")
Back to Archived Journals » Hypoxia » Volume 2
Hypoxia and the modulation of the actin cytoskeleton – emerging interrelations
Authors Zieseniss A
Received 17 December 2013
Accepted for publication 29 January 2014
Published 25 March 2014 Volume 2014:2 Pages 11—21
DOI https://doi.org/10.2147/HP.S53575
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Anke Zieseniss
Institute of Cardiovascular Physiology, University Medical Center, Georg-August University, Göttingen, Germany
Abstract: Recent progress in understanding the influence of hypoxia on cell function has revealed new information about the interrelationship between the actin cytoskeleton and hypoxia; nevertheless, details remain cloudy. The dynamic regulation of the actin cytoskeleton during hypoxia is complex, varies in different cells and tissues, and also depends on the mode of hypoxia. Several molecular players and pathways are emerging that contribute to the modulation of the actin cytoskeleton and that affect the large repertoire of actin-binding proteins in hypoxia. This review describes and discusses the accumulated knowledge about actin cytoskeleton dynamics in hypoxia, placing special emphasis on the Rho family of small guanosine triphosphatases (Rho GTPases). Given that RhoA, Rac and Cdc42 are very well characterized, the review is focused on these family members of Rho GTPases. Notably, in several cell types and tissues, hypoxia, presumably via Rho GTPase signaling, induces actin rearrangement and actin stress fiber assembly, which is a prevalent modulation of the actin cytoskeleton in hypoxia.
Keywords: Rho GTPases, actin dynamics, stress fibers, RhoA
Introduction
A myriad of changes are necessary for cells and tissues to adapt to hypoxia. These hypoxic changes are conveyed by altering gene expression patterns, which for the most part are controlled by hypoxia-inducible factor (HIF) transcription factors. Lately, it has become increasingly evident that hypoxia also prompts cytoskeletal alterations in different cell types. The cytoskeleton is indispensable for any eukaryotic cell, as it governs many cellular processes. A main component of the cytoskeleton is the highly conserved protein actin, which contributes to the shape, motility, and polarity of the cells, to cell division, and the maintenance of multicellular tissue organization, including the maintenance of epithelial barriers.1 All of these aspects are involved in morphogenesis, and might go awry in pathological disorders, such as fibrosis and cancer. Also, other hypoxia-related diseases, such as pulmonary hypertension, wound healing, and brain edema, are associated with a dysregulation in the cytoskeleton. Therefore, a better understanding of hypoxia-induced changes in the cytoskeletal regulation might lead to better insight into these pathologies. Force production by actin filaments is the basis for the aforementioned cellular processes, and is achieved by two mechanisms. First, actin polymerization into networks drives protrusive forces, and second, actin and myosin II filaments together form contractile structures in cells.2 In both scenarios, actin polymerization and dynamics are controlled by actin-binding proteins. More than 100 actin-binding proteins are known to date. They act to maintain a pool of monomeric G-actin, promote F-actin polymerization, restrict the length of actin filaments, regulate the assembly and disassembly of filaments, and organize actin filaments into networks and bundles.1
The precise regulation of the actin-binding proteins and consequently actin cytoskeleton dynamics is fundamental for many physiological processes, such as cell locomotion and cell morphology, which are involved in the aforementioned disease pathologies. Since hypoxia influences actin dynamics, it is a straightforward finding that cell shape, adhesion, and movement are greatly altered in hypoxic cells compared to cells grown in normoxia.3,4 However, depending on the origin of the cells and the experimental setup, the literature shows at least in part heterogeneous effects of hypoxia on the actin cytoskeleton and actin-dependent cellular processes. Acute hypoxia, for example, has been reported to enhance the migration of MDA-MB-231 breast cancer cells,5,6 whereas in L929 murine fibrosarcoma cells the migration is impaired under hypoxic conditions.4 Apart from cell type-dependent effects, different modes of migration, eg, different serum conditions and single-cell versus gap-closure experiments, might be one possible explanation for the existence of these conflicting data.
In this review, the findings concerning actin cytoskeleton rearrangements in hypoxia are gathered and the involved signaling pathways are summarized. Examples of pathways and proteins involved in the molecular mechanisms that add to hypoxic actin changes are listed. The comprehensive description of the up to now presented findings should help to understand common and cell type-dependent divergent hypoxia-induced cytoskeletal changes.
The regulation of actin dynamics in hypoxia
Expression and activation of Rho GTPases in hypoxia
The mechanisms by which hypoxia and/or HIF-1α stabilization impact the actin cytoskeleton are undoubtedly complex, and several signaling pathways might be activated by hypoxia in different tissues. Overall evidence continues to mount that a hypoxic cell environment changes the activity of Rho guanosine triphosphatases (Rho GTPases), especially RhoA, and downstream signaling molecules that regulate the actin lattice. The Rho GTPase family of signaling molecules includes over 20 different members, and plays an important role in the regulation of the actin cytoskeleton.7 Rho GTPases exert their influence on the actin cytoskeleton by modulating the activity of actin-binding proteins. Rho GTPases cycle between a guanosine diphosphate-bound inactive form and a GTP-bound active form. The switch between these two states is mediated by three groups of proteins: guanine nucleotide exchange factors (GEFs),8 GTPase-activating proteins (GAPs),9 and guaninge nucleotide-dissociation inhibitors (GDIs).10 Several types of plasma-membrane receptors, such as G-protein-coupled receptors (GPCRs) and receptor tyrosine kinases, influence actin dynamics by modulating Rho GTPase activity. Some GPCRs have been shown to be HIF-1 target genes,11,12 and various GPCR ligands are induced in hypoxia and stimulate GEFs.13 Also, among the receptor tyrosine kinases so far known, many are known to stimulate GEFs and subsequently activate Rho GTPases, and are furthermore involved in hypoxic signaling, including, eg, vascular endothelial growth-factor (VEGF) receptor.14 Historically, RhoA, Rac, and Cdc42 have by far been the best characterized family members of the Rho GTPases.7,15–17 The activation of RhoA leads to the assembly of contractile actin-myosin filaments, and is thus required for generating contractile forces, eg, for cell translocation. Lamellipodium and membrane ruffle formation is mostly dependent on Rac1-dependent actin polymerization, and Cdc42 plays a role in the actin cytoskeleton-dependent regulation of cell polarity and filopodium formation.
RhoA in hypoxia
Cross talk between the hypoxia/HIF-1 and RhoA pathways has been widely discussed in the context of several cell types and tissues, including fibroblasts4 and cancer cells,6,18,19 smooth-muscle cells, endothelial cells,20–26 isolated trophoblast cells,27 and mesenchymal stem cells.28–30 However, the effects of hypoxia on RhoA levels and activation vary greatly between different cell types and culturing conditions, and are summarized in Table 1.
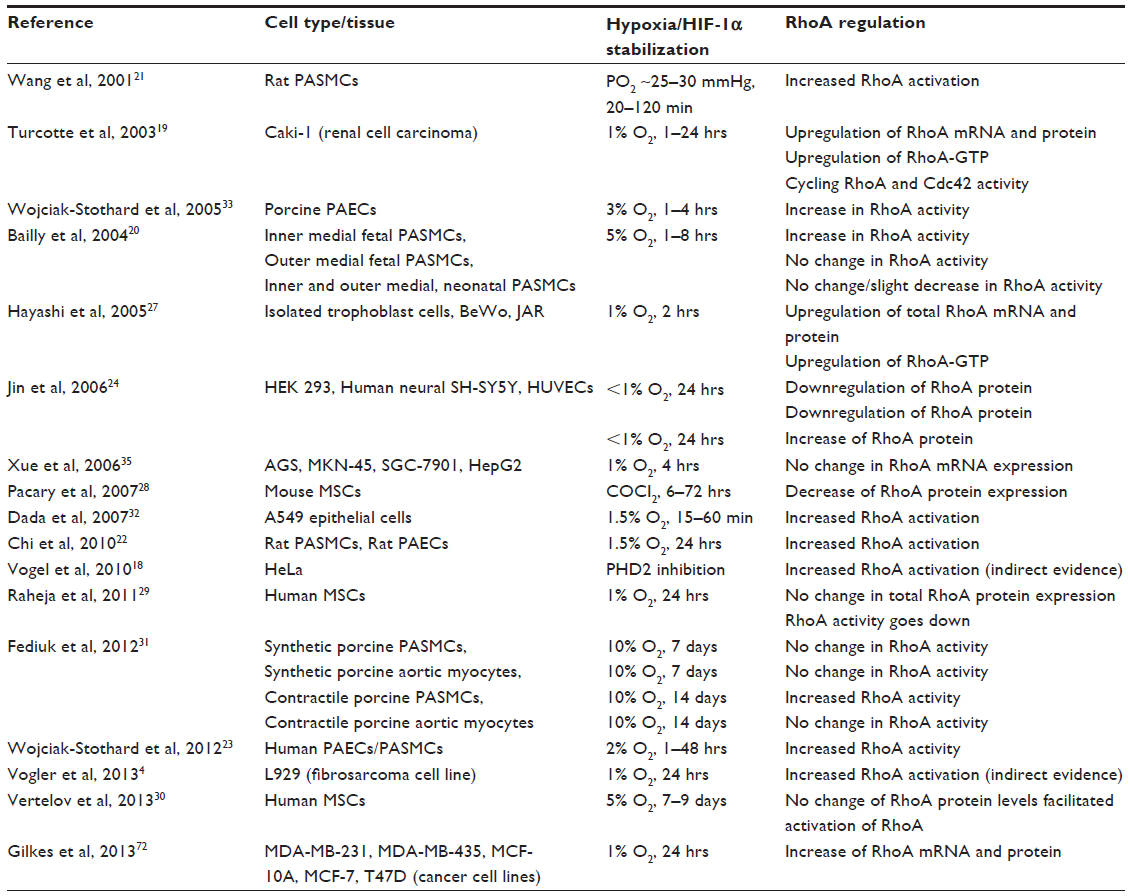 | Table 1 RhoA regulation in different cell types in hypoxia |
There is direct and indirect evidence that chronic and acute hypoxia activate RhoA in pulmonary vascular smooth-muscle25,26 and pulmonary arterial smooth muscle cells (PASMCs).20–24,31 However, both the origin and age of cells seem to influence hypoxic activation: RhoA activity increases in hypoxia in piglet fetal PASMCs, isolated from the inner layer of the tunica media. On the other hand, no hypoxic RhoA activation was seen in smooth-muscle cells isolated from the outer medial layer or in smooth-muscle cells isolated from neonatal piglets.20 Furthermore, Fediuk et al showed that the mechanism that drives actin polymerization in PASMCs also depends on the cell phenotype. In contractile porcine pulmonary artery myocytes, the RhoA pathway is involved in actin remodeling, whereas in synthetic pulmonary artery myocytes, the remodeling of the actin cytoskeleton is not Rho-dependent.31 In many human cell cultures, the activity of RhoA increases in hypoxia. In human endothelial cells, an increase in expression of RhoA can be noted after 4 hours of hypoxic incubation,24 and in human trophoblast cells and renal and epithelial carcinoma cells, activated RhoA was elevated in hypoxia.19,32 Also, very recently it has been shown in human breast cancer cell lines that RhoA is a direct target gene of HIF-1 and HIF-2.6 Nonetheless, it cannot be claimed generally that hypoxia causes an increase in RhoA activity, as a decrease of RhoA expression has been reported in epidermal and neuronal cells.24 Moreover, conflicting data exist concerning the influence of hypoxia on RhoA activity in human mesenchymal stem cells. Vertelov et al reported that human mesenchymal stem cells show enhanced RhoA activation under hypoxic conditions,30 whereas Raheja et al showed that hypoxia decreased RhoA activation.29 It should be noted that the time of hypoxic incubation diverged vastly between the studies (Table 1), and increasing evidence suggests that RhoA activation during hypoxia is rapid and time course-regulated.19,22,23,32,33 Conflicting results, therefore, might very well be due to differences in experimental timing.
Rac1 and Cdc42 in hypoxia
The expression and activity of the two other prominent members of the Rho GTPases – Rac1 and Cdc42 – are hypoxia-inducible in some cancer cell lines,19,34,35 whereas in porcine pulmonary artery endothelial cells, Cdc42 is activated and Rac1 activity is slightly downregulated.33 Similar to RhoA, the activation of Rac1 and Cdc42 is time course-dependently regulated and reaches peak activity within hours after hypoxic induction. Experiments with dominant-negative forms of Rac1 and Cdc42 show a significant decrease of HIF-1α stabilization and HIF-1 target-gene expression.34,35 Therefore, Rac1 and Cdc42 contribute to the regulation of HIF-1. However, it should be mentioned here that prolyl hydroxylase domain enzymes (PHDs) play the central role in the regulation of HIFα stability,36 whereas Rac1 and Cdc42 just modulate the regulation under specific circumstances.
How is Rac1 involved in a signal-transduction pathway leading to the induction of HIF-1 activity? In addition to its role in actin cytoskeleton regulation, specifically lamellipodia formation, Rac1 participates in reactive oxygen species (ROS) formation via nicotinamide adenine dinucleotide phosphate oxidases in several cell types.37,38 ROS impair the enzymatic function of PHDs,39 and subsequently HIF-1α is not hydroxylated and not marked for proteasomal degradation and is stabilized even under normoxic conditions. Therefore, Rac1 might indirectly contribute to the stabilization of HIF-1α (Figure 1). Interestingly, chemical stabilization of HIF-1α by dimethyloxalylglycine (DMOG)-induced inhibition of PHDs results in reduced Rac activity and places Rac downstream of HIF-1α, suggesting the existence of a positive-feedback loop.40
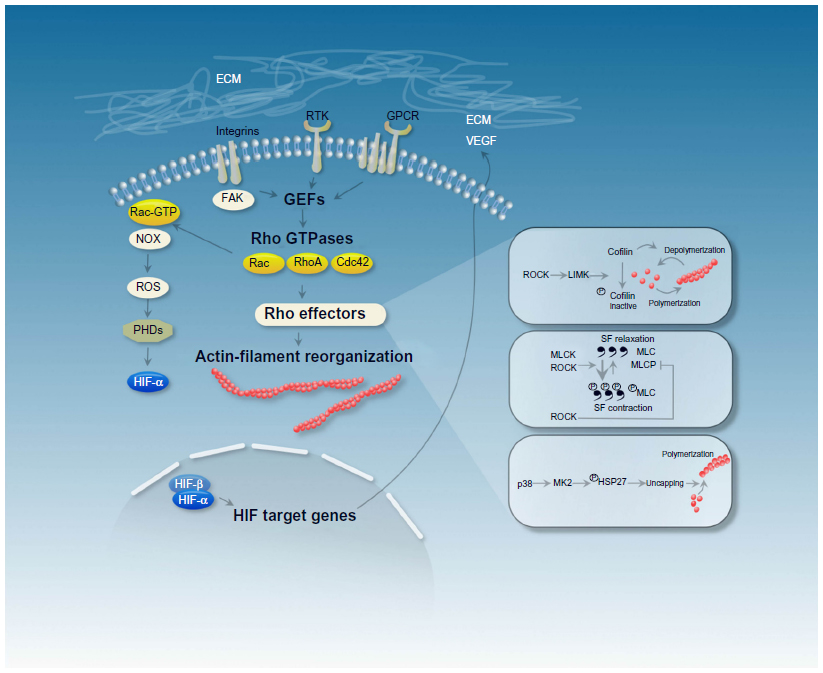 | Figure 1 The schema depicts mechanisms that are involved in actin filament reorganization, including stress fiber polymerization and stress fiber contraction in hypoxia. Cytoskeletal actin dynamics are affected by different classes of plasma membrane receptors, among them G-protein-coupled receptors (GPCRs) and receptor tyrosine kinases (RTKs). The plasma-membrane receptors modulate the activity of Rho guanosine triphosphatases (Rho GTPases) through Rho guanine nucleotide-exchange factors (GEFs), which then orchestrate changes to the actin cytoskeleton via a variety of downstream effectors. HIF target genes (eg, VEGF) bind to plasma-membrane receptors, which could provide a way of hypoxic regulation of the actin cytoskeleton. |
Signaling downstream of the Rho GTPases in hypoxia and the regulation of structural remodeling, cell contractility, and cellular barrier permeability
Rho GTPases control actin dynamics by activating downstream signaling modules. Downstream effectors of the Rho GTPases involve, for example, the Rho-associated protein kinase (ROCK)–LIM-domain kinase/cofilin pathway41 (Figure 1). Cofilin is an actin-binding and -severing protein; its activity is controlled by phosphorylation. Phosphorylation by LIM kinase 1 inactivates cofilin, leading to enhanced actin filament formation.42,43 As the regulation of cofilin is downstream of the Rho GTPases, it is not further surprising that hypoxia/the HIF-pathway can have an impact on cofilin activation. This has indeed been shown in several cell types,4,18,44,45 eg, in primary human endothelial and cancer cells. The lungs of mice that had been subjected to hypoxia also showed pronounced upregulation of cofilin protein levels44,45 and increased phosphorylation of cofilin.45 This elevated cofilin phosphorylation was seen in chronic (3 weeks) hypoxic mice as well, and might contribute to cytoskeletal reorganization and pulmonary vascular remodeling, resulting in pulmonary hypertension. More studies support the notion of elevated phospho-cofilin levels as a result of HIF-pathway modulations. For example, the knockdown of PHD2 in HeLa cells resulted in the enhanced phosphorylation of cofilin.18 Mechanistically, the PHD2 knockdown in HeLa cells and the subsequent HIF-1α stabilization leads to activation of the RhoA/ROCK/cofilin signal-transduction pathway and results in the modulation of the actin cytoskeleton with increased accumulation of actin filaments. The inhibition of PHD activity with the oxoglutarate analog DMOG had a similar outcome with regard to phospho-cofilin levels and actin rearrangement.4,18
Rho GTPases are also involved in the modulation of actin–myosin interaction. In nonmuscle and smooth-muscle cells, the actin–myosin interaction is regulated by the phosphorylation and dephosphorylation of myosin light chain (MLC).46–48 The phosphorylation of MLC favors actin–myosin interaction and the development of actomyosin-based contractile forces; dephosphorylation by MLC phosphatase (MLCP) relaxes the cytoskeleton. Several kinases have been reported to phosphorylate MLC, including the calcium/calmodulin-dependent MLC kinase (MLCK), which seems to be specific for MLC, and the Rho-associated kinase ROCK, which is a downstream target of RhoA. ROCK is not specific for MLC, and phosphorylates several targets. Although it can phosphorylate MLC directly, it mainly acts as an inhibitor of MLCP, which consequently results in increased MLC phosphorylation and activation (Figure 1).
This pathway and consequently the actin–myosin interaction has been shown to be triggered in hypoxia in metastatic MDA-MB-231 and MDA-MB-435 breast cancer cell lines,6 PASMCs,20,21,23,49,50 and endothelial cells.51,52 In MDA-MB and PASMCs, hypoxia activates ROCK and inactivates MLCP. This results in hypoxia-induced MLC phosphorylation and induces actin stress fiber formation.6,23,49 In metastatic breast cancer cell lines, actin filament generation could add to increased cell motility in hypoxia.6 In PASMCs, increased actin stress fiber formation regulates the contractile state of the PASMCs, and apparently contributes to the structural remodeling of pulmonary arteries and to pulmonary hypertension seen after chronic hypoxic exposure.13 It should be noted, however, that the origin and age of the cultured PASMCs influences hypoxic signaling, as experiments with cells isolated at different developmental stages showed that PASMCs increasingly lose the capacity to activate RhoA in response to hypoxia.20 Interestingly, in both normoxia and hypoxia, HIF-1α decreases MLC phosphorylation in PASMCs and the smooth muscle specific deletion of HIF-1α in mice increases the pulmonary vascular tone, suggesting that HIF-1α takes part in reducing hypoxic pulmonary hypertension.50 The mechanistic link between hypoxia/HIF-1α stabilization and MLC phosphorylation in PASMCs remains unknown and may very well be cell type-specific, as in some endothelial cells, a slightly different regulation of the actin–myosin interaction in hypoxia has been described.51,52 In endothelial cells, the actin–myosin-based contractile machinery plays an important role in maintaining the integrity of the endothelial barrier. The forces generated by the actin–myosin interaction tend to pull cells apart, whereas at the same time actin cytoskeleton-mediated endothelial adherens junctions are critical structures that maintain barrier function. Changes in actin cytoskeleton dynamics therefore affect the stability and integrity of the barrier function and adherens junctions.53,54 It has been shown that hypoxia induces endothelial barrier dysfunction through active reorganization of the cytoskeleton, which has been linked to changes in MLC phosphorylation. In endothelial cells increased MLC phosphorylation has also been linked to a mechanism involving ROCK;55,56 however, in the human pulmonary vein endothelial cell line VE, the phosphorylation of MLC has been attributed to increased HIF-1α-dependent MLCK protein expression.51,52 In brain vascular endothelial cells, the signaling cascade that results in MLC phosphorylation is thought to include increased intracellular Ca2+ levels, enhanced ROS generation, and oxidative stress, which then activate MLCK.57,58
In hypoxic rat pulmonary microvascular endothelial cells, it was noted that the mechanism that stabilizes the actin cytoskeleton at or near focal adhesions is different from the actin–myosin-mediated cell-contractility response. In these cells, the increased accumulation of filamentous actin in response to hypoxia is thought to be triggered via the p38 mitogen-activated protein-kinase pathway.55,56,59 Activation of p38 causes phosphorylation and activation of its downstream effector MK2 (mitogen-activated protein kinase-activated protein kinase 2) (Figure 1). MK2 in turn phosphorylates the 27 kDa heat-shock protein (HSP27). In its nonphosphorylated form, HSP27 is an actin-capping protein and prevents actin filament polymerization. Phosphorylation of HSP27 releases this inhibition,60,61 which is thought to stabilize the actin cytoskeleton near cell-adhesion sites in order to counteract the increased actin–myosin contractility and strain.
Hypoxic control of the expression of actin-binding proteins
In addition to changes in the activity of actin-binding proteins, the expression level of actin-binding proteins can also be modified in hypoxia. The actin-binding protein vasodilator-stimulated phosphoprotein (VASP) binds to actin filaments close to their barbed ends, prevents the interaction of capping proteins with the actin filament, and thus promotes filament elongation. Besides its anticapping activity, VASP also functions in filament clustering, bundling, and antibranching of actin filaments.62 Therefore, VASP can regulate the assembly of the actin filament network, support the formation of filopodia and lamellipodia, and play an important role in cell motility. VASP also assembles multiprotein complexes and stabilizes cell–cell contacts.62,63 Hypoxia represses VASP mRNA and protein expression in endothelial cells and tissues through an HIF-1α-dependent mechanism, and this repression presumably adds to a reduction in cellular barrier function. Due to the multitude of proposed biochemical activities of VASP, the exact underlying mechanism is still vague (Figure 2).64,65
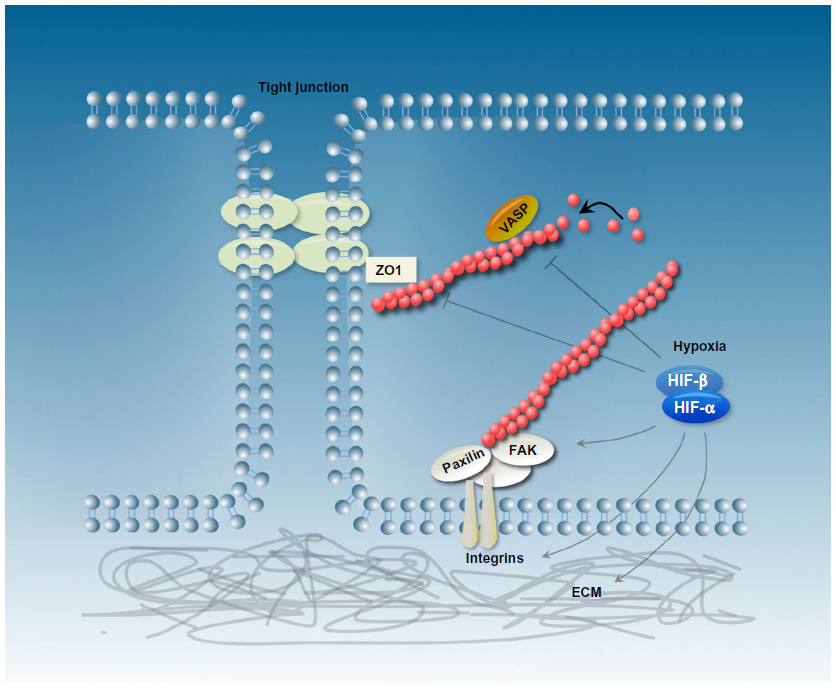 | Figure 2 Schematic representation of epithelial cells and the actin cytoskeleton reorganization in response to hypoxia/hypoxia-inducible factor (HIF)-α stabilization. HIF-1 is a critical regulator of the extracellular matrix (ECM) and changes integrin signaling and focal adhesion-complex formation. Furthermore, hypoxia influences the expression and activity of several actin-binding proteins like vasodilator-stimulated phosphoprotein (VASP) and zonula occludens (ZO)-1. |
To further add to the complexity, cell permeability and cell-barrier function in hypoxia can also be regulated by changes in tight-junction architecture. The zona occludens (ZO) family of tight-junction proteins binds directly to F-actin and is necessary for the assembly of tight junctions.66 In alveolar epithelial cells, prolonged hypoxia causes a decrease and mislocalization of the tight junction proteins ZO1 and occludin and a breakdown of the cytoskeletal network with a disruption and clumping of actin stress fibers67 (Figure 2). Hypoxia also affects ZO1 and F-actin distribution in brain-derived endothelial cells.68
One example of an actin-binding protein that is upregulated in hypoxia is the junction-mediating and regulatory protein (JMY). JMY can drive actin filament nucleation, and is involved in actin-dependent cellular adhesion and motility.69 JMY expression increases during hypoxia in human cancer cells in an HIF-1α-dependent manner,70 and hypoxia-responsive elements within the JMY promoter are required for this induction. Depleting JMY under hypoxic conditions results in decreased cell motility.
Hypoxia modulates ECM and focal adhesion composition
The behavior of a cell, eg, its cell morphology, adhesion, and migration, is undoubtedly affected by its surrounding extracellular matrix (ECM). The ECM directs cellular organization by interacting with cell-surface receptors and by the subsequent activation of intracellular signal-transduction pathways. Hypoxia induces changes of the ECM,71 and HIF-1 has been shown to be a critical player for hypoxic ECM remodeling in fibroblasts (Figure 2).72 Important ECM modulators are collagen hydroxylases, which are induced in hypoxia and affect the composition and mechanical properties of the ECM.72,73
The transmembrane protein family of integrins provides a link between the ECM and the intracellular cytoskeleton at focal adhesions. Several reports have described an upregulation of integrin cell-surface expression in various cell lines in hypoxia.4,74–78 Integrins initiate intracellular signaling events through the recruitment and activation of signaling molecules, and are prototypical activators of the focal adhesion kinase (FAK), a non-receptor cytoplasmic tyrosine kinase. The activation of FAK signaling can promote the remodeling of the actin cytoskeleton and the modulation of both cell motility and cell shape.79,80 In addition to integrin clustering at the cell surface, the activation of FAK has been described in hypoxia. In hypoxic human mesenchymal stem cells, FAK is phosphorylated and changes cell migration in an HIF-1α and VEGF-dependent fashion,81 and also in metastatic breast cancer cells the activity of FAK is altered in hypoxia in an HIF-1α and HIF-2 α-dependent manner.6 However, FAK might also lie upstream of HIF-1, as the absence of FAK reduces HIF-1α levels and VEGF induction in hypoxia. This suggests the existence of a regulatory loop.82 The integrin–FAK signaling complex is linked to the actin cytoskeleton via binding to talin and paxillin. It is therefore not surprising that evidence suggests a hypoxia-dependent paxillin regulation. Under hypoxic conditions, paxillin intensity increases in focal adhesions, and paxillin has been shown to be a direct HIF-1 target gene.83 Taken together, these reports suggest that the ECM and focal adhesion complexes are important components in hypoxia-induced intracellular signaling and cytoskeletal modulation.
Implications for cellular physiology
This review is based on evidence from studies where often different hypoxic conditions and cell types were used. Since cytoskeletal changes are highly cell type-dependent the big question for the actin system in hypoxia – “How is it controlled?” – cannot be answered in a simplified or generalized manner. Overall, the current data show that in hypoxia, numerous regulatory mechanisms – ranging from ECM modifications via Rho GTPases to the modulation of the expression and activity of actin-binding proteins – impact the actin cytoskeleton, and the list of confounders is most likely far from being complete. In tumor cells, many aspects of cell behavior that are controlled by the actin cytoskeleton, such as cell–cell and cell–matrix interaction and cell migration, get out of control. This is at least in part the basis for metastasis and tumor progression. Both processes are highly affected by hypoxia. Therefore, it is not surprising that some key proteins that have been described herein are regulated in hypoxia, like LIM-domain kinase and cofilin.84 Consequently, understanding hypoxic actin-cytoskeleton signaling will likely contribute to our insight into tumor metastasis and progression.
A very prominent cytoskeletal effect of hypoxic incubation that is seen in several cell types, and that in contrast to the aforementioned regulations might even be a general principle, is an increase of stress fibers (Figure 3).4,6,20,23,32,59,85 The question of whether this remodeling is actin isoform-specific cannot be answered conclusively. The remodeling might very well be cell type- and context-specific. For example, it has been shown that in L929 fibroblasts, hypoxia induces the formation of β- and γ-actin-containing stress fibers.4 In animal tissues, stress fibers are especially abundant in endothelial cells, myofibroblasts, and epithelial cells. It is important to note that different types of stress fibers exist. They vary in their morphology and association with focal contacts and myosin.2 The actin–myosin bundles are the major contractile structures, and as discussed before, hypoxia influences the actin–myosin interaction through various mechanisms. Accordingly, hypoxia can trigger cell contractility, resulting in the junctional disruption of cellular barriers in the case of epithelial and endothelial cells. Disruption in the epithelial barrier has indeed been shown to coincide with hypoxia-induced alterations of the actin cytoskeleton in several diseases,66 eg, a detrimental effect of hypoxia on the permeability of the blood–brain barrier has been described.57 In another cell type, the PASMCs, hypoxia-induced remodeling of the actin–myosin interaction contributes to pulmonary vasoconstriction.86
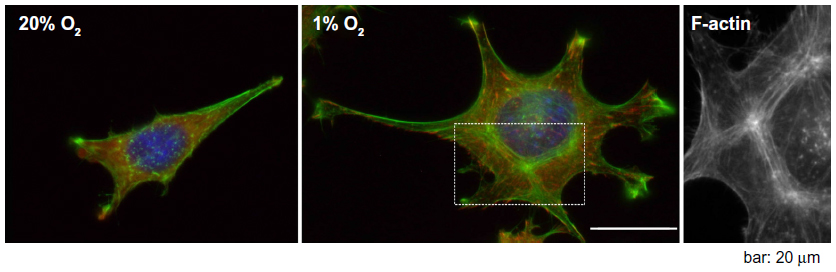 | Figure 3 Immunofluorescence images of L929 fibroblasts cultured under normoxic (20% O2) and hypoxic conditions (1% O2). |
Apart from adding to actin–myosin-mediated cell contractility, hypoxia also boosts the actin network polymerization that drives cellular protrusions. This presumably adds to the increased motility of some cell lines seen under hypoxic conditions.5,6,70,87–89 Wound healing occurs in a relatively hypoxic environment. Balanced cell migration within and adjacent to the hypoxic wound is an important part of the repair process, and hypoxia-driven actin rearrangement and cell motility could be a critical component of the repair process. However, it is not possible to generalize that increased actin filament content due to hypoxia undoubtedly initiates cell migration. Depending on the cell type and culture conditions, the increased amount of actin filaments together with overall actin-filament reorganization can also diminish cell movement in hypoxia.4
Summary and future directions
Although the role of hypoxia and HIF signaling for cytoskeletal dynamics is becoming increasingly evident, interpretation of the relation is immature, and additional information is clearly essential to understand the interconnection. Based on the current stage of knowledge, within the broad range of actin-regulatory pathways and mechanisms, the Rho GTPases take center stage in modulating actin dynamics in hypoxia. Conflicting results exist with regard to Rho GTPase and especially RhoA activation in hypoxia. However, considerable evidence exists that RhoA is activated during hypoxia, and the data inconsistency might be explained by different experimental setups, eg, hypoxic environment- and cell type-dependent effects. Moreover, data point to a rapid activation of Rho GTPases during hypoxia, and the brief time frame of Rho activation might have been missed in some experiments. In some reports, RhoA protein expression was analyzed but not RhoA activation. One big mystery is still how the activity of Rho GTPases is regulated in hypoxia, and a pressing challenge is to analyze the Rho GEFs, GAPs and GDIs in hypoxia. Besides being regulated by Rho GTPases, actin dynamics are also modulated by changes of the ECM and the expression of actin-binding proteins during hypoxia. This is partly achieved by HIF-1-dependent changes in the expression profile of proteins. Also, the question of to what extent HIF-1 and HIF-2 are involved in cytoskeletal dynamics has not been analyzed in great detail, and even though there is some indication that HIF-1α and HIF-2α have distinct roles in actin modulation,40 the details remain sketchy.
Defining the key molecular players, however, continues to be challenging, as they differ according to the cell type and the cell environment. Therefore, dissecting general principles like stress fiber induction from more cell type-dependent cytoskeletal effects helps to clear the picture of the emerging interrelations between hypoxia and the actin cytoskeleton.
Disclosure
The author reports no conflicts of interest in this work.
References
Pollard TD, Cooper JA. Actin, a central player in cell shape and movement. Science. 2009;326(5957):1208–1212. | |
Tojkander S, Gateva G, Lappalainen P. Actin stress fibers – assembly, dynamics and biological roles. J Cell Sci. 2012;125(Pt 8):1855–1864. | |
Misra A, Pandey C, Sze SK, Thanabalu T. Hypoxia activated EGFR signaling induces epithelial to mesenchymal transition (EMT). PLoS One. 2012;7(11):e49766. | |
Vogler M, Vogel S, Krull S, et al. Hypoxia modulates fibroblastic architecture, adhesion and migration: a role for HIF-1α in cofilin regulation and cytoplasmic actin distribution. PLoS One. 2013;8(7):e69128. | |
Nagelkerke A, Bussink J, Mujcic H, et al. Hypoxia stimulates migration of breast cancer cells via the PERK/ATF4/LAMP3-arm of the unfolded protein response. Breast Cancer Res. 2013;15(1):R2. | |
Gilkes DM, Xiang L, Lee SJ, et al. Hypoxia-inducible factors mediate coordinated RhoA-ROCK1 expression and signaling in breast cancer cells. Proc Natl Acad Sci U S A. 2014;111(3):E384–E393. | |
Heasman SJ, Ridley AJ. Mammalian Rho GTPases: new insights into their functions from in vivo studies. Nat Rev Mol Cell Biol. 2008;9(9):690–701. | |
Rossman KL, Der CJ, Sondek J. GEF means go: turning on RHO GTPases with guanine nucleotide-exchange factors. Nat Rev Mol Cell Biol. 2005;6(2):167–180. | |
Tcherkezian J, Lamarche-Vane N. Current knowledge of the large RhoGAP family of proteins. Biol Cell. 2007;99(2):67–86. | |
Dovas A, Couchman JR. RhoGDI: multiple functions in the regulation of Rho family GTPase activities. Biochem J. 2005;390(Pt 1):1–9. | |
Recchia AG, De Francesco EM, Vivacqua A, et al. The G protein-coupled receptor 30 is up-regulated by hypoxia-inducible factor-1a (HIF-1a) in breast cancer cells and cardiomyocytes. J Biol Chem. 2011;286(12):10773–10782. | |
Ronkainen VP, Tuomainen T, Huusko J, et al. Hypoxia-inducible factor 1-induced G protein-coupled receptor 35 expression is an early marker of progressive cardiac remodelling. Cardiovasc Res. 2014;101(1):69–77. | |
Stenmark KR, Fagan KA, Frid MG. Hypoxia-induced pulmonary vascular remodeling: cellular and molecular mechanisms. Circ Res. 2006;99(7):675–691. | |
Schiller MR. Coupling receptor tyrosine kinases to Rho GTPases – GEFs what’s the link. Cell Signal. 2006;18(11):1834–1843. | |
Disanza A, Steffen A, Hertzog M, Frittoli E, Rottner K, Scita G. Actin polymerization machinery: the finish line of signaling networks, the starting point of cellular movement. Cell Mol Life Sci. 2005;62(9):955–970. | |
Hall A. Rho family GTPases. Biochem Soc Trans. 2012;40(6):1378–1382. | |
Jaffe AB, Hall A. Rho GTPases: biochemistry and biology. Annu Rev Cell Dev Biol. 2005;21:247–269. | |
Vogel S, Wottawa M, Farhat K, et al. Prolyl hydroxylase domain (PHD) 2 affects cell migration and F-actin formation via RhoA/rho-associated kinase-dependent cofilin phosphorylation. J Biol Chem. 2010;285(44):33756–33763. | |
Turcotte S, Desrosiers RR, Beliveau R. HIF-1a mRNA and protein upregulation involves Rho GTPase expression during hypoxia in renal cell carcinoma. J Cell Sci. 2003;116(Pt 11):2247–2260. | |
Bailly K, Ridley AJ, Hall SM, Haworth SG. RhoA activation by hypoxia in pulmonary arterial smooth muscle cells is age and site specific. Circ Res. 2004;94(10):1383–1391. | |
Wang Z, Jin N, Ganguli S, Swartz DR, Li L, Rhoades RA. Rho-kinase activation is involved in hypoxia-induced pulmonary vasoconstriction. Am J Respir Cell Mol Biol. 2001;25(5):628–635. | |
Chi AY, Waypa GB, Mungai PT, Schumacker PT. Prolonged hypoxia increases ROS signaling and RhoA activation in pulmonary artery smooth muscle and endothelial cells. Antioxid Redox Signal. 2010;12(5):603–610. | |
Wojciak-Stothard B, Zhao L, Oliver E, et al. Role of RhoB in the regulation of pulmonary endothelial and smooth muscle cell responses to hypoxia. Circ Res. 2012;110(11):1423–1434. | |
Jin HG, Yamashita H, Nagano Y, et al. Hypoxia-induced upregulation of endothelial small G protein RhoA and Rho-kinase/ROCK2 inhibits eNOS expression. Neurosci Lett. 2006;408(1):62–67. | |
Jernigan NL, Walker BR, Resta TC. Reactive oxygen species mediate RhoA/Rho kinase-induced Ca2+ sensitization in pulmonary vascular smooth muscle following chronic hypoxia. Am J Physiol Lung Cell Mol Physiol. 2008;295(3):L515–L529. | |
Broughton BR, Jernigan NL, Norton CE, Walker BR, Resta TC. Chronic hypoxia augments depolarization-induced Ca2+ sensitization in pulmonary vascular smooth muscle through superoxide-dependent stimulation of RhoA. Am J Physiol Lung Cell Mol Physiol. 2010;298(2):L232–L242. | |
Hayashi M, Sakata M, Takeda T, et al. Hypoxia up-regulates hypoxia-inducible factor-1a expression through RhoA activation in trophoblast cells. J Clin Endocrinol Metab. 2005;90(3):1712–1719. | |
Pacary E, Tixier E, Coulet F, Roussel S, Petit E, Bernaudin M. Crosstalk between HIF-1 and ROCK pathways in neuronal differentiation of mesenchymal stem cells, neurospheres and in PC12 neurite outgrowth. Mol Cell Neurosci. 2007;35(3):409–423. | |
Raheja LF, Genetos DC, Wong A, Yellowley CE. Hypoxic regulation of mesenchymal stem cell migration: the role of RhoA and HIF-1a. Cell Biol Int. 2011;35(10):981–989. | |
Vertelov G, Kharazi L, Muralidhar MG, Sanati G, Tankovich T, Kharazi A. High targeted migration of human mesenchymal stem cells grown in hypoxia is associated with enhanced activation of RhoA. Stem Cell Res Ther. 2013;4(1):5. | |
Fediuk J, Gutsol A, Nolette N, Dakshinamurti S. Thromboxane-induced actin polymerization in hypoxic pulmonary artery is independent of Rho. Am J Physiol Lung Cell Mol Physiol. 2012;302(1):L13–L26. | |
Dada LA, Novoa E, Lecuona E, Sun H, Sznajder JI. Role of the small GTPase RhoA in the hypoxia-induced decrease of plasma membrane Na,K-ATPase in A549 cells. J Cell Sci. 2007;120(Pt 13):2214–2222. | |
Wojciak-Stothard B, Tsang LY, Haworth SG. Rac and Rho play opposing roles in the regulation of hypoxia/reoxygenation-induced permeability changes in pulmonary artery endothelial cells. Am J Physiol Lung Cell Mol Physiol. 2005;288(4):L749–L760. | |
Hirota K, Semenza GL. Rac1 activity is required for the activation of hypoxia-inducible factor 1. J Biol Chem. 2001;276(24):21166–21172. | |
Xue Y, Bi F, Zhang X, et al. Role of Rac1 and Cdc42 in hypoxia induced p53 and von Hippel-Lindau suppression and HIF1a activation. Int J Cancer. 2006;118(12):2965–2972. | |
Myllyharju J. Prolyl 4-hydroxylases, master regulators of the hypoxia response. Acta Physiol (Oxf). 2013;208(2):148–165. | |
Bosco EE, Mulloy JC, Zheng Y. Rac1 GTPase: a “Rac” of all trades. Cell Mol Life Sci. 2009;66(3):370–374. | |
Moldovan L, Mythreye K, Goldschmidt-Clermont PJ, Satterwhite LL. Reactive oxygen species in vascular endothelial cell motility. Roles of NAD(P)H oxidase and Rac1. Cardiovasc Res. 2006;71(2):236–246. | |
Acker T, Fandrey J, Acker H. The good, the bad and the ugly in oxygen-sensing: ROS, cytochromes and prolyl-hydroxylases. Cardiovasc Res. 2006;71(2):195–207. | |
Weidemann A, Breyer J, Rehm M, et al. HIF-1a activation results in actin cytoskeleton reorganization and modulation of Rac-1 signaling in endothelial cells. Cell Commun Signal. 2013;11(1):80. | |
Olson EN, Nordheim A. Linking actin dynamics and gene transcription to drive cellular motile functions. Nat Rev Mol Cell Biol. 2010;11(5):353–365. | |
Arber S, Barbayannis FA, Hanser H, et al. Regulation of actin dynamics through phosphorylation of cofilin by LIM-kinase. Nature. 1998;393(6687):805–809. | |
Bernard O. Lim kinases, regulators of actin dynamics. Int J Biochem Cell Biol. 2007;39(6):1071–1076. | |
Ostergaard L, Simonsen U, Eskildsen-Helmond Y, et al. Proteomics reveals lowering oxygen alters cytoskeletal and endoplasmatic stress proteins in human endothelial cells. Proteomics. 2009;9(19):4457–4467. | |
Veith C, Schmitt S, Veit F, et al. Cofilin, a hypoxia-regulated protein in murine lungs identified by 2-dimensional gel electrophoresis: The role of the cytoskeletal protein cofilin in pulmonary hypertension. Proteomics. 2013;13(1):75–88. | |
Murthy KS. Signaling for contraction and relaxation in smooth muscle of the gut. Annu Rev Physiol. 2006;68:345–374. | |
Vicente-Manzanares M, Ma X, Adelstein RS, Horwitz AR. Non-muscle myosin II takes centre stage in cell adhesion and migration. Nat Rev Mol Cell Biol. 2009;10(11):778–790. | |
Shen Q, Rigor RR, Pivetti CD, Wu MH, Yuan SY. Myosin light chain kinase in microvascular endothelial barrier function. Cardiovasc Res. 2010;87(2):272–280. | |
Wang Z, Lanner MC, Jin N, Swartz D, Li L, Rhoades RA. Hypoxia inhibits myosin phosphatase in pulmonary arterial smooth muscle cells: role of Rho-kinase. Am J Respir Cell Mol Biol. 2003;29(4):465–471. | |
Kim YM, Barnes EA, Alvira CM, Ying L, Reddy S, Cornfield DN. Hypoxia-inducible factor-1a in pulmonary artery smooth muscle cells lowers vascular tone by decreasing myosin light chain phosphorylation. Circ Res. 2013;112(9):1230–1233. | |
Qi H, Wang P, Liu C, et al. Involvement of HIF-1a in MLCK-dependent endothelial barrier dysfunction in hypoxia. Cell Physiol Biochem. 2011;27(3–4):251–262. | |
Wang P, Qi H, Sun C, et al. Overexpression of hypoxia-inducible factor-1a exacerbates endothelial barrier dysfunction induced by hypoxia. Cell Physiol Biochem. 2013;32(4):859–870. | |
Waschke J, Curry FE, Adamson RH, Drenckhahn D. Regulation of actin dynamics is critical for endothelial barrier functions. Am J Physiol Heart Circ Physiol. 2005;288(3):H1296–H1305. | |
Aslam M, Schluter KD, Rohrbach S, et al. Hypoxia-reoxygenation-induced endothelial barrier failure: role of RhoA, Rac1 and myosin light chain kinase. J Physiol. 2013;591(Pt 2):461–473. | |
An SS, Pennella CM, Gonnabathula A, et al. Hypoxia alters biophysical properties of endothelial cells via p38 MAPK- and Rho kinase-dependent pathways. Am J Physiol Cell Physiol. 2005;289(3):C521–C530. | |
Liu T, Guevara OE, Warburton RR, Hill NS, Gaestel M, Kayyali US. Modulation of HSP27 alters hypoxia-induced endothelial permeability and related signaling pathways. J Cell Physiol. 2009;220(3):600–610. | |
Kuhlmann CR, Tamaki R, Gamerdinger M, et al. Inhibition of the myosin light chain kinase prevents hypoxia-induced blood-brain barrier disruption. J Neurochem. 2007;102(2):501–507. | |
Hicks K, O’Neil RG, Dubinsky WS, Brown RC. TRPC-mediated actin-myosin contraction is critical for BBB disruption following hypoxic stress. Am J Physiol Cell Physiol. 2010;298(6):C1583–C1593. | |
Kayyali US, Pennella CM, Trujillo C, Villa O, Gaestel M, Hassoun PM. Cytoskeletal changes in hypoxic pulmonary endothelial cells are dependent on MAPK-activated protein kinase MK2. J Biol Chem. 2002;277(45):42596–42602. | |
Rouse J, Cohen P, Trigon S, et al. A novel kinase cascade triggered by stress and heat shock that stimulates MAPKAP kinase-2 and phosphorylation of the small heat shock proteins. Cell. 1994;78(6):1027–1037. | |
Guay J, Lambert H, Gingras-Breton G, Lavoie JN, Huot J, Landry J. Regulation of actin filament dynamics by p38 map kinase-mediated phosphorylation of heat shock protein 27. J Cell Sci. 1997;110(Pt 3):357–368. | |
Bear JE, Gertler FB. Ena/VASP: towards resolving a pointed controversy at the barbed end. J Cell Sci. 2009;122(Pt 12):1947–1953. | |
Krause M, Dent EW, Bear JE, Loureiro JJ, Gertler FB. Ena/VASP proteins: regulators of the actin cytoskeleton and cell migration. Annu Rev Cell Dev Biol. 2003;19:541–564. | |
Rosenberger P, Khoury J, Kong T, Weissmüller T, Robinson AM, Colgan SP. Identification of vasodilator-stimulated phosphoprotein (VASP) as an HIF-regulated tissue permeability factor during hypoxia. FASEB J. 2007;21(10):2613–2621. | |
Schmit MA, Mirakaj V, Stangassinger M, König K, Köhler D, Rosenberger P. Vasodilator phosphostimulated protein (VASP) protects endothelial barrier function during hypoxia. Inflammation. 2012;35(2):566–573. | |
Rodgers LS, Fanning AS. Regulation of epithelial permeability by the actin cytoskeleton. Cytoskeleton (Hoboken). 2011;68(12):653–660. | |
Bouvry D, Planes C, Malbert-Colas L, Escabasse V, Clerici C. Hypoxia-induced cytoskeleton disruption in alveolar epithelial cells. Am J Respir Cell Mol Biol. 2006;35(5):519–527. | |
Fischer S, Wobben M, Marti HH, Renz D, Schaper W. Hypoxia-induced hyperpermeability in brain microvessel endothelial cells involves VEGF-mediated changes in the expression of zonula occludens-1. Microvasc Res. 2002;63(1):70–80. | |
Rottner K, Hanisch J, Campellone KG. WASH, WHAMM and JMY: regulation of Arp2/3 complex and beyond. Trends Cell Biol. 2010;20(11):650–661. | |
Coutts AS, Pires IM, Weston L, et al. Hypoxia-driven cell motility reflects the interplay between JMY and HIF-1a. Oncogene. 2011;30(48):4835–4842. | |
Myllyharju J, Schipani E. Extracellular matrix genes as hypoxia-inducible targets. Cell Tissue Res. 2010;339(1):19–29. | |
Gilkes DM, Bajpai S, Chaturvedi P, Wirtz D, Semenza GL. Hypoxia-inducible factor 1 (HIF-1) promotes extracellular matrix remodeling under hypoxic conditions by inducing P4HA1, P4HA2, and PLOD2 expression in fibroblasts. J Biol Chem. 2013;288(15):10819–10829. | |
Hofbauer KH, Gess B, Lohaus C, Meyer HE, Katschinski D, Kurtz A. Oxygen tension regulates the expression of a group of procollagen hydroxylases. Eur J Biochem. 2003;270(22):4515–4522. | |
Blaschke F, Stawowy P, Goetze S, et al. Hypoxia activates beta(1)-integrin via ERK 1/2 and p38 MAP kinase in human vascular smooth muscle cells. Biochem Biophys Res Commun. 2002;296(4):890–896. | |
Skuli N, Monferran S, Delmas C, et al. Alphavbeta3/alphavbeta5 integrins-FAK-RhoB: a novel pathway for hypoxia regulation in glioblastoma. Cancer Res. 2009;69(8):3308–3316. | |
Cowden Dahl KD, Robertson SE, Weaver VM, Simon MC. Hypoxia-inducible factor regulates alphavbeta3 integrin cell surface expression. Mol Biol Cell. 2005;16(4):1901–1912. | |
Yoon SO, Shin S, Mercurio AM. Hypoxia stimulates carcinoma invasion by stabilizing microtubules and promoting the Rab11 trafficking of the alpha6beta4 integrin. Cancer Res. 2005;65(7):2761–2769. | |
Keely S, Glover LE, MacManus CF, et al. Selective induction of integrin beta1 by hypoxia-inducible factor: implications for wound healing. FASEB J. 2009;23(5):1338–1346. | |
Mitra SK, Hanson DA, Schlaepfer DD. Focal adhesion kinase: in command and control of cell motility. Nat Rev Mol Cell Biol. 2005;6(1):56–68. | |
Wehrle-Haller B. Structure and function of focal adhesions. Curr Opin Cell Biol. 2012;24(1):116–124. | |
Lee SH, Lee YJ, Song CH, Ahn YK, Han HJ. Role of FAK phosphorylation in hypoxia-induced hMSCS migration: involvement of VEGF as well as MAPKS and eNOS pathways. Am J Physiol Cell Physiol. 2010;298(4):C847–C856. | |
Zhu J, Wang YS, Zhang J, et al. Focal adhesion kinase signaling pathway participates in the formation of choroidal neovascularization and regulates the proliferation and migration of choroidal microvascular endothelial cells by acting through HIF-1 and VEGF expression in RPE cells. Exp Eye Res. 2009;88(5):910–918. | |
Veith C, Marsh LM, Wygrecka M, et al. Paxillin regulates pulmonary arterial smooth muscle cell function in pulmonary hypertension. Am J Pathol. 2012;181(5):1621–1633. | |
Yamaguchi H, Condeelis J. Regulation of the actin cytoskeleton in cancer cell migration and invasion. Biochim Biophys Acta. 2007;1773(5):642–652. | |
Shen WG, Peng WX, Shao Y, et al. Localization and activity of calmodulin is involved in cell-cell adhesion of tumor cells and endothelial cells in response to hypoxic stress. Cell Biol Toxicol. 2007;23(5):323–335. | |
Ward JP, McMurtry IF. Mechanisms of hypoxic pulmonary vasoconstriction and their roles in pulmonary hypertension: new findings for an old problem. Curr Opin Pharmacol. 2009;9(3):287–296. | |
Eul B, Rose F, Krick S, et al. Impact of HIF-1a and HIF-2a on proliferation and migration of human pulmonary artery fibroblasts in hypoxia. FASEB J. 2006;20(1):163–165. | |
Li W, Li Y, Guan S, et al. Extracellular heat shock protein-90a: linking hypoxia to skin cell motility and wound healing. EMBO J. 2007;26(5):1221–1233. | |
O’Toole EA, Marinkovich MP, Peavey CL, et al. Hypoxia increases human keratinocyte motility on connective tissue. J Clin Invest. 1997;100(11):2881–2891. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.