")
Back to Journals » OncoTargets and Therapy » Volume 13
Ibrutinib in Chronic Lymphocytic Leukemia: Clinical Applications, Drug Resistance, and Prospects
Authors Zhou H , Hu P, Yan X, Zhang Y , Shi W
Received 13 February 2020
Accepted for publication 28 April 2020
Published 29 May 2020 Volume 2020:13 Pages 4877—4892
DOI https://doi.org/10.2147/OTT.S249586
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Yao Dai
Hong Zhou,1,* Pan Hu,1,* Xiyue Yan,1 Yaping Zhang,2 Wenyu Shi1,2
1Department of Oncology, Affiliated Hospital of Nantong University, Nantong 226001, Jiangsu, People’s Republic of China; 2Department of Hematology, Affiliated Hospital of Nantong University, Nantong 226001, Jiangsu, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Wenyu Shi; Yaping Zhang
Department of Oncology, Affiliated Hospital of Nantong University, Nantong 226001, Jiangsu, People’s Republic of China
Tel +86-513-81161052; +86-513-81161052 Fax +86-513-85519820
Email [email protected]; [email protected]
Abstract: Bruton’s tyrosine kinase (BTK), a pivotal component of B-cell receptor (BCR) signaling, has been recognized as an important driver of the pathogenesis of chronic lymphocytic leukemia. Ibrutinib is a highly active and selectively irreversible inhibitor of BTK, which has been approved to be effective in both frontline and recurrent therapy of CLL. Acquired resistance has become a greater problem than initially anticipated with the widespread use of ibrutinib. An ongoing exploration of the mechanisms of ibrutinib resistance (IR) in CLL has revealed potentially useful therapeutic targets. New drugs expected to overcome IR in CLL are in the early stages of clinical development. This study aimed to summarize the possible mechanisms of IR and retrospectively analyze promising therapies that might have superior efficacy in overcoming IR.
Keywords: ibrutinib, chronic lymphocytic leukemia, clinical applications, novel therapeutic agents, resistance mechanism
Introduction
Chronic lymphocytic leukemia (CLL) is one of the most prevalent lymphocytic cancers in the Western world, accounting for approximately 11% of all hematological malignancies.1,2 Some patients with CLL have an indolent course and do not require any treatment.3 However, those with unmutated IGHV, chromosomal aberrations of del(17p) and del(11q), and loss or mutation of the TP53 gene generally have an aggressive disease course and poor prognosis.4 The median survival (MS) of the total CLL population is approximately 10 years after diagnosis. Chemoimmunotherapy (CIT), such as bendamustine plus rituximab (BR) and the fludarabine, cyclophosphamide, rituximab (FCR) regimens, showed promising results in most patients.5 Taking into account toxic side effects and tolerance of organ function, FCR can be used as a standard first-line treatment for young patients with CLL (aged less than 65 years), except for physical discomfort and/or complications. Furthermore, FCR protects approximately half of all patients with CLL having IGHV mutations from progression for up to 8 years after initial treatment.6–8 However, CIT treatment is not suitable for high-risk patients with del(17p) or TP53 mutation. It is often accompanied by significant toxic effects, such as infection, second primary malignancies, myelosuppression, or even death, limiting its widespread use throughout the population.9–11
BTK is a key component of BCR signaling, which mediates the interaction of CLL cells with the tumor microenvironment (TME) and promotes their survival and progression.12 Ibrutinib was the first bioavailable irreversible BTK inhibitor (BTKi) to inhibit the function of BTK kinase by covalently binding a cysteine residue at position 481 in the BTK active site.13 The emergence of ibrutinib has completely revolutionized the treatment of CLL. In Phase III CLL/SLL trials, ibrutinib monotherapy showed better efficacy than chlorambucil in the first-line treatment of elderly patients (RESONATE-2).14 Recent results from E1912, iLLUMINATE, and ALLIANCE trials on patients with previously untreated CLL shifted the importance of ibrutinib to frontline therapy.8,15,16 Likewise, ibrutinib monotherapy was more effective than ofatumumab in previously treated adults in a phase III study (RESONATE),17 and the combination of ibrutinib and BR (IBR) showed better efficacy than BR alone in treatment-naïve adults in the HELIOS trial.18 Additionally, the benefits of ibrutinib regimens were not influenced by poor prognostic factors such as del(11q), TP53 mutations, and del(17p).19 Considering that ibrutinib has comprehensively covered the treatment stage of patients with CLL, this study aimed to conduct a comprehensive analysis of the clinical application and drug resistance mechanisms of ibrutinib in CLL, and describe recent research strategies for overcoming IR.
Mechanism of Action of Ibrutinib in the Pathogenesis of CLL
The key sites for the expansion of malignant CLL cells are the lymph node (LN) proliferation centers (also known as pseudofollicles), in which the chemokines, BCR signaling, TLR ligands, and TME provide survival advantages for CLL cells and stimulate tumor clonal proliferation and migration.20,21 Figure 1 depicts the signaling pathways involved in the mechanism of action of ibrutinib in CLL. Among them, BCR signaling is the most prominent mechanism responsible for activating CLL cells isolated from the proliferation center of LNs.21 Previous studies elucidated ligand-independent (“tonic”) and ligand-dependent BCR signaling in CLL.22,23 The former is mainly produced by the self-identification of the intrinsic epitope of BCR through the heavy-chain complementarity-determining region,23 while the latter is mainly triggered by antigens derived from apoptotic cells or specific autoantigens present in the microenvironment.22 BTK signaling is also the initiator of CLL development, and BTK phosphorylation levels are significantly elevated in CLL B cells.24 Furthermore, BTK deficiency reduces the tumor burden in mice with CLL, and the overexpression of BTK is related to the selection of nonstereotypical BCR into malignant clones and increased mortality.25
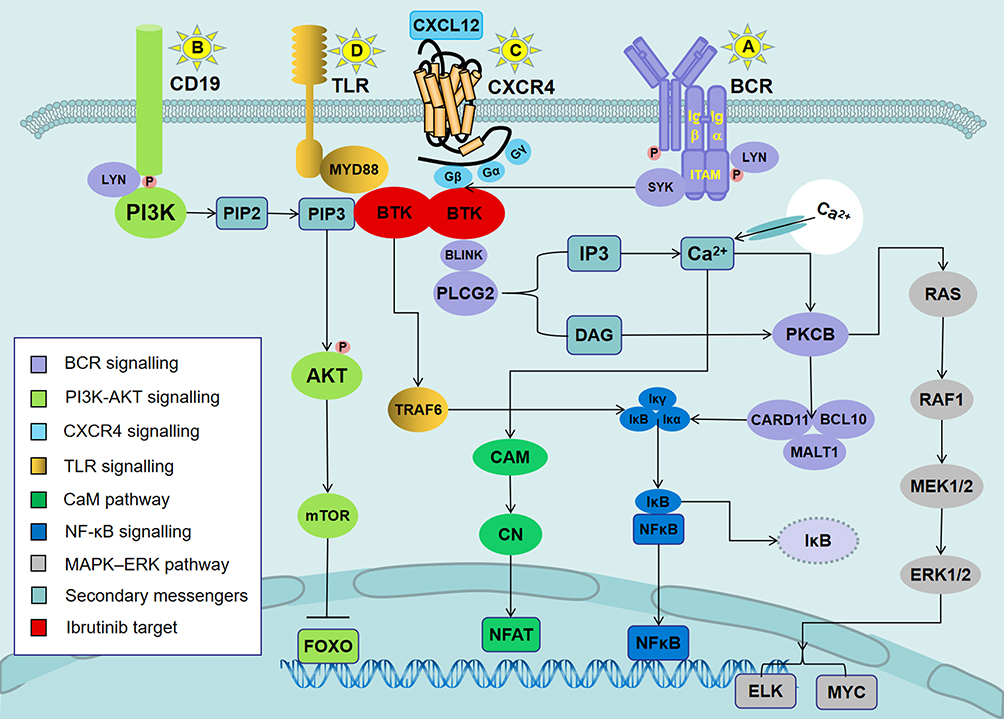 |
Figure 1 Signaling pathways involved in the mechanisms of action of ibrutinib in CLL. Notes: (A) Antigen binding to the BCR triggers the activation of SYK and BTK. BTK is mostly responsible for the activation of PLCG2. PLCG2 is involved in inducing intracellular calcium release and extracellular calcium influx, followed by the activation of ERK1/ERK2 and NF-κB, as well as NFAT. (B) In parallel, LYN phosphorylates the BCR co-receptor CD19, which activates PI3K. Akt is activated via PI3K. PI3K catalyzes membrane-associated PIP2 to generate PIP3. PIP3 attracts the amino-terminal PH lipid-interaction module of BTK, which in turn allows SYK and LYN to completely activate the BTK enzyme. (C) BTK is essential for CXCR4- and CXCR5-mediated signaling pathways. CXCL12 most probably induces BTK activation through the interaction of heterotrimeric G protein subunits with BTK. (D) Ligands binding to TLR induce the MYD88-mediated activation of NF-κB. BTK has been shown to contribute to TLR signaling by interacting with the intercellular domains of most TLRs. Abbreviations: ERK1/2, extracellular signal-regulated kinase 1/2 (also termed MAPK3/MAPK1); NF-κB, nuclear factor-κB; PI3K, phosphatidylinositol 3-kinase; NFAT, nuclear factor of activated T cells; PIP2, phosphatidylinositol 4,5-diphosphate; PIP3, phosphatidylinositol 3,4,5-trisphosphate; CXCR4, CXC-chemokine receptor 4; CXCL12, CXC chemokine ligand 12; TLR, Toll-like receptor; MYD88, myeloid differentiation primary response 88. |
The treatment of CLL cells with BTKi in vitro reduces cell proliferation and survival.26,27 Also, it eliminates BCR-stimulated Akt and ERK phosphorylation.26 Additionally, the function of C-X-C motif chemokine ligand (CXCL) 12 and CXCL13 responsible for B-cell trafficking and tissue homing is also effectively blocked after the use of BTKi.26 Therefore, BTK signaling plays a supporting role in the migration of CLL cells to LN pseudofollicles. Another interesting aspect is the C-C motif chemokine ligand (CCL) 3 and CCL4, which are linearly associated with the prognosis of CLL;26 their expression levels in the plasma are rapidly reduced in patients with BTKi-treated CLL. This reduction is consistent with the level of inhibition of nurse-like cell induction of CCL3 and CCL4 secreted by CLL cells in LN.28 Therefore, the inhibition of BTK may destroy co-stimulatory feedback in the LN microenvironment. The activity of CLL cells co-cultured with interleukin (IL)-4, CD40L, IL-6, tumor necrosis factor, and B cell–activating factor significantly decreased in vitro with ibrutinib.29 Therefore, the function of prosurvival factors in the TME might be abolished by BTKi.
Additionally, the early stages of CLL may be related to oxidative stress caused by the imbalance between the antioxidant defense mechanisms and the levels of pro-oxidants.30 Infectious factors such as bacteria and viruses trigger inflammatory responses by attracting neutrophils and release reactive oxygen species (ROS) in an activated state. ROS can produce pro-inflammatory cytokines to initiate B-cell activation and cause DNA oxidative damage and clonal expansion of tumors, thereby transforming normal B cells into a malignant phenotype.31 Subsequently, in the late stages of the disease, aggressive chemotherapy can exacerbate leukemia cells to produce ROS more actively compared with normal cells, thus forming a vicious cycle.30 Oxidative stress induces an increase in the production of immunoglobulin kappa light chains in B cells at different stages in patients with CLL, leading to antibody deficiency and hypogammaglobulinemia in them.30 Interestingly, Sun et al found that treatment with ibrutinib allowed a partial reconstruction of normal B cells in patients with CLL and improved total IgA and IgM levels, and that increased IgA levels were associated with a decreased risk of infection.32 Therefore, the unique effect of ibrutinib on humoral immunity may be synergistic with exogenous antioxidants and a healthy lifestyle to reduce the incidence of complications such as infection, and it may become the preferred treatment method for patients with hypogammaglobulinemia.
Clinical Applications of Ibrutinib in CLL
Previously Untreated CLL
Monotherapy
The RESONATE-2 study compared the efficacy of ibrutinib with that of chlorambucil in 269 treatment-naïve patients, aged 65 years or older, with CLL/SLL (Table 1).14 After a median follow-up (MF) of 18.4 months, ibrutinib improved the overall response rate (ORR), regardless of the inclusion of a partially responding patient with lymphocytosis. Patients treated with ibrutinib also had significantly longer progression-free survival (PFS) compared with chlorambucil (median, not reached vs 18.9 months) and an 84% reduction in the risk of disease progression (DP) or death.14 Furthermore, the subgroup analysis showed that the superiority of ibrutinib in PFS was independent of factors such as age, bulky disease, Rai stage, ECOG score, IGHV mutation status, or del(11q).14
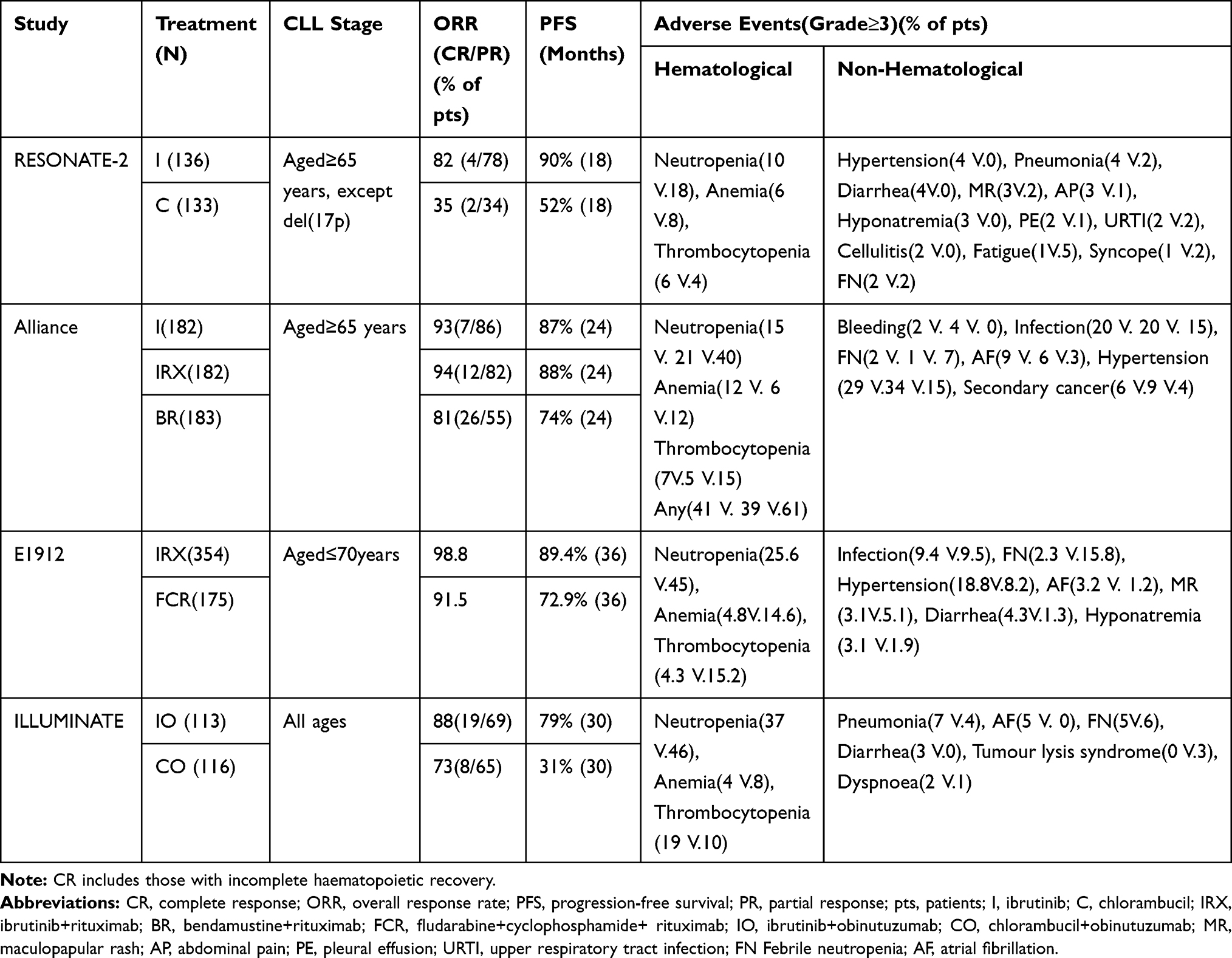 |
Table 1 Ibrutinib in Previously Untreated CLL Patients |
In addition, ibrutinib improved patient hematologic parameters (hemoglobin, absolute neutrophil count, and platelet count) for more than 56 days. Therefore, ibrutinib was more conducive to the recovery of bone marrow hematopoietic function.14
With a median follow-up of 60 months, the PFS benefits obtained by ibrutinib versus chlorambucil continued to exist (70% vs 12%).33 Patients with a high prognostic risk, regardless of unmutated IGHV, del(11q), and TP53 mutations, also benefitted from ibrutinib. A study on the ibrutinib group demonstrated a total response rate of 92% with a complete response (CR) rate of 30% (11% in the primary analysis). The incidence of adverse events (AEs) of grade 3 or higher was lower in patients treated with ibrutinib than in those treated with chlorambucil (58% vs 83%), and the most common AE was diarrhea (50%).33
Combination Therapy
The ALLIANCE study involving 547 treatment-naïve patients with CLL, aged 65 years, showed that the 2-year PFS in the ibrutinib monotherapy group or the ibrutinib plus rituximab (IRX) group was superior to that in the BR group (Table 1).15 However, no significant difference was found between the ibrutinib monotherapy group and the ibrutinib plus rituximab (IRX) group. The incidence of grade 3, 4, or 5 nonhematologic AEs in patients treated with both regimens containing ibrutinib (74% for each group) was higher than that in the BR group (63%). The rate of hematologic AEs was significantly lower in the ibrutinib or IRX group than in the BR group (41%, 39%, and 61%, respectively).15 All patients in the three treatment groups had infections. Furthermore, the benefits of ibrutinib in terms of PFS were not affected by a complex karyotype or IGVH mutation status.15
In the E1912 study, 529 patients with CLL (initial treatment at the age of ≤70 years and without the chromosome 17p13 deletion) were randomly assigned to IRX and FCR regimens in a 2:1 ratio (Table 1).8 After 3 years, the PFS and OS favored the IRX group over the FCR group. Remarkably, in the subgroup of patients with unmutated IGHV, the PFS in the IRX group was significantly higher than that in the FCR group (90.7% and 62.5%, respectively).8 The incidence of AEs of grade 3 or higher was similar in both groups (regardless of attribution), except for the lower incidence of complications in the IRX group.8
The iLLUMINATE study on patients with previously untreated CLL/SLL, aged either 65 years or older or less than 65 years with co-morbidities, compared ibrutinib plus obinutuzumab (IO) group with chlorambucil plus obinutuzumab (CO) group (Table 1).16 The PFS benefit in the IO group was exceptionally prominent in high-risk patients (mainly with TP53 mutation, unmutated IGHV, del 11q, or del 17p); the 30-month PFS was not significantly different from that in the overall population. Furthermore, the minimal residual disease (MRD)-negative rate in bone marrow (BM) or peripheral blood (PB) after treatment was lower in the CO group (25%) than in the IO group (35%); the corresponding CR rate in the two groups was 16% and 41%, respectively.16 This finding suggested that the depth and frequency of responses were more pronounced in the IO group than in the CO group. In terms of AEs, the incidence of AEs was higher in the IO group (58%) than in the CO group (35%). Among them, the grade 3 or 4 AEs most commonly seen in the IO group were thrombocytopenia, neutropenia, pneumonia, and atrial fibrillation (AF). Overall, the safety of the IO regimen was within the controllable range, and most patients could tolerate up to 3 years of treatment.16
Relapsed/Refractory CLL
Monotherapy
The RESONATE study compared the efficacy of ibrutinib with that of the anti-CD20 antibody ofatumumab in previously treated patients (Table 2).17 During an MF of 9.4 months, ibrutinib was found to exceed ofatumumab in terms of PFS and reduced the risk of DP or death by 78%. In addition, patients who progressed with ofatumumab treatment and crossed over to ibrutinib treatment achieved 12 months of OS benefit; the ORR was notably better in the ibrutinib group than in the ofatumumab group.17 The toxic effects associated with ibrutinib did not result in treatment interruption or frequent dose reduction, but the median duration of AEs was longer in the ibrutinib group than in the ofatumumab group by more than 3 months (8.6 vs 5.3). The subgroup analysis results showed that ibrutinib exhibited significant PFS superior to that for ofatumumab regardless of baseline characteristics. Compared with ofatumumab, ibrutinib decreased the risk of death or DP by 81% and 75%, irrespective of del(17p). With an MF of 65.3 months, ibrutinib prolonged the median PFS in patients with high-risk genomic features (44.1 vs 8.1 months).34 The ORR in the ibrutinib group reached 91%, and 11% of patients achieved CR (with or without normal blood count recovery). For patients in the ofatumumab treatment group who crossed over to ibrutinib, the overall survival (OS) was extended by 71 months (median 41 months).14,34
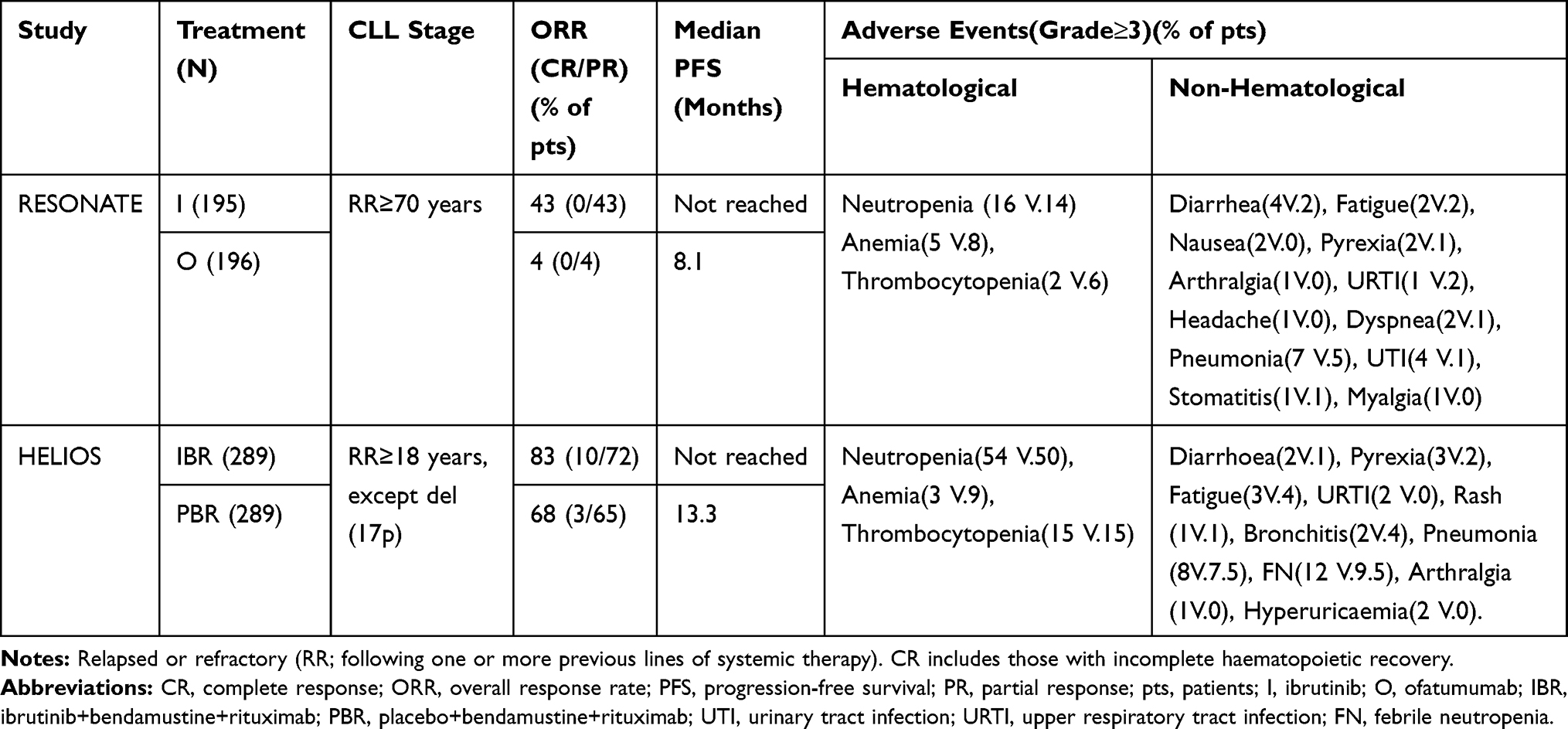 |
Table 2 Ibrutinib in Previously Treated CLL Patients |
Combination Therapy
The HELIOS was a placebo-controlled trial assessing the efficacy of ibrutinib in combination with BR in adult patients with previously treated RR-CLL/SLL (aged ≥18 years) (Table 2).18 Among them, patients with del(17p) were eliminated because they were recognized to have poor outcomes for BR. In an MF of 17 months, the proportion of patients who achieved 18-month PFS was significantly lower in the placebo group than in the ibrutinib group (24% vs 79%). The addition of ibrutinib reduced the risk of DP or death in patients by 80%. The subgroup analysis showed that patients could benefit from the ibrutinib regimen irrespective of their baseline characteristics. The CR rate in patients receiving ibrutinib was remarkably higher than that in the placebo group (21% and 6%, respectively).18 With an MF of 34.8 months, the median PFS was not reached in the ibrutinib group and was 14.3 months in the placebo group. The 36-month PFS in the two groups was 68.0% and 13.9%, respectively.35 A median OS was not achieved in any of the treatment groups, and no significant difference in OS was found between the two regimens. This phenomenon was potentially attributed to 31% of patients in the placebo group who switched to ibrutinib at the time of DP. The negative response rate for MRD (a potent predictor of PFS) was 26.3% in the ibrutinib group compared with 6.2% in the placebo group.35 The addition of ibrutinib provided substantial benefits to patients with RR-CLL.
Adverse Events
At present, low-dose long-term maintenance of ibrutinib is considered to provide clinical benefit to patients.36 However, three follow-up studies over 5 years reported that more than 40% of patients with CLL discontinued treatment with ibrutinib.33,37,38 Correspondingly, a retrospective study of the “real world” also showed that 41% of patients discontinued treatment with ibrutinib. Up to 51% of these patients discontinued treatment due to the occurrence of AEs.39 In summary, common AEs for ibrutinib treatment were nonhematological (diarrhea, bleeding, upper respiratory infections, fatigue, and musculoskeletal pain) and hematological (anemia, thrombocytopenia, neutropenia, and bleeding).40 Depending on the tolerability profile across indications, ibrutinib-related severe adverse events, including AF, bleeding, infection, hypertension, cytopenia, interstitial lung disease (ILD), and second primary malignancies, in particular AF and bleeding probably associated with AEs of grade 3 or higher, required strict monitoring and timely prevention.19
Real-world results show that the incidence of AF in patients with RR-CLL treated with ibrutinib is as high as 8%, and 25% of treatment-naïve and 12% of previously treated patients had to discontinue treatment with ibrutinib due to AF.39 The median duration of discontinuation of ibrutinib due to AF was 7 months.39 Increased diameter and area of the left atrium detected by echocardiography and pre-existing comorbidities indicated a tendency of patients treated with ibrutinib to develop AF.41 The proposed mechanism was that the downregulation of the PI3K–Akt signaling pathway in the heart might lead to increased susceptibility of patients to AF, which might be related to the molecular mechanism of ibrutinib-induced AF.42
The findings on ibrutinib recipients in clinical trials and clinical practice indicate that around 66% of patients have minor bleeding risks, such as epistaxis, petechiae, bruising, and contusions, Up to 6% of patients have major hemorrhage risks, such as gastrointestinal bleeding and intracranial hematoma.17,43-45 In real-world analysis, the incidence of minor bleeding in 70 patients was 56%, while the risk of major bleeding was as high as 19%, which was significantly higher than the clinical trial rate.46 The aforementioned phenomenon may be attributed to the combination of ibrutinib and anticoagulants/antithrombotics in the real world; patients who need other vitamin K antagonists or warfarin in clinical trials are excluded. The former may increase the risk of major bleeding and require rigorous monitoring and particular care.47 In conclusion, for adverse reactions that require special treatment, the benefits and risks of switching to an alternate CLL therapy or continuing with ibrutinib should be carefully weighed, and the best decision should be made for each patient.
Molecular Mechanisms Underlying IR
Previous studies reported that about 19% of patients with CLL experienced disease progression or relapse within 4 years of treatment with ibrutinib. This often caused accelerated disease, especially when ibrutinib was discontinued.48–50 Jain et al found that patients with CLL had poor prognoses after treatment failure with ibrutinib, with a median OS of 3.1 months.48 Patients with CLL and early disease progression during treatment with ibrutinib were usually associated with Richter’s transformation (RT), a highly invasive and destructive lymphoma most frequently presenting as diffuse large B-cell lymphoma (DLBCL).51 Maddocks et al found that 18 of the 232 patients with CLL treated with ibrutinib discontinued treatment due to RT.49 RT occurs earlier than the progressive disease; the estimated prevalence after 12 months of treatment was approximately 4.5%, and patients with RT who continued to receive other treatments had a poor prognosis, with an MS of only 3.5 months.49 Therefore, understanding the evolution of resistance mechanisms may help to seamlessly interface subsequent treatments and prevent the rapid progression of the disease. The mechanism of IR in patients with CLL mainly includes about 13–30% of primary resistance (lack of initial response to ibrutinib) and about 8–13% of acquired resistance (generated after the initial response).14,18,52 However, the sequencing results of the study by Maddocks et al showed that only two patients with RT had BTK mutations in the PB, but no mutations in the LN, suggesting that the mechanism of IR in this patient population was significantly different from that in patients with CLL.49 Furthermore, the clonal evolution studies by Kadri et al on six patients with RT found that the transformed lymphoma cells in tissues were derived from the cloned progeny of circulating leukemia cells undergoing evolution and drift; it was possible to acquire BTK mutations different from those in CLL leukemia cells.53
Molecular Mechanisms Underlying Acquired IR in CLL
BTK and PLCG2 Mutations
Woyach et al reported that approximately 85% of patients who relapsed after treatment with ibrutinib acquired mutations in BTK or PLCG2, which were detectable approximately 9.3 months before relapse.54 In the relapsed patient population, these two gene mutations may exist in a separate form or in a synergistic manner in the same individual.55 BTK mutation is the most frequently described mutation that confers resistance to ibrutinib.54,56,57 In terms of structure, Hamasy et al described the other three scenarios of cysteine substitution: (1) serine replacement has catalytic activity and becomes dominant in drug-resistant patients; (2) threonine replacement also has catalytic activity, but its substitution requires two nucleotides and is therefore relatively rare; and (3) BTK mutants substituted with phenylalanine, arginine, tryptophan, or tyrosine can abolish the catalytic properties of BTK.58 The other two new mutations include V537I located in the BTK gene kinase domain and T316A in the SH2 domain of BTK.53,59 Unlike other mutant genes (such as EGFR, BCR-ABL, and ALK), the BTK mutation is not a secondary mutation of the mutated gene, but a primary mutation present in the gene that is not repeatedly mutated.57
Functionally speaking, the consequence of the BTKC481S mutation is a gradual evolutionary process primarily manifested by affecting cellular signaling, gene expression, and cellular behavior.60 The BTKC481S mutation reduced the potency of ibrutinib by 500-fold and attenuated its irreversible binding. Eventually, the ability of ibrutinib to inhibit the phosphorylation of downstream PLCG2, Akt, and ERK signaling molecules diminished (Figure 2).57 At the genetic level, patients with CLL having BTK mutations were often associated with deletions of preexisting mutations, such as TP53 mutation, BIRC3 mutation, del(17p), and trisomy 12. This finding suggested that these mutant clones could be eliminated by BTKi therapy, and their inactivation might provide a suitable environment for the production or progression of BTK mutant clones.53,59 At the cellular level, asymptomatic resistant mutant clones were already present in the cell before the disease recurred. The inhibitory effect of ibrutinib provided strong selection pressure for these resistant clones, ultimately allowing the mutant cells to break through the pressure to obtain sustained cumulative amplification and lead to disease recurrence.60,61 The diversity of the allele frequencies of the mutant cells was often associated with the progression of LN or PB recurrence, with low allele frequencies predicting LN progression without corresponding PB progression. This finding suggested that most of the mutant cells were present in LN before recurrence.54 In the presence of PB disease, the allele frequency tended to be higher, but if the results were reversed, it suggested that a synergistic effect of the two mutations might exist. Therefore, the presence of these aberrations in the PB might serve as an early molecular marker for future clinical recurrence.54
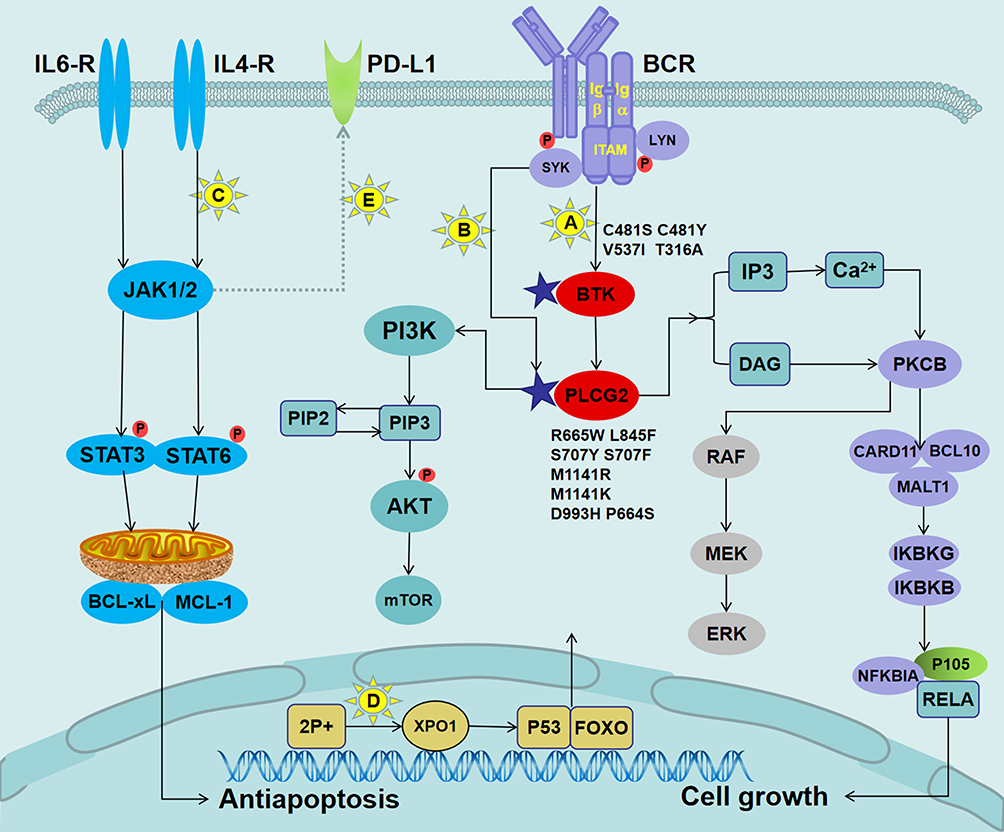 |
Figure 2 Mechanisms of ibrutinib resistance in CLL and mutations in pathways governing BCR signaling. Notes: (A) The BTK mutation attenuated its binding to ibrutinib, resulting in a reduced ability to inhibit the phosphorylation of downstream molecules, thereby allowing the BCR signal to continue to be passed down. (B) LYN and SYK kinase bypassed BTK to directly activate the mutant PLCG2, which caused increased Ca2+ influx to activate different oncogenic pathways, including PIK3–Akt, NF-κB survival signaling, and the MAP kinase pathway. (C) The binding of IL-4 and IL-6 released from the microenvironment to their corresponding receptors activated JAK kinase, followed by the phosphorylation of the STAT6 or STAT3. The activated STAT upregulated the expression levels of anti-apoptotic proteins MCL-1 and BCL-xL. (D) The acquired short arm of chromosome 2 (2p+) induced the overexpression of XPO1. XPO1 promoted the export of tumor suppressor proteins (such as p53 and FOXO) to the cytoplasm, thereby relieving their inhibitory effect on the cell cycle. (E) Activation of the JAK/STAT signaling pathways might lead to overexpression of PD-L1/PD-L2 in patients with RT.Abbreviations: IL-4, interleukin 4; JAK, Janus kinase; STAT6, signal transducer and activator of transcription factor 6; MCL-1, myeloid cell leukemia-1; BCL-xL, B-cell lymphoma-xL; XPO1, exportin-1. |
Some patients with resistance to ibrutinib were reported to possess the PLCG2 mutation but no coexisting BTK mutation, suggesting that the PLCG2 mutation could stand alone in driving IR.57 The PLCG2 gene mainly included the following potential function-acquired mutations: mutation of serine to tyrosine at position 707 (S707Y), leucine to phenylalanine at position 845 (L845F), arginine to tryptophan at position 665 (R665W), and five other newly discovered mutations, including P664S, S707F, M1141R, M1141K, and D993H.55,57 In addition, Landau et al found that repetitive small deletions in the C2 terminal domain of the PLCG2 gene might also be associated with resistance.55 This finding might provide a reference for clinically unexplained drug resistance. Unlike the BTK mutation, the mutation of PLCG2 was located downstream from BTK and continued to downregulate the survival-promoting signal independently of BTK activity (Figure 2).57 After stimulation of the BCR antigen on the surface of PLCG2R665W or PLCG2L845F mutant cells with anti-IgM antibody, the increased calcium ion flux of the cells was still observed, and the phosphorylation levels of the downstream signaling molecules ERK and Akt tended to increase.57 Among them, the PLCG2S707Y mutation was shown to disrupt the self-inhibiting SH2 domain of PLCG2 in vitro.62 Moreover, Liu et al found that the R665W mutant was functionally dependent on LYN and SYK, which collaborated with PLCG2 to comprise a signal bypass independent of BTK.63 As mentioned earlier, the PLCG2 mutation at the genetic level had a similar evolutionary history as the BTK mutation.55 In general, repeated mutations in PLCG2 and BTK were at least the primary cause of IR in some patients with CLL. Interestingly, compared with patients with CLL progression, patients with RT had a higher frequency of SF3B1 mutations with the disappearance of BTK mutations, suggesting that BTK mutations might not be a factor in the development of RT.59 However, patients with RT might carry driver mutations independent of CLL leukemia cells, such as TP53 mutation expansion, MYC abnormality, 8q gain, and loss of trisomy 12.53
Deletion 8p and ITPKB Mutations
Burger et al found signs of clonal expansion of del(8p) clones in three patients with CLL.64 The del(8p) clones also showed a slow decline after initial treatment with ibrutinib, but as time progressed, this mutation synergized with other driver mutations (eg, MLL2, SF3B1, and EP300) to confer tumor proliferation advantage and provide a potential avenue to bypass BTK signaling, ultimately leading to IR.55,64 The deletion of chromosome 8p was consistent with the levels of downregulation of the TRAIL-R1 and TRAIL-R2 genes. Binding of TRAIL to TRAIL-R1 or TRAIL-R2 induced apoptosis, and the level of apoptosis depended on the dose of the receptor.65 Del(8p) resulted in haploid insufficiency of the TRAIL-R, and patients with del(8p) had a significant reduction in TRAIL-R mRNA and protein expression levels.64 The concentration of TRAIL in circulating blood was significantly higher than that in the LN microenvironment.64 However, del(8p) CLL cells released from LN were insensitive to high levels of TRAIL, leading to unbridled growth of cloned cancer cells. Hence, the del(8p) clone might be an important factor driving IR.
Landau et al’s study on the evolutionary trend of patients with CLL found that a patient with ITPKB mutation-driven CLL experienced RT after 9 months of treatment with ibrutinib and progressed at a rate of up to 5.89% per day. As an inhibitor of BCR survival signal feedback to the central nervous system, ITPKB mutation might enhance the transmission of BCR signaling downstream of BTK.55,66 Furthermore, the expression of the ITPKB mutation in patients with DLBCL was also significantly elevated.67
Gain of Chromosome 2p and Overexpression of Exportin-1
Cosson et al detected an acquired short arm of chromosome 2 (2p+) in roughly 16% of patients with CLL.68 This chromosomal abnormality was closely related to the poor prognostic factors in patients with CLL, including unmutated IGHV and del(11q).68 In addition, they also confirmed that 2p+ could induce the overexpression of exportin-1 (XPO1). The upregulation of XPO1 protein led to abnormal cytoplasmic localization and degradation of tumor suppressor factors such as FOXO and p53, thereby promoting the constitutive activation of tumor proliferation signaling pathways.68,69 An increased number of clones carrying XPO1 were detected in eight patients with recurrent 2p+ CLL. Moreover, XPO1 was confirmed to play a central role in driving IR in patients with 2p+ CLL through the aforementioned effects.68
Molecular Mechanisms Underlying Primary IR in CLL
IL-4-Mediated Signaling
Ibrutinib reduced the plasma levels of various cytokines except for IL-4 and IL-6. The expression levels of IL-4 and IL-6 receptors on CLL cells were still higher than those on naïve B cells.70 Guo et al found that IL-4 was a microenvironmental factor enriched in LN pseudofollicles.71 When tumor cells that migrated to PB returned to the proliferation center, the resident IL-4 could rescue CD79b protein to increase the expression level of surface IgM and counterbalance the internalization of BCR complexes after antigen binding, thereby enhancing BCR-mediated signaling.71 Moreover, after IL-4 treatment, the ability of ibrutinib or idelalisib to inhibit BCR survival signaling was remarkably attenuated.71
In addition, the binding of IL-4R and IL-6R on the surface of CLL cells to IL-4 and IL-6 produced by T cells in the microenvironment activated the downstream Janus protein tyrosine kinases JAK1 and JAK3, which in turn promoted the phosphorylation of signal transducer and activator of transcription factor 6 (STAT) 6 and STAT3.72–74 The activation of this signal increased the expression levels of the anti-apoptotic proteins myeloid cell leukemia-1 (MCL-1) and B-cell lymphoma-xL (BCL-xL) to promote sustained survival of the ibrutinib-insensitive CLL cell subset in the microenvironment.72–74 Previous studies also found that IL-4 and IL-6 survival signals transmitted by JAK1 and JAK3 could attenuate the activity of ibrutinib in vitro, while the JAK1/3 inhibitors tofacitinib and ruxolitinib could restore the sensitivity of resistant CLL cells to apoptotic signals.72,75 Thus, the alternative pathway via the IL4/IL4R and IL6/IL6R signaling modules conferred resistance to ibrutinib in CLL cells (Figure 2).
The use of ibrutinib in patients with CLL provided encouraging data. However, recurrent patients often developed progressive disease and had a poor prognosis, with short survival of only a few months. Several new drugs to overcome resistance to ibrutinib are currently under study (Figure 3).
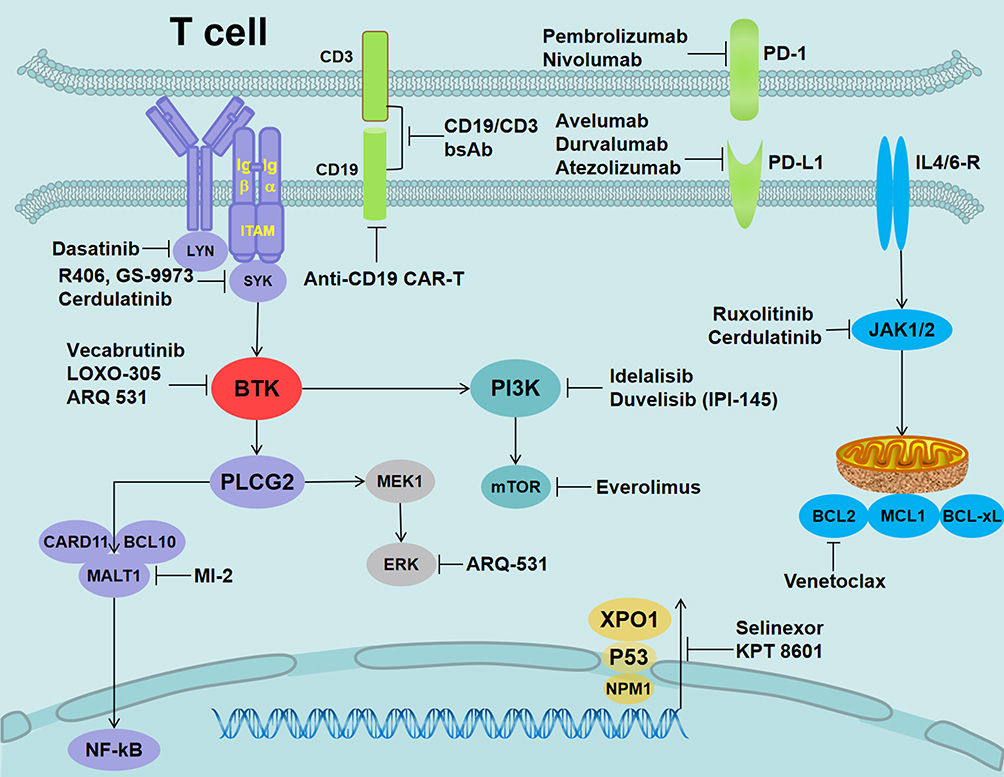 |
Figure 3 Alternative target inhibitors predicted to overcome ibrutinib resistance. Notes: LYN inhibition (dasatinib), SYK inhibition (R406, GS-9973, and cerdulatinib), reversible BTKi (vecabrutinib and LOXO-305), nonselective reversible BTKi (ARQ-531), Bcl-2 inhibition (venetoclax), JAK1/2 (ruxolitinib and cerdulatinib), mTOR inhibition (everolimus), MALT1 inhibition (MI-2), XPO1 inhibition (selinexor and KPT 8601), PD-1 inhibition for RT (nivolumab and pembrolizumab), PD-L1 inhibition (avelumab, durvalumab, and atezolizumab), PI3K inhibition (duvelisib/IPI-145 and idelalisib), and CD19 (anti-CD19 CAR-T and CD19/CD3-scFv-Fc bsAb). |
Reversible BTK Inhibitors
Reversible BTK inhibitors, such as vecabrutinib, LOXO-305, GDC-0853, and ARQ-531, can effectively block the downstream signaling of BCR, thereby conferring cytotoxicity and inhibiting cell proliferation.76–78 A preclinical study by Reiff et al showed that ARQ-531 inhibited the activation of BTK and SRC family members LYN, Akt, and ERK without interaction with C481.78 ARQ-531 was significantly cytotoxic in the acquired IR model rich in recurrent BTKC481S mutations and inhibited the transmission of downstream prosurvival signals in a model expressing activation of the PLCG2 mutation. The inhibitory effect of ARQ-531 on the MEK1 signal (key survival signal for DLBCL) and its distal target also resulted in superior efficacy in the RT mouse model.78 Another study found that vecabrutinib potently inhibited BTK and ITK and also the transmission of BTK signals in vitro in the presence of C481S mutations.76 GDC-0853 is another reversible BTKi with a unique BTK-binding configuration. A previously discontinued Phase I study found that the plasma CCL3 levels were significantly lower in patients with CLL after GDC-0853 treatment, and one of the five patients with RR-CLL carrying the C481S mutation achieved PR.79 These drugs are still in the early stages of clinical research, and their exact efficacy in patients with IR CLL requires further exploration.
BCL2 Inhibitors
Venetoclax acts as a BCL-2 homology 3 (BH3) mimetic, independent of exogenous apoptosis caused by BCR signal blockade and intrinsic apoptotic pathways induced by TP53. It directly antagonizes the function of BCL-2 in mitochondria.80 Jones et al found that among 91 patients with RR-CLL treated with ibrutinib, 30 (33%) discontinued treatment due to AEs and 50 (55%) due to disease progression.81 These patients with historically poor outcomes achieved an OR of 65% after treatment with venetoclax. A total of 12 of the 17 patients who progressed with ibrutinib-resistant mutations responded well to venetoclax. Among them, the allele frequency of the BTKC481S mutation in eight patients had observable decreases for up to 72 weeks.81 This finding suggested that venetoclax opened up new avenues for overcoming IR clones. Recently, Jain et al reported the efficacy of combination therapy with ibrutinib and venetoclax (IV) in 80 treatment-naïve, high-risk, and elderly patients with CLL. The MF was 14.8 months. After 12 cycles of combination therapy, 88% of patients achieved CR and a rate of undetectable MRD of 61%.82 The CLARITY study found that 54 patients with RR-CLL (mainly including chromosomal del(17p) or patients who progressed after conventional CIT treatment) well tolerated IV therapy. After 12 months of treatment, the number of patients with MRD negativity in the bone marrow and PB was 19 (36%) and 28 (53%), respectively. High MRD eradication rates in this combination therapy were expected to permanently eradicate the disease in patients with CLL.83 Therefore, the application prospects of venetoclax monotherapy or combination therapy with ibrutinib in overcoming IR or in patients with RR-CLL are encouraging.
XPO1 Inhibitors
XPO1 is responsible for the nuclear export of proteins and RNA. It is highly expressed in many cancers and is one of the leading contributors to poor prognosis.84 Selinexor (KPT-330) is a first-generation oral XPO1 inhibitor, which specifically and reversibly interacts with XPO1. Previous clinical studies found that selinexor significantly blocked the transport of CLL cells to the protective matrix microenvironment and cell-surface BCR-induced survival, proliferation, and migration.85 Hing et al found that selinexor combined with ibrutinib was significantly superior to ibrutinib monotherapy in the CLL mouse model. In addition, selinexor overcame the resistance of the CLL cell line carrying the BTKC481S mutation to ibrutinib and significantly improved the survival rate of ibrutinib-resistant mice.86 These data suggested that selinexor enabled patients with CLL to show a stronger response and had the potential to become a new treatment modality for IR CLL populations.
Inhibitors of BTK Upstream Signaling Molecules
SYK is a cytoplasmic tyrosine kinase protein upstream of BTK in the BCR signaling pathway. Previous studies explored selective SYK inhibitors, including R406, cerdulatinib, and the SRC and LYN inhibitor dasatinib, to overcome CLL resistance to ibrutinib.87 Liu et al found that GS-9973, R406, and dasatinib were sufficient to prevent the release of hypermorphic calcium from CLL cells in patients with IR expressing PLCG2R665W, and significantly reversed the activation of downstream p-ERK and other signals.63 When BTK function was suppressed, SYK and LYN bypassed BTK and directly initiated the proximal kinase of the PLCG2 mutant to activate the signal. Thus, targeted inhibitors of SYK and LYN played a significant role in repairing the sensitivity of IR cells to apoptosis. Blunt et al showed that IL-4-mediated upregulation of IgM and inhibition of CXCR4 might enhance BCR signaling and reduce the effectiveness of ibrutinib.87 The dual SYK and JAK inhibitor cerdulatinib significantly reduced the expression level of anti-apoptotic proteins in MCL-1 and BCL-XL (except BCL-2). Treatment with cerdulatinib helped overcome the protective effects of nurse-like cells, IL-4/CD40L, and anti-IgM on tumor cells.87,88 Blunt et al also suggested that the combination of cerdulatinib and venetoclax might reverse IL-4-mediated IR.87 Subsequently, Guo et al further demonstrated that cerdulatinib had stronger antitumor activity than ibrutinib, which significantly inhibited the proliferation of BTKC481S-transfected cell lines and primary ibrutinib-resistant CLL cells.74
MALT1 Inhibitors
MALT1 is an enzymatically active member of the CARD11–BCL10–MALT1 signal complex. As mentioned earlier, IR–related mutations could reactivate BCR signals upstream of MALT1. Hence, targeting MALT1 could effectively overcome tumor re-proliferation caused by drug-resistant mutations such as BTK and PLCG2. MI-2, a small-molecule inhibitor of caspase MALT1, could significantly inhibit the proteolytic activity of MALT1 in CLL cells and inhibit NF-κB by reducing the expression levels of anti-apoptotic proteins Bcl-xL, NF-κB regulatory molecules, and nuclear p50 and RelB.89 In addition, MI-2 could inhibit Ras, interferon, and JAK–STAT signaling pathways, and effectively eliminate the survival signals provided by BCR and the cross-linking of lymph node stromal cells.90 Therefore, Nakhle et al found that MI-2 had a significant killing effect on ibrutinib-resistant CLL cells and CLL cells with poor prognosis (such as unmutated IGHV and 17p deletion).91
Combination Therapy
PI3K Inhibitors
Recent studies found that dynamic feedback between mantle cell lymphoma cells and TME promoted the mutual activation of PI3K–Akt–mTOR and integrin b1 signals in vitro. The development of this evolutionary process drove IR.92,93 The results suggested that the combination of the PI3K inhibitor and ibrutinib could simultaneously disrupt BCR signaling and the PI3K-AKT signaling axis to promote tumor cell release in the microenvironment.92 Treatment with the combination therapy reversed the body’s resistance to ibrutinib and enhanced the transmission of antitumor signals.92 Currently, various PI3K inhibitors have been developed, among which idelalisib is a leading player.94 Spaargaren et al studied the effect of idelalisib (alone or in combination with ibrutinib) on the apoptosis of ibrutinib-resistant CLL cells. The addition of idelalisib prevented acquired IR or overcame primary resistance.95 Later, Visentin et al also confirmed an encouraging effect of idelalisib in two patients with IR CLL and patients who progressed to RT after treatment with ibrutinib.96 Although PI3Kδ inhibitors displayed promising results in patients with CLL, few studies were conducted on the efficacy of idelalisib in IR CLL. Hence, large-sample follow-up studies are urgently needed.
JAK1/2 Inhibitors
As described earlier, ibrutinib cannot inhibit JAK-mediated cytokine signaling and allows a subpopulation of CLL cells to continue to grow and proliferate.87 Therefore, the combination of a JAK inhibitor and ibrutinib can restore the sensitivity of CLL cells to apoptosis, thus rendering a promising alternative treatment for patients with IR.97 Spaner et al explored the efficacy of the JAK1/2 inhibitor ruxolitinib plus ibrutinib in 12 patients with CLL who did not achieve remission after treatment with ibrutinib, including splenomegaly or persistent lymphadenopathy after 12 months or abnormally elevated serum β-2 microglobulin (β2-MG) levels following 6 months of ibrutinib therapy.75 Two of the patients achieved PR, and six had diminution of splenomegaly and residual lymphadenopathy; the level of β2-MG decreased during each treatment cycle but was recovered after 2 weeks of interruption of ruxolitinib therapy.75 This finding might be attributed to the addition of ruxolitinib to allow the flushing out of drug-resistant cells from their protective microenvironment for exposure to the cytotoxic effects of ibrutinib, thereby reversing the therapeutic effect.75
Immunotherapy
Functional defects in healthy effector or effector memory T cells caused by direct contact with tumor cells are major factors in the pathogenesis of CLL.98 The gradually exhausted T cells overexpressing programmed cell death protein 1 (PD-1) induce defective actin polymerization and fail to form an immune synapse at the junction site with antigen-presenting cells. However, they bind to PD-L1 overexpressed on the surfaces of leukemia B cells to promote unlimited tumoral proliferation and escape from T-cell immune surveillance.99,100
CD19-Related Therapy
Ibrutinib not only repairs endogenous T-cell compartments but also decreases the number of immunosuppressive regulatory (Treg) T cells in CLL and increases antitumor T-cell immunity.101 Thus, ibrutinib can enhance adoptively transferred chimeric antigen receptor modified–T cell (CAR-T)-mediated antitumor immunity and improve its therapeutic efficacy in patients resistant to ibrutinib.99 Turtle et al observed that anti-CD19 CAR-T cell therapy was safe and feasible in 24 patients with CLL who experienced treatment failure with ibrutinib.102 After 4 weeks of CAR-T infusion, 17 patients achieved remission (CR/PR of 71%), and residual tumors in the bone marrow were not detected in approximately half of the patients evaluated by IGH sequencing. Out of the other six patients who had sustained or relapsed disease after an initial CAR-T cell infusion, two achieved CR after a second CAR-T cell infusion.102 Recently, the CD19/CD3 single-chain Fv-Fc bispecific antibody (bsAb) developed by Robinson et al was shown to efficiently recruit autologous T cells and rapidly kill CLL cells in vitro and in vivo. Encouragingly, this bsAb-mediated cytotoxicity was improved after treatment with ibrutinib, which had significant antitumor activity in several patients with IR having classical BTK and PLCG2 mutations.103 The results of this study indicated that bsAb could be used in combination with ibrutinib as effective immunotherapy and also overcame the resistance to ibrutinib to become a salvage therapy.
PD-1/PD-L1 Inhibitors
The PD-1/PD-L1 pathway is the primary immune checkpoint of the immune regulatory system, and leukemia B cells evade T-cell immune surveillance through the expression of PD-L1.100 The co-culture of peripheral T cells from patients with CLL with PD-1 inhibitors can repair healthy immune synapses between leukemia B cells and T cells.104 In vivo experiments showed that the use of PD-L1 inhibitors for the treatment of the EμTCL1-CLL mouse model effectively corrected leukemia-induced immune function defects and eliminated the development of mouse CLL.99,105 Based on these preclinical data, Ding et al tested the efficacy and safety of pembrolizumab in 25 patients with CLL (16 with RR-CLL and 9 with developing RT, where 60% of patients previously received ibrutinib).106 An OR was observed in 44% of patients with RT, and confirmed clinical responses were also observed in 66% (four out of six patients) of patients who had previously received ibrutinib, while 0% of patients with RR-CLL had OR. After a MF of 11 months, the median OS in the RT group was 10.7 months, which was not achieved in patients developing RT after receiving previous ibrutinib treatment.106 The results of this study showed that blocking the expression of PD-1/PD-L1 in tumors and the TME showed selective efficacy in patients with CLL and RT progression.106 Thus, the treatment paradigm may be changed in the future for patients with RT.
Acknowledgments
This study was funded by the National Natural Science Foundation international cooperation (81570184), the Science and Technology Project of Nantong City (MS22018008), China Postdoctoral Science Foundation (2019M660127), Jiangsu Province Postdoctoral Science Foundation (2019K062), Jiangsu Province Postdoctoral Foundation (2019Z146). The authors thank Trent Rogers, PhD, from Liwen Bianji, Edanz Editing China, for editing the English text of a draft of this manuscript.
Disclosure
The authors report no conflicts of interest in this study.
References
1. O’Reilly A, Murphy J, Rawe S, Garvey M. chronic lymphocytic leukemia: a review of front-line treatment options, with a focus on elderly CLL patients. Clin Lymphoma Myeloma Leuk. 2018;18(4):249–256. doi:10.1016/j.clml.2018.02.003
2. Goede V, Fischer K, Busch R, et al. Obinutuzumab plus chlorambucil in patients with CLL and coexisting conditions. N Engl J Med. 2014;370(12):1101–1110. doi:10.1056/NEJMoa1313984
3. Wierda WG, Byrd JC, Abramson JS, et al. NCCN guidelines insights: chronic lymphocytic leukemia/small lymphocytic lymphoma, version 2.2019. J Natl Compr Canc Netw. 2019;17(1):12–20. doi:10.6004/jnccn.2019.0002
4. Burger JA, O’Brien S. Evolution of CLL treatment - from chemoimmunotherapy to targeted and individualized therapy. Nat Rev Clin Oncol. 2018;15(8):510–527. doi:10.1038/s41571-018-0037-8
5. Parikh SA. Chronic lymphocytic leukemia treatment algorithm 2018. Blood Cancer J. 2018;8(10):93. doi:10.1038/s41408-018-0131-2
6. Gocke CD, Gladstone DE. The absolute percent deviation of IGHV mutation rather than a 98% cut-off predicts survival of chronic lymphocytic leukaemia patients treated with fludarabine, cyclophosphamide and rituximab. Br J Haematol. 2018;180(1):7–8. doi:10.1111/bjh.15015
7. Thompson PA, Tam CS, O’Brien SM, et al. Fludarabine, cyclophosphamide, and rituximab treatment achieves long-term disease-free survival in IGHV-mutated chronic lymphocytic leukemia. Blood. 2016;127(3):303–309. doi:10.1182/blood-2015-09-667675
8. Shanafelt TD, Wang XV, Kay NE, et al. Ibrutinib-rituximab or chemoimmunotherapy for chronic lymphocytic leukemia. N Engl J Med. 2019;381(5):432–443. doi:10.1056/NEJMoa1817073
9. Hallek M. Chronic lymphocytic leukemia: 2020 update on diagnosis, risk stratification and treatment. Am J Hematol. 2019;94(11):1266–1287. doi:10.1002/ajh.25595
10. Brieghel C, Kinalis S, Yde CW, et al. Deep targeted sequencing of TP53 in chronic lymphocytic leukemia: clinical impact at diagnosis and at time of treatment. Haematologica. 2019;104(4):789–796. doi:10.3324/haematol.2018.195818
11. Eichhorst B, Fink AM, Bahlo J, et al. First-line chemoimmunotherapy with bendamustine and rituximab versus fludarabine, cyclophosphamide, and rituximab in patients with advanced chronic lymphocytic leukaemia (CLL10): an international, open-label, randomised, Phase 3, non-inferiority trial. Lancet Oncol. 2016;17(7):928–942. doi:10.1016/s1470-2045(16)30051-1
12. Byrd JC, Furman RR, Coutre SE, et al. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. N Engl J Med. 2013;369(1):32–42. doi:10.1056/NEJMoa1215637
13. Patel V, Balakrishnan K, Bibikova E, et al. Comparison of acalabrutinib, a selective bruton tyrosine kinase inhibitor, with ibrutinib in chronic lymphocytic leukemia cells. Clin Cancer Res. 2017;23(14):3734–3743. doi:10.1158/1078-0432.ccr-16-1446
14. Burger JA, Tedeschi A, Barr PM, et al. Ibrutinib as initial therapy for patients with chronic lymphocytic leukemia. N Engl J Med. 2015;373(25):2425–2437. doi:10.1056/NEJMoa1509388
15. Woyach JA, Ruppert AS, Heerema NA, et al. Ibrutinib regimens versus chemoimmunotherapy in older patients with untreated CLL. N Engl J Med. 2018;379(26):2517–2528. doi:10.1056/NEJMoa1812836
16. Moreno C, Greil R, Demirkan F, et al. Ibrutinib plus obinutuzumab versus chlorambucil plus obinutuzumab in first-line treatment of chronic lymphocytic leukaemia (iLLUMINATE): a multicentre, randomised, open-label, phase 3 trial. Lancet Oncol. 2019;20(1):43–56. doi:10.1016/s1470-2045(18)30788-5
17. Byrd JC, Brown JR, O’Brien S, et al. Ibrutinib versus ofatumumab in previously treated chronic lymphoid leukemia. N Engl J Med. 2014;371(3):213–223. doi:10.1056/NEJMoa1400376
18. Chanan-Khan A, Cramer P, Demirkan F, et al. Ibrutinib combined with bendamustine and rituximab compared with placebo, bendamustine, and rituximab for previously treated chronic lymphocytic leukaemia or small lymphocytic lymphoma (HELIOS): a randomised, double-blind, phase 3 study. Lancet Oncol. 2016;17(2):200–211. doi:10.1016/s1470-2045(15)00465-9
19. Gribben JG, Bosch F, Cymbalista F, et al. Optimising outcomes for patients with chronic lymphocytic leukaemia on ibrutinib therapy: European recommendations for clinical practice. Br J Haematol. 2018;180(5):666–679. doi:10.1111/bjh.15080
20. Dürr C, Hanna BS, Schulz A, et al. Tumor necrosis factor receptor signaling is a driver of chronic lymphocytic leukemia that can be therapeutically targeted by the flavonoid wogonin. Haematologica. 2018;103(4):688–697. doi:10.3324/haematol.2017.177808
21. Szymańska A, Bojarska-Junak A, Drobiecki A, et al. TLR2 expression on leukemic B cells from patients with chronic lymphocytic leukemia. Arch Immunol Ther Exp (Warsz). 2019;67(1):55–65. doi:10.1007/s00005-018-0523-9
22. Burger JA, Chiorazzi N. B cell receptor signaling in chronic lymphocytic leukemia. Trends Immunol. 2013;34(12):592–601. doi:10.1016/j.it.2013.07.002
23. Dühren-von Minden M, Übelhart R, Schneider D, et al. Chronic lymphocytic leukaemia is driven by antigen-independent cell-autonomous signalling. Nature. 2012;489(7415):309–312. doi:10.1038/nature11309
24. Ten Hacken E, Burger JA. Microenvironment interactions and B-cell receptor signaling in chronic lymphocytic leukemia: implications for disease pathogenesis and treatment. Biochim Biophys Acta. 2016;1863(3):401–413. doi:10.1016/j.bbamcr.2015.07.009
25. Kil LP, de Bruijn MJ, van Hulst JA, Langerak AW, Yuvaraj S, Hendriks RW. Bruton’s tyrosine kinase mediated signaling enhances leukemogenesis in a mouse model for chronic lymphocytic leukemia. Am J Blood Res. 2013;3(1):71–83.
26. Ponader S, Chen SS, Buggy JJ, et al. The Bruton tyrosine kinase inhibitor PCI-32765 thwarts chronic lymphocytic leukemia cell survival and tissue homing in vitro and in vivo. Blood. 2012;119(5):1182–1189. doi:10.1182/blood-2011-10-386417
27. Herman SE, Gordon AL, Hertlein E, et al. Bruton tyrosine kinase represents a promising therapeutic target for treatment of chronic lymphocytic leukemia and is effectively targeted by PCI-32765. Blood. 2011;117(23):6287–6296. doi:10.1182/blood-2011-01-328484
28. Sivina M, Hartmann E, Kipps TJ, et al. CCL3 (MIP-1alpha) plasma levels and the risk for disease progression in chronic lymphocytic leukemia. Blood. 2011;117(5):1662–1669. doi:10.1182/blood-2010-09-307249
29. Hoogeboom R, Wormhoudt TA, Schipperus MR, et al. A novel chronic lymphocytic leukemia subset expressing mutated IGHV3-7-encoded rheumatoid factor B-cell receptors that are functionally proficient. Leukemia. 2013;27(3):738–740. doi:10.1038/leu.2012.238
30. Gaman AM, Buga AM, Gaman MA, Popa-Wagner A. The role of oxidative stress and the effects of antioxidants on the incidence of infectious complications of chronic lymphocytic leukemia. Oxid Med Cell Longev. 2014;2014:158135. doi:10.1155/2014/158135.
31. Zinzani PL. The many faces of marginal zone lymphoma. Hematology Am Soc Hematol Educ Program. 2012;2012:426–432. doi:10.1182/asheducation-2012.1.426.
32. Sun C, Tian X, Lee YS, et al. Partial reconstitution of humoral immunity and fewer infections in patients with chronic lymphocytic leukemia treated with ibrutinib. Blood. 2015; 126(19):2213–9. DOI:10.1182/blood-2015-04-639203
33. Burger JA, Barr PM, Robak T, et al. Long-term efficacy and safety of first-line ibrutinib treatment for patients with CLL/SLL: 5 years of follow-up from the phase 3 RESONATE-2 study. Leukemia. 2019. doi:10.1038/s41375-019-0602-x
34. Merli M, Passamonti F. A final listening about ibrutinib in relapsed or refractory CLL: conclusive results from RESONATE sound definitely good! Am J Hematol. 2019. doi:10.1002/ajh.25662
35. Fraser G, Cramer P, Demirkan F, et al. Updated results from the phase 3 HELIOS study of ibrutinib, bendamustine, and rituximab in relapsed chronic lymphocytic leukemia/small lymphocytic lymphoma. Leukemia. 2019;33(4):969–980. doi:10.1038/s41375-018-0276-9
36. Tresckow JV, Eichhorst B, Bahlo J, Hallek M. The Treatment of Chronic Lymphatic Leukemia. Dtsch Arztebl Int. 2019;116(4):41–46. doi:10.3238/arztebl.2019.0041
37. Munir T, Brown JR, O’Brien S, et al. Final analysis from RESONATE: up to six years of follow-up on ibrutinib in patients with previously treated chronic lymphocytic leukemia or small lymphocytic lymphoma. Am J Hematol. 2019;94(12):1353–1363. doi:10.1002/ajh.25638
38. O’Brien S, Furman RR, Coutre S, et al. Single-agent ibrutinib in treatment-naïve and relapsed/refractory chronic lymphocytic leukemia: a 5-year experience. Blood. 2018;131(17):1910–1919. doi:10.1182/blood-2017-10-810044
39. Mato AR, Nabhan C, Thompson MC, et al. Toxicities and outcomes of 616 ibrutinib-treated patients in the United States: a real-world analysis. Haematologica. 2018;103(5):874–879. doi:10.3324/haematol.2017.182907
40. Paydas S. Management of adverse effects/toxicity of ibrutinib. Crit Rev Oncol Hematol. 2019;136:56–63. doi:10.1016/j.critrevonc.2019.02.001.
41. Reda G, Fattizzo B, Cassin R, et al. Predictors of atrial fibrillation in ibrutinib-treated CLL patients: a prospective study. J Hematol Oncol. 2018;11(1):79. doi:10.1186/s13045-018-0626-0
42. McMullen JR, Boey EJ, Ooi JY, Seymour JF, Keating MJ, Tam CS. Ibrutinib increases the risk of atrial fibrillation, potentially through inhibition of cardiac PI3K-Akt signaling. Blood. 2014;124(25):3829–3830. doi:10.1182/blood-2014-10-604272
43. Brown JR, Hillmen P, O’Brien S, et al. Extended follow-up and impact of high-risk prognostic factors from the phase 3 RESONATE study in patients with previously treated CLL/SLL. Leukemia. 2018;32(1):83–91. doi:10.1038/leu.2017.175
44. Barrientos JC, O’Brien S, Brown JR, et al. Improvement in parameters of hematologic and immunologic function and patient well-being in the Phase III resonate study of ibrutinib versus ofatumumab in patients with previously treated chronic lymphocytic leukemia/small lymphocytic lymphoma. Clin Lymphoma Myeloma Leuk. 2018;18(12):803–13.e7. doi:10.1016/j.clml.2018.08.007
45. Barr PM, Robak T, Owen C, et al. Sustained efficacy and detailed clinical follow-up of first-line ibrutinib treatment in older patients with chronic lymphocytic leukemia: extended phase 3 results from RESONATE-2. Haematologica. 2018;103(9):1502–1510. doi:10.3324/haematol.2018.192328
46. Mock J, Kunk PR, Palkimas S, et al. Risk of major bleeding with ibrutinib. Clin Lymphoma Myeloma Leuk. 2018;18(11):755–761. doi:10.1016/j.clml.2018.07.287
47. Deeks ED. Ibrutinib: a review in chronic lymphocytic leukaemia. Drugs. 2017;77(2):225–236. doi:10.1007/s40265-017-0695-3
48. Jain P, Keating M, Wierda W, et al. Outcomes of patients with chronic lymphocytic leukemia after discontinuing ibrutinib. Blood. 2015;125(13):2062–2067. doi:10.1182/blood-2014-09-603670
49. Maddocks KJ, Ruppert AS, Lozanski G, et al. Etiology of ibrutinib therapy discontinuation and outcomes in patients with chronic lymphocytic leukemia. JAMA Oncol. 2015;1(1):80–87. doi:10.1001/jamaoncol.2014.218
50. Mato AR, Nabhan C, Barr PM, et al. Outcomes of CLL patients treated with sequential kinase inhibitor therapy: a real world experience. Blood. 2016;128(18):2199–2205. doi:10.1182/blood-2016-05-716977
51. Hleuhel MH, Ben-Dali Y, Da Cunha-bang C, et al. Risk factors associated with Richter’s transformation in patients with chronic lymphocytic leukaemia: protocol for a retrospective population-based cohort study. BMJ Open. 2019;9(3):e023566. doi:10.1136/bmjopen-2018-023566
52. Furman RR, Cheng S, Lu P, et al. Ibrutinib resistance in chronic lymphocytic leukemia. N Engl J Med. 2014; 370(24):2352–4. DOI:10.1056/NEJMc1402716.
53. Kadri S, Lee J, Fitzpatrick C, et al. Clonal evolution underlying leukemia progression and Richter transformation in patients with ibrutinib-relapsed CLL. Blood Adv. 2017;1(12):715–727. doi:10.1182/bloodadvances.2016003632
54. Woyach JA, Ruppert AS, Guinn D, et al. BTK(C481S)-mediated resistance to ibrutinib in chronic lymphocytic leukemia. J Clin Oncol. 2017;35(13):1437–1443. doi:10.1200/jco.2016.70.2282
55. Landau DA, Sun C, Rosebrock D, et al. The evolutionary landscape of chronic lymphocytic leukemia treated with ibrutinib targeted therapy. Nat Commun. 2017;8(1):2185. doi:10.1038/s41467-017-02329-y
56. Quinquenel A, Fornecker LM, Letestu R, et al. Prevalence of BTK and PLCG2 mutations in a real-life CLL cohort still on ibrutinib after 3 years: a FILO group study. Blood. 2019;134(7):641–644. doi:10.1182/blood.2019000854
57. Woyach JA, Furman RR, Liu TM, et al. Resistance mechanisms for the Bruton’s tyrosine kinase inhibitor ibrutinib. N Engl J Med. 2014;370(24):2286–2294. doi:10.1056/NEJMoa1400029
58. Hamasy A, Wang Q, Blomberg KE, et al. Substitution scanning identifies a novel, catalytically active ibrutinib-resistant BTK cysteine 481 to threonine (C481T) variant. Leukemia. 2017;31(1):177–185. doi:10.1038/leu.2016.153
59. Kanagal-Shamanna R, Jain P, Patel KP, et al. Targeted multigene deep sequencing of Bruton tyrosine kinase inhibitor-resistant chronic lymphocytic leukemia with disease progression and Richter transformation. Cancer. 2019;125(4):559–574. doi:10.1002/cncr.31831
60. Cheng S, Guo A, Lu P, Ma J, Coleman M, Wang YL. Functional characterization of BTK(C481S) mutation that confers ibrutinib resistance: exploration of alternative kinase inhibitors. Leukemia. 2015;29(4):895–900. doi:10.1038/leu.2014.263
61. Ahn IE, Underbayev C, Albitar A, et al. Clonal evolution leading to ibrutinib resistance in chronic lymphocytic leukemia. Blood. 2017;129(11):1469–1479. doi:10.1182/blood-2016-06-719294
62. Zhou Q, Lee GS, Brady J, et al. A hypermorphic missense mutation in PLCG2, encoding phospholipase Cgamma2, causes a dominantly inherited autoinflammatory disease with immunodeficiency. Am J Hum Genet. 2012;91(4):713–720. doi:10.1016/j.ajhg.2012.08.006
63. Liu TM, Woyach JA, Zhong Y, et al. Hypermorphic mutation of phospholipase C, gamma2 acquired in ibrutinib-resistant CLL confers BTK independency upon B-cell receptor activation. Blood. 2015;126(1):61–68. doi:10.1182/blood-2015-02-626846
64. Burger JA, Landau DA, Taylor-Weiner A, et al. Clonal evolution in patients with chronic lymphocytic leukaemia developing resistance to BTK inhibition. Nat Commun. 2016;7:11589. doi:10.1038/ncomms11589
65. Rubio-Moscardo F, Blesa D, Mestre C, et al. Characterization of 8p21.3 chromosomal deletions in B-cell lymphoma: TRAIL-R1 and TRAIL-R2 as candidate dosage-dependent tumor suppressor genes. Blood. 2005;106(9):3214–3222. doi:10.1182/blood-2005-05-2013
66. Landau DA, Carter SL, Stojanov P, et al. Evolution and impact of subclonal mutations in chronic lymphocytic leukemia. Cell. 2013;152(4):714–726. doi:10.1016/j.cell.2013.01.019
67. Mareschal S, Dubois S, Viailly PJ, et al. Whole exome sequencing of relapsed/refractory patients expands the repertoire of somatic mutations in diffuse large B-cell lymphoma. Genes Chromosomes Cancer. 2016;55(3):251–267. doi:10.1002/gcc.22328
68. Cosson A, Chapiro E, Bougacha N, et al. Gain in the short arm of chromosome 2 (2p+) induces gene overexpression and drug resistance in chronic lymphocytic leukemia: analysis of the central role of XPO1. Leukemia. 2017;31(7):1625–1629. doi:10.1038/leu.2017.100
69. Senapedis WT, Baloglu E, Landesman Y. Clinical translation of nuclear export inhibitors in cancer. Semin Cancer Biol. 2014;27;74–86. doi:10.1016/j.semcancer.2014.04.005.
70. Niemann CU, Herman SE, Maric I, et al. Disruption of in vivo chronic lymphocytic leukemia tumor-microenvironment interactions by ibrutinib–findings from an investigator-initiated Phase II Study. Clin Cancer Res. 2016;22(7):1572–1582. doi:10.1158/1078-0432.ccr-15-1965
71. Guo B, Zhang L, Chiorazzi N, Rothstein TL. IL-4 rescues surface IgM expression in chronic lymphocytic leukemia. Blood. 2016;128(4):553–562. doi:10.1182/blood-2015-11-682997
72. Lu K, Fang XS, Feng LL, et al. The STAT3 inhibitor WP1066 reverses the resistance of chronic lymphocytic leukemia cells to histone deacetylase inhibitors induced by interleukin-6. Cancer Lett. 2015;359(2):250–258. doi:10.1016/j.canlet.2015.01.021
73. Steele AJ, Prentice AG, Cwynarski K, et al. The JAK3-selective inhibitor PF-956980 reverses the resistance to cytotoxic agents induced by interleukin-4 treatment of chronic lymphocytic leukemia cells: potential for reversal of cytoprotection by the microenvironment. Blood. 2010;116(22):4569–4577. doi:10.1182/blood-2009-09-245811
74. Guo A, Lu P, Coffey G, Conley P, Pandey A, Wang YL. Dual SYK/JAK inhibition overcomes ibrutinib resistance in chronic lymphocytic leukemia: cerdulatinib, but not ibrutinib, induces apoptosis of tumor cells protected by the microenvironment. Oncotarget. 2017;8(8):12953–12967. doi:10.18632/oncotarget.14588
75. Spaner DE, Wang G, McCaw L, et al. Activity of the Janus kinase inhibitor ruxolitinib in chronic lymphocytic leukemia: results of a phase II trial. Haematologica. 2016;101(5):e192–5. doi:10.3324/haematol.2015.135418
76. Thompson PA, Burger JA. Bruton’s tyrosine kinase inhibitors: first and second generation agents for patients with Chronic Lymphocytic Leukemia (CLL). Expert Opin Investig Drugs. 2018;27(1):31–42. doi:10.1080/13543784.2018.1404027
77. Bond DA, Woyach JA. Targeting BTK in CLL: beyond ibrutinib. Curr Hematol Malig Rep. 2019;14(3):197–205. doi:10.1007/s11899-019-00512-0
78. Reiff SD, Mantel R, Smith LL, et al. The BTK inhibitor ARQ 531 targets ibrutinib-resistant CLL and richter transformation. Cancer Discov. 2018;8(10):1300–1315. doi:10.1158/2159-8290.cd-17-1409
79. Byrd JC, Smith S, Wagner-Johnston N, et al. Correction: first-in-human Phase 1 study of the BTK inhibitor GDC-0853 in relapsed or refractory B-cell NHL and CLL. Oncotarget. 2019;10(38):3827–3830. doi:10.18632/oncotarget.27011
80. Adams JM, Cory S. The BCL-2 arbiters of apoptosis and their growing role as cancer targets. Cell Death Differ. 2018;25(1):27–36. doi:10.1038/cdd.2017.161
81. Jones JA, Mato AR, Wierda WG, et al. Venetoclax for chronic lymphocytic leukaemia progressing after ibrutinib: an interim analysis of a multicentre, open-label, Phase 2 trial. Lancet Oncol. 2018;19(1):65–75. doi:10.1016/s1470-2045(17)30909-9
82. Jain N, Keating M, Thompson P, et al. Ibrutinib and venetoclax for first-line treatment of CLL. N Engl J Med. 2019;380(22):2095–2103. doi:10.1056/NEJMoa1900574
83. Hillmen P, Rawstron AC, Brock K, et al. Ibrutinib plus venetoclax in relapsed/refractory chronic lymphocytic leukemia: the CLARITY Study. J Clin Oncol. 2019;37(30):2722–2729. doi:10.1200/JCO.19.00894
84. Syed YY. Selinexor: first global approval. Drugs. 2019;79(13):1485–1494. doi:10.1007/s40265-019-01188-9
85. Zhong Y, El-Gamal D, Dubovsky JA, et al. Selinexor suppresses downstream effectors of B-cell activation, proliferation and migration in chronic lymphocytic leukemia cells. Leukemia. 2014;28(5):1158–1163. doi:10.1038/leu.2014.9
86. Hing ZA, Mantel R, Beckwith KA, et al. Selinexor is effective in acquired resistance to ibrutinib and synergizes with ibrutinib in chronic lymphocytic leukemia. Blood. 2015;125(20):3128–3132. doi:10.1182/blood-2015-01-621391
87. Blunt MD, Koehrer S, Dobson RC, et al. The dual Syk/JAK inhibitor cerdulatinib antagonizes B-cell receptor and microenvironmental signaling in chronic lymphocytic leukemia. Clin Cancer Res. 2017;23(9):2313–2324. doi:10.1158/1078-0432.ccr-16-1662
88. Hamlin PA, Flinn IW, Wagner-Johnston N, et al. Efficacy and safety of the dual SYK/JAK inhibitor cerdulatinib in patients with relapsed or refractory B-cell malignancies: results of a phase I study. Am J Hematol. 2019;94(4):E90–e3. doi:10.1002/ajh.25387
89. Wu G, Wang H, Zhou W, et al. Synthesis and structure-activity relationship studies of MI-2 analogues as MALT1 inhibitors. Bioorg Med Chem. 2018;26(12):3321–3344. doi:10.1016/j.bmc.2018.04.059
90. Olias P, Etheridge RD, Zhang Y, Holtzman MJ, Sibley LD. Toxoplasma effector recruits the Mi-2/NuRD complex to repress STAT1 transcription and block IFN-γ-dependent gene expression. Cell Host Microbe. 2016;20(1):72–82. doi:10.1016/j.chom.2016.06.006
91. Saba NS, Wong DH, Tanios G, et al. MALT1 inhibition is efficacious in both naïve and ibrutinib-resistant chronic lymphocytic leukemia. Cancer Res. 2017;77(24):7038–7048. doi:10.1158/0008-5472.CAN-17-2485
92. Zhao X, Lwin T, Silva A, et al. Unification of de novo and acquired ibrutinib resistance in mantle cell lymphoma. Nat Commun. 2017;8:14920. doi:10.1038/ncomms14920
93. Zhou H, Yang L, Dang Q, et al. Ibrutinib resistance in a patient with transformed diffuse large B-cell lymphoma from primary pulmonary mucosa-associated lymphoid tissue lymphoma. Cancer Biol Ther. 2020;21(4):303–308. doi:10.1080/15384047.2019.1700743
94. Kapoor I, Li Y, Sharma A, et al. Resistance to BTK inhibition by ibrutinib can be overcome by preventing FOXO3a nuclear export and PI3K/AKT activation in B-cell lymphoid malignancies. Cell Death Dis. 2019;10(12):924. doi:10.1038/s41419-019-2158-0
95. de Rooij MFM, Kuil A, Kater AP, Kersten MJ, Pals ST, Spaargaren M. Ibrutinib and idelalisib synergistically target BCR-controlled adhesion in MCL and CLL: a rationale for combination therapy. Blood. 2015;125(14):2306–2309. doi:10.1182/blood-2014-12-619163
96. Visentin A, Imbergamo S, Scomazzon E, et al. BCR kinase inhibitors, idelalisib and ibrutinib, are active and effective in Richter syndrome. Br J Haematol. 2019;185(1):193–197. doi:10.1111/bjh.15440
97. Spaner DE, McCaw L, Wang G, Tsui H, Shi Y. Persistent janus kinase-signaling in chronic lymphocytic leukemia patients on ibrutinib: results of a phase I trial. Cancer Med. 2019;8(4):1540–1550. doi:10.1002/cam4.2042
98. Shanafelt TD, Ramsay AG, Zent CS, et al. Long-term repair of T-cell synapse activity in a phase II trial of chemoimmunotherapy followed by lenalidomide consolidation in previously untreated chronic lymphocytic leukemia (CLL). Blood. 2013;121(20):4137–4141. doi:10.1182/blood-2012-12-470005
99. McClanahan F, Hanna B, Miller S, et al. PD-L1 checkpoint blockade prevents immune dysfunction and leukemia development in a mouse model of chronic lymphocytic leukemia. Blood. 2015;126(2):203–211. doi:10.1182/blood-2015-01-622936
100. Miao Y, Medeiros LJ, Li Y, Li J, Young KH. Genetic alterations and their clinical implications in DLBCL. Nat Rev Clin Oncol. 2019;16(10):634–652. doi:10.1038/s41571-019-0225-1
101. Fraietta JA, Beckwith KA, Patel PR, et al. Ibrutinib enhances chimeric antigen receptor T-cell engraftment and efficacy in leukemia. Blood. 2016;127(9):1117–1127. doi:10.1182/blood-2015-11-679134
102. Turtle CJ, Hay KA, Hanafi LA, et al. Durable molecular remissions in chronic lymphocytic leukemia treated with cd19-specific chimeric antigen receptor-modified T cells after failure of ibrutinib. J Clin Oncol. 2017;35(26):3010–3020. doi:10.1200/jco.2017.72.8519
103. Robinson HR, Qi J, Cook EM, et al. A CD19/CD3 bispecific antibody for effective immunotherapy of chronic lymphocytic leukemia in the ibrutinib era. Blood. 2018;132(5):521–532. doi:10.1182/blood-2018-02-830992
104. Ramsay AG, Clear AJ, Fatah R, Gribben JG. Multiple inhibitory ligands induce impaired T-cell immunologic synapse function in chronic lymphocytic leukemia that can be blocked with lenalidomide: establishing a reversible immune evasion mechanism in human cancer. Blood. 2012;120(7):1412–1421. doi:10.1182/blood-2012-02-411678
105. McClanahan F, Riches JC, Miller S, et al. Mechanisms of PD-L1/PD-1-mediated CD8 T-cell dysfunction in the context of aging-related immune defects in the Emicro-TCL1 CLL mouse model. Blood. 2015;126(2):212–221. doi:10.1182/blood-2015-02-626754
106. Ding W, LaPlant BR, Call TG, et al. Pembrolizumab in patients with CLL and Richter transformation or with relapsed CLL. Blood. 2017;129(26):3419–3427. doi:10.1182/blood-2017-02-765685
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.