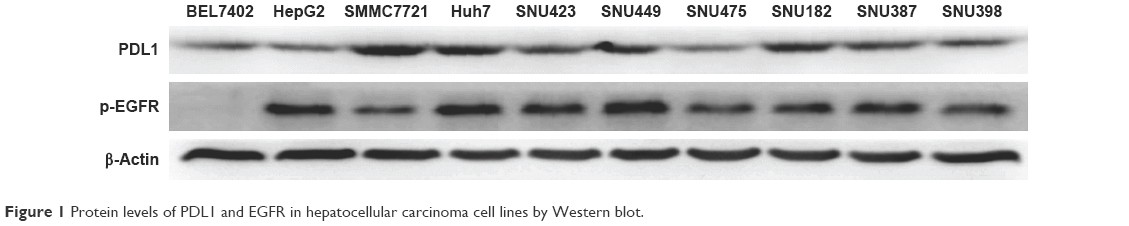 | Figure 1 Protein levels of PDL1 and EGFR in hepatocellular carcinoma cell lines by Western blot. |
Icotinib had differing inhibitory capacity on HCC cells
By MTT assay, the results demonstrated that icotinib had different inhibitory effects on different HCC cells. All IC50 values are shown in Table 1. Based on MTT results and Western blot of p-EGFR expression and PDL1, we found that icotinib had the best inhibitory capability on HCC cells, which have higher p-EGFR and PDL1 expression, such as in SMMC7721 and Huh7. Further, if PDL1 is lower, p-EGFR is higher, and inhibitory effect of icotinib is still not high, such as on HepG2 and SNU475.
Downregulation of PDL1 reduced inhibitory capability of icotinib on HCC
SMMC7721 and Huh7, which had relatively lower IC50 and relatively higher PDL1, were selected for siRNA experiments. By siRNA, PDL1 expression was significantly decreased, which was testified by RT-PCR and Western blot (Figure 2A). On MTT assays, after PDL1 expression had been downregulated, icotinib’s proliferation-inhibitory effect was significantly reduced and IC50 decreased to 12.8 and 16.3 μM respectively (Figure 2B).
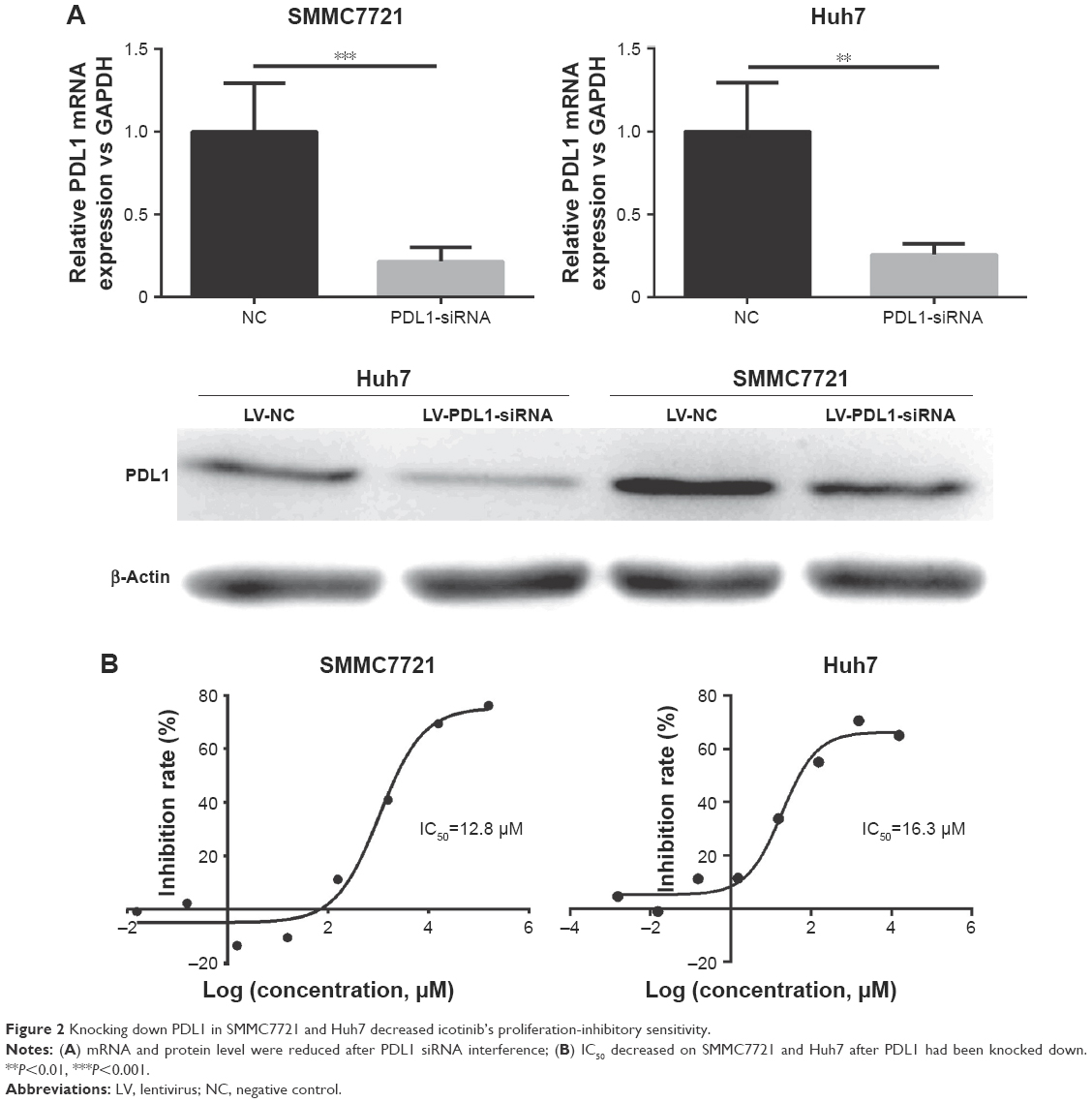 | Figure 2 Knocking down PDL1 in SMMC7721 and Huh7 decreased icotinib’s proliferation-inhibitory sensitivity. |
PDL1 exogenous expression enhanced icotinib sensitivity for HCC
BEL7402 and HepG2 had lower PDL1 protein expression and showed resistance to icotinib, although HepG2 had higher p-EGFR expression. Therefore, in the current study, the PDL1 exogenous expression vector was transfected into BEL7402 and HepG2. By RT-PCR and Western blot, the exogenous expression of PDL1 was verified (Figure 3A). After transfection, MTT assays demonstrated that sensitivity to icotinib was significantly enhanced for HepG2 (IC50 decreased to 6.41 μM), but not BEL7402 (IC50 changed to 75.3 μM), which means that higher expression of both p-EGFR and PDL1 was necessary to retain icotinib chemosensitivity for HCC (Figure 3B).
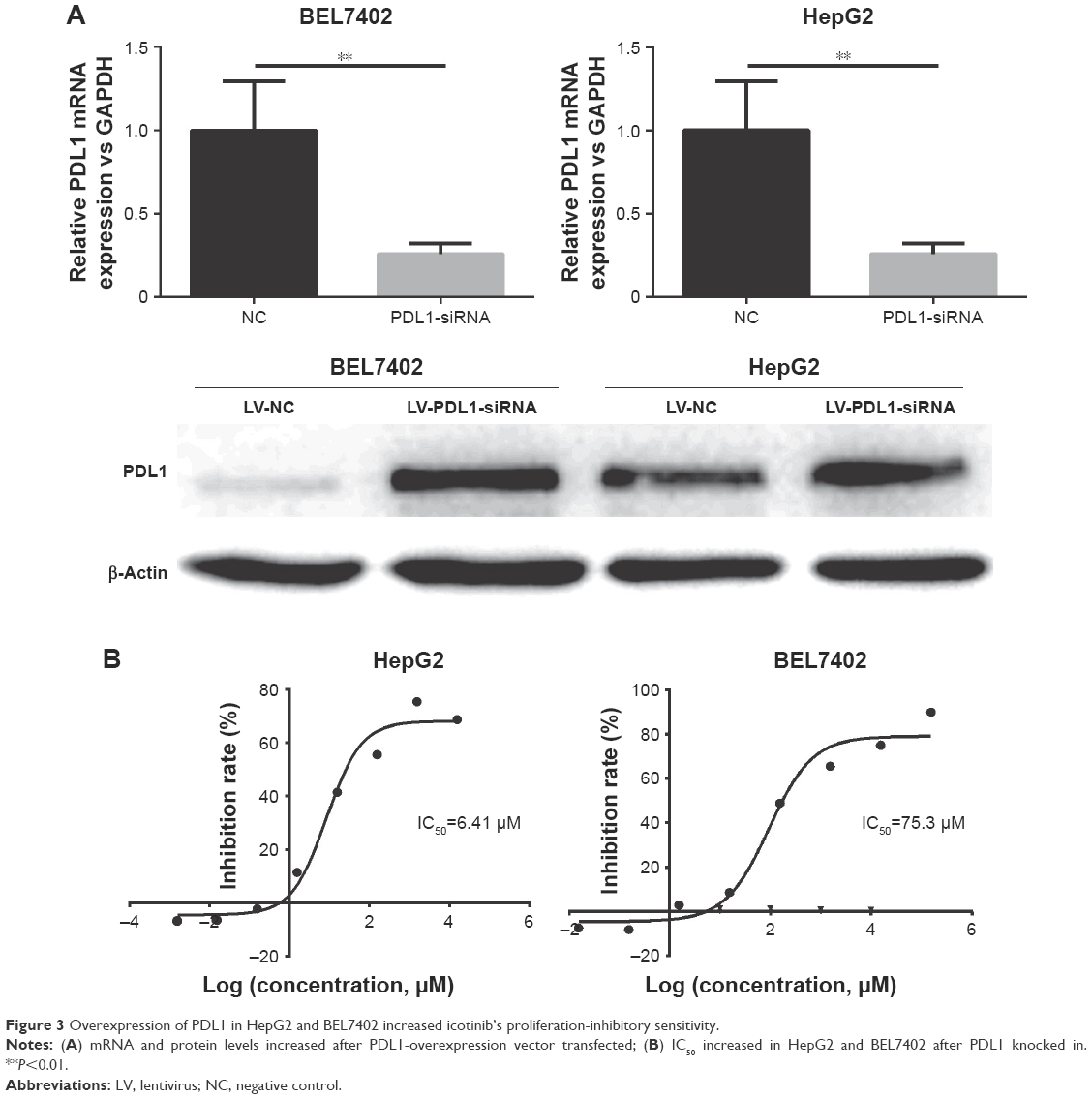 | Figure 3 Overexpression of PDL1 in HepG2 and BEL7402 increased icotinib’s proliferation-inhibitory sensitivity. |
Icotinib induced HCC apoptosis by activating caspase-family members and downregulated PDL1 expression
Previous research has demonstrated that the proliferation-inhibitory capability of icotinib is due to its induced apoptosis.17 On fluorescence microscopy, apoptosis induced by icotinib in SMMC7721 and Huh7 was observed to be in a dose dependent manner (Figure 4). By Western blot, icotinib significantly increase cleaved caspase 3 production, and meanwhile reduced the BCL2, p-EGFR, and PDL1 expression in SMMC7721 and Huh7 cells (Figure 5).
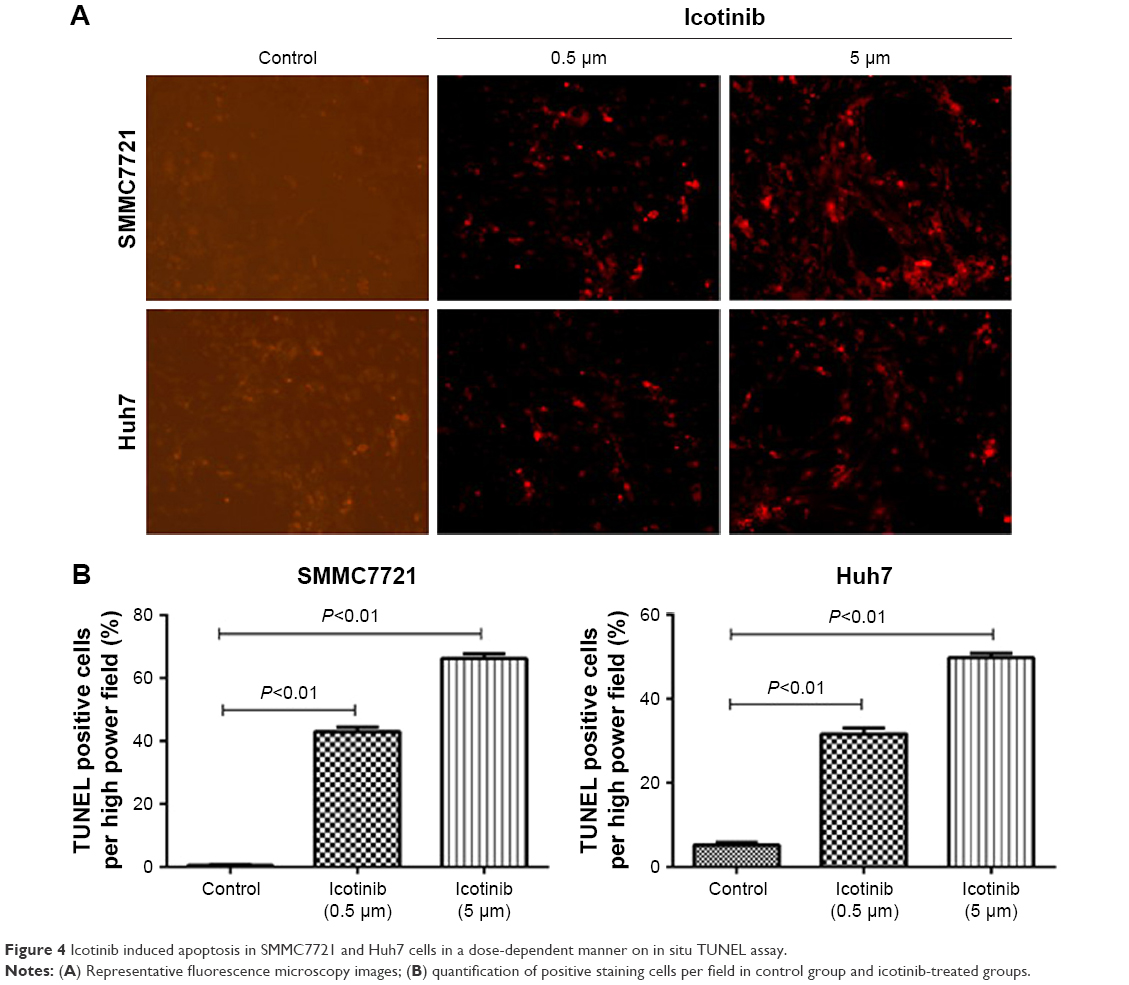 | Figure 4 Icotinib induced apoptosis in SMMC7721 and Huh7 cells in a dose-dependent manner on in situ TUNEL assay. |
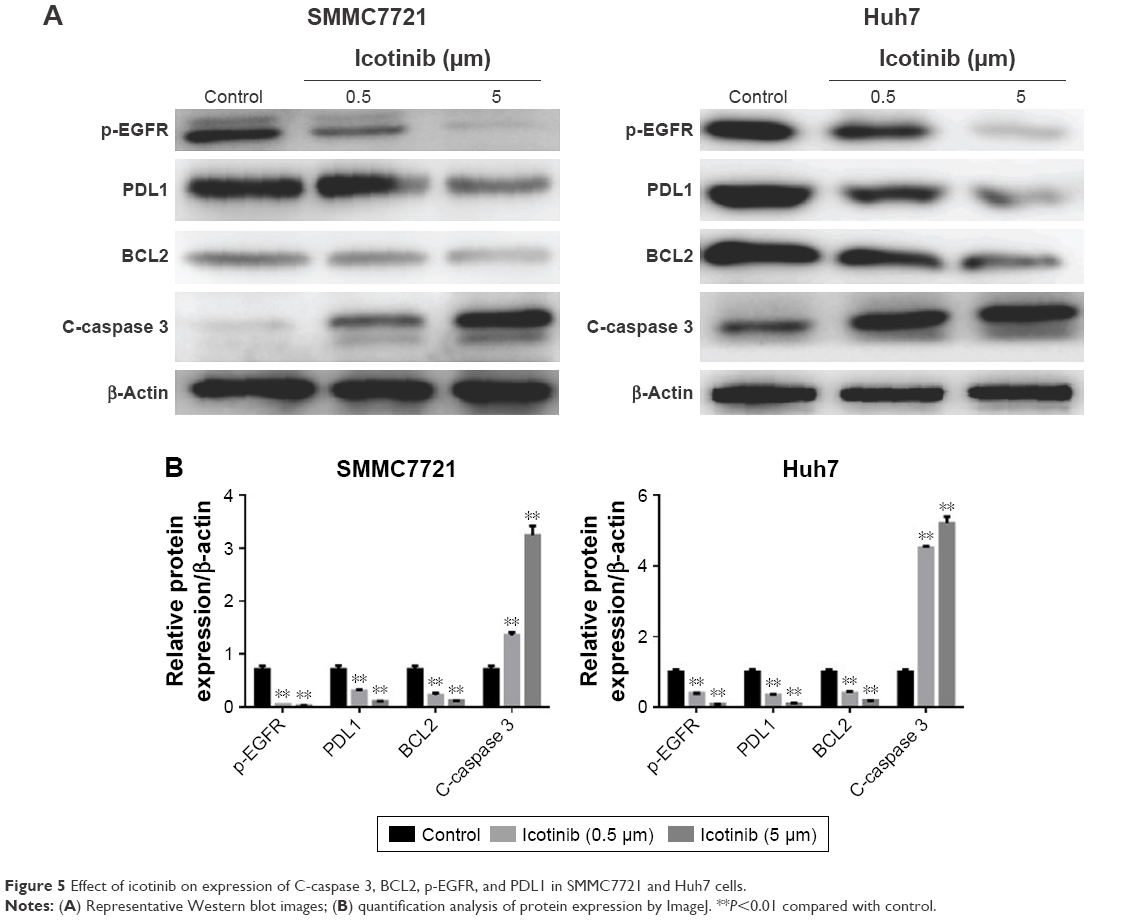 | Figure 5 Effect of icotinib on expression of C-caspase 3, BCL2, p-EGFR, and PDL1 in SMMC7721 and Huh7 cells. |
Icotinib significantly inhibited exogenous Huh7 tumor growth in vivo
Huh7 cells were injected subcutaneously into the right limb, and when tumors had reached 100 mm3, the mice began to receive icotinib administration every 2 days. The whole treatment lasted 3 weeks. Tumor sizes in the control group increased significantly. Compared with the control group, tumor volumes in the icotinib treated group decreased dose-dependently, and the difference was statistically significant (Figure 6), which suggested that icotinib reduced sensitive HCC-tumor growth in vivo.
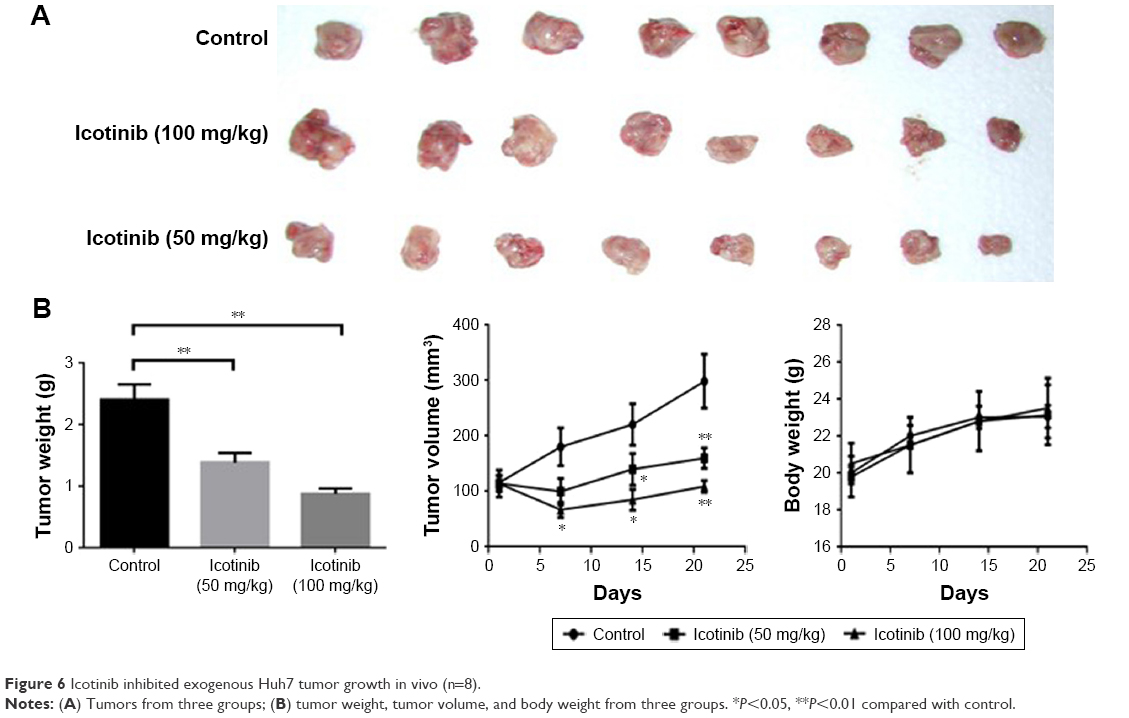 | Figure 6 Icotinib inhibited exogenous Huh7 tumor growth in vivo (n=8). |
Discussion
EGFR TKIs, such as gefitinib and erlotinib, have been the first-line treatment regimens in EGFR-mutated NSCLC patients, but are not indicated for liver cancer. However, basic research and several preclinical trials have suggested that gefitinib can inhibit the proliferation of human liver cancer cells only at the EGFR-activation stage.18 Meanwhile, a phase II trial of the combination of bevacizumab and erlotinib in patients who have advanced HCC has showed that significant, clinically meaningful antitumor activity,19 suggesting that EGFR TKI might also be beneficial for HCC, could be especially helpful for some specific subgroups who are sensitive to TKIs. However, which subgroup of HCC is more sensitive to TKIs is still not clear. Icotinib is another effective TKI, developed by the Chinese Bei Da pharmaceutical company, and has been widely used for NSCLC chemotherapy,20,21 and research has suggested that icotinib can sensitize radiotherapy.22 Several Chinese research projects aim to expand its indications and find more cancer types that are sensitive to icotinib treatment. In the current study, HCC was selected to test whether icotinib could be effective against HCC. Ten HCC cell lines were used to test icotinib’s inhibitory effects, and icotinib showed a discrepancy in inhibitory effect. To understand the molecular background of each cell line, p-EGFR expression was tested for each cell line and combined with MTT proliferation assays, and we found that although HCC cells with higher p-EGFR expression were more sensitive to icotinib, expression level of p-EGFR did not predict the sensitivity of icotinib to HCC with 100% certainty.
With a boom in immunocheckpoint therapy, PDL1 expression in tumor cells was found to be correlated with EGFR activation in some tumors,23,24 which provides a clue for us to investigate whether the activation of EGFR in HCC is related to PDL1, and further investigate TKI effects on HCC cells with higher PDL1 expression. In the current study, PDL1 expression was examined among ten HCC cell lines, and results suggested that HCC cell lines with higher PDL1 protein levels had more sensitivity to icotinib, with lower IC50 values. Research has demonstrated that EGFR TKIs can not only directly inhibit tumor cell-viability but also enhance antitumor immunity through the downregulation of PDL1.13 Takada et al suggested that PDL1 positivity was associated with wild-type EGFR in primary lung adenocarcinoma,25 and that although EGFR mutation is very common in NSCLC, it is mutated less in HCC, which may suggest that EGFR activation in HCC is more possibly correlated with or regulated by PDL1, instead of mutation-induced activation.
In this study, the inhibitory effect of icotinib on the proliferation of human HCC cells was observed by MTT methods. Icotinib had the strongest inhibitory effect on HCC cells that had higher levels of both p-EGFR and PDL1, such as in SMMC7721 and Huh7. For HCC cells with lower PDL1, p-EGFR was higher, showing resistance to icotinib. To further prove that PDL1 protein level was associated with icotinib sensitivity against HCC cells, the knock-in and knockdown experiments were performed. Knocking down PDL1 significantly decreased icotinib sensitivity to original sensitive HCC cells. On the contrary, exogenously increasing PDL1 expression in insensitive HCC cells enhanced their sensitivity to icotinib antiproliferation activity. All these results demonstrate that PDL1 protein level is positively correlated with icotinib inhibitory efficiency.
Programmed cell death, or apoptosis, is an important way to control tumor proliferation.26 In the current study, TUNEL assays showed that icotinib significantly induced apoptosis on SMMC7721 and Huh7 cells in vitro by fluorescence microscopy. Further Western blot results suggested that this apoptosis was possibly due to activation of caspase 3 and blockage of the BCL2 antiapoptosis effect. Western blot results also demonstrated that icotinib significantly downregulated PDL1 and p-EGFR expression in SMMC7721 and Huh7 cells in vitro.
Our in vivo research shows that icotinib has inhibitory effects on Huh7 xenograft tumor mass and tumor volume in male tumor-bearing mice. Compared with the model group, icotinib significantly inhibited tumor growth. The tumor-growth-inhibition rate reached 70%, and more importantly no notable toxic effect was observed by animal body weight. In a recent study performed in tumor-bearing mice, icotinib exerted antitumor effects by enhancing the immunoadhesion ability of erythrocytes to tumor cells,27 which demonstrated erythrocyte immunity might be one of the mechanisms of icotinib. These in vivo data also suggest icotinib might be beneficial for HCC patients, and sensitive groups need to be verified.
Studies have suggested that epithelial–mesenchymal transition is a determinant of sensitivity of HCC to EGFR inhibitors.28 Mesenchymal cells resistant to EGFR inhibitors increased Akt and STAT3 activation through elevated expression of ILK. Mesenchymal cell lines were thus experimentally transformed with kinase-inactive ILK with a resultant decrease in ILK activity and activation of Akt. Kinase-inactive ILK overexpression increased sensitivity to EGFR inhibitors both in vitro and in an in vivo xenograft model. Similarly, our study also suggests that PDL1 is another possible sensitive biomarker for EGFR-TKI utilization against HCC.
Conclusion
In summary, our research shows that icotinib can selectively inhibit HCC cell proliferation via inducing apoptosis, but its inhibitory effect is partially dependent on PDL1 expression in HCC cells. In vivo study also indicated that icotinib can inhibit the growth of HCC xenografts in nude mice. Icotinib is rarely reported to inhibit HCC, and thus, based on our knowledge, it might be time to study icotinib’s effects on HCC and clarify that PDL1 expression is one of the key determinants of its sensitivity.
Disclosure
The authors report no conflicts of interest in this work.
References
Balogh J, Victor D 3rd, Asham EH, et al. Hepatocellular carcinoma: a review. J Hepatocell Carcinoma. 2016;3:41–53. | ||
El-Serag HB. Hepatocellular carcinoma. N Engl J Med. 2011;365(12):1118–1127. | ||
Globocan Ciafro. Estimated Cancer Incidence, Mortality and Prevalence Worldwide in 2012. 2014 [eb/ol]. 2012;2014:12–16. http://globocaniarcfr/Defaultaspx | ||
Zuo T, Zheng R, Zeng H, Zhang S, Chen W. Analysis of liver cancer incidence and trend in China. Zhonghua Zhong Liu Za Zhi. 2015;37(9):691–696. | ||
Fan ST, Mau Lo C, Poon RT, et al. Continuous improvement of survival outcomes of resection of hepatocellular carcinoma: a 20-year experience. Ann Surg. 2011;253(4):745–758. | ||
Xia H, Dai X, Yu H, et al. EGFR-PI3K-PDK1 pathway regulates YAP signaling in hepatocellular carcinoma: the mechanism and its implications in targeted therapy. Cell Death Dis. 2018;9(3):269. | ||
Höpfner M, Sutter AP, Huether A, Schuppan D, Zeitz M, Scherübl H. Targeting the epidermal growth factor receptor by gefitinib for treatment of hepatocellular carcinoma. J Hepatol. 2004;41(6):1008–1016. | ||
Schiffer E, Housset C, Cacheux W, et al. Gefitinib, an EGFR inhibitor, prevents hepatocellular carcinoma development in the rat liver with cirrhosis. Hepatology. 2005;41(2):307–314. | ||
Fuchs BC, Hoshida Y, Fujii T, et al. Epidermal growth factor receptor inhibition attenuates liver fibrosis and development of hepatocellular carcinoma. Hepatology. 2014;59(4):1577–1590. | ||
Erstad DJ, Fuchs BC, Tanabe KK. Molecular signatures in hepatocellular carcinoma: A step toward rationally designed cancer therapy. Cancer. 2018;124(15):3084–3104. | ||
Llovet JM, Villanueva A, Lachenmayer A, Finn RS. Advances in targeted therapies for hepatocellular carcinoma in the genomic era. Nat Rev Clin Oncol. 2015;12(7):408–424. | ||
Tebbutt N, Pedersen MW, Johns TG. Targeting the ERBB family in cancer: couples therapy. Nat Rev Cancer. 2013;13(9):663–673. | ||
Chen N, Fang W, Zhan J, et al. Upregulation of PD-L1 by EGFR Activation Mediates the Immune Escape in EGFR-Driven NSCLC: Implication for Optional Immune Targeted Therapy for NSCLC Patients with EGFR Mutation. J Thorac Oncol. 2015;10(6):910–923. | ||
Akbay EA, Koyama S, Carretero J, et al. Activation of the PD-1 pathway contributes to immune escape in EGFR-driven lung tumors. Cancer Discov. 2013;3(12):1355–1363. | ||
Lin K, Cheng J, Yang T, Li Y, Zhu B. EGFR-TKI down-regulates PD-L1 in EGFR mutant NSCLC through inhibiting NF-κB. Biochem Biophys Res Commun. 2015;463(1–2):95–101. | ||
Tan F, Shen X, Wang D, et al. Icotinib (BPI-2009H), a novel EGFR tyrosine kinase inhibitor, displays potent efficacy in preclinical studies. Lung Cancer. 2012;76(2):177–182. | ||
Zhang Y, Meng X, Shi H, et al. The role of JAK/STAT3 signaling pathway on apoptosis of lung adenocarcinoma cell line PC-9 induced by icotinib. Am J Transl Res. 2016;8(4):1730–1737. | ||
Okano J, Matsumoto K, Nagahara T, Murawaki Y. Gefitinib and the modulation of the signaling pathways downstream of epidermal growth factor receptor in human liver cancer cells. J Gastroenterol. 2006;41(2):166–176. | ||
Moore MJ, Goldstein D, Hamm J, et al. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol. 2007;25(15):1960–1966. | ||
Wang Y, Li L, Han R, Jiao L, Zheng J, He Y. Clinical analysis by next-generation sequencing for NSCLC patients with MET amplification resistant to osimertinib. Lung Cancer. 2018;118:105–110. | ||
Fan T, Zhou JY. Marked Response to Icotinib in Lung Squamous Cell Carcinoma with EGFR Wild-Type. J Coll Physicians Surg Pak. 2018;28(6):S120–S122. | ||
Zhang S, Fu Y, Wang D, Wang J. Icotinib enhances lung cancer cell radiosensitivity in vitro and in vivo by inhibiting MAPK/ERK and Akt activation. Clin Exp Pharmacol Physiol. 2018:969–977. | ||
Azuma K, Ota K, Kawahara A, et al. Association of PD-L1 overexpression with activating EGFR mutations in surgically resected nonsmall-cell lung cancer. Ann Oncol. 2014;25(10):1935–1940. | ||
Zhang W, Pang Q, Yan C, et al. Induction of PD-L1 expression by epidermal growth factor receptor-mediated signaling in esophageal squamous cell carcinoma. Onco Targets Ther. 2017;10:763–771. | ||
Takada K, Toyokawa G, Tagawa T, et al. PD-L1 expression according to the EGFR status in primary lung adenocarcinoma. Lung Cancer. 2018;116:1–6. | ||
Monian P, Jiang X. The Cellular Apoptosis Susceptibility Protein (CAS) Promotes Tumor Necrosis Factor-related Apoptosis-inducing Ligand (TRAIL)-induced Apoptosis and Cell Proliferation. J Biol Chem. 2016;291(5):2379–2388. | ||
Xu RD, Shao TY, Jia SH. Effect of Aesculetin on function of erythrocyte membrane in tumor-bearing mice. Journal of Harbin University of Commerce (Natural Sciences Edition). 2016;32:146–153. | ||
Fuchs BC, Fujii T, Dorfman JD, et al. Epithelial-to-mesenchymal transition and integrin-linked kinase mediate sensitivity to epidermal growth factor receptor inhibition in human hepatoma cells. Cancer Res. 2008;68(7):2391–2399. | ||
National Research Council (US) Committee for the Update of the Guide for the Care and Use of Laboratory Animals. Guide for the care and use of laboratory animals. 8th edition. Washington (DC): National Academies Press (US); 2011. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.