")
Back to Journals » OncoTargets and Therapy » Volume 12
Hypermethylation Of ADHFE1 Promotes The Proliferation Of Colorectal Cancer Cell Via Modulating Cell Cycle Progression
Authors Hu YH, Ma S, Zhang XN, Zhang ZY, Zhu HF, Ji YH, Li J, Qian XL, Wang YX
Received 16 July 2019
Accepted for publication 18 September 2019
Published 4 October 2019 Volume 2019:12 Pages 8105—8115
DOI https://doi.org/10.2147/OTT.S223423
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Takuya Aoki
Yu-Han Hu,1,* Shuai Ma,1,* Xiang-Nan Zhang,2 Zhe-Ying Zhang,1 Hui-Fang Zhu,1,2 Ying-Hua Ji,3 Jian Li,4 Xin-Lai Qian,1,2 Yong-Xia Wang1,2
1Department of Pathology, School of Basic Medical Sciences, Xinxiang Medical University, Xinxiang, Henan 453000, People’s Republic of China; 2Department of Pathology, The Third Affiliated Hospital of Xinxiang Medical University, Xinxiang, Henan 453000, People’s Republic of China; 3Department of Oncology, The First Affiliated Hospital of Xinxiang Medical University, Xinxiang, Henan 453000, People’s Republic of China; 4Department of Surgery, The First Affiliated Hospital of Xinxiang Medical University, Xinxiang, Henan 453000, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Yong-Xia Wang; Xin-Lai Qian
Department of Pathology, School of Basic Medical Sciences, Xinxiang Medical University, Xinxiang, Henan 453000, People’s Republic of China
Tel +86-15090325842; +86-13839073820
Email [email protected]; [email protected]
Background: Colorectal cancer (CRC) is one of the most common malignancies worldwide. Studies have demonstrated that epigenetic modifications play essential roles in the development of CRC. ADHFE1 is a differentially expressed gene that has been reported to be hypermethylated in CRC. However, the role and mechanism of ADHFE1 in the proliferation of CRC remain unclear.
Materials and methods: ADHFE1 expression was analyzed in CRC tissues by IHC and qRT-PCR, and the relationship between ADHFE1 expression and the clinicopathological parameters was analyzed. Cell proliferation were assessed by the in vitro and in vivo experimental models. GSEA assay was performed to explore the mechanism of ADHFE1 in the proliferation of CRC. Flow cytometry and Western blot were used to detect the activation of the cell cycle signaling. Bisulfite genomic sequence (BSP) assay was used to test the methylation degree of ADHFE1 gene promoter in CRC tissues.
Results: Here, we verified that ADHFE1 was down-regulated and hypermethylated in CRC tissues. The down-regulation of ADHFE1 was correlated with poor differentiation and advanced TNM stage of CRC patients. And ADHFE1 expression restored when the CRC cell line SW620 was treated with the demethylating agent 5-Aza-CdR. Overexpression of ADHFE1 inhibited the proliferation of CRC, while ADHFE1 knockdown promoted the proliferation of CRC cells in vitro and in vivo. Moreover, ADHFE1 overexpression could induce a significant G1-S cell cycle arrest in CRC cells and vice versa.
Conclusion: Hypermethylation of ADHFE1 might promote cell proliferation by modulating cell cycle progression in CRC, potentially providing a new therapeutic target for CRC patients.
Keywords: ADHFE1, colorectal cancer, hypermethylation, proliferation, cell cycle
Introduction
Colorectal cancer (CRC) is one of the most lethal malignancies worldwide.1 The initiation of CRC is a complex process characterized by activation of oncogenes, inactivation of tumor suppressor genes and multiple risk factors.2 However, the precise molecular mechanism underlying the initiation and progression of CRC remain unclear. Therefore, it is essential to explore the function and molecular mechanisms in the development of CRC.
Numerous studies have revealed that genetic alterations and mutations are involved in the tumorigenesis and progression of CRC.3 Recently, epigenetic modifications are also demonstrated to be required for the initiation and development of CRC.4–6 Aberrant DNA methylation is the most common type of epigenetic alterations, in which a methyl group is added to the cytosine base in the dinucleotide sequence CpG islands, which are often associated with the promoter.4,7 The abnormal methylation in a gene promoter generally correlates with an aberrant expression of itself. For example, hypermethylation of a gene promoter may cause the inactivation of a tumor suppressor gene and will further lead to tumor initiation and metastasis.8–10
Alcohol Dehydrogenase Iron Containing 1 (ADHFE1) is a member of the iron-activated alcohol dehydrogenase family.11 It is reported that ADHFE1 has a conserved NAD-binding site and it is related to bacterial γ-hydroxybutyrate dehydrogenase.12 Investigations have demonstrated that ADHFE1 promoter is significantly hypermethylated and down-regulated in CRC, and ADHFE1 expression is correlated with the differentiation of CRC.13–15 However, the precise functions and molecular mechanisms of ADHFE1 in CRC still need further exploration.
Therefore, in this study, we detected ADHFE1 expression in CRC tissues, illustrated the role and molecular mechanism of ADHFE1 in the proliferation of CRC, and verified the upstream regulatory mechanisms underlying the down-regulation of ADHFE1.
Materials And Methods
Tissue Specimens
All CRC tissues and matched adjacent normal mucosa samples were collected from the First Affiliated Hospital of Xinxiang Medical University (Xinxiang, China) with the written informed consent of patients from 2016 to 2017. The clinical research was performed according to the written approval obtained from the Ethics Committee of Xinxiang Medical University (Xinxiang, China). For each tissue, one portion was snap-frozen in liquid nitrogen and stored at −80°C for quantitative real-time PCR (qRT-PCR), and another portion was fixed in 10% formalin within 1 h after surgical removal, embedded in paraffin, and sectioned consecutively at 3 μm thickness using a rotary microtome for immunohistochemical assays. All the patients did not receive radiotherapy or chemotherapy before surgery.
Cell Cultures And 5-Aza-CdR Treatment
Human CRC cell lines SW620, HCT116, SW480, HT29, LS174T, LOVO, CACO2 and RKO were obtained from the American Type Culture Collection (Manassas, VA, USA). The cells were cultured in RPMI1640 (HyClone; GE Healthcare Life Sciences, Logan, UT, USA) supplemented with 10% fetal bovine serum (FBS) (Gibco; Thermo Fisher Scientific, Inc.) at 37 °C in a humidified atmosphere with 5% CO2. SW620 cells were treated with 0, 5, 10 and 20μM 5-Aza-CdR (Sigma-Aldrich, St. Louis, Mo, USA) for 48 hrs.
Vector Construction And Lentivirus Infection
The ADHFE1 construct was generated by subcloning PCR amplified full-length human ADHFE1 cDNA into plasmid PSIN-EF2-puro. For deletion of ADHFE1, shRNA sequence was cloned into the pSuper-retro-puro vector. The primers used to generate the shRNA nucleotide sequences for repressing ADHFE1 are listed in Supplementary Table S1. Retroviral production and infection were performed as previously described.16 Stable cell lines expressing ADHFE1 or shADHFE1 were selected for 10 days with 0.5 mg/mL puromycin.
Immunohistochemistry (IHC)
The 3 μm-thick tissue sections were deparaffinized and rehydrated, and incubated with primary antibody at 4°C overnight. Prior to incubation with anti-ADHFE1 (1:200 dilution; GeneTex Inc, Texas, St. Antonio, USA), and anti-Ki67 antibodies (1:100 dilution; Bioworld Technology Inc., St. Louis Park, MN, USA), the sections were heated in 0.01M sodium citrate buffer, pH6.0 for antigen retrieval, and incubated in 3% H2O2 to inhibit endogenous peroxidase activity. On the next day, the sections were washed and incubated with the secondary antibody for 30 mins at room temperature. Finally, the slides were developed using a DAB chromogen kit and counterstained with Mayer’s hematoxylin. The total ADHFE1 and Ki67 immunostaining score was calculated as the sum of the percentage positivity of stained tumor cells and the staining intensity as previously described.10
RNA Extraction And qRT-PCR
Total RNA was extracted using TRIzol reagent (Takara) according to the manufacturer’s instructions. cDNAs were synthesized using a Reverse Transcription Kit (Takara). Quantitative real-time PCR was carried out using SYBR Green I (Takara) in triplicate. The results were normalized to the expression of GAPDH. The primer sequences used for qRT-PCR are listed in Supplementary Table S2. Relative quantification of mRNA expression was calculated using the 2−ΔΔCt method.
Western Blot Assay
Protein lysates were prepared using a lysis buffer and quantified using a bicinchoninic acid (BCA) protein quantification kit (KeyGen Biotech, China). The proteins were subjected to SDS-PAGE and transferred onto a PVDF membrane. The PVDF membrane was subsequently blocked in PBST solution containing 5% non-fat milk and incubated at 4°C overnight with specific antibodies anti-ADHFE1 (1:1000 dilution; GeneTex Inc, Texas, St. Antonio, USA), anti-CyclinD1 (1:500 dilution; ProteinTech Group, Chicago, IL, USA), anti-p21 (1:500 dilution; ProteinTech Group, Chicago, IL, USA), anti-p27 (1:1000 dilution; ProteinTech Group, Chicago, IL, USA), anti-p53 (1:2000 dilution; ProteinTech Group, Chicago, IL, USA) and anti-GAPDH (1:5000 dilution; ProteinTech Group, Chicago, IL, USA). The next day, membranes were incubated with the HRP-conjugated goat anti-rabbit secondary antibody (dilution 1:5,000; ABclonal Biotech Co., Ltd., Woburn, MA, USA). Subsequently, the membranes were detected using Pierce ECL Western Blotting Substrate (Thermo Scientific, USA).
CCK-8 Cell Proliferation Assay
1×103 cells were seeded at 96-well plates and cultured for 24 h. The cell proliferation assay was performed on days 1, 2, 3, 4, and 5 using Cell Counting Kit-8 (CCK-8) (Dojindo Laboratories, Japan). 10 μl CCK-8 reagent was added to each well and then the plate was incubated for 2 h at 37°C. At the endpoint of incubation, the absorbance was measured at 450 nm using a Vmax microplate spectrophotometer (Molecular Devices, Sunnyvale, CA). Each sample was assayed in triplicate and repeated 3 times independently.
Colony Formation Assay
200 cells were seeded on 6-well plates and cultured for 2 weeks. The colonies were fixed with 4% paraformaldehyde for 10 min and then stained with hematoxylin and eosin (H&E) for 30 min. The number of colonies, defined as > 50 cells/colony, was counted. Three independent experiments were performed.
Xenograft Growth Assay
Xenograft tumors were generated by subcutaneous injection of 2 × 106 stable cells of SW480/ShNC, SW480/ShADHFE1 (n = 6 per group) on the hind limbs of female athymic BALB/c nude mice between 4 and 6 weeks of age. All animal experiments were conducted according to the National Institutes of Health (NIH) Guidelines for Laboratory Animal Care and approved by the Xinxiang Medical University Institutional Animal Care and Use Committee. The primary tumor growth was measured using a slide caliper and the tumor volume was determined using the formula (length × width2)/2. After euthanasia, the tumors were surgically removed, fixed in formalin (neutral buffered 10%), embedded in paraffin, and prepared into 3-μm sections for staining with hematoxylin and eosin (H&E) or Ki67 immunohistochemical staining.17
Flow Cytometry
The stable transfected cells were harvested by trypsinization and flow cytometry assays were performed as previously described.18,19 1x106 indicated cells were collected and fixed with 70% cold ethanol. After treated with RNase A (10 μg/mL) for 30 mins at 37°C, the cells were resuspended in 0.5 mL propidium iodide (PI) solution (50 μg/mL in 0.1% sodium citrate with 0.1% NP-40). Cell cycle distribution was analyzed by FACScan cytometry (Becton-Dickinson, San Jose, CA, USA).
Bisulfite Genomic Sequence (BSP) Assay
The methylation status of the CpG dinucleotides in the ADHFE1 promoter was analyzed. A bisulfate-sequencing assay was performed using 1.0 mg of bisulfite-treated genomic DNA from the clinical samples. Bisulfite conversion was performed using the EpiTect Bisulfite Kit (QIAGEN, German) according to the manufacturer’s instructions. The fragments of interest were amplified using the following specific primer pairs designed with the MethPrimer software47: forward, 5′- GGAGATGGAGTGAAGGTTGTTT-3′; reverse, 5′- ATTAACTCAAAACCCCTCTCTCTC −3′. The PCR products were gel purified and cloned into the pMD19-T vectors using pMD19-T vector cloning kit (TAKARA, Japan). Individual bacterial colonies were picked and sequenced to analyze DNA methylation19
Accession Numbers For Data Sets
The different expression of ADHFE1 generated in the study came from the GEO database (GSE32323 and GSE21510) and the TCGA data. The methylation percentage of ADHFE1 in the normal and colorectal cancer tissues reanalyzed in the study came from the GEO database (GSE17648, GSE25062 and GSE40055). The GEO databases (GSE13294 and GSE13067) were used for the GSEA analysis of the “REACTOME_CELL CYCLE” and “REACTOME_G1_S_TRANSITION” gene sets in the study.
Statistical Analysis
All statistical analyses were performed using the Statistical Package for the Social Sciences version 19.0 software (SPSS Inc., Chicago, IL, USA). The differences between groups were examined using one-way ANOVA or two-tailed Student’s t-test. The relationships between ADHFE1 expression and clinicopathological characteristics were determined using the χ2 test. Data are presented as the means ± SD of at least 3 independent experiments. p< 0.05 was considered statistically significant.
Results
ADHFE1 Is Down-Regulated In CRC
The TCGA database was first used to analyze the expression of ADHFE1 in CRC tissues. It revealed that the expression of ADHFE1 was significantly down-regulated in CRC tissues compared with that in normal colorectal tissues (Figure 1A). The analysis from GEO datasets (GSE32323 and GSE21510) also showed the same results (Figure 1B and C).
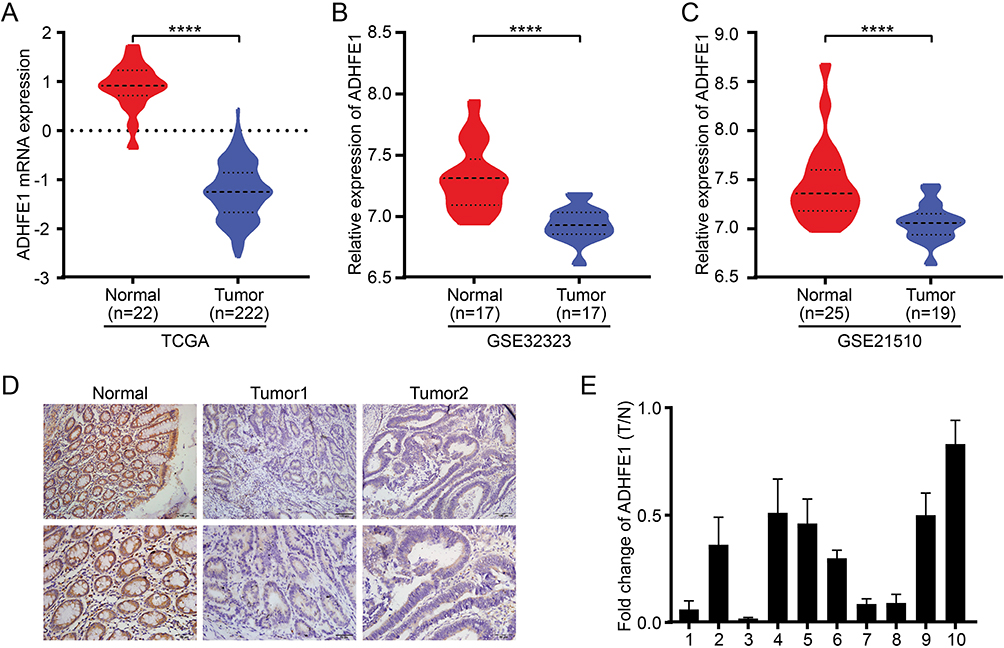 |
Figure 1 ADHFE1 is down-regulated in CRC. (A, B and C) The expression of ADHFE1 in CRC tissues and normal colorectal tissues in the TCGA mRNAArray data and GEO datasets (GSE32323 and GSE21510). ****p<0.0001. (D) Immunostaining of ADHFE1 protein in 80 CRC tissues and the adjacent normal colorectal tissues. (E) qRT-PCR analysis of ADHFE1 expression in 10 paired CRC tissues; ADHFE1 was quantified relative to the matched adjacent no tumor tissues. Error bars represent means ± SD calculated from three parallel experiments. |
Next, IHC was performed to detect the expression of ADHFE1 in 80 paraffin-embedded CRC samples. The result showed that the expression of ADHFE1 in CRC tissue was down-regulated by 81.3% (65/80) in CRC patients. The positive ADHFE1 expression was mainly localized in cytoplasm, as marked by yellow-brown staining (Figure 1D). And the low expression level of ADHFE1 protein was significantly correlated with poor differentiation and advanced TNM stage (p<0.05) (Table 1). Moreover, the mRNA expression of ADHFE1 was significantly down-regulated in 10 CRC tissues compared to their adjacent normal mucosa using qRT-PCR (Figure 1E).
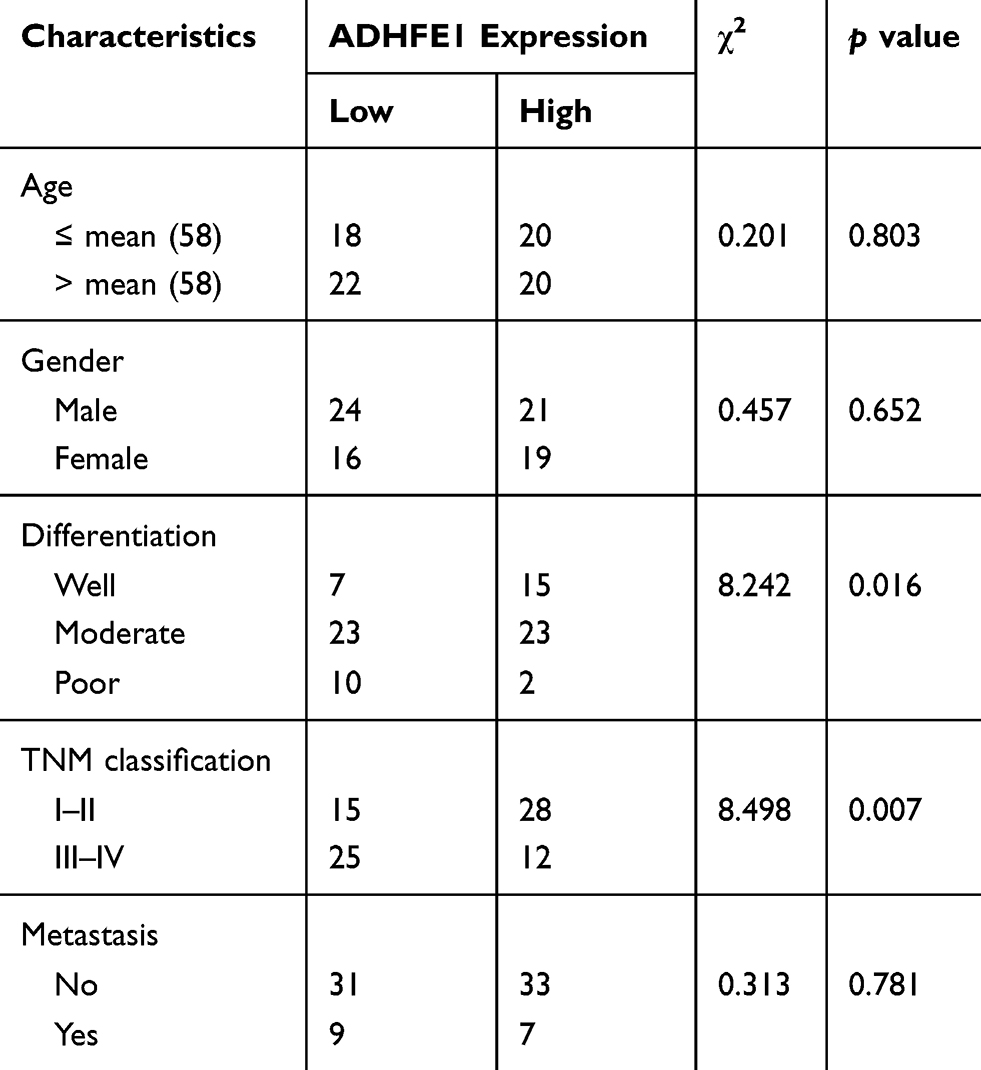 |
Table 1 The Relationship Between The Expression Of ADHFE1 And Clinicopathological Parameters |
Up-Regulation Of ADHFE1 Inhibits The Proliferation Of CRC Cells And Vice Versa
The expression of ADHFE1 was detected in different CRC cell lines (Figure 2A). Then we established stable expression cell lines according to its expression. The result of Western blot suggested that stable CRC cell lines with ADHFE1 over-expression and knockdown were successfully established (Figure 2B).
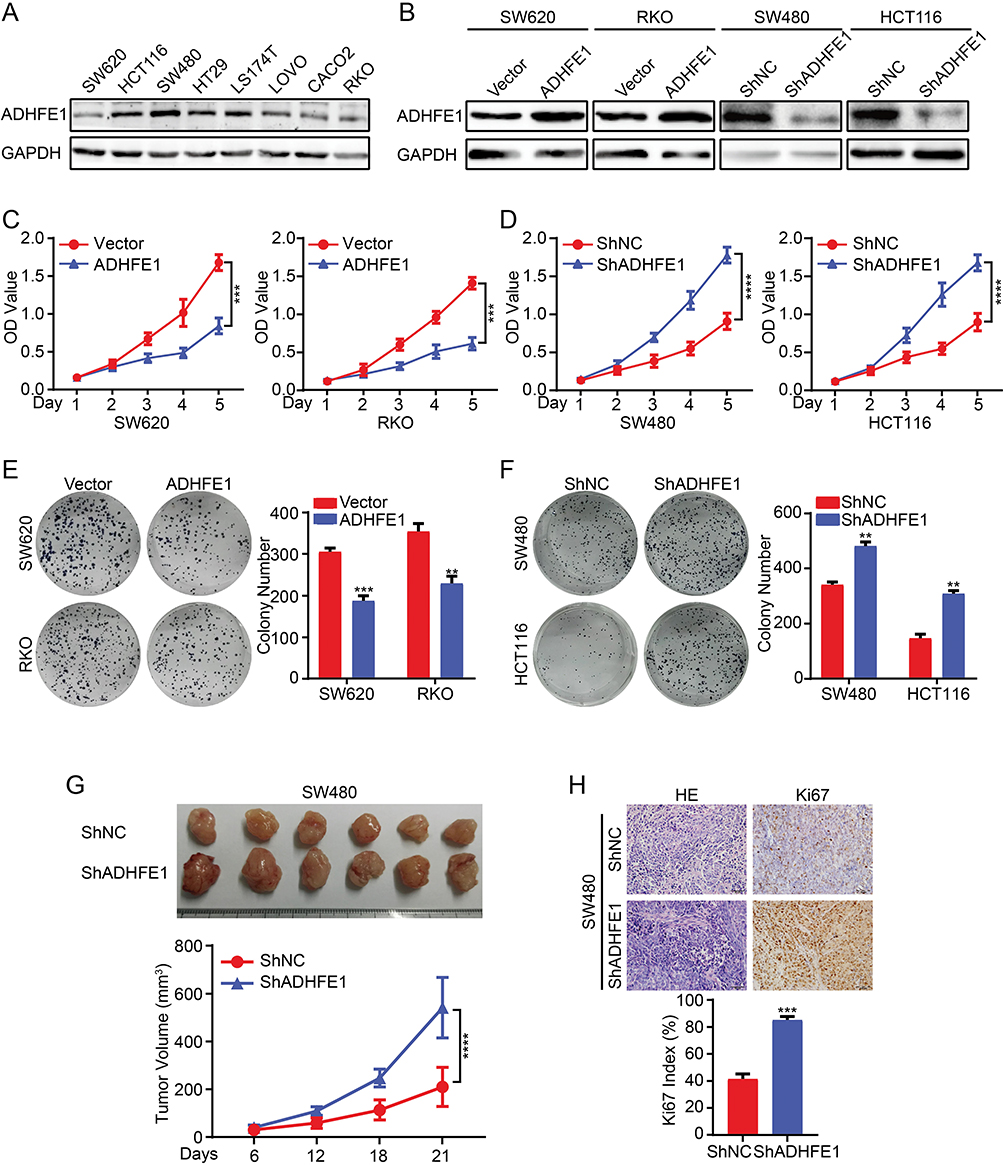 |
Figure 2 Exogenous ADHFE1 knockdown promotes the proliferation of CRC cells, and the upregulation of ADHFE1 inhibits the proliferation of CRC cells. (A) Expression analyses of ADHFE1 protein in different CRC cells using western. GAPDH was used as a loading control. (B) Western blot analysis of the overexpression and knockdown of ADHFE1 in CRC cell lines. GAPDH was used as a loading control. (C and D) CCK8 analyses of the CRC cell proliferation with ADHFE1 overexpression or knockdown. (E and F) Colony formation analyses of the CRC cell proliferation with ADHFE1 overexpression or knockdown. (G) The xenograft models were generated after injecting SW480/ShNC and SW480/ShADHFE1 cells in nude mice (n = 6/group). The tumor volumes were measured on the indicated days. The data points represent the mean tumor volumes ± SD. (H) The sections of tumor were subjected to H&E staining or IHC staining using an antibody against Ki-67. Error bars represent the means ± SD from three independent experiments. **p<0.01, ***p<0.001, ****p<0.0001. |
Subsequently, we detected the effects of ADHFE1 on the proliferation of CRC cells in vitro. The results of CCK8 cell proliferation assay showed that the proliferative ability of CRC cells was significantly inhibited when ADHFE1 was overexpressed, while the proliferation of CRC cells increased dramatically when ADHFE1 was knockdown (Figure 2C and D). The results of colony formation assay illustrated that the colony numbers were much less in ADHFE1 stably expressed cell lines compared with the control group, while the colony numbers were much more when ADHFE1 was stably knockdown (Figure 2E and F). Moreover, we performed xenograft growth assay to detect the effects of ADHFE1 in vivo. Consistent with in vitro investigations, the subcutaneous tumors generated from SW480/ShADHFE1 cells were larger than those derived from SW480/ShNC cells (Figure 2G). IHC staining confirmed that a higher Ki-67 index could be found in the tumors formed by SW480/ShADHFE1 CRC cells than that from the control group (Figure 2H).
ADHFE1 Modulates Cell Cycle Progression In CRC
GSEA (Gene Set Enrichment Analysis) was performed to investigate the enrichment of gene signatures in CRC with low ADHFE1 expression (GSE13067 and GSE13294). The results suggested that cell cycle related gene sets “REACTOME_CELL_CYCLE” and “REACTOME_G1_S_TRANSTION” was significantly enriched in CRC tissues when the expression of ADHFE1 was low (Figure 3A).
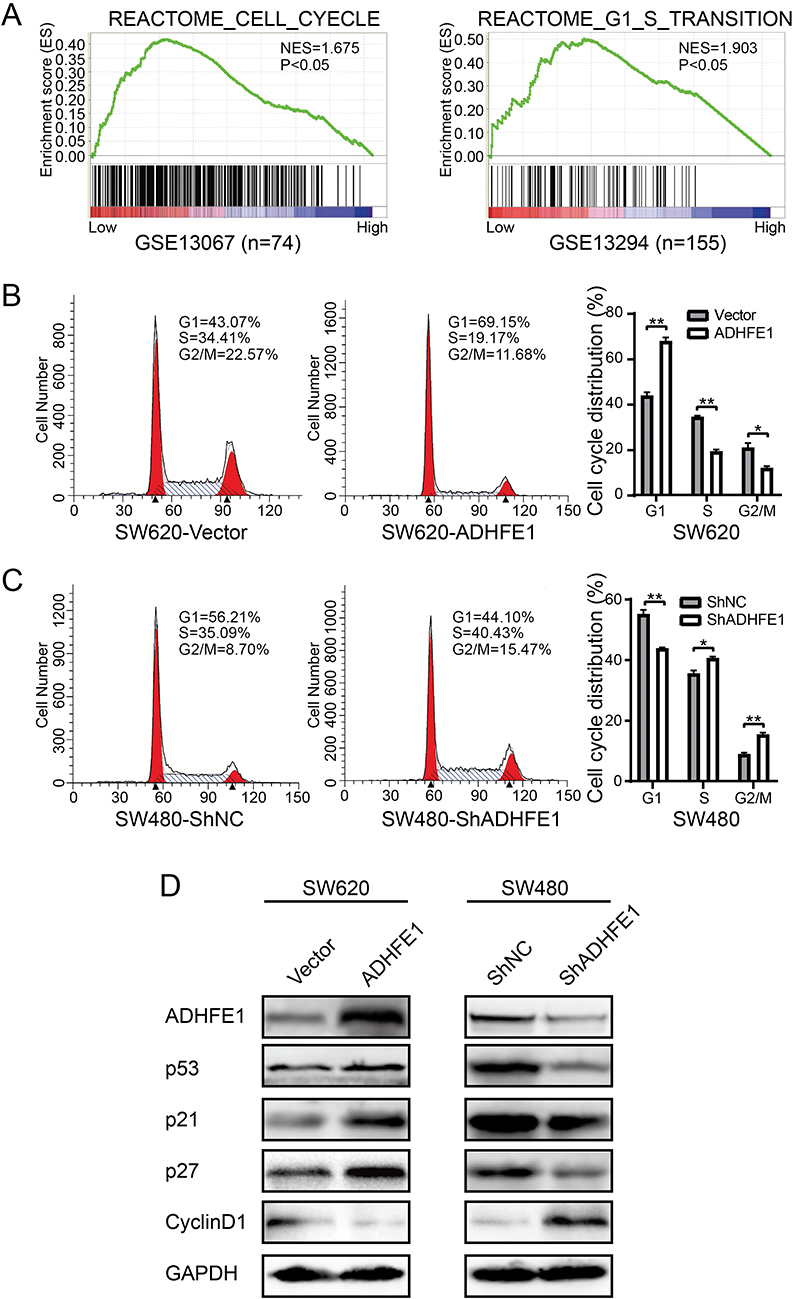 |
Figure 3 ADHFE1 modulates cell cycle progression. (A) GSEA analyses of the “REACTOME_CELL_CYCLE” and “REACTOME_G1_S_TRANSITION” gene sets in the low versus high expression group of ADHFE1 in CRC. (B and C) Flow-cytometry analyses of the cell cycle progression in the indicated CRC cells. Error bars represent the means ± SD from three independent experiments. *p<0.05, **p<0.01. (D) Western blot analyses of p53, p21, p27 and CyclinD1 in the indicated cells. GAPDH was used as a loading control. |
Next, flow cytometry was utilized to assess the numbers of cells in G1, S or G2/M phase. And we found that SW620/ADHFE1 cells had a larger proportion in G1 phase and had a smaller proportion in S and G2/M phases, which was an indication of a significant G1-S cell cycle arrest compared with the control group (Figure 3B). However, the percentage of cells in the G1 phase reduced evidently and the percentage of cells in S and G2/M phase increased significantly with the depletion of ADHFE1 (Figure 3C). The result of Western blot also showed that CyclinD1 was significantly down-regulated, whereas p53, p21 and p27 were strikingly up-regulated when ADHFE1 was overexpressed and vice versa (Figure 3D).
ADHFE1 Is Down-Regulated In CRC Contributing To Methylation Changes In The Gene Promoter
Mutation, deletion, and epigenetic alterations are the most common factors in the inactivation of tumor suppressors. We analyzed the mutation and CNV (copy, number, variation) status of ADHFE1 in CRC using cBioPortal for Cancer Genomics (http://www.cbioportal.org/), and observed that there were no significant mutation and CNV of ADHFE1 in CRC (Supplementary Figure S1). These results suggested other factors might participate in the inactivation of ADHFE1.
Therefore, we analyzed the methylation status of ADHFE1 promoter. We analyzed CpG islands in the promoter of ADHFE1 using the UCSC database (http://genome.ucsc.edu/). Next, we detected the methylation status of the CpG island in 5 paired fresh CRC tissues using the bisulfite genomic sequence (BSP) assay. The results illustrated that a higher degree of CpG island methylation could be found in ADHFE1 promoter in CRC tissues compared with the normal colorectal tissues (Figure 4A and B).
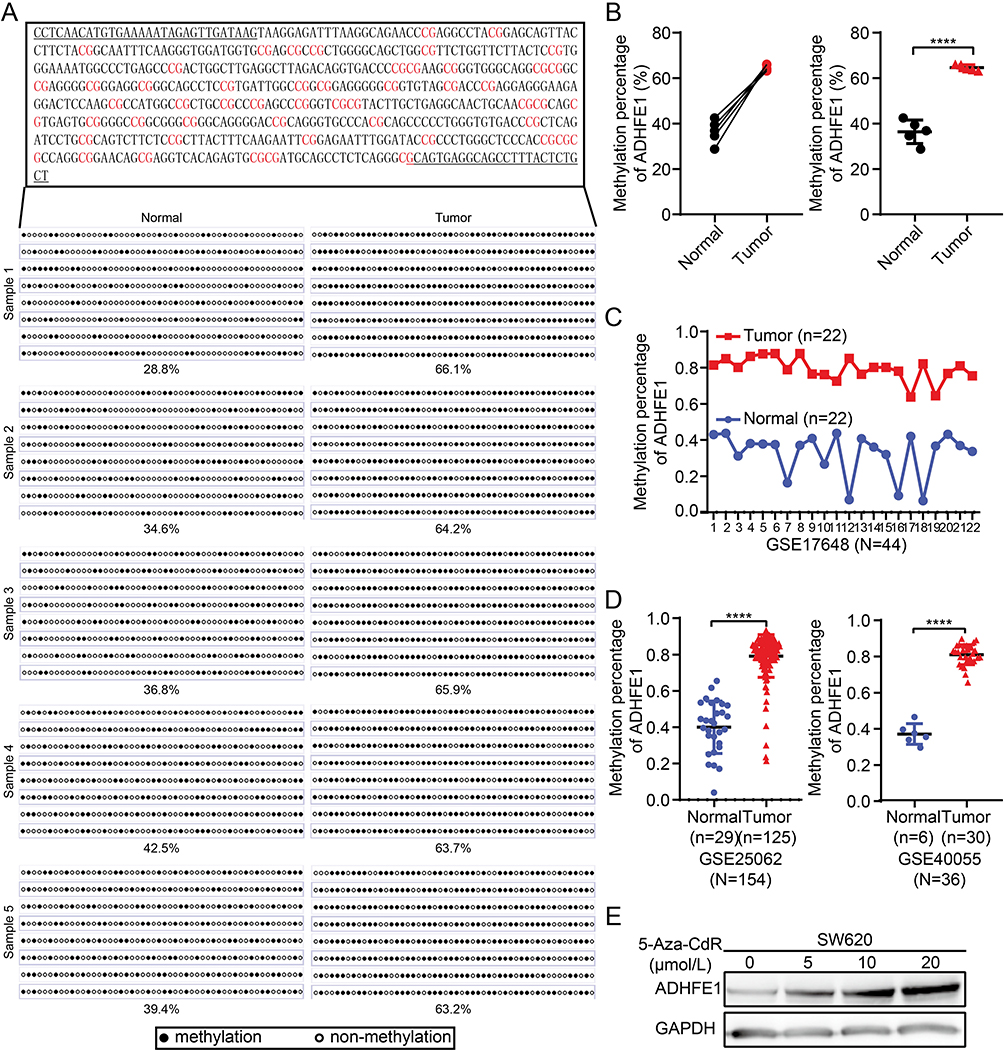 |
Figure 4 ADHFE1 is hypermethylated in CRC. (A and B) The degree of CpG island methylation of ADHFE1 promoter in 5 paired CRC tissues. (C and D) The results of GEO analyses of the methylation level in the promoter of ADHFE1 in CRC compared with that in the normal intestinal mucosa. (E) Treatment with a DNA demethylation agent 5-Aza-CdR increased ADHFE1 expression at different levels in the CRC cell line SW620. ****p<0.0001. |
The analysis results of GEO data also showed that the methylation status of ADHFE1 promoter was significantly higher in CRC tissues than that in the normal colorectal tissues (Figure 4C and D). Furthermore, we used demethylation drug 5-Aza-CdR to treat the CRC cells and observed the expression of ADHFE1. The result of Western blot revealed that the expression of ADHFE1 increased in CRC cells treated with 5-Aza-CdR in a concentration-dependent manner (Figure 4E). Moreover, CyclinD1 was down-regulated, p53, p21 and p27 were up-regulated significantly in CRC cells after demethylation treatment (Supplementary Figure S2).
Discussion
CRC is one of the most common malignant tumors with high mobility and mortality.20 Numerous studies reveal that many gene alterations are involved in the initiation and progression of CRC.4,21,22 ADHFE1 is a transcriptional differentially expressed gene (DEG) that was identified using CRC mRNA sequencing data from The Cancer Genome Atlas (TCGA) and GEO. ADHFE1 is mapped to human chromosome 8q13.1 and encodes the protein which belongs to the iron-activated alcohol dehydrogenase family.11 It is reported that ADHFE1 is down-regulated in CRC and its expression is correlated with the differentiation of CRC.15 Our results showed that ADHFE1 was significantly down-regulated in CRC tissues by using IHC, and the down-regulation of ADHFE1 was correlated with poor differentiation and advanced TNM stage of CRC patients. which is consistent with the previous studies. Therefore, ADHFE1 has the potential to be a valuable tumor suppressor in CRC.
Abnormal and uncontrolled cell proliferation is a hallmark of cancer and is caused by the misregulation of various crucial protein expressions.23,24 The sustaining proliferation of cells will ultimately cause the initiation and progression of CRC.25,26 It is demonstrated that ADHFE1 knockdown promotes CRC cell proliferation in vitro by transiently transfecting siRNAs.27 Here, we showed that stable overexpression of ADHFE1 inhibited CRC cell proliferation in vitro, while permanent ADHFE1 knockdown promoted CRC cell proliferation in vitro and in vivo. However, the underlying mechanisms of ADHFE1 in CRC remain poorly identified.
Cell cycle is usually dysregulated in various cancer cells, thereby breaking homeostasis of cell number and causing uncontrolled cell proliferation.28 In our investigations, we found an enrichment of gene signatures which promote the cell cycle progression with low expression of ADHFE1 by using the GSEA (Gene Set Enrichment Analysis). Our results also illustrated that enforced expression of ADHFE1 decelerated the G1–S transition, while ADHFE1 knockdown promoted the G1-S transition with the down-regulation of p53, p21 and p27 and up-regulation of Cyclin D1. Thus, down-regulation of ADHFE1 may promote the proliferation of CRC cells via promoting cell cycle progression. However, the detailed underlying mechanisms still need to be further investigated.
Recently, studies reveal that the inactivation of tumor suppressor genes may be caused by epigenetic modifications, such as promoter hypermethylation.9,29,30 And ADHFE1 promoter is aberrantly hypermethylated in CRC by a genome-wide methylation profiling screen using an array-based chip assay.13,31 The result of GEO CRC microarray dataset analysis also suggested that ADFHE1 promoter was hypermethylated in CRC compared to the normal tissues. In consistent with the previous studies and the bioinformatics analysis, we observed that the CpG island methylation status of ADHFE1 promoter is higher in CRC tissues than that in the adjacent normal mucosa, and the loss expression of ADHFE1 was associated with the degree of methylation in CRC. These results suggested that promoter hypermethylation of the gene may be a major factor in the inactivation of ADHFE1.
In conclusion, we found that the expression of ADHFE1 was significantly down-regulated in CRC tissues compared with that in adjacent normal mucosa. And ADHFE1 overexpression inhibited the proliferation of CRC cells, while ADHFE1 knockdown promoted CRC cell proliferation. Moreover, hypermethylation and the deletion of ADHFE1 promoted cell cycle progression, and finally facilitated the proliferation of CRC cells (Figure 5). Restoration of ADHFE1 might provide a new therapeutic approach enabling cancer precision medicine for CRC patients.
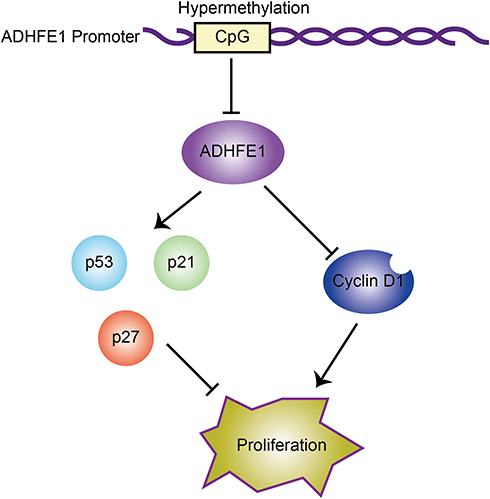 |
Figure 5 Proposed model: hypermethylation of ADHFE1 facilitates the proliferation of CRC cells via modulating the proteins which are essential in cell cycle progression. |
Disclosure
The authors report no conflicts of interest in this work.
References
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. doi:10.3322/caac.21492
2. Migliore L, Migheli F, Spisni R, Coppede F. Genetics, cytogenetics, and epigenetics of colorectal cancer. J Biomed Biotechnol. 2011;2011:792362.
3. Ogino S, Lochhead P, Giovannucci E, Meyerhardt JA, Fuchs CS, Chan AT. Discovery of colorectal cancer PIK3CA mutation as potential predictive biomarker: power and promise of molecular pathological epidemiology. Oncogene. 2014;33(23):2949–2955. doi:10.1038/onc.2013.244
4. Cancer Genome Atlas N. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487(7407):330–337. doi:10.1038/nature11252
5. Zhang B, Wang J, Wang X, et al. Proteogenomic characterization of human colon and rectal cancer. Nature. 2014;513(7518):382–387. doi:10.1038/nature13438
6. Liu Y, Sethi NS, Hinoue T, et al. Comparative molecular analysis of gastrointestinal adenocarcinomas. Cancer Cell. 2018;33(4):721–735 e728. doi:10.1016/j.ccell.2018.03.010
7. Robertson KD, Jones PA. DNA methylation: past, present and future directions. Carcinogenesis. 2000;21(3):461–467. doi:10.1093/carcin/21.3.461
8. Yang X, Han H, De Carvalho DD, Lay FD, Jones PA, Liang G. Gene body methylation can alter gene expression and is a therapeutic target in cancer. Cancer Cell. 2014;26(4):577–590. doi:10.1016/j.ccr.2014.07.028
9. Pfeifer GP. Defining driver DNA methylation changes in human cancer. Int J Mol Sci. 2018;19(4):1166.
10. Ye YP, Jiao HL, Wang SY, et al. Hypermethylation of DMTN promotes the metastasis of colorectal cancer cells by regulating the actin cytoskeleton through Rac1 signaling activation. J Exp Clin Cancer Res. 2018;37(1):299.
11. Deng Y, Wang Z, Gu S, et al. Cloning and characterization of a novel human alcohol dehydrogenase gene (ADHFe1). DNA Sequence. 2002;13(5):301–306.
12. Lyon RC, Johnston SM, Panopoulos A, Alzeer S, McGarvie G, Ellis EM. Enzymes involved in the metabolism of gamma-hydroxybutyrate in SH-SY5Y cells: identification of an iron-dependent alcohol dehydrogenase ADHFe1. Chem Biol Interact. 2009;178(1–3):283–287. doi:10.1016/j.cbi.2008.10.025
13. Kim YH, Lee HC, Kim SY, et al. Epigenomic analysis of aberrantly methylated genes in colorectal cancer identifies genes commonly affected by epigenetic alterations. Ann Surg Oncol. 2011;18(8):2338–2347. doi:10.1245/s10434-011-1573-y
14. Naumov VA, Generozov EV, Zaharjevskaya NB, et al. Genome-scale analysis of DNA methylation in colorectal cancer using Infinium HumanMethylation450 BeadChips. Epigenetics. 2013;8(9):921–934. doi:10.4161/epi.25577
15. Tae CH, Ryu KJ, Kim SH, et al. Alcohol dehydrogenase, iron containing, 1 promoter hypermethylation associated with colorectal cancer differentiation. BMC Cancer. 2013;13:142.
16. Lu YX, Yuan L, Xue XL, et al. Regulation of colorectal carcinoma stemness, growth, and metastasis by an miR-200c-Sox2-negative feedback loop mechanism. Clin Cancer Res. 2014;20(10):2631–2642. doi:10.1158/1078-0432.CCR-13-2348
17. Liao WT, Li TT, Wang ZG, et al. microRNA-224 promotes cell proliferation and tumor growth in human colorectal cancer by repressing PHLPP1 and PHLPP2. Clin Cancer Res. 2013;19(17):4662–4672. doi:10.1158/1078-0432.CCR-13-0244
18. Zhang Z, Zhou C, Chang Y, et al. Long non-coding RNA CASC11 interacts with hnRNP-K and activates the WNT/beta-catenin pathway to promote growth and metastasis in colorectal cancer. Cancer Lett. 2016;376(1):62–73. doi:10.1016/j.canlet.2016.03.022
19. Hu YH, Chen Q, Lu YX, et al. Hypermethylation of NDN promotes cell proliferation by activating the Wnt signaling pathway in colorectal cancer. Oncotarget. 2017;8(28):46191–46203. doi:10.18632/oncotarget.17580
20. Siegel RL, Miller KD, Fedewa SA, et al. Colorectal cancer statistics, 2017. CA Cancer J Clin. 2017;67(3):177–193. doi:10.3322/caac.21395
21. Jiao HL, Ye YP, Yang RW, et al. Downregulation of SAFB sustains the NF-kappaB pathway by targeting TAK1 during the progression of colorectal cancer. Clin Cancer Res. 2017;23(22):7108–7118. doi:10.1158/1078-0432.CCR-17-0747
22. Wang S, Xiao Z, Hong Z, et al. FOXF1 promotes angiogenesis and accelerates bevacizumab resistance in colorectal cancer by transcriptionally activating VEGFA. Cancer Lett. 2018;439:78–90. doi:10.1016/j.canlet.2018.09.026
23. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. doi:10.1016/j.cell.2011.02.013
24. Gu Y, Wang Q, Guo K, et al. TUSC3 promotes colorectal cancer progression and epithelial-mesenchymal transition (EMT) through WNT/beta-catenin and MAPK signalling. J Pathol. 2016;239(1):60–71. doi:10.1002/path.4697
25. Lin C, Zhang J, Lu Y, et al. NIT1 suppresses tumour proliferation by activating the TGFbeta1-Smad2/3 signalling pathway in colorectal cancer. Cell Death Dis. 2018;9(3):263. doi:10.1038/s41419-018-1111-y
26. Cui YM, Jiang D, Zhang SH, et al. FOXC2 promotes colorectal cancer proliferation through inhibition of FOXO3a and activation of MAPK and AKT signaling pathways. Cancer Lett. 2014;353(1):87–94. doi:10.1016/j.canlet.2014.07.008
27. Moon JW, Lee SK, Lee YW, et al. Alcohol induces cell proliferation via hypermethylation of ADHFE1 in colorectal cancer cells. BMC Cancer. 2014;14:377.
28. Evan GI, Vousden KH. Proliferation, cell cycle and apoptosis in cancer. Nature. 2001;411(6835):342–348. doi:10.1038/35077213
29. Fatemi M, Paul TA, Brodeur GM, Shokrani B, Brim H, Ashktorab H. Epigenetic silencing of CHD5, a novel tumor-suppressor gene, occurs in early colorectal cancer stages. Cancer. 2014;120(2):172–180. doi:10.1002/cncr.28316
30. Zhang L, Li W, Cao L, et al. PKNOX2 suppresses gastric cancer through the transcriptional activation of IGFBP5 and p53. Oncogene. 2019.
31. Oster B, Thorsen K, Lamy P, et al. Identification and validation of highly frequent CpG island hypermethylation in colorectal adenomas and carcinomas. Int J Cancer. 2011;129(12):2855–2866. doi:10.1002/ijc.25951
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.