")
Back to Journals » Diabetes, Metabolic Syndrome and Obesity » Volume 13
Hydrogen Sulfide, Adipose Tissue and Diabetes Mellitus
Authors Zhu L, Yang B, Ma D, Wang L, Duan W
Received 13 February 2020
Accepted for publication 9 May 2020
Published 3 June 2020 Volume 2020:13 Pages 1873—1886
DOI https://doi.org/10.2147/DMSO.S249605
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Ming-Hui Zou
Lin Zhu,1 Bo Yang,2 Dongxia Ma,3 Lan Wang,4 Wu Duan5
1Department of Pediatrics, Tongji Hospital, Tongji Medicine College, Huazhong University of Science and Technology, Wuhan 430030, People’s Republic of China; 2Institute of Organ Transplantation, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan 430030, People’s Republic of China; 3Department of Allergy, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, People’s Republic of China; 4Reproductive Medicine Center, Tongji Hospital, Tongji Medicine College, Huazhong University of Science and Technology, Wuhan 430030, People’s Republic of China; 5Division of Endocrinology, Department of Internal Medicine, Qilu Hospital of Shandong University, Jinan 250012, People’s Republic of China
Correspondence: Wu Duan
Division of Endocrinology, Department of Internal Medicine, Qilu Hospital of Shandong University, Jinan 250012, People’s Republic of China
Tel/ Fax +86-531-8692-7544
Email [email protected]
Abstract: Hydrogen sulfide (H2S) is now increasingly considered to be the third gasotransmitter alongside other gaseous signaling molecules, nitric oxide (NO) and carbon monoxide (CO). H2S is produced by a variety of endogenous enzymatic and non-enzymatic pathways and acts as a modulator of the physiological and pathological events of the body. Adipocytes express the cystathionine γ lyase (CSE)/H2S system, which modulates a variety of biological activities in adipose tissue (AT), including inflammation, apoptosis, insulin resistance, adipokine secretion and adipocyte differentiation. Abnormalities in the physiological functions of AT play an important role in the process of diabetes mellitus. Therefore, this review provides an overview of the general aspects of H2S biochemistry, the effect of H2S on AT function and diabetes mellitus and its molecular signalling mechanisms as well as the potential application of H2S in pharmacotherapy.
Keywords: hydrogen sulfide, adipose tissue, diabetes mellitus, drug therapy
Introduction
H2S, a toxic gas with a rotten egg flavour used as a chemical reagent, was first proposed by Abe and Kimura in 1996 to be an endogenous neuromodulator.1 Considerable evidence suggests that H2S is produced endogenously in many parts of the body and participates in various physiological and pathological functions in mammals.2 Altered levels of endogenous H2S and H2S synthetase expression have been observed in diabetic patients and animals. The blood H2S levels in diabetic patients were found to be significantly lower than those in age-matched normal control subjects.3 However, YusufM4 investigated the increased H2S production rate in the pancreatic islets and livers of diabetic rats. Zucker diabetic fatty (ZDF) rats have significantly higher levels of pancreatic CSE expression and H2S production when compared with Zucker fatty (ZF) or Zucker lean (ZL) rats.5 Increasing numbers of studies have shown that H2S is involved in the development of diabetes,6 however, its impact on diabetes now appears to be contradictory. The dysfunction of AT plays an important role in the development of diabetes. We will briefly review the effect of H2S on diabetes by discussing the influence of H2S on the function of AT.
Hydrogen Sulfide Biosynthesis and Catabolism
H2S is a colourless, flammable gas with a strong odour of rotten eggs. It is produced by cystathionine β-synthetase (CBS) in the mammalian brain, selectively enhances NMDA (N-methyl-D-aspartate) receptor-mediated responses and facilitates the induction of long-term potentiation in the hippocampus, which was the first proposed biological role of H2S as a neuromodulator.1 Several lines of evidence have suggested that H2S is naturally synthesized by four enzymes: CBS, CSE, 3-mercaptopyruvate sulfurtransferase (3-MST) and cysteine aminotransferase (CAT). It is produced in vivo from L-cysteine (LC) by those enzymes.6,7 Those enzymes are constitutively present and can be found widely in cells and tissues, and their expression can be induced by a number of disease states.8
Four enzymatic pathways for H2S production have been described thus far. Two of them are catalysed by CBS and CSE, which are pyridoxal 5ʹ-phosphate (vitamin B6)-dependent cytoplasmic enzymes of the trans-sulfuration pathway. The third pathway for H2S generation requires two enzymes, CAT and 3-MST. In contrast with CBS and CSE, this pathway operates primarily in the mitochondria6 (Figure 1). Finally, H2S in the cerebellum and the kidney can be synthesized from D-cysteine by the synergism of D-amino acid oxidase (which is highly expressed in the peroxisomes of these organs) and 3-MST.9 The degradation pathways involved in H2S consumption are mediated by ethylmalonic encephalopathy protein 1 (ETHE1), mitochondrial sulfide quinone oxidoreductase (SQR) and cysteine dioxygenase (CDO).10,11
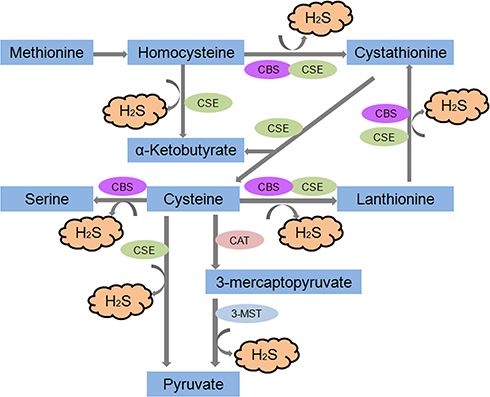 |
Figure 1 Generalised overview of H2S production in the cell. The endogenous production of H2S occurs via two main pathways-reverse transsulfuration and cysteine oxidation which take place partly inside mitochondria. |
Changes in the levels of H2S or H2S synthetase expression may contribute to the pathogenesis of many pathophysiological processes, such as neurological systems, inflammation, apoptosis, vascular function, energy metabolism and biogenesis, obesity and ageing.6 The pharmacological or genetic inhibition or activation of H2S production is protective in some models. Therefore, the application of H2S donors and/or the augmentation of endogenous H2S or H2S inhibitors and/or the reduction in endogenous H2S has attracted much attention as possible therapeutic approaches.
Hydrogen Sulfide and Diabetes
Endogenous H2S can be synthesized by multiple systems, with the highest rates occurring in the brain, cardiovascular system, liver and kidney.11 At present, studies have shown that almost every mammalian tissue cell can produce a certain amount of H2S. Under physiological conditions, the H2S concentration in rat brain tissue is 50–160 μmol/L, and the serum H2S concentration is approximately 46 μmol/L.12 Human blood contains a significant amount (10–100 μmol/L) of H2S.13
Altered endogenous H2S levels, and the expression of H2S synthetases have been observed in diabetic animals and patients.6 YusufM4 first reported that a marked increase in H2S synthesis from exogenous LC in the pancreas and liver is associated with streptozotocin (STZ)-induced diabetes in rat, and the liver pancreatic CBS and CSE mRNA levels were concomitantly increased, with no change in the pancreatic CSE mRNA levels in diabetic rats. However, the plasma H2S concentrations were similar in diabetic and non-diabetic animals. Therefore, the enhanced formation of H2S is likely to be a local tissue response to diabetes, which is not reflected in increased circulating plasma levels. Other researchers5 have also indicated that the expression of CSE and the production rate of H2S in pancreatic islets were significantly higher in ZDF rats than in ZL or ZF rats. The serum H2S levels of ZDF rats were also significantly higher than those of ZF or ZL rats. Increased pancreatic H2S production underlies the reduced insulin release from the pancreatic β cells of ZDF rats. The activation of K+ATP channels by high levels of endogenous H2S in pancreatic β cells may be the molecular basis underlying H2S-suppressed insulin release.5 It is well-known that H2S functions as an endogenous activator of K+ATP channels in β cells.14 Further research15 has found that the CSE/H2S system plays a critical role in regulating cell functions by stimulating β cell apoptosis and inducing K+ATP channel activity and that STZ-induced diabetes is largely mediated by the effects of pancreas-produced H2S on β cell mass and K+ATP channel activity. A CSE deficiency significantly delayed the development of T1DM induced by STZ.16
Interestingly, the results of other studies are diametrically opposite. Brancaleone et al17 found that the plasma H2S value in NDR (non-obese resistant) mice was approximately 50 μmol/L, whereas NOD (non-obese diabetic) mice exhibited a progressive decrease in plasma H2S levels that paralleled the disease severity, reaching a 50% reduction in NOD III mice and indicating that endogenous H2S production is significantly impaired under hyperglycaemic conditions. A further study found that the blood H2S levels of diabetic patients were significantly decreased when compared with age-matched normal control subjects, which is in accordance with their study of rats.3 Recently, Kunihiro et al13 investigated the plasma H2S levels in Japanese individuals with type 2 diabetes mellitus and found that the patients showed a progressive decrease in the plasma H2S levels (45.1±15.5 μmol/L versus 54.0±26.4 μmol/L), which is in accordance with poor glycaemic control. There was a significant correlation between the reduction in plasma H2S levels and the level of HbA1c. They also found that the reduction in plasma H2S levels was associated with a history of cardiovascular disease in type 2 diabetic patients.
The above study shows that H2S may play a role in the development of diabetes. Researchers have observed different changes in H2S levels in different diabetic patients and animals, possibly due to the different stages of diabetes in the diabetic patients studied by different researchers, the different methods of inducing diabetic animals and the use of different animal species.
Hydrogen Sulfide and Adipose Tissue
Diabetes-related obesity and general obesity are characterized by an increase in the AT mass, which can be defined as an increase in the BMI. In addition to the increase in AT mass (eg, increase in the size and number of adipocytes), histological and macroscopic changes and AT dysfunction, including hypersecretion of proinflammatory, proatherogenic and prodiabetic adipocytokines, occurs in obesity.18–20 Feng et al21 first identified the endogenous CSE/H2S pathway in AT. They detected the rate of H2S production from visceral AT, including perirenal fat (2.93 ± 0.27 nmol/min/mg protein), epididymal fat (4.76 ± 0.92 nmol/min/mg protein) and brown fat tissue (4.65 ± 0.81 nmol/min/protein). RT-PCR results revealed that CBS and CSE mRNA were expressed in those fat tissues and illuminated the CSE-mediated primary pathway of H2S generation in AT. Meanwhile, they found H2S-generation rates in adipocytes and preadipocytes of 2.89±1.34 and 2.17±1.14 nmol/min/mg protein, respectively, and demonstrated that adipocytes also endogenously produce H2S through a CSE catalytic pathway. Furthermore, they found that H2S production and CSE protein expression in the epididymal fat pad and in perirenal fat increased in an age-dependent manner. The same year, Fang et al22 first demonstrated that CSE protein expression and endogenous H2S production in rat PVAT (perivascular adipose tissue) were detectable and that the endogenous H2S generated by PVAT was predominantly CSE-catalysed.
In recent years, as increasing research has focused on AT-derived H2S and explored the pathophysiological roles of H2S in AT,23 we have uncovered the effects of H2S on AT inflammation, apoptosis, adipokine secretion, glucose and lipid metabolism and vascular tension.
Hydrogen Sulfide and Inflammation of Adipose Tissue
A series of epidemiological studies have shown that circulating inflammatory markers are closely related to type 2 diabetes mellitus (T2DM) and are risk factors for the development of future T2DM.24 Numerous preclinical and clinical studies now strongly support the view that obesity-induced inflammation plays an important role in the development of insulin resistance and T2DM.25,26 Although obesity-induced inflammation is similar in many ways to inflammation observed in classical immunity, it is a low-grade form of inflammation that produces much lower levels of circulating cytokines.27 It is also considered to be chronic inflammation, as it requires relatively long dietary therapy before it becomes clearly discernible in AT, which displays the most severe obesity-induced inflammation of all insulin-responsive tissues (as characterized by the increased cytokine/chemokine expression and immune cell infiltration).28 Indeed, AT produces high levels of many inflammatory cytokines in obesity and is therefore considered to be the major inflammatory organ that mediates obesity-induced inflammation. Chronic inflammation and metabolic detrimental factors released by AT into the circulation are associated with some metabolic complications of obesity, such as T2DM and atherosclerosis.29 AT inflammation is frequently observed in obesity and diabetes, is associated with the infiltration of macrophages, and may be caused by the death of adipocytes, the secretion of adipokines, such as TNF-α and IL-6, and adipocyte chemokines, such as monocyte chemo-attractant protein-1 (MCP-1) (Figure 2A).30,31
The gaseous signalling molecule H2S has been identified as an important inflammatory mediator. At present, several research groups worldwide are focused on determining the role of H2S in inflammation. H2S has different effects on inflammation in different models.32–34 The observation that an H2S donor significantly attenuates the production of cytokines triggered by the pro-inflammatory stimuli of primary macrophages35 and macrophage cell lines36 supports the anti-inflammatory action of H2S in macrophages. These findings are particularly intriguing for data that point to impaired H2S signalling in type 2 diabetes3,17,37–39 and show a decrease in the plasma concentration of H2S in human diabetes and obesity.40
The overexpression of CSE and exogenous application of NaHS both increased H2S concentrations and significantly inhibited high glucose (HG)-induced MCP-1 secretions in mature 3T3-LI adipocytes, indicating that H2S exhibited anti-inflammatory effects in mature adipocytes.41 A further study42 found that obesity increased the production of H2S while also increasing the consumption of H2S likely due to the promotion of H2S catabolism by radical species (the reactive oxygen and nitrogen species). In addition, H2S depletion was observed in chronic inflammation models [AT macrophages (ATMs) from obese mice]. In obesity, store-operated Ca2+ entry (SOCE) increased concurrently with the consumption of H2S, and a reduction in the concentration of cellular H2S promotes an increase in SOCE.43 The activation of the SOCE pathway during pro-inflammatory stimulation provides the Ca2+ signals required for the activation of nuclear factor of activated T cells (NFAT) and nuclear factor κB (NFκB), the transcription factors responsible for the expression of cytokine encoding genes.43,44 During obesity, an increased abundance of M1-associated markers, reduced H2S bioavailability and increased SOCE were observed in ATMs. The inhibition of SOCE or the prevention of H2S loss from these cells also limited pro-inflammatory cytokine production. In parallel with the study by Pan,41 this research implied that H2S plays an anti-inflammatory role in the ATMs from obese mice (Figure 2B).
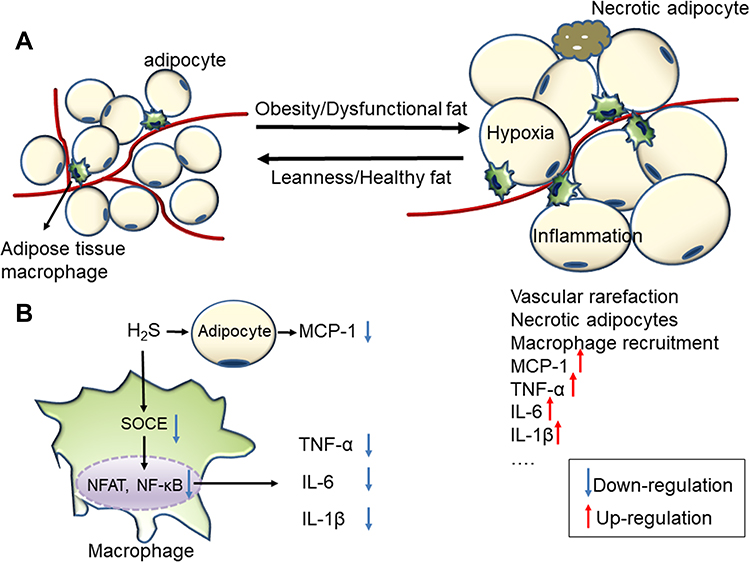 |
Figure 2 (A) Healthy and obese adipose tissue. Obese adipose tissue is characterized by inflammation and progressive infiltration by macrophages as obesity develops. (B) The anti-inflammation effect of H2S on obese adipose tissue. |
Those reports suggested that therapeutic strategies designed to maintain H2S bioavailability may be effective in a number of inflammatory pathological contexts. However, further studies are required to specifically examine the effects of H2S on AT inflammation in diabetes, obesity and other pathological conditions.
Hydrogen Sulfide and Insulin Resistance of Adipose Tissue
T2DM consists of two independent but related alterations: insulin resistance and dysfunction of the β-cells in the pancreas. AT is an insulin-sensitive organ that mediates glucose uptake and metabolism45 and is an important source of methionine metabolism for the synthesis of fatty acids.46,47 Increasing evidence has shown that excess AT accumulation alters AT secretion profiles,48 and only a few adipokines have been shown to regulate insulin sensitivity, including TNF-α, adiponectin, interleukin-6, resistin and leptin (Figure 3A). TNF-α provides a molecular basis for insulin resistance and contributes by inhibiting the expression of genes that are essential for insulin signalling and adipocyte differentiation, including adiponectin.49 Single-nucleotide polymorphisms (SNPs) in the promoter region of the adiponectin gene may be associated with the development of insulin resistance, obesity, and T2DM.50,51
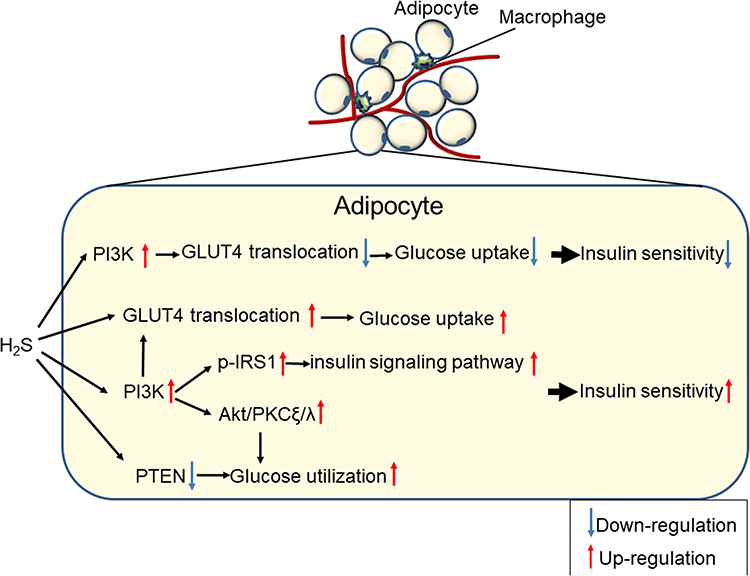 |
Figure 3 The bilateral regulation of H2S on glucose metabolism and insulin sensitivity in adipocytes. |
Researchers have found H2S-reduced insulin sensitivity in adipocytes, and CSE inhibitors might reverse this effect. A high level of glucose (20 mmol/L) inhibited the H2S production of adipocytes in a time- and concentration-dependent manner. Under basal and insulin stimulating conditions, H2S inhibited glucose transport into adipocytes in a concentration-dependent manner. The CSE inhibitors PAG (propargylglycine) and BCA (β-cyano-L-alanine) increased basal and insulin-stimulated adipocyte glucose uptake.21 Further research52 indicated that the upregulation of the endogenous CSE/H2S system contributes to TNF-α induced insulin resistance in 3T3-L1 adipocytes, and exogenous H2S causes a decrease in insulin-stimulated glucose consumption and uptake, which can be blocked by pretreatment with LY294002, a PI3K inhibitor, but not with glibenclamide, a non-selective K+ATP channel inhibitor. Pinacidil, a non-selective K+ATP channel opener, also has no effect on the glucose uptake of adipocytes. Therefore, H2S might inhibit GLUT4 (glucose transporter 4) translocation through the PI3K (phosphatidylinositol 3-kinase) pathway, rather than through the K+ATP channel pathway, to inhibit glucose uptake by adipocytes.21
However, the effect of H2S on the insulin resistance of adipocytes is controversial. Prasenjit et al53 argued that H2S plays an important role in vitamin D-induced GLUT4 translocation to the cell surface for insulin-stimulated glucose uptake and utilization in 3T3-L1 adipocytes. Interestingly, they also indicated that LC and H2S caused an increase in PIP3 (phosphatidylinositol-3,4,5 trisphosphate, a positive regulator of glucose metabolism), Akt (serine/threonine protein kinase) phosphorylation, and glucose utilization in HG (25 mM) treated 3T3-L1 adipocytes.54 In addition, H2S mediated the above effect of LC. H2S or PIP3 increased GLUT4 activation, glucose utilization and the phosphorylation of IRS1 (insulin receptor substrate 1), Akt and PKCξ/λ (protein kinase C ξ/λ) in HG-treated cells. They provide evidence for a molecular mechanism by which H2S can upregulate the insulin-signalling pathways by increasing PIP3 and glucose utilization by inhibiting PTEN (Phosphatase and Tensin Homolog) protein and activating the PI3K/AKT/PKCξ/λ pathway in 3T3-L1 adipocytes.55
Moreover, Rong et al56 found that NaHS treatment may ameliorate insulin resistance by sensitizing insulin receptor (IR)-mediated signals. The phosphorylation of IR, PI3K and Akt increased in adipocytes treated with NaHS. However, the ability of NaHS to activate PI3K was blocked by the IR inhibitor HNMPA (C11H11O4P). In addition, NaHS directly activated the IRs in a cell-free system, indicating that IRs may act as a direct target molecule for NaHS and that PI3K may act as a downstream element of IR in the actions of NaHS. This hypothesis was further supported by the notion that increased NaHS-induced glucose uptake was blocked by siRNA-mediated IR knockdown or by pretreatment with the IR inhibitor HNMPA or the PI3K inhibitor LY294002. NaHS may react with some amino acid residues of IRs, leading to certain chemical modifications of the residues and to subsequent conformational changes in the IRs. Changes in IR conformation induced by NaHS may promote the trans-autophosphorylation of IRs and, thus, enhance IR activation. The stimulating effects of NaHS on the IRs may have potential clinical relevance. Fasting blood glucose levels were decreased and glucose tolerance was increased in GK rats chronically treated with NaHS at a dose of 30 μmol/kg•day. The activation of the PI3K/Akt pathway was also observed in some tissues in GK rats treated with NaHS. Increased insulin sensitivity and reduced plasma fasting glucose levels were also observed in Wistar rats treated with NaHS.56 These data further confirm that the insulin sensitizing effects of exogenous H2S treatment are also present in subjects without diabetes. Another study57 found that H2S ameliorated insulin resistance by upregulating IRS1 protein, and the CSE/H2S system might regulate the gene transcription of glucose metabolic enzymes or transporter proteins through a nuclear receptor.
The bilateral regulation of the CSE/H2S system in glucose metabolism and insulin sensitivity suggested that CSE/H2S might be an energy balancer58 (Figure 3B). Under physiological conditions, the CSE/H2S system tended to reduce energy consumption, resulting in a slight decrease in glucose utilization, whereas under stress or inflammation, the CSE/H2S system antagonized injury and increased glucose utilization, resulting in increasing insulin sensitivity. Clarifying the regulation of CSE/H2S during energy metabolism may help elucidate the complex interactions among glucose, fat and sulfur-containing amino acids in physiology and diseases.
Hydrogen Sulfide and Adipocyte Lipid Metabolism
During AT expansion, adipocytes become either hyperplastic, when their number increases through adipogenesis, or hypertrophic, when their size increases through lipogenesis.59 The increased number of adipocytes is primarily determined by the process of adipocyte differentiation, termed adipogenesis, which refers to the conversion of undifferentiated preadipocytes to mature adipocytes. In contrast, lipolysis is one of the most important processes for reducing adipose mass in which triglycerides stored in adipocytes are broken down into glycerol and fatty acids.60 The dysregulation of AT metabolism (including lipogenesis and lipolysis) is related to obesity, insulin resistance and diabetes in human and experimental animal models.19,61
The role of H2S in the regulation of AT metabolism is complex. Geng et al57 found that the H2S precursor LC or the donor-GYY4137 (morpholin-4-ium 4 methoxyphenyl (morpholino) phosphinodithioate) suppressed lipolysis while PAG induced basal and isoproterenol-stimulated lipolysis in rat epididymal adipocytes. PAG also increased lipolysis in normal chow and high-fat diet (HFD) fed mice by increasing serum glycerol levels without affecting food consumption and then blunting fat deposition and weight gain. GYY4137 reduced lipolysis in HFD mice without increasing fat mass and body weight. Moreover, they found that the PKA-HSL/perilipin 1 pathway may be involved in the lipolytic regulation of H2S.
Endogenous H2S was increased after 3T3-L1 differentiation.62 The expression levels of the H2S-synthesizing enzymes CSE, CBS and 3-MST were increased in a time-dependent manner during 3T3-L1 differentiation. GYY4137 and NaHS increased the expression of genes involved in adipogenesis, which include fatty acid binding protein 4 (FABP4/aP2), a key regulator of adipogenesis, and significantly increased the size and number of lipid droplets in mature adipocytes, which also compromised the ability of CL316,243 (a β3-agonist) to promote lipolysis in these cells. In contrast, aminooxyacetic acid (AOAA) and PAG had the opposite effect. Moreover, the inhibition of H2S production by CBS/CSE inhibitors seriously prevented the induction of adipogenic marker genes,63 PPARγ2 (peroxisome proliferator-activated receptor gamma 2) and its target gene perlipin1, Fabp4 and stearoyl-CoA desaturase-1 (Scd1). In addition, Cai et al64 confirmed the upregulation of an endogenous CSE/H2S system during adipocyte differentiation. Increased H2S promoted adipogenesis and inhibited lipolysis, resulting in the storage of triglycerides in lipid droplets. More interestingly, using H2S instead of 3-isobutyl-1-methylxanthine (IBMX) in the differentiation cocktail also upregulated adipogenesis markers and slightly increased the intracellular triglyceride levels. Based on the IBMX mechanism of action, H2S might directly promote PPARγ expression in part by inhibiting PDE (phosphodiesterase) activity. Therefore, H2S sulfhydrates PPARγ, enhancing its activity to promote adipogenesis, increase insulin sensitivity and inhibit basal lipolysis, causing a decrease in the circulating free fatty acid level and thereby ameliorating target injury in obesity.65
In summary, H2S increases the expression of adipogenesis-related proteins, increases lipid synthesis, promotes the differentiation of preadipocytes into mature adipocytes and reduces the lipolysis of adipocytes (Figure 4). H2S synthase inhibitors or siRNAs have the opposite effect.
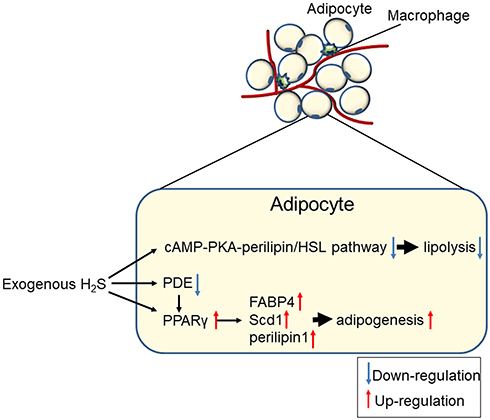 |
Figure 4 Generalised overview of the effects of H2S on adipocyte lipolysis and adipogenesis. |
Hydrogen Sulfide Is a Vascular Tone Modifier
The contractile force of aortas induced by norepinephrine66 and other vasoconstrictors was mitigated by PVAT in a paracrine manner.67,68 The anti-contractile effect of PVAT was also observed for both large and small vessels in mice, pigs and humans.69,70 Bioassay experiments showed that the adipocyte derived relaxing factor (ADRF), produced by PVAT, activates the K+ channels in vascular smooth muscle cells, exerting its anti-contractile effect on systemic peripheral arteries.68
Fang et al22 first demonstrated that the CSE protein was expressed in both the PVAT and aorta and in the adipocytes of PVAT and vascular smooth muscle cells (VSMCs) of aorta media. The rate of endogenous H2S generation was similar to that of aortic tissues and was primarily catalysed by CSE. The H2S donor NaHS produces dose-dependent vasorelaxation effects, which are blocked by XE991 (the KCNQ inhibitor). Therefore, H2S may be a candidate or modulator of ADRF, which activates the Kv channels encoded by KCNQ genes to achieve the paracrine control of vascular tone by PVAT. It is unclear how the PVAT derived H2S activates KCNQ channels, where it activates the channel directly or produces sulfhydryl radicals [HS(*)/S(*-)], which, coupled to the formation of superoxide radical anions,71 are powerful KCNQ channel activators.72 Further research confirmed that H2S generated by PVAT was a releasable vascular relaxation factor, acting in a paracrine manner by opening the K+ATP channel in a NO-, endothelium- and Ca2+ channel-independent manner. Inhibitors of CSE, such as BCA (5 mmol/l) and PAG (10 mmol/l), inhibit the anti-contractile effects of PVAT.73
Moreover, the anti-contractile effect of PVAT on rat aortic rings has been demonstrated to be blunted not only by XE991 or 4-AP (4-aminopyridine, the Kv channel inhibitor) but also by glibenclamide. In addition, the dilation of serotonin-preconstricted aortic rings without PVAT induced by NaHS was also abolished by glibenclamide, 4-AP or XE991.74 It is unclear whether this phenomenon results from the nonspecific effects of these inhibitors on different types of K+ channels or from the fact that both K+ATP and KCNQ channels are necessary for vasodilation induced by PVAT-derived H2S. Interestingly, the expression levels of CSE in PVAT surrounding mouse aortas is much lower than that in rats, and PVAT also has an anti-contractile effect on mouse aortic rings; this effect is not mediated by H2S, but it is still inhibited by XE991. These data indicate that H2S is the ADRF in rat but not mouse aorta, but the H2S-independent anti-contractile effects of PVAT on mouse aorta are also mediated by KCNQ channels.74
The contractions of arteries induced by 5-HT (5-hydroxytryptamine) in diabetic and healthy rats with intact PVAT were significantly different from those in arteries without PVAT. At the highest studied concentrations of 5-HT (10−5 mol/L), the arteries of diabetic rats with PVAT had significantly stronger contractions than those without PVAT. The different regulatory roles of PVAT in diabetic and control rats could be explained by the decreased levels of H2S caused by diabetes.75
Almost all of the blood vessels are surrounded by a different amount of PVAT. PVAT may have a paracrine function in the regulation of arterial tone, vascular reactivity, and more.76 This paracrine adipose-vascular coupling is achieved by the production and function of various ADRFs, which might be a volatile, gaseous mediator, namely, H2S.75 There is evidence that the opening of myocyte K+ channels plays a critical role in the paracrine regulation of arterial tone by H2S. KCNQ (Kv7) channels could represent, at least in part, the subtypes of the voltage-dependent K+ (Kv) channels involved.77 Additional ion channels, such as Ca2+-activated K+ (K+-Ca2+) channels, and cellular mechanisms appear to be involved in the vasoactive effects.78 Therefore, PVAT-H2S-smooth muscle crosstalk in the artery wall (Figure 5) may constitute a therapeutic approach against the harmful effects of diabetes and obesity in different vascular beds.
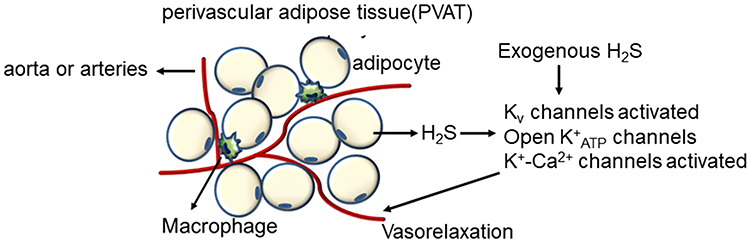 |
Figure 5 The mechanisms of the vasorelaxation effect of adipocytes-derived and exogenous H2S. |
Hydrogen Sulfide and Adipokines
Adipocytes and AT produce a wide range of hormones and cytokines involved in glucose metabolism (eg, adiponectin, resistin), lipid metabolism (eg, cholesteryl ester transfer protein, CETP; stearoyl-CoA desaturase-1, adipose triglyceride lipase, hormone-sensitive lipase), inflammation [eg, interleukin (IL)-1β, IL-6, IL-8, IL-10, tumour growth factor (TGF)-β, TNF-α, osteopontin (OPN)], the acute-phase and immune response (eg, serum amyloid A, adipsin, PAI-1), blood pressure (eg, angiotensinogen, angiotensin II) and feeding behaviour (eg, leptin), thus affecting the metabolism and function of many organs and tissues, including muscle, liver, vascular and brain tissues79–84 (Table 1). Approximately 20% of all genes in subcutaneous adipose tissue (SAT) and approximately 30% of genes in visceral adipose tissue (VAT) encode adipokine secretion.85 AT also expresses many of the receptors for most of these factors.86
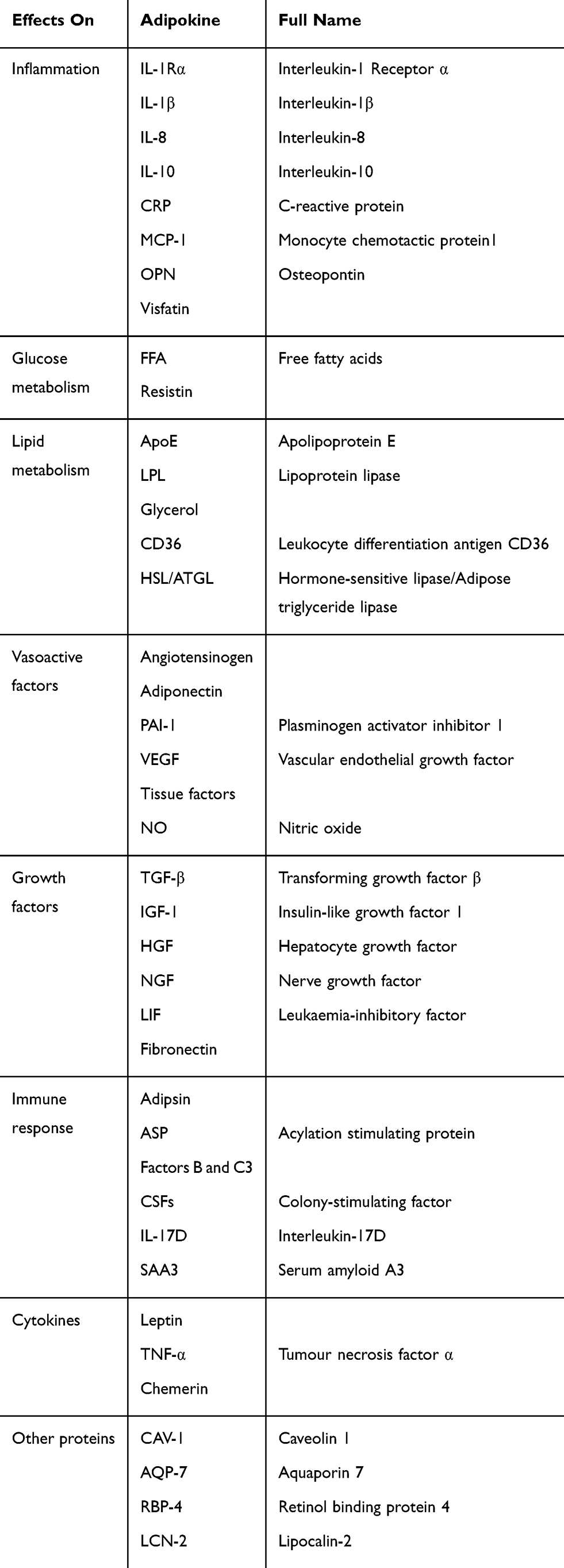 |
Table 1 Multiple Products Called Adipokines Involving Different Biological Processes Expressed and Secreted by White Adipose Tissue May Act at Both the Systemic (Endocrine) and Local (Autocrine and/Or Paracrine) Levels |
Adiponectin is an anti-diabetic and anti-atherogenic adipokine. Although the plasma adiponectin levels are reduced in obesity, other plasma adipocytokine levels increase with increases in AT and adipocyte volume.87,88 H2S is a recently identified endogenous gasotransmitter. Pan et al41 indicated that the overexpression of CSE and exogenously supplied NaHS, which both increase the concentration of H2S could significantly increase the adiponectin secretion that had been depressed by HG (25 mM) in mature 3T3-LI adipocytes, whereas reduced H2S production may contribute to deregulated adipokine secretion under HG conditions. H2S may exert anti-diabetic effects by increasing adiponectin levels.
Hydrogen Sulfide and Adipocytes Apoptosis
In several disease states, including autoimmune lipodystrophy,89 tumour cachexia,90 and HIV patients under highly active anti-retroviral therapy,91 the apoptosis of adipocytes is accompanied by a loss of AT. In obese individuals, AT is poorly oxygenated,92 which may lead to localized hypoxia, promote free fatty acid (FFA) release and inhibit glucose uptake in adipocytes by inhibiting the insulin signalling pathway.93 Hypoxia is associated with ER (endoplasmic reticulum) stress in 3T3-L1 adipocytes, as demonstrated by elevated levels of GRP78 (glucose-regulated protein, 78 kDa) and CHOP (C/EBP homologous protein).94 Excessive ER stress induces apoptosis. Adipocyte apoptosis is still poorly understood, as adipocytes are known to be resistant to apoptosis. Studies have reported that weight loss in obese models could be attributed to apoptosis in adipocytes.95–97 Adipokines such as leptin and TNF-α mediate apoptosis through caspase-dependent pathways and regulate body fat mass.98,99 In addition, a variety of natural compounds, including xanthohumol, isoxanthohumol and ajoene, have been reported to induce apoptosis in adipocytes.100,101 Interestingly, these compounds have been independently studied for their anti-obesity effects,102,103 which means that the anti-obesity effects of some natural compounds may be achieved by inducing the brown fat-like phenotype and apoptosis of white adipocytes. Controlling apoptosis in mature white AT appears to be an ideal strategy for combating obesity and related metabolic syndromes.104
H2S has become an effective cytoprotective mediator in various models of tissue and cellular injuries.105,106 After the ischaemic period, mature adipocytes were treated with NaHS (0, 0.1, 1, 10, 100, and 1000 μmol/L as an H2S donor), and the late apoptosis level of mature adipocytes decreased in every dose group.107 In addition, the gene expression level of caspase-3 decreased while that of Bcl-2 increased. These results imply that H2S has a protective effect on mature adipocytes under ischaemic conditions that is mediated by elevating anti-apoptotic gene expression (Figure 6).
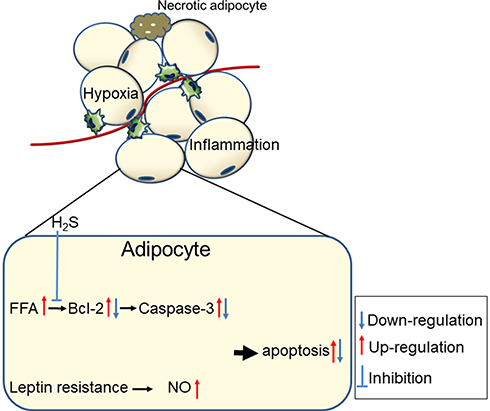 |
Figure 6 The mechanisms of adipocytes apoptosis in obese adipose tissue and the anti-apoptosis effect of H2S. |
The current knowledge regarding the effects of H2S on adipocyte apoptosis is inadequate, and it is necessary to explore it further and elucidate the roles of hypoxia, ER stress and pro-apoptotic and anti-apoptotic molecules in this process. H2S may become a new anti-obesity strategy.
The H2S/CSE System as a Target for Pharmacotherapy
The anti-inflammatory and insulin-sensitizing properties, the protective effects on mature adipocytes, and the beneficial effects on vascular tone of H2S suggest that increasing H2S signaling could be a potential new therapeutic strategy for the treatment of diabetes. The application of H2S donors and the augmentation of endogenous H2S has attracted the most attention as possible therapeutic approaches for diabetes, even if reducing H2S could be protective in some instances. However, the available CBS and CSE inhibitors have limited membrane permeability and are not very specific. In addition, there is no 3-MST inhibitor available. CBS, CSE or 3-MST knockout mice exhibit many abnormalities, which precludes considering such an approach in humans. Therefore, in the subsequent section, we will focus on only the approaches aimed at increase H2S signaling.
In most studies on H2S, inorganic sulfide salts such as sodium hydrosulfide (NaHS) and sodium sulfide (Na2S) were used, as they increase the H2S concentration rapidly and in the short term.108 NaHS may activate K+ATP channels, protein kinase B/Akt or Nrf2-induced signalling109 to exert its protective effects. To overcome the limitations of sulfide salts, several organic, slow-releasing H2S compounds have been synthesized, with GYY4137 being the most popular; however, they may possess H2S-independent activities mediated by the parent compound.110 LC is widely used in experimental studies to augment endogenous H2S production. However, LC is not a good H2S precursor because it is metabolized in many other pathways. In recent years, a new group of H2S donors has been developed. This group consists of a traditional NSAID that has been structurally modified to release H2S (S-NSAID). S-NSAIDs possess anti-inflammatory effects and are potentially protective for digestive and circulatory systems.108 Garlic, originally grown in central Asia, has many medicinal properties based on its antibacterial, antifungal, and antiviral activities and usually acts through the cardiovascular system. Garlic contains approximately 2,000 active compounds, including various sulfur-containing chemicals that may release H2S. It reduces blood pressure, suppresses blood platelet aggregation and has anti-atherosclerotic properties.111 Moreover, garlic lowers cholesterol. Nevertheless, the development of H2S donors is critical for understanding the biological functions of H2S. There will be more interesting work arising in this field.
Conclusions and Future Perspectives
The altered expression of H2S-synthesizing enzymes, as well as endogenous H2S levels, were observed in diabetic and obese animals. H2S is synthesized in AT and participates in the regulation of adipose tissue metabolism and function (Table 2). Although increasing research been performed in this field, some effects of H2S on AT, such as its role in the regulation of insulin sensitivity, are controversial, and its roles in adipokine secretion and adipocyte apoptosis have been incompletely demonstrated. Moreover, the mechanisms through which H2S acts on AT metabolism, such as glycolipid metabolism, have not been fully clarified; therefore, much work remains to be performed. Because of these controversies, the notion of using H2S donors or enhancing H2S signaling to alter AT dysfunction in common metabolic pathologies should be treated with caution.
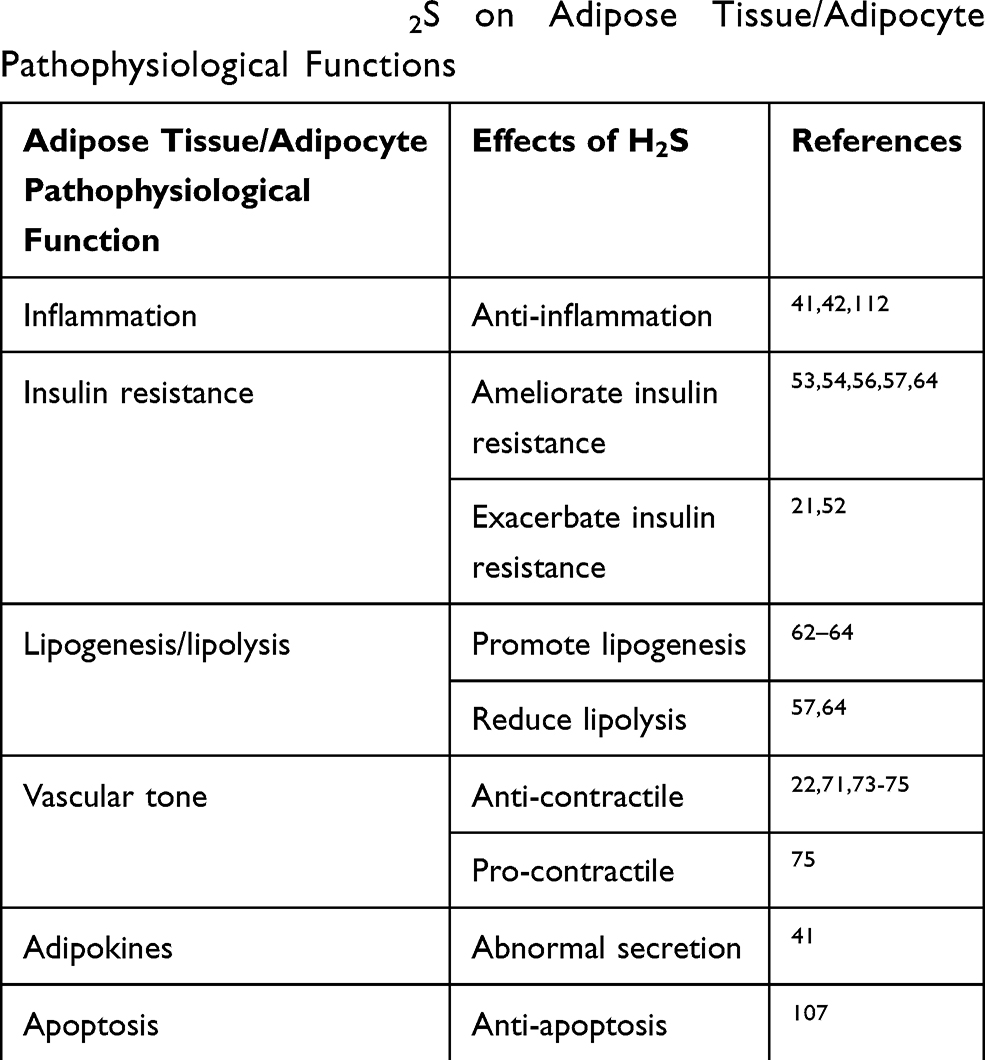 |
Table 2 Effects of H2S on Adipose Tissue/Adipocyte Pathophysiological Functions |
Data Sharing Statement
All data included in this study are available upon request by contact with the corresponding author.
Author Contributions
All authors contributed to data analysis, drafting or revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Abe K, Kimura H. The possible role of hydrogen sulfide as an endogenous neuromodulator. J Neurosci. 1996;16(3):1066. doi:10.1523/JNEUROSCI.16-03-01066.1996
2. Sies H, Jones DP. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat Rev Mol Cell Biol. 2020.
3. Jain SK, Bull R, Rains JL, et al. Low levels of hydrogen sulfide in the blood of diabetes patients and streptozotocin-treated rats causes vascular inflammation? Antioxid Redox Signal. 2010;12(11):1333–1337. doi:10.1089/ars.2009.2956
4. Yusuf M, Kwong Huat BT, Hsu A, Whiteman M, Bhatia M, Moore PK. Streptozotocin-induced diabetes in the rat is associated with enhanced tissue hydrogen sulfide biosynthesis. Biochem Biophys Res Commun. 2005;333(4):1146–1152. doi:10.1016/j.bbrc.2005.06.021
5. Wu L, Yang W, Jia X, et al. Pancreatic islet overproduction of H2S and suppressed insulin release in Zucker diabetic rats. Lab Invest. 2009;89(1):59–67. doi:10.1038/labinvest.2008.109
6. Gheibi S, Samsonov AP, Gheibi S, Vazquez AB, Kashfi K. Regulation of carbohydrate metabolism by nitric oxide and hydrogen sulfide: implications in diabetes. Biochem Pharmacol. 2020;113819.
7. Zhang Y, Yang J, Wang T, et al. Decreased endogenous hydrogen sulfide generation in penile tissues of diabetic rats with erectile dysfunction. J Sex Med. 2016;13(3):350–360. doi:10.1016/j.jsxm.2016.01.002
8. Kimura H. Signalling by hydrogen sulfide and polysulfides via protein S-sulfuration. Br J Pharmacol. 2020;177:720–733. doi:10.1111/bph.14579
9. Shibuya N, Koike S, Tanaka M, et al. A novel pathway for the production of hydrogen sulfide from D-cysteine in mammalian cells. Nat Commun. 2013;4(1):1366. doi:10.1038/ncomms2371
10. Kabil O, Banerjee R. Enzymology of H2S biogenesis, decay and signaling. Antioxid Redox Signal. 2014;20(5):770–782. doi:10.1089/ars.2013.5339
11. Huang CW, Moore PK. H2S synthesizing enzymes: biochemistry and molecular aspects. Handb Exp Pharmacol. 2015;230:3–25.
12. Ahmad FU, Sattar MA, Rathore HA, et al. Exogenous Hydrogen Sulfide (H2S) reduces blood pressure and prevents the progression of diabetic nephropathy in spontaneously hypertensive rats. Ren Fail. 2012;34(2):203–210. doi:10.3109/0886022X.2011.643365
13. Suzuki K, Sagara M, Aoki C, Tanaka S, Aso Y. Clinical implication of plasma hydrogen sulfide levels in Japanese patients with Type 2 diabetes. Inter Med. 2017;56(1):17–21. doi:10.2169/internalmedicine.56.7403
14. Ali MY, Whiteman M, Low CM, Moore PK. Hydrogen sulphide reduces insulin secretion from HIT-T15 cells by a KATP channel-dependent pathway. J Endocrinol. 2007;195(1):105–112. doi:10.1677/JOE-07-0184
15. Yang G, Yang W, Wu L, Wang R. H2S, endoplasmic reticulum stress, and apoptosis of insulin-secreting beta cells. J Biol Chem. 2007;282(22):16567–16576. doi:10.1074/jbc.M700605200
16. Yang G, Tang G, Zhang L, Wu L, Wang R. The pathogenic role of cystathionine gamma-lyase/hydrogen sulfide in streptozotocin-induced diabetes in mice. Am J Pathol. 2011;179(2):869–879. doi:10.1016/j.ajpath.2011.04.028
17. Brancaleone V, Roviezzo F, Vellecco V, De Gruttola L, Bucci M, Cirino G. Biosynthesis of H2S is impaired in non-obese diabetic (NOD) mice. Br J Pharmacol. 2008;155:673–680. doi:10.1038/bjp.2008.296
18. Coppack SW. Adipose tissue changes in obesity. Biochem Soc Trans. 2005;33(5):1049. doi:10.1042/BST0331049
19. Hajer GR, van Haeften TW, Visseren FLJ. Adipose tissue dysfunction in obesity, diabetes, and vascular diseases. Eur Heart J. 2008;29(24):2959–2971. doi:10.1093/eurheartj/ehn387
20. Unamuno X, Gómez-Ambrosi J. Adipokine dysregulation and adipose tissue inflammation in human obesity. 2018;48:e12997.
21. Feng X, Chen Y, Zhao J, Tang C, Jiang Z, Geng B. Hydrogen sulfide from adipose tissue is a novel insulin resistance regulator. Biochem Biophys Res Commun. 2009;380(1):153–159. doi:10.1016/j.bbrc.2009.01.059
22. Fang L, Zhao J, Chen Y, et al. Hydrogen sulfide derived from periadventitial adipose tissue is a vasodilator. J Hypertens. 2009;27(11):2174–2185. doi:10.1097/HJH.0b013e328330a900
23. Bełtowski J, Jamroz-Wiśniewska A. Hydrogen sulfide in the adipose tissue-physiology, pathology and a target for pharmacotherapy. Molecules (Basel, Switzerland). 2016;22.
24. Akash MS, Rehman K, Chen S. Role of inflammatory mechanisms in pathogenesis of type 2 diabetes mellitus. J Cell Biochem. 2013;114(3):525–531. doi:10.1002/jcb.24402
25. Richardson VR, Smith KA, Carter AM. Adipose tissue inflammation: feeding the development of type 2 diabetes mellitus. Immunobiology. 2013;218(12):1497–1504. doi:10.1016/j.imbio.2013.05.002
26. Deng J, Wang M, Guo Y, et al. Activation of α7nAChR via vagus nerve prevents obesity-induced insulin resistance via suppressing endoplasmic reticulum stress-induced inflammation in Kupffer cells. Med Hypotheses. 2020;140:109671. doi:10.1016/j.mehy.2020.109671
27. Crujeiras AB, Cordero P. Molecular basis of the inflammation related to obesity. Oxid Med Cell Longevity. 2019;2019:5250816.
28. Reilly SM, Saltiel AR. Adapting to obesity with adipose tissue inflammation. Nat Rev Endocrinol. 2017;13(11):633–643. doi:10.1038/nrendo.2017.90
29. Petrus P, Lecoutre S, Dollet L, et al. Glutamine links obesity to inflammation in human white adipose tissue. Cell Metab. 2020;31(2):375–90.e11. doi:10.1016/j.cmet.2019.11.019
30. Saxton SN, Clark BJ, Withers SB, Eringa EC, Heagerty AM. Mechanistic links between obesity, diabetes, and blood pressure: role of perivascular adipose tissue. Physiol Rev. 2019;99(4):1701–1763. doi:10.1152/physrev.00034.2018
31. Zhu Q, Scherer PE. Immunologic and endocrine functions of adipose tissue: implications for kidney disease. Nat Rev Nephrol. 2018;14(2):105–120. doi:10.1038/nrneph.2017.157
32. Comas F, Latorre J, Cussó O, et al. Hydrogen sulfide impacts on inflammation-induced adipocyte dysfunction. Food Chem Toxicol. 2019;131:110543.
33. Chi Q, Chi X, Hu X, Wang S, Zhang H, Li S. The effects of atmospheric hydrogen sulfide on peripheral blood lymphocytes of chickens: perspectives on inflammation, oxidative stress and energy metabolism. Environ Res. 2018;167:1–6. doi:10.1016/j.envres.2018.06.051
34. Barton M, Meyer MR. HuR-ry up: how hydrogen sulfide protects against atherosclerosis. Circulation. 2019;139(1):115–118. doi:10.1161/CIRCULATIONAHA.118.036854
35. Xie L, Gu Y, Wen M, et al. Hydrogen sulfide induces Keap1 s-sulfhydration and suppresses diabetes-accelerated atherosclerosis via Nrf2 activation. Diabetes. 2016;65(10):3171–3184. doi:10.2337/db16-0020
36. Luo ZL, Ren JD, Huang Z, et al. The role of exogenous hydrogen sulfide in free fatty acids induced inflammation in macrophages. Cell Physiol Biochem. 2017;42:1635–1644. doi:10.1159/000479405
37. Suzuki K, Olah G, Modis K, et al. Hydrogen sulfide replacement therapy protects the vascular endothelium in hyperglycemia by preserving mitochondrial function. Proc Natl Acad Sci U S A. 2011;108(33):13829–13834. doi:10.1073/pnas.1105121108
38. Cheng Z, Shen X, Jiang X, et al. Hyperhomocysteinemia potentiates diabetes-impaired EDHF-induced vascular relaxation: role of insufficient hydrogen sulfide. Redox Biol. 2018;16:215–225. doi:10.1016/j.redox.2018.02.006
39. Cheng Z, Garikipati VN, Nickoloff E, et al. Restoration of hydrogen sulfide production in diabetic mice improves reparative function of bone marrow cells. Circulation. 2016;134(19):1467–1483. doi:10.1161/CIRCULATIONAHA.116.022967
40. Whiteman M, Gooding KM, Whatmore JL, et al. Adiposity is a major determinant of plasma levels of the novel vasodilator hydrogen sulphide. Diabetologia. 2010;53(8):1722–1726. doi:10.1007/s00125-010-1761-5
41. Pan Z, Wang H, Liu Y, et al. Involvement of CSE/H2S in high glucose induced aberrant secretion of adipokines in 3T3-L1 adipocytes. Lipids Health Dis. 2014;13(1):155. doi:10.1186/1476-511X-13-155
42. Velmurugan GV, Huang H, Sun H, et al. Depletion of H2S during obesity enhances store-operated Ca2+entry in adipose tissue macrophages to increase cytokine production. Sci Signal. 2015;8(407):ra128. doi:10.1126/scisignal.aac7135
43. Zhou X, Yang W, Li J. Ca2+- and protein kinase C-dependent signaling pathway for nuclear Factor-κB activation, inducible nitric-oxide synthase expression, and tumor necrosis factor-α production in lipopolysaccharide-stimulated rat peritoneal macrophages. J Biol Chem. 2006;281(42):31337–31347. doi:10.1074/jbc.M602739200
44. Kim Y, Moon JS, Lee KS, et al. Ca2+/calmodulin-dependent protein phosphatase calcineurin mediates the expression of iNOS through IKK and NF-kappaB activity in LPS-stimulated mouse peritoneal macrophages and RAW 264.7 cells. Biochem Biophys Res Commun. 2004;314(3):695–703. doi:10.1016/j.bbrc.2003.12.153
45. Barazzoni R, Gortan Cappellari G, Ragni M, Nisoli E. Insulin resistance in obesity: an overview of fundamental alterations. Eating Weight Disord. 2018;23(2):149–157. doi:10.1007/s40519-018-0481-6
46. Feller DD, Feist E. Conversion of methionine and threonine to fatty acids by adipose tissue. Can J Biochem Physiol. 1963;41(1):269–273. doi:10.1139/y63-034
47. Heeren J, Scheja L. Brown adipose tissue and lipid metabolism. Curr Opin Lipidol. 2018;29(3):180–185. doi:10.1097/MOL.0000000000000504
48. Antonopoulos AS, Tousoulis D. The molecular mechanisms of obesity paradox. Cardiovasc Res. 2017;113(9):1074–1086. doi:10.1093/cvr/cvx106
49. Kim WJ, Lee W, Jung Y, Jang HJ, Kim YK, Kim SN. PPARβ/δ agonist GW501516 inhibits TNFα-induced repression of adiponectin and insulin receptor in 3T3-L1 adipocytes. Biochem Biophys Res Commun. 2019;510(4):621–628. doi:10.1016/j.bbrc.2019.02.013
50. Palit SP, Patel R, Jadeja SD, Rathwa N, Mahajan A, Ramachandran AV. A genetic analysis identifies a haplotype at adiponectin locus: association with obesity and type 2 diabetes. Sci Rep. 2020;10:2904.
51. Bermúdez VJ, Rojas E, Toledo A, et al. Single-nucleotide polymorphisms in adiponectin, AdipoR1, and AdipoR2 genes: insulin resistance and type 2 diabetes mellitus candidate genes. Am J Ther. 2013;20(4):414–421. doi:10.1097/MJT.0b013e318235f206
52. Huang CY, Yao WF, Wu WG, Lu YL, Wan H, Wang W. Endogenous CSE/H2 S system mediates TNF-alpha-induced insulin resistance in 3T3-L1 adipocytes. Cell Biochem Funct. 2013;31:468–475. doi:10.1002/cbf.2920
53. Manna P, Jain SK. Vitamin D up-regulates glucose transporter 4 (GLUT4) translocation and glucose utilization mediated by cystathionine-gamma-lyase (CSE) activation and H2S formation in 3T3L1 adipocytes. J Biol Chem. 2012;287:42324–42332. doi:10.1074/jbc.M112.407833
54. Manna P, Jain SK. Hydrogen sulfide and L-cysteine increase phosphatidylinositol 3,4,5-trisphosphate (PIP3) and glucose utilization by inhibiting phosphatase and tensin homolog (PTEN) protein and activating phosphoinositide 3-kinase (PI3K)/serine/threonine protein kinase (AKT)/protein kinase Czeta/lambda (PKCzeta/lambda) in 3T3l1 adipocytes. J Biol Chem. 2011;286:39848–39859.
55. Manna P, Jain SK. Hydrogen sulfide and L-cysteine increase phosphatidylinositol 3,4,5-trisphosphate (PIP3) and glucose utilization by inhibiting phosphatase and tensin homolog (PTEN) protein and activating phosphoinositide 3-kinase (PI3K)/serine/threonine protein kinase (AKT)/protein kinase Cζ/λ (PKCζ/λ) in 3T3l1 adipocytes. J Biol Chem. 2011;286(46):39848–39859.
56. Xue R, Hao DD, Sun JP, et al. Hydrogen sulfide treatment promotes glucose uptake by increasing insulin receptor sensitivity and ameliorates kidney lesions in type 2 diabetes. Antioxid Redox Signal. 2013;19:5–23. doi:10.1089/ars.2012.5024
57. Geng B, Cai B, Liao F, et al. Increase or decrease hydrogen sulfide exert opposite lipolysis, but reduce global insulin resistance in high fatty diet induced obese mice. PLoS One. 2013;8(9):e73892. doi:10.1371/journal.pone.0073892
58. Geng B. [Adipocytic endogenous hydrogen sulfide-function,regulation and diseases]. Sheng li Ke Xue Jin Zhan [Progress in Physiology]. 2017;48(1):37–41. Chinese.
59. Lee WH, Rho JG, Han HS, et al. Self-assembled hyaluronic acid nanoparticle suppresses fat accumulation via CD44 in diet-induced obese mice. Carbohydr Polym. 2020;237:116161. doi:10.1016/j.carbpol.2020.116161
60. Duncan RE, Ahmadian M, Jaworski K, Sarkadi-Nagy E, Sul HS. Regulation of lipolysis in adipocytes. Annu Rev Nutr. 2007;27(1):79–101. doi:10.1146/annurev.nutr.27.061406.093734
61. Abranches MV, Oliveira FC, Conceicao LL, Peluzio MD. Obesity and diabetes: the link between adipose tissue dysfunction and glucose homeostasis. Nutr Res Rev. 2015;28:121–132. doi:10.1017/S0954422415000098
62. Tsai CY, Peh MT, Feng W, Dymock BW, Moore PK. Hydrogen sulfide promotes adipogenesis in 3T3L1 cells. PLoS One. 2015;10:e0119511. doi:10.1371/journal.pone.0119511
63. Haj-Yasein NN, Berg O, Jerneren F, Refsum H, Nebb HI, Dalen KT. Cysteine deprivation prevents induction of peroxisome proliferator-activated receptor gamma-2 and adipose differentiation of 3T3-L1 cells. Biochim Biophys Acta. 2017;1862:623–635.
64. Cai J, Shi X, Wang H, et al. Cystathionine gamma lyase-hydrogen sulfide increases peroxisome proliferator-activated receptor gamma activity by sulfhydration at C139 site thereby promoting glucose uptake and lipid storage in adipocytes. Biochim Biophys Acta. 2016;1861:419–429. doi:10.1016/j.bbalip.2016.03.001
65. Haj-Yasein NN, Berg O, Jernerén F, Refsum H, Nebb HI, Dalen KT. Cysteine deprivation prevents induction of peroxisome proliferator-activated receptor gamma-2 and adipose differentiation of 3T3-L1 cells. Biochimica Et Biophysica Acta Molecular and Cell Biology of Lipids. 2017;1862(6):623–635.
66. Soltis EE, Cassis LA. Influence of perivascular adipose tissue on rat aortic smooth muscle responsiveness. Clin Exp Hypertension Part A. 1991;13(2):277–296. doi:10.3109/10641969109042063
67. Kassam SI, Lu C, Buckley N, Gao YJ, Lee RM. Modulation of thiopental-induced vascular relaxation and contraction by perivascular adipose tissue and endothelium. Br J Anaesth. 2012;109(2):177–184. doi:10.1093/bja/aes127
68. Oriowo MA. Perivascular adipose tissue, vascular reactivity and hypertension. Med Principl Pract. 2015;24(Suppl 1):29–37. doi:10.1159/000356380
69. Szasz T, Webb RC. Perivascular adipose tissue: more than just structural support. Clin sci. 2012;122(1):1–12. doi:10.1042/CS20110151
70. Gollasch M. Vasodilator signals from perivascular adipose tissue. Br J Pharmacol. 2012;165(3):633–642. doi:10.1111/j.1476-5381.2011.01430.x
71. Stasko A, Brezova V, Zalibera M, Biskupic S, Ondrias K. Electron transfer: a primary step in the reactions of sodium hydrosulphide, an H2S/HS−donor. Free Radic Res. 2009;43(6):581–593. doi:10.1080/10715760902977416
72. Gamper N, Zaika O, Li Y, et al. Oxidative modification of M-type K+ channels as a mechanism of cytoprotective neuronal silencing. EMBO J. 2006;25(20):4996–5004. doi:10.1038/sj.emboj.7601374
73. Schleifenbaum J, Kohn C, Voblova N, et al. Systemic peripheral artery relaxation by KCNQ channel openers and hydrogen sulfide. J Hypertens. 2010;28(9):1875–1882. doi:10.1097/HJH.0b013e32833c20d5
74. Kohn C, Schleifenbaum J, Szijarto IA, et al. Differential effects of cystathionine-gamma-lyase-dependent vasodilatory H2S in periadventitial vasoregulation of rat and mouse aortas. PLoS One. 2012;7(8):e41951.
75. Emilova R, Dimitrova D, Mladenov M, Daneva T, Schubert R, Gagov H. Cystathionine gamma-lyase of perivascular adipose tissue with reversed regulatory effect in diabetic rat artery. Biotechnol Biotechnol Equip. 2015;29(1):147–151. doi:10.1080/13102818.2014.991565
76. Queiroz M, Sena CM. Perivascular adipose tissue in age-related vascular disease. Ageing Res Rev. 2020;59:101040. doi:10.1016/j.arr.2020.101040
77. Köhn C, Schleifenbaum J, Szijártó IA, et al. Differential effects of cystathionine-γ-lyase-dependent vasodilatory H2S in periadventitial vasoregulation of rat and mouse aortas. PLoS One. 2012;7(8):e41951.
78. Orlov SN, Gusakova SV, Smaglii LV, Koltsova SV, Sidorenko SV. Vasoconstriction triggered by hydrogen sulfide: evidence for Na(+),K(+),2Cl(-)cotransport and L-type Ca(2+) channel-mediated pathway. Biochem Biophys Rep. 2017;12:220–227. doi:10.1016/j.bbrep.2017.09.010
79. Yamauchi T, Kamon J, Waki H, et al. The fat-derived hormone adiponectin reverses insulin resistance associated with both lipoatrophy and obesity. Nat Med. 2001;7(8):941–946. doi:10.1038/90984
80. Ran J, Hirano T, Fukui T, et al. Angiotensin II infusion decreases plasma adiponectin level via its type 1 receptor in rats: an implication for hypertension-related insulin resistance. Metabolism. 2006;55(4):478–488. doi:10.1016/j.metabol.2005.10.009
81. Chu NF, Spiegelman D, Hotamisligil GS, Rifai N, Stampfer M, Rimm EB. Plasma insulin, leptin, and soluble TNF receptors levels in relation to obesity-related atherogenic and thrombogenic cardiovascular disease risk factors among men. Atherosclerosis. 2001;157(2):495–503. doi:10.1016/S0021-9150(00)00755-3
82. Fruhbeck G, Catalan V, Gomez-Ambrosi J, Rodriguez A. Aquaporin-7 and glycerol permeability as novel obesity drug-target pathways. Trends Pharmacol Sci. 2006;27(7):345–347. doi:10.1016/j.tips.2006.05.002
83. Gomez-Ambrosi J, Catalan V, Ramirez B, et al. Plasma osteopontin levels and expression in adipose tissue are increased in obesity. J Clin Endocrinol Metab. 2007;92(9):3719–3727. doi:10.1210/jc.2007-0349
84. Furuhashi M, Fucho R, Gorgun CZ, Tuncman G, Cao H, Hotamisligil GS. Adipocyte/macrophage fatty acid-binding proteins contribute to metabolic deterioration through actions in both macrophages and adipocytes in mice. J Clin Invest. 2008;118:2640–2650. doi:10.1172/JCI34750
85. Matsuzawa Y. Therapy Insight: adipocytokines in metabolic syndrome and related cardiovascular disease. Nat Clin Pract Cardiovasc Med. 2006;3(1):35–42. doi:10.1038/ncpcardio0380
86. Fruhbeck G. The Sir David Cuthbertson Medal Lecture. Hunting for new pieces to the complex puzzle of obesity. Proc Nutr Soc. 2006;65:329–347.
87. Zhang Y, Zitsman JL, Hou J, et al. Fat cell size and adipokine expression in relation to gender, depot, and metabolic risk factors in morbidly obese adolescents. Obesity (Silver Spring, Md). 2014;22(3):691–697. doi:10.1002/oby.20528
88. Mihu D, Ciortea R, Mihu CM. Abdominal adiposity through adipocyte secretion products, a risk factor for endometrial cancer. Gynecol Endocrinol. 2013;29(5):448–451. doi:10.3109/09513590.2012.752452
89. Fischer-Posovszky P, Hebestreit H, Hofmann AK, et al. Role of CD95-mediated adipocyte loss in autoimmune lipodystrophy. J Clin Endocrinol Metab. 2006;91(3):1129–1135. doi:10.1210/jc.2005-0737
90. Prins JB, Walker NI, Winterford CM, Cameron DP. Human adipocyte apoptosis occurs in malignancy. Biochem Biophys Res Commun. 1994;205(1):625–630. doi:10.1006/bbrc.1994.2711
91. Domingo P, Matias-Guiu X, Pujol RM, et al. Subcutaneous adipocyte apoptosis in HIV-1 protease inhibitor-associated lipodystrophy. AIDS (London, England). 1999;13(16):2261–2267. doi:10.1097/00002030-199911120-00008
92. Ortiz VE, Vidal-Melo MF, Walsh JL. Strategies for managing oxygenation in obese patients undergoing laparoscopic surgery. Surg Obesity Related Dis. 2015;11(3):721–728. doi:10.1016/j.soard.2014.11.021
93. Gong L, Zou Z, Huang L, Guo S, Xing D. Photobiomodulation therapy decreases free fatty acid generation and release in adipocytes to ameliorate insulin resistance in type 2 diabetes. Cell Signal. 2020;67:109491. doi:10.1016/j.cellsig.2019.109491
94. Hosogai N, Fukuhara A, Oshima K, et al. Adipose tissue hypoxia in obesity and its impact on adipocytokine dysregulation. Diabetes. 2007;56(4):901–911. doi:10.2337/db06-0911
95. Prins JB, Walker NI, Winterford CM, Cameron DP. Apoptosis of human adipocytes in vitro. Biochem Biophys Res Commun. 1994;201(2):500–507. doi:10.1006/bbrc.1994.1730
96. Papineau D, Gagnon A, Sorisky A. Apoptosis of human abdominal preadipocytes before and after differentiation into adipocytes in culture. Metabolism. 2003;52(8):987–992. doi:10.1016/S0026-0495(03)00165-3
97. Chaiittianan R, Sutthanut K, Rattanathongkom A. Purple corn silk: a potential anti-obesity agent with inhibition on adipogenesis and induction on lipolysis and apoptosis in adipocytes. J Ethnopharmacol. 2017;201:9–16. doi:10.1016/j.jep.2017.02.044
98. Hamrick MW, Della Fera MA, Choi YH, Hartzell D, Pennington C, Baile CA. Injections of leptin into rat ventromedial hypothalamus increase adipocyte apoptosis in peripheral fat and in bone marrow. Cell Tissue Res. 2007;327(1):133–141. doi:10.1007/s00441-006-0312-3
99. Jung TW, Kim ST, Lee JH, et al. Phosphatidylcholine causes lipolysis and apoptosis in adipocytes through the tumor necrosis factor alpha-dependent pathway. Pharmacology. 2018;101(3–4):111–119. doi:10.1159/000481571
100. Yang J-Y, Della-Fera MA, Rayalam S, Baile CA. Effect of xanthohumol and isoxanthohumol on 3T3-L1 cell apoptosis and adipogenesis. Apoptosis. 2007;12(11):1953–1963. doi:10.1007/s10495-007-0130-4
101. Yang JY, Della-Fera MA, Nelson-Dooley C, Baile CA. Molecular mechanisms of apoptosis induced by ajoene in 3T3-L1 adipocytes. Obesity (Silver Spring, Md). 2006;14(3):388–397. doi:10.1038/oby.2006.52
102. Rayalam S, Yang JY, Ambati S, Della-Fera MA, Baile CA. Resveratrol induces apoptosis and inhibits adipogenesis in 3T3-L1 adipocytes. Phytother Res. 2008;22:1367–1371. doi:10.1002/ptr.2503
103. Ambati S, Yang JY, Rayalam S, Park HJ, Della-Fera MA, Baile CA. Ajoene exerts potent effects in 3T3-L1 adipocytes by inhibiting adipogenesis and inducing apoptosis. Phytother Res. 2009;23(4):513–518. doi:10.1002/ptr.2663
104. Qi R, Huang J, Wang Q, et al. MicroRNA-224-5p regulates adipocyte apoptosis induced by TNFalpha via controlling NF-kappaB activation. J Cell Physiol. 2018;233:1236–1246. doi:10.1002/jcp.25992
105. Henderson PW, Singh SP, Weinstein AL, et al. Therapeutic metabolic inhibition: hydrogen sulfide significantly mitigates skeletal muscle ischemia reperfusion injury in vitro and in vivo. Plast Reconstr Surg. 2010;126(6):1890–1898. doi:10.1097/PRS.0b013e3181f446bc
106. Henderson PW, Singh SP, Belkin D, et al. Hydrogen sulfide protects against ischemia-reperfusion injury in an in vitro model of cutaneous tissue transplantation. J Surg Res. 2010;159(1):451–455. doi:10.1016/j.jss.2009.05.010
107. Aykan A, Ozturk S, Sahin I, Avcu F, Sagkan RI, Isik S. The effects of hydrogen sulfide on adipocyte viability in human adipocyte and adipocyte-derived mesenchymal stem cell cultures under ischemic conditions. Ann Plast Surg. 2015;75(6):657–665. doi:10.1097/SAP.0000000000000595
108. Powell CR, Dillon KM, Matson JB. A review of hydrogen sulfide (H(2)S) donors: chemistry and potential therapeutic applications. Biochem Pharmacol. 2018;149:110–123. doi:10.1016/j.bcp.2017.11.014
109. Zhao S, Song T, Gu Y, et al. Hydrogen sulfide alleviates liver injury via S-sulfhydrated-Keap1/Nrf2/LRP1 pathway. Hepatology (Baltimore, Md). 2020.
110. Nin DS, Idres SB, Song ZJ, Moore PK, Deng LW. Biological effects of morpholin-4-Ium 4 Methoxyphenyl (Morpholino) phosphinodithioate and other phosphorothioate-based hydrogen sulfide donors. Antioxid Redox Signal. 2020;32(2):145–158. doi:10.1089/ars.2019.7896
111. Bayan L, Koulivand PH, Gorji A. Garlic: a review of potential therapeutic effects. Avicenna J Phytomed. 2014;4(1):1–14.
112. Stein A, Bailey SM. Redox biology of hydrogen sulfide: implications for physiology, pathophysiology, and pharmacology. Redox Biol. 2013;1(1):32–39. doi:10.1016/j.redox.2012.11.006
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.