")
Back to Journals » International Journal of Nanomedicine » Volume 18
Hybrid Hydrogel Loaded with Chlorhexidine⊂β-CD-MSN Composites as Wound Dressing
Authors Lin J, Shi T, Wang Y, He Z, Mu Z, Cai X, Deng H, Shen J, Liu F
Received 30 December 2022
Accepted for publication 21 February 2023
Published 31 March 2023 Volume 2023:18 Pages 1725—1740
DOI https://doi.org/10.2147/IJN.S401705
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Yan Shen
Jian Lin,1,* Tianpeng Shi,2,* Yi Wang,1 Zhiqi He,1 Zhixiang Mu,1 Xiaojun Cai,1 Hui Deng,1 Jianliang Shen,3– 5 Fen Liu6
1School & Hospital of Stomatology, Wenzhou Medical University, Wenzhou, People’s Republic of China; 2Department of Stomatology, PLA Strategic Support Force Medical Center, Beijing, People’s Republic of China; 3State Key Laboratory of Ophthalmology, Optometry and Vision Science, School of Ophthalmology & Optometry, School of Biomedical Engineering, Wenzhou Medical University, Wenzhou, People’s Republic of China; 4Wenzhou Institute, University of Chinese Academy of Sciences, Wenzhou, People’s Republic of China; 5Department of Regenerative Medicine, Vision, and Brain Health, Oujiang Laboratory (Zhejiang Lab for Regenerative Medicine, Vision, and Brain Health), Wenzhou, People’s Republic of China; 6Department of Histology and Embryology, Wenzhou Medical University, Wenzhou, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Hui Deng; Fen Liu, Email [email protected]; [email protected]
Background: Much attention has been paid to sustained drug release and anti-infection in wound management. Hydrogels, which are biocompatible materials, are promising tools for controlled drug release and infective protection during wound healing. However, hydrogels also demonstrate limitations in the highly efficient treatment of wounds because of the diffusion rate. In this work, we explored pH-sensitive hydrogels that enable ultra-long-acting drug release and sustained antibacterial properties.
Methods: We constructed a hybrid gelatin methacrylate (GelMA) system with sustainable antibacterial properties combining hyaluronic acid (HA)-coated mesoporous silica nanoparticles (MSN), which loaded host-guest complexes of chlorhexidine (CHX) with β-cyclodextrins (β-CD) (CHX⊂CD-MSN@HA@GelMA). The release mechanism of CHX was explored using UV-vis spectra after intermittent diffusion of CHX. The hybrid hydrogels were characterized, and the drug content in terms of the release profile, bacterial inhibition, and in vivo experiments were investigated.
Results: Except for dual protection from both hydrogels, MSN in the HA improved the drug loading efficiency to promote the local drug concentration. It showed that complicated CHX-loaded MSN releases CHX more gradually and over a longer duration than CHX-loaded MSNs. This demonstrated a 12-day CHX release time and antibacterial activity, primarily attributable to the capacity of β-CD to form an inclusion complex with CHX. Meanwhile, in vivo experiments revealed that the hydrogels safely promote skin wound healing and enhance therapeutic efficacy.
Conclusion: We constructed pH-sensitive CHX⊂CD-MSN@HA@GelMA hydrogels that enable ultra-long-acting drug release and sustained antibacterial properties. The combination of β-CD and MSN would be better suited to release a reduced rate of active molecules over time (slow delivery), making them great candidates for wound dressing anti-infection materials.
Keywords: mesoporous silica, cyclodextrin, pH-responsive, antibacterial property, wound healing
Introduction
Traumatic infections, caused by bacterial pathogens, are common causes of patient morbidity and a major global healthcare challenge, leading to wound infection and even life-threatening illnesses.1–3 Infected wounds, as one of the most significant fatality issues, delay the healing cycles and even cause deterioration, resulting in serious tissue damage as microorganisms compete with the host immune system and invade viable tissue, especially Staphylococcus aureus.4–6 Currently, a few types of biocompatible hydrogels, such as hyaluronic acid (HA) and gelatin methacryloyl (GelMA), have been applied as wound dressings which can effectively isolate further wound infection.7,8 The wound healing of surface infection normally lasts long. Simple physical defense can only alleviate further wound infection but not heal.9,10 Therefore, some antibacterial drugs are loaded into hydrogels to improve healing. However, the morphological structure of hydrogels limits the capacity of drug loading, and no strong interactions can be formed between drug molecules and internal structures.11–13 Increasing the loading capacity of hydrogels is key to achieving long-term wound healing. Nanoparticles and nanofibers have high porosity and high specific surface area, which allowed drug to permeate and facilitates the growth of cell tissues; it is similar to the scale and morphology of extracellular matrix (ECM), which is facilitate antibacterial and proliferation. It can meet the special environment of the Infected wounds.14,15 In the past two decades, much attention has been paid to mesoporous silica nanoparticles (MSN) resulting from the high surface area and tunable pores, demonstrating significant advantages in drug delivery systems.16–19 Previous studies based on surface modification of MSN can increase the ion-dipole interactions between drugs and MSN surfaces, further achieving both the high capacity of drug loading and slow release.20–23 After that, stimulus-response structures, such as nanovalves and polymers, are installed and coated on the surface of MSN. These promote a series of activations that can operate to process the targeted and controlled release.24–27
We currently combine GelMA and drug-loaded MSN to prepare a new type of wound dressing, which achieves physical isolation and chemical treatment (Scheme 1). Chlorhexidine (CHX), a typical antibacterial drug, is selected to be loaded into the pores of MSN.28 Moreover, MSN is coated with HA, which can be activated by different pH values to control the CHX release accurately.29,30 In addition, host-guest inclusions, rather than only drug molecules, can provide extra control of CHX to prolong the release period further.31,32 In the host-guest system, β-cyclodextrins (β-CD) have been chosen as host molecules because of their biocompatibility, which possesses hydrophilic outers and hydrophobic cavities.33,34 The cavities can form host-guest inclusions with CHX molecules (CHX⊂CD) through dipole-dipole and hydrophobic interactions.35–37 After the inclusions are released from MSN, CHX will slowly dissociate from the host-guest complex to achieve the long-lasting antibacterial activity. To successfully load the inclusions, swollen MSN is synthesized using a co-template for enlarging the pore sizes of MSN to about 5 nm, which can quickly load the inclusions to obtain CHX⊂CD loaded MSN (CHX⊂CD-MSN). After coating with HA, CHX⊂CD-MSN@HA is mixed with GelMA (CHX⊂CD-MSN@HA@GelMA) to form wound dressings by radical polymerization.
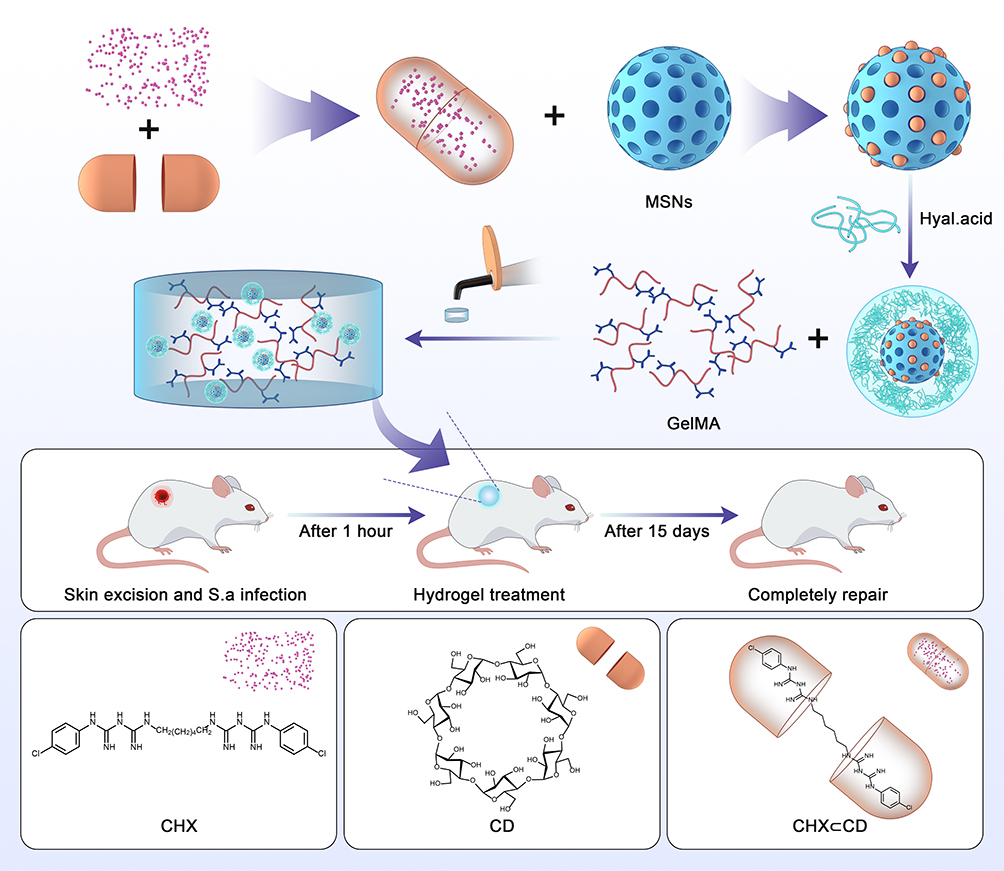 |
Scheme 1 Schematic illustration of the preparation of CHX⊂CD-MSN@HA@GelMA for the bacteria-infected wound healing. |
Compared with previous research, the wound dressing in the design blocks prevents wound infection from the outside and integrates chemotherapy. Furthermore, this design uses host-guest inclusions to replace drug molecules for the first time, which significantly prolongs the release period of the drug, thus promoting a stable and long-lasting repair effect on wound healing. Additionally, the HA coating on the surface of MSN enhances the binding force between CHX⊂CD-MSN@HA and GelMA, which also brings another property of controlled release.
Materials and Methods
Materials
All the reagents were used as received unless noted otherwise. Hexadecyltrimethylammonium bromide (CTAB, 98%) and Irgacure 2959 were purchased from Sigma-Aldrich. Chlorhexidine, Tetraethyloxysilane (TEOS), and gelatin methacryloyl (GelMA) were obtained from Aladdin Reagent Co. Ltd. Sodium hydroxide (NaOH), acetone and methanol (MeOH) were obtained from Sinopharm Chemical Reagent Limited Corporation and used as received. Trypsin-EDTA (0.25%), fetal bovine serum (FBS), Dulbecco’s modified Eagle medium (DMEM), Hank’s Balanced Salt Solution, LIVE/DEAD™ Viability/Cytotoxicity Kit, LIVE/DEADTM BacLightTM Bacterial Viability Kit, and CCK-8 kit were purchased from Life Technologies. Trypticase soy broth (TSB), β-cyclodextrin (β-CD), and chlorhexidine were purchased from Solarbio. Hyaluronic acid (HA, MW = 30–50 kDa) was purchased from Bloomage Biotechnology Corporation Limited. All glassware and Teflon-coated magnetic stirring bars were carefully cleaned with aqua regia, followed by profuse washing with pure DI water using a Millipore system.
Preparation of CHX⊂CD and CHX⊂CD-MSN@HA
Generally, a solution of CHX (2.5 mg) dissolved in acetone (10 mL) was mixed into a β-CD aqueous solution (10 mg/10 mL) and stirred for 24 h. The inclusion complex, CHX⊂CD, was placed in an aqueous solution when the organic solvent was evaporated. However, the free CHX molecules were precipitated and removed by filtration. The inclusion complex of CHX⊂CD was collected after the removal of the aqueous solution.
Bare MSN was synthesized using a modified approach from the previous literature.38 CTAB (1 g) was dissolved in 480 mL of H2O and 3.5 mL of NaOH aqueous solution (2 mol/L). To activate the template, 7 mL of mesitylene was added to the solution and vigorously agitated for 2 h at 80°C. Sequentially, dropwise additions of TEOS (5 mL) were added to the solution, which was then rapidly agitated for another 2 h at 80°C to form a white precipitate. The precipitate was collected using hot filtration, then washed with excess MeOH and dried under vacuum overnight at room temperature. A suspension of the as-synthesized material (1 g) in MeOH (100 mL) was agitated for 6 h at 50°C in the presence of concentrated HCl to remove the template (37%, 0.75 mL). Filtration was used to capture the bare MSN, which was then dried under vacuum at room temperature overnight.
Bare MSN (20 mg) was soaked in an aqueous solution of CHX⊂CD (0.5 mmol/L in PBS, 10 mL, pH = 7.4) for 5 h at room temperature to obtain CHX⊂CD-MSN. Then, HA (15 mg) in PBS (1 mL, pH = 7.4) was added to the loading solution, and the mixture was then stirred for 15 min. The solution was centrifuged for 1 min at 8000 rpm, and the precipitate was redispersed into a new batch of PBS (11 mL) containing HA (15 mg). CHX⊂CD-MSN@HA was obtained by repeating the previous steps three times, then lyophilized after three washes in PBS (10 mL, pH = 7.4).
Preparation of CHX⊂CD-MSN@HA@GelMA Hydrogels
Irgacure 2959 aqueous solution (0.05%, w/v) was prepared by dissolving 50 mg of Irgacure 2959 in DI water (10 mL). The GelMA (10 g) was then dissolved in the prepared Irgacure 2959 aqueous solution (5 mL) and stirred for 10 min at 60°C to obtain a 20% GelMA aqueous solution (w/v). To form CHX⊂CD-MSN@HA@GelMA, a few amounts (1, 0.5, 0.25, 0.125, 0.0625, 0.03125 mg/mL) of CHX⊂CD-MSN@HA were centrifuged and distributed in GelMA solution (20%, w/v). Under UV light (LED light: 385–515 nm), this mixture was applied to a PMMA mold to obtain disc-shaped CHX⊂CD-MSN@HA@GelMA.
Characterization
All the samples were vacuum freeze-dried before material characterization. The surface chemical structures were evaluated using Fourier Transform Infrared (FTIR, Tensor II, BRUKER, Germany) spectroscopy. The sampling range was adjusted at 500–4000 cm−1. The elemental composition of the surface structure was determined using an X-ray photoelectron spectroscopy (XPS, Thermo Fisher Scientific, U.K.), which used a monochromatic source of Al Kα radiation as the x-ray source. Thermal gravimetric analysis (TGA) was collected on a TGA thermal analyzer (TGA8000, PerkinElmer, USA). The samples were heated at a rate of 10°C/min from 100–900°C under a nitrogen atmosphere. NMR spectra were obtained by a Bruker AvanceTM 400 spectrometer with a Bruker Ultra-Shield 9.4 T (proton Larmor frequency of 400.33 MHz), and a BBI probe was used to record the experiments (1H, X). The morphology of nanoparticles was studied using scanning electron microscopy (SEM) and transmission electron microscopy (TEM). SEM images were taken with Hitachi (SU8010) equipment. Samples were put on silica wafers and sprayed with platinum for 60s. Next, TEM images were collected using FEI Talos 200s equipment with a 200 kV operating voltage. X-ray diffraction (XRD) was used to determine the mesostructured ordering. Cu radiation was used to record XRD data on a Rigaku D/Max-2550 V diffractometer. Micromeritics Tristar 3000 analyzer was used to conduct nitrogen adsorption-desorption isotherm tests at 77 K under a continuous adsorption condition. Brunauer-Emmett-Teller (BET) and Barrette-Joynere-Halenda (BJH) were collected to investigate the specific surface area and pore size distribution curve, respectively. The mechanical characteristics were confirmed using a stress-controlled rheometer (DHR-2, TA, USA). All the samples were made with a height of 1 mm and a diameter of 40 mm. In a shear strain of 0.5%, the energy storage and loss modulus distribution of the samples were measured at a frequency range from 0.1 to 10 Hz. To determine the loading efficiency of CHX⊂CD, CHX⊂CD (10 mg) was degraded with hydrochloric acid, and the quantity of chlorhexidine in the solution was then measured by HPLC. Hydrogels were submerged in PBS until equilibrium swelling was reached. Each hydrogel was blotted gently with KimWipes to remove the surface liquid, and its original mass was measured. Each disc-shaped hydrogel was destroyed at 37°C in a 2 mL PBS (pH = 7.4) solution containing Proteinase K enzyme (0.1 mg/mL). Every day, the enzyme solution was refreshed. At certain intervals, the surface liquid of each hydrogel was wiped away, and the remaining mass was measured.
In vitro Drug Release Profiles
CHX⊂CD-MSN (10 mg) or CHX-MSN (10 mg) suspension in H2O (2 mL, pH 7.4) were dialyzed against 20 mL of H2O in a dialysis bag (MWCO=1 KDa). At predetermined intervals, 1.0 mL of release solution was withdrawn and replaced with an equivalent amount of fresh medium. The quantity of CHX released was measured using a UV-vis spectroscopy curve. Then, CHX⊂CD-MSN@HA (10 mg) and CHX-MSN@HA (10 mg) suspension in H2O (2 mL, pH 7.4) were also dialyzed against 20 mL of various buffer media (pH 7.5 and pH 5.5) in the dialysis bag. The amount of emitted CHX was measured using UV-vis spectroscopy at intervals of 1, 2, 4, 6, 12, 24, 48, 72, 96, 144, 192, 240, 288, and 360 h. Hydrogel samples (8 mm diameter × 3 mm thick) were submerged in various buffer mediums (pH 7.5 and 5.5) to assess the triggered release behavior of CHX from the CHX⊂CD-MSN@HA@GelMA hydrogels. The amount of emitted CHX was measured using UV-vis spectroscopy at intervals of 1, 2, 4, 6, 12, 24, 48, 72, 96, 144, 192, 240, 288, and 360 h. The procedure for measuring the quantity of CHX released was reported in previous research,39 and the amount of CHX released was estimated using the calibration curve and measuring the absorbance at 254 nm.
In vitro Cytotoxicity Studies
In vitro cytotoxicity of CHX⊂CD-MSN@HA@GelMA hydrogels against L929 cells (American Type Culture Collection, ATCC, Rockville, MD, United States) was determined by the CCK-8 and LIVE/DEAD assay. Briefly, L929 cells were cultured in DMEM with different concentrations of CHX⊂CD-MSN@HA embedded in GelMA hydrogels at 37°C for 24 h. After that, 10 μL of CCK-8 solution was added and incubated in the dark. After 2 h of culture, 200 μL of supernatant was transferred to a 96-well plate, and the OD of the culture plate solution at 450 nm was determined using a spectrophotometer (1530, Thermo Fisher, Finland). Then, L929 cells were incubated with MSN@GelMA, CHX-MSN@HA@GelMA, and CHX⊂CD-MSN@HA@GelMA for 3, 5, 7, and 9 days under the optimal concentrations indicated above. The CCK-8 solution (10 μL) was added and measured using a spectrophotometer at 450 nm. On days 5, 7, and 9, the LIVE/DEADTM Viability/Cytotoxicity Kit was also utilized to determine cell viability following the specified therapy. L929 cells were assessed using a ZEISS Axio Vert.A1 fluorescent microscope imaging system and stained with the LIVE/DEADTM Viability/Cytotoxicity Kit.
In vitro Antibacterial Effects
In vitro antibacterial effects of CHX⊂CD-MSN@HA@GelMA hydrogels were assessed against S. aureus and E. coli, respectively. At first, either 100 μL of S. aureus or E. coli (106 CFU/mL) was incubated with different concentrations of CHX⊂CD-MSN@HA embedded in GelMA hydrogels at 37°C for 24 h. Then, 100 μL of bacterial suspension was incubated with MSN@GelMA hydrogels, CHX-MSN@HA@GelMA hydrogels, and CHX⊂CD-MSN@HA@GelMA hydrogels for 1, 3, 5, 7, and 9 days in TSB medium. S. aureus and E. coli culture without any materials served as control. The growth of S. aureus and E. coli was then validated using a SpectraMax M2 microplate reader to measure the optical density (OD) at a wavelength of 600 nm (OD600nm) (Molecular Devices, USA). The bacteria were washed twice with PBS (1 mL, pH 7.4) after removing the supernatants before staining with the LIVE/DEADTM BacLightTM Bacterial Viability Kit. Using the ZEISS Axio Vert. A1 fluorescent microscope imaging equipment, the bacterial viability was observed. S. aureus and E. coli were collected in centrifuge tubes and rinsed three times with PBS solution after treatment with prepared materials. The bacterial precipitates were then extracted using centrifugation, fixed with 2.5% glutaraldehyde, and dehydrated with ethanol at various concentrations. Finally, the bacteria were preserved in ethanol (99.5%) and examined by SEM. Agar plate diffusion tests were also employed to evaluate the antibacterial activity. On each agar plate, 100 μL of bacterial suspension was added to agar plates, and hydrogels were placed onto these agar plates and incubated for 24 h at 37°C. In addition, at 5, 7, and 9 days, bacterial suspensions were serially diluted in PBS (1 mL, 0.01 M, pH 7.4) and plated on blood agar plates to quantify live bacterial cells by counting CFU (CFU/mL).
In vivo Antibacterial Properties
The Laboratory Animal Ethics Committee of Wenzhou Medical University & Laboratory Animal Centre of the Wenzhou Medical University allowed in vivo animal research. All animal experiments are conducted in strict accordance with the guidelines for Ethical Review of Experimental Animal Welfare of the People’s Republic of China (GB/T35892-2018). After a conventional anesthetic approach, a full-thickness round skin incisional wound (5 mm) was produced in male BALB/c mice (Beijing Vital River Laboratory Animal Technology Co, Ltd) to establish an infected wound model for in vivo research. Then, the wounds were infected for 1 hour with 20 L of 108 CFU/mL S. aureus to form the infected wound. The mice were separated into four treatment groups, with the wounds being coated with PBS (50 μL, pH 7.4), MSN@GelMA, CHX-MSN@HA@GelMA and CHX⊂CD-MSN@HA@GelMA (n=5). After one day, rats were sacrificed, and the bacteria amounts in wounds were determined using an agar plate counting method. All the treated mice were kept in separate cages during the trial and constantly observed. The wound areas were assessed on treatment at 1, 5, 10, and 15 days, and the wound closure rate was estimated using Image-Pro Plus. Wound samples were collected after 15 days and preserved in a 4% formaldehyde solution before being embedded in paraffin. For histological investigation, tissue slices were placed on slides. Using normal techniques, hematoxylin and eosin (H&E) staining (and a Nikon microscope) were used to visualize the pathological alterations of the wounds and the vital organs. In this study, a hemolysis assessment was carried out to evaluate the blood compatibility of the hydrogel. Typically, PBS was the negative control group, while DW was the positive control group. After coculturing with the PBS group, CHX⊂CD-MSN@HA@GelMA hydrogels groups, and DW group for 6 h, all supernatants were collected, and their OD values at 545 nm were recorded.
Statistical Analysis
The results were expressed as mean ± standard deviation (SD) from at least three independent experiments. One-way analysis of variance with Tukey’s test (Origin 8.0) was used for statistical analysis. *p < 0.05 was statistically significant.
Results and Discussion
Characterization of the Inclusion: CHX⊂CD
In the preparation of CHX⊂CD, no new chemical bonds were formed in the inclusion. Therefore, the FTIR spectrum of CHX⊂CD presents the vibrations of the bonds from both CHX and β-CD. The aromatic rings of CHX are represented by the wide and solitary N-H vibration at ~3400 cm−1 and the C=C stretching bands at 1602, 1537, and 1480 cm−1. At 3500–3300 cm−1, the OH groups of the β-CD ring overlap with the NH band of CHX. The C-O-C stretch vibration of the glucose unit of β-CD causes the band at 1032 cm−1. After complicated development, the β-CD bands at 3500–3300 cm−1 and 1032 cm−1 are sharpened. Physical combinations of free CHX and free β-CD have been reported in the literature to have wide IR bands around 3500–3300 cm−1 and about 1100 cm−1, which are sharpened following complexation resulting from the breakdown of hydrogen bonding upon inclusion.40 The C=C bands of CHX at 1602 cm−1 change to higher wavenumbers because of complexation, whereas the band at 1537 cm−1 shifts to lower wavenumbers. In contrast to the β-CD bands becoming sharper, the typical CHX bands get larger (Figure S1 and Figure 1A). In this study, CHX⊂CD showed 23.07% CHX loading efficiency. As a result, by comparing the proton chemical shifts obtained for the guest-host combination with those identified for the separate species, 1H NMR can offer direct information on the complex structure. Two pairs of linked doublets in the aromatic area (between 7 and 7.3 ppm), three multiplets for the biguanide portion (at 3.0, 1.3, and 1.1 ppm), and a multiplet for the digluconate ion (between 3.5 and 4 ppm) are seen in the CHX spectrum (Figure 1B and C). The signal of β-CD protons is also visible, with H1 at 4.91 ppm, internal protons H3 and H5, and exterior protons H6, H4, and H2 at 3.40 and 3.85 ppm. Nevertheless, a shift of the CHX aromatic protons is observed on the spectra. The aromatic region of CHX⊂CD was shifted from 7.0 to 5.0 ppm. The signal of the β-CD protons also altered, especially in the 3.7 to 3.9 ppm range, which corresponds to the internal protons (H3 and H5). Conversely, the signals of the external protons (H2 and H4) did not change or only marginally changed. These findings align with Denadai et al, who demonstrated the host–guest-type assembly between CHX and β-CD in detail.41 X-ray photoelectron spectroscopy (XPS) also proved that the CHX⊂CD had been successfully prepared (Figures S2–S4). Figure 1D–I shows the results of XPS analysis of N1s and Cl2p of β-CD, CHX, and CHX⊂CD. First, the β-CD did not detect the spectra of N1s and Cl2p as expected (Figure 1D and G), and the CHX (Figure 1E and H) and CHX⊂CD (Figure 1F and I) showed the N1s and Cl2p spectra at 394–410 eV and 192–210 eV. In addition, compared with CHX, the relative intensity of the C=N of the CHX⊂CD at 398 eV is reduced, which proves that the content of C=N bonds in the system is reduced, which means that the hydroxyl group in β-CD is more than CHX. The amino group of the complex is successfully covalently linked through hydroxylamination to generate CHX⊂CD. In addition, the Cl2p spectra of β-CD, CHX, and CHX⊂CD were analyzed to clarify the changes in chlorine groups. In the Cl2p spectrum (Figure 1H and I), there are two typical characteristic peaks at 197.45 and 199 eV corresponding to the chlorine group in the chloride, and two typical characteristic peaks at 200.5 and 202.5 eV correspond to the chloride. The chlorine group and the Cl2p spectra of CHX and CHX⊂CD are very similar. Therefore, it can be concluded that the valence state and chemical environment of Cl have not undergone any changes after recombination, so we can think that the CHX is compounding with the β-CD. During the process, except for hydroxylation, no other side reactions occurred. CHX maintained the structural integrity in the CHX⊂CD. Incorporating a guest molecule into the hydrophobic cavity of a cyclodextrin molecule changes the environment of the inserted guest moiety’s protons and the host cavity’s protons. The TG curves of the β-CD and CHX⊂CD (Figure S5) showed similar trends with a significant weight loss stage, and the CHX showed two major weight loss stages. The TGA and DTG curves of CHX revealed two thermic processes at 214°C and 475°C, respectively. The weight loss at 294°C (weight loss of 22%) and 475°C (weight loss of 58%) was attributed to pyrolytic breakdown followed by burning the biopolymer.
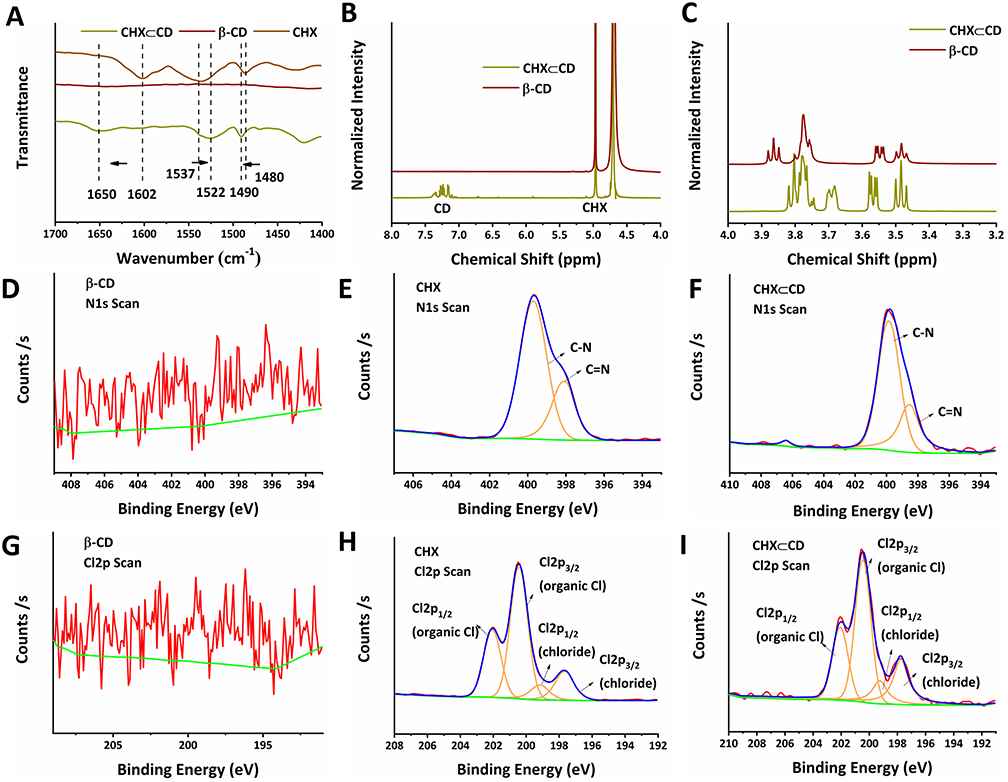 |
Figure 1 The characterization of CHX⊂CD. (A) FTIR spectra of the β-CD, CHX, and the CHX⊂CD complex are represented at 1700–1400 cm−1. (B) 1H NMR spectra in D2O of the β-CD, CHX⊂CD, and 1H NMR spectra in DMSO-d6 of the CHX between 8 and 4 ppm. (C) 1H NMR spectra of β-CD, CHX⊂CD, and CHX between 4 and 3.2 ppm. The N1s spectrum of the β-CD (D), CHX (E), and CHX⊂CD (F). The Cl2p spectrum of the β-CD (G), CHX (H), and CHX⊂CD (I). |
Characterization of CHX⊂CD-MSN@HA
The morphology of several MSN samples was studied using TEM and SEM. The generated MSN, CHX⊂CD-MSN CHX⊂CD-MSN@HA, were usually spherical and had a well-defined porous structure (Figure 2A–C and Figure S6). Blank MSN had an average diameter of 110 nm. Various procedures were used to examine the generated products to disclose the effective modification of HA molecules onto MSN step-by-step. The low-angle XRD pattern was also used to analyze the homogenous mesoporous structure of the as-obtained MSN. The as-synthesized MSN had low-angle diffraction peaks, with reflections as Bragg peaks (Figure S7). After modification with HA, the intensity of the diffraction peaks of CHX⊂CD-MSN@HA decreases sharply. Figure 2D–F shows the XPS analysis of the N1s spectrum of MSN, CHX⊂CD-MSN, and CHX⊂CD-MSN@HA. First, MSN delivered NSi3 and NSiO2 characteristic peaks at 399.5 and 403 eV, respectively (Figure 2D). Still, after MSN was loaded with CHX⊂CD, the N1S characteristic peaks of MSN had disappeared, while the CN (399.5 eV) and C=N (403 eV) characteristic peaks were detected in the CHX⊂CD-MSN (Figure 2E), which proves that the CHX⊂CD was successfully loaded into the mesoporous silica. Additionally, after the surface of CHX⊂CD-MSN is modified with HA, the binding energy of the C-N and C=N bond remains the same as unmodified. So, the HA is not only well adjusted and coated with CHX⊂CD-MSN, and does not destroy the uniformity and integrity (Figure 2F). The BET measurement of MSN indicated that the as-synthesized MSN had a typical type-IV isotherm curve (Figure 2G), indicating that MSN had a mesoporous structure. More importantly, the surface areas of MSN, CHX⊂CD-MSN, and CHX⊂CD-MSN@HA were decreased with the incorporation of CHX⊂CD and modification of HA. Pore size and volumes decreased slightly upon CHX⊂CD incorporation and HA modification in MSN (Figure S8). The surface chemistry of MSN was monitored using FTIR spectra during the alteration process (Figure 2H and Figure S9). The spectra’s most essential elements are enlarged, and the distinctive absorption band at 1580 cm−1 in the FT-IR spectra of CHX⊂CD-MSN@HA can be attributed to the deformation mode of the N-H+ group originating from the protonation of the biguanide moiety of chlorhexidine. In the infrared spectra of chlorhexidine base, this absorption band is undetectable. Furthermore, the absorption bands at 1540/1490 cm−1 in the spectra of CHX⊂CD-MSN@HA were ascribed to the C-C skeletal vibrations of aromatic rings. The stretching and deformation vibrations of the Si-O-Si framework caused large absorption bands in all mesoporous materials at 1080/805 cm−1 and 460 cm−1. This may be attributed to the stretching and deformation vibrations of the Si-O-Si framework, respectively. Figure 2I shows the thermal gravimetric analysis (TGA) curves, which demonstrate a gradual weight drop after each alteration, suggesting that CHX⊂CD and HA were conjugated to MSN. The TGA also showed a 30.2% CHX⊂CD loading efficiency, and the molecular dimensions of the β-CD cavity (width 0.60–0.65 nm and height 0.78 nm) are thought to make it the best host for the inclusion of complex formation among the three native CDs.42 In this study, the pore size of MSN was about 5 nm, which can effectively load the CHX⊂CD.
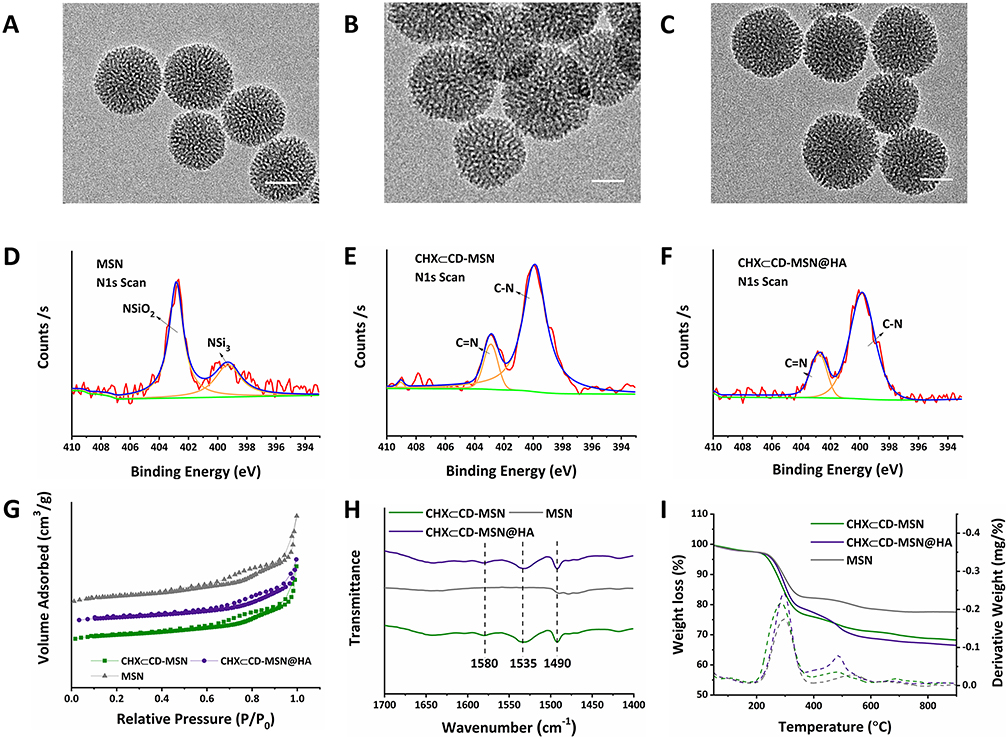 |
Figure 2 The characterization of CHX⊂CD-MSN@HA, CHX⊂CD-MSN, and MSN. TEM image of MSN (A), CHX⊂CD-MSN (B), CHX⊂CD-MSN@HA (C). Scale bar = 50 nm. The N1s spectrum of MSN (D), CHX⊂CD-MSN (E), and CHX⊂CD-MSN@HA (F). (G) BET nitrogen adsorption/desorption isotherms of CHX⊂CD-MSN@HA, CHX⊂CD-MSN, and MSN. (H) FTIR spectra of CHX⊂CD-MSN@HA, CHX⊂CD-MSN, and MSN. (I) TGA curves of CHX⊂CD-MSN@HA, CHX⊂CD-MSN, and MSN. |
Characterization of CHX⊂CD-MSN@HA@GelMA and in vitro Release Behavior
Hydrogels are significantly easier to manufacture and administer as semi-solid nanomaterials than most solid nanomaterials, and they have strong biocompatibility and adhesion, allowing them to adhere to infected wounds quickly. Figure 3A shows that CHX⊂CD-MSN@HA@GelMA hydrogels have a pore size distribution ranging from 45–67 nm on the surface. Compared with the pure GelMA, the rheometer of CHX⊂CD-MSN@HA@GelMA hydrogels reduces to 35% after adding CHX⊂CD-MSN@HA (Figure 3B). The elastic modulus and adhesive strength of GelMA may be adjusted by varying the prepolymer and photoinitiator concentrations. The 10% w/v GelMA prepolymers have outstanding adhesion capabilities, allowing the hydrogel to efficiently cling to the substrate’s surface and allowing for long-term survival. For in vivo applications of biomaterials, tunable biodegradation is also of utmost importance. When implanted, biomaterials should deteriorate in vivo at the same rate as newly-formed tissues. This degradation study utilized Proteinase K, a broad-spectrum protease whose potent proteolytic activity on native proteins may be observed in moderate neutral conditions. It required four days for GelMA and CHX⊂CD-MSN@HA@GelMA to be degraded up to 82.95 ± 4.24% and 94.37 ± 3.53%, respectively, CHX⊂CD-MSN@HA@GelMA was degraded entirely within 6 days (Figure 3C).
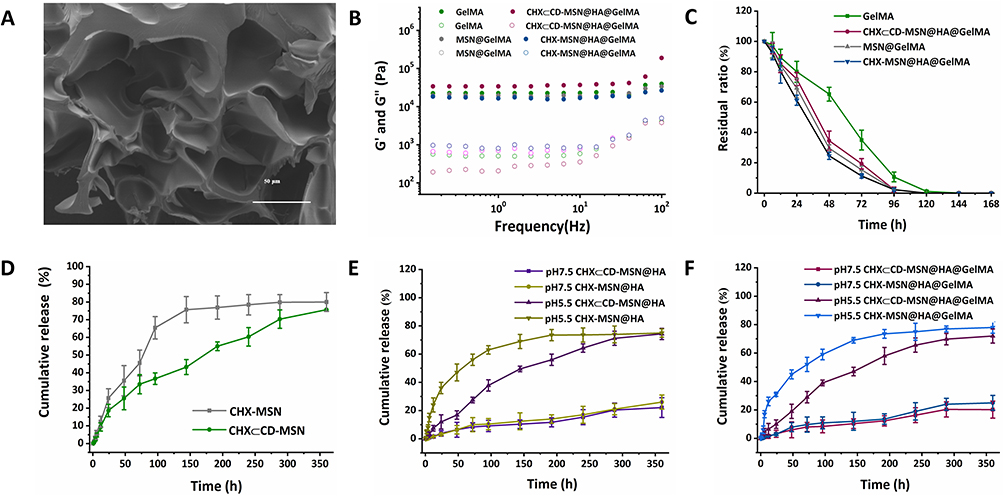 |
Figure 3 The characterization of CHX⊂CD-MSN@HA@GelMA hydrogels and the release curves of CHX in vitro. (A) SEM image of the internal structure. Scale bar = 50 μm. (B) Rheological behavior of CHX⊂CD-MSN@HA@GelMA hydrogels. (C) Biodegradation rate of the remaining hydrogels at designated time points for CHX⊂CD-MSN@HA@GelMA hydrogels. (D) Cumulative amounts of CHX were released from CHX⊂CD-MSN and CHX-MSN. (E) Cumulative amounts of CHX were released from CHX⊂CD-MSN@HA and CHX-MSN@HA at different pH conditions (pH 7.5 and 5.5). (F) Cumulative amounts of CHX were released from CHX⊂CD-MSN@HA@GelMA hydrogels and CHX-MSN@HA@GelMA hydrogels at different pH conditions (pH 7.5 and 5.5). The data is presented as Mean/SD with n = 3 independent samples. Data are expressed as mean ± SD. |
Figure 3D shows that CHX⊂CD release from the MSN (CHX⊂CD-MSN) was compared with CHX-MSN, where CHX⊂CD was directly loaded in MSN. The goal was to determine what function CHX segments and β-CD units had in modulating CHX⊂CD release characteristics. During the 70 h, CHX-MSN released CHX quicker than CHX⊂CD-MSN, showing better release behavior, and the release behavior of CHX may be affected by the high inclusion complex. The capping molecule HA agents were modified on the surface of MSN to prevent premature leakage of loaded drug molecules from the nanocarriers and realize pH-control release by the acid labile hydrazine bonds (Figure 3E and F). Therefore, the in vitro drug release experiments were performed by observing the release behavior of CHX⊂CD-MSN@HA@GelMA hydrogels at different pH conditions to simulate human physiological and inflammatory environments (pH 7.5 and 5.5). Figure 3E shows that the CHX release curves differed significantly depending on the environment. Within 24 h at pH 7.5, the accumulative release of CHX was only around 7.53%. Nonetheless, after 24 h at pH 5.5, the accumulative release quantity of CHX reached 19.87%. Because pH-sensitive Schiff base bonds were able to maintain stability at pH 7.5 but easily hydrolyzed in an acidic environment, the minimal drug release ratio at pH 7.5 showed that the tunnels of MSN were still firmly capped by HA, and the more acidic situation made it easier to be hydrolyzed. CHX⊂CD-MSN@HA@GelMA hydrogels were shown to be capable of pH-responsive drug release during in vitro drug release tests.
Moreover, in vitro release of CHX from the CHX⊂CD-MSN@HA@GelMA hydrogels was evaluated under experimental conditions. During the first 6 h, the CHX-MSN@HA@GelMA hydrogels released CHX quickly and uncontrollably (Figure 3F). This phenomenon is called the burst effect, when the polymeric matrix fails to hold drug molecules within its MSN. The CHX⊂CD-MSN@HA@GelMA hydrogels, conversely, showed the reverse behavior. Even after 240 h in the release medium, the matrix could not release more than 59.2% of the encapsulated drug. The high stability of the CHX⊂CD inclusion complex and the MSN may provide a hurdle to drug diffusion and release in this scenario. The favorable release profiles that differentiate this system are based on the improved hydrophilic nature of β-CD units and their ability to form stable complexes with CHX.
In vitro Cytotoxicity Studies
The biomaterials used in clinical applications must be both antibacterial and biocompatible. The CCK-8 test was used to determine the cytotoxicity of the current hydrogels, and coculture with L929 cells was used to determine biocompatibility. Figure 4A shows that hydrogels containing varying concentrations of embedded CHX⊂CD-MSN@HA were cocultured for 24 h with L929 cells and compared with the control group, which all inhibited the cells marginally. The CHX⊂CD-MSN@HA@GelMA hydrogels at a concentration of 1 mg/mL were the most visible; however, their inhibition rate was just 4%. Additionally, the cell survival of CHX⊂CD-MSN@HA@GelMA hydrogels was determined after 3, 5, 7, and 9 days of culture and the blank group. After 9 days of culture, the cell viability of the CHX⊂CD-MSN@HA@GelMA hydrogels decreased slightly below 97%, while the cell viability of the CHX-MSN@HA@GelMA hydrogels decreased below 95% (Figure 4B). The findings indicated that the toxicity of CHX-MSN@HA@GelMA hydrogels was somewhat more significant than that of CHX⊂CD-MSN@HA@GelMA hydrogels because CHX was released from the hydrogel more rapidly than it was from CHX⊂CD-MSN@HA@GelMA hydrogels. Further biocompatibility testing was performed utilizing a Live/Dead staining technique. Figure 4C shows that the control group comprises around 99% viable cells labeled green and approximately 1% dead cells stained red. The CHX⊂CD-MSN@HA@GelMA hydrogels contain roughly 98% live cells and only 2% dead cells, demonstrating biocompatibility. GelMA is a visually appealing photocurable substance made from gelatin chemically modified with methacrylic anhydride (MAA) and preserves its inherent biological features, such as enzymatic degradation and cell adhesion enhancement. Additionally, the findings revealed that the gradual release kinetics of CHX as a wound dressing helped to lower the danger of cells, which was compatible with the wound management approach.
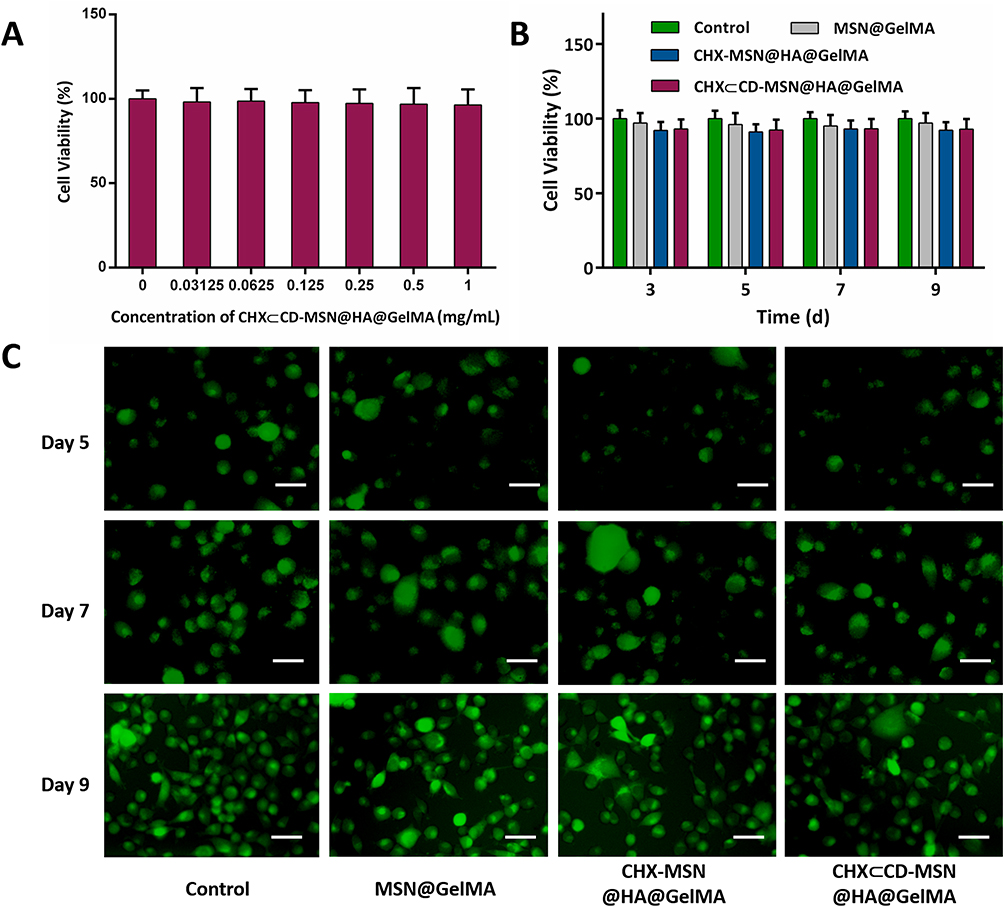 |
Figure 4 The cell viability and biocompatibility of CHX⊂CD-MSN@HA@GelMA hydrogels. (A) Cell viability of L929 fibroblast cells after treatment with different concentrations of CHX⊂CD-MSN@HA@GelMA hydrogels at 24 h (B) Cell viability of L929 fibroblast cells after treatment at 3, 5, 7, and 9 days. (C) Live/dead fluorescence images of L929 fibroblast cells after treatment with CHX⊂CD-MSN@HA@GelMA hydrogels on days 5, 7, and 9. Scale bar = 100 μm. n = 3 independent samples. Data are expressed as mean ± SD. A significant difference was detected by the multiple t-test comparisons test. |
In vitro Antibacterial Effects
Since the entire wound healing process is typically accompanied by a significant risk of infection, which delays the healing process, current dressings are intended to have an anti-infective function. The antibacterial activities of MSN@GelMA hydrogels, CHX-MSN@HA@GelMA hydrogels, and CHX⊂CD-MSN@HA@GelMA hydrogels were first evaluated qualitatively, with a blank group serving as a control. The CHX⊂CD-MSN@HA@GelMA hydrogels inhibited the growth of S. aureus and E. coli in a dose-dependent manner (Figures S10 and S11). Moreover, the hydrogels without loading CHX⊂CD-MSN@HA were not antibacterial against S. aureus and E. coli, respectively. Figure S12 and S13 showed images of the CHX⊂CD-MSN@HA@GelMA hydrogels against S. aureus and E. coli, respectively. MSN@GelMA hydrogels did not generate an inhibition zone, but CHX-MSN@HA@GelMA and CHX⊂CD-MSN@HA@GelMA hydrogels formed prominent inhibition zones against S. aureus and E. coli for 24 h. Therefore, 0.25 mg/mL CHX⊂CD-MSN@HA@GelMA hydrogels were selected for the treatment concentration of subsequent experiments. Figure 5A and E show that all mesoporous silica systems containing the CHX or CHX⊂CD inclusion compounds showed satisfactory inhibition against S. aureus and E. coli, as well as the antimicrobial agents. Although the CHX content in CHX-MSN@HA@GelMA hydrogels was similar to the CHX⊂CD-MSN@HA@GelMA hydrogels, CHX⊂CD-MSN@HA@GelMA hydrogels exhibited a more lasting antibacterial property. To compare the two drug loading methods, bacterial suspensions were cultivated extensively with CHX-MSN@HA@GelMA hydrogels and CHX⊂CD-MSN@HA@GelMA hydrogels on days 1, 3, 5, 7, and 9. Figure 5A and E show that over 90% of bacteria (S. aureus and E. coli) were almost destroyed on days 1 and 3 by CHX-MSN@HA@GelMA and CHX⊂CD-MSN@HA@GelMA hydrogels. On day 5, the two hydrogels above demonstrated a distinct and beneficial antibacterial impact. After a week, both hydrogels lost their capacity to kill S. aureus and E. coli, most notably the CHX-MSN@HA@GelMA hydrogels. After 9 days, the CHX-MSN@HA@GelMA hydrogels lost their antibacterial activity, but the CHX⊂CD-MSN@HA@GelMA hydrogels retained an antibacterial ratio of 59.71% and 47.56% against S. aureus and E. coli, respectively, confirming the hydrogels’ stable release profile. On day 9, bacterial colonies were cultivated using CHX-MSN@HA@GelMA hydrogels and CHX⊂CD-MSN@HA@GelMA hydrogels (Figure 5B and F, respectively). Additionally, live/dead staining of bacteria revealed a high number of live bacteria in the control group and MSN@GelMA hydrogels, while dead bacteria were identified in the CHX-MSN@HA@GelMA and CHX⊂CD-MSN@HA@GelMA hydrogels (Figure 5C and G). The morphologies of bacteria following various treatments were observed using SEM imaging. In the control group, S. aureus and E. coli maintained their original round and rod-like form with a smooth surface (Figure 5D and H). However, after treatment with CHX⊂CD-MSN@HA@GelMA hydrogels, the bacterial morphology was altered, and the cell walls became entirely wrinkled and perforated. On day 5, both groups had few visible bacterial colonies against S. aureus and E. coli, respectively, without dilution (Figures S14 and S15). On day 7, CHX-MSN@HA@GelMA hydrogels developed considerably fewer bacterial colonies compared with CHX⊂CD-MSN@HA@GelMA hydrogels. The antibacterial activity of CHX-MSN@HA@GelMA hydrogels declined abruptly and significantly on day 5 against S. aureus and E. coli, like the cumulative release profile (Figure 3F). In contrast, CHX⊂CD-MSN@HA@GelMA hydrogels exhibited a better persistent and consistent antibacterial activity against E. coli and S. aureus over 9 days. In that sense, the regulated release of CHX from the β-CD inclusion complex resulted in an antibacterial impact that was better maintained during the antimicrobial period because of the inclusion complex’s lengthier CHX release. The antibacterial activity of CHX-MSN@HA@GelMA was affected by the release of CHX, and the release rate was faster than CHX⊂CD-MSN@HA@GelMA. The antibacterial effect in the early stage is similar to that of the CHX⊂CD-MSN@HA@GelMA, but its antibacterial efficiency gradually decreases at 5 days. Additionally, the antibacterial effect was completely lost at 7 days, which was far from the long-term antibacterial effect. The findings indicated that CHX⊂CD-MSN@HA@GelMA hydrogels had the most significant and permanent antibacterial effect, which was beneficial for wound infection and required fewer replacements owing to its long-lasting antibacterial characteristics.
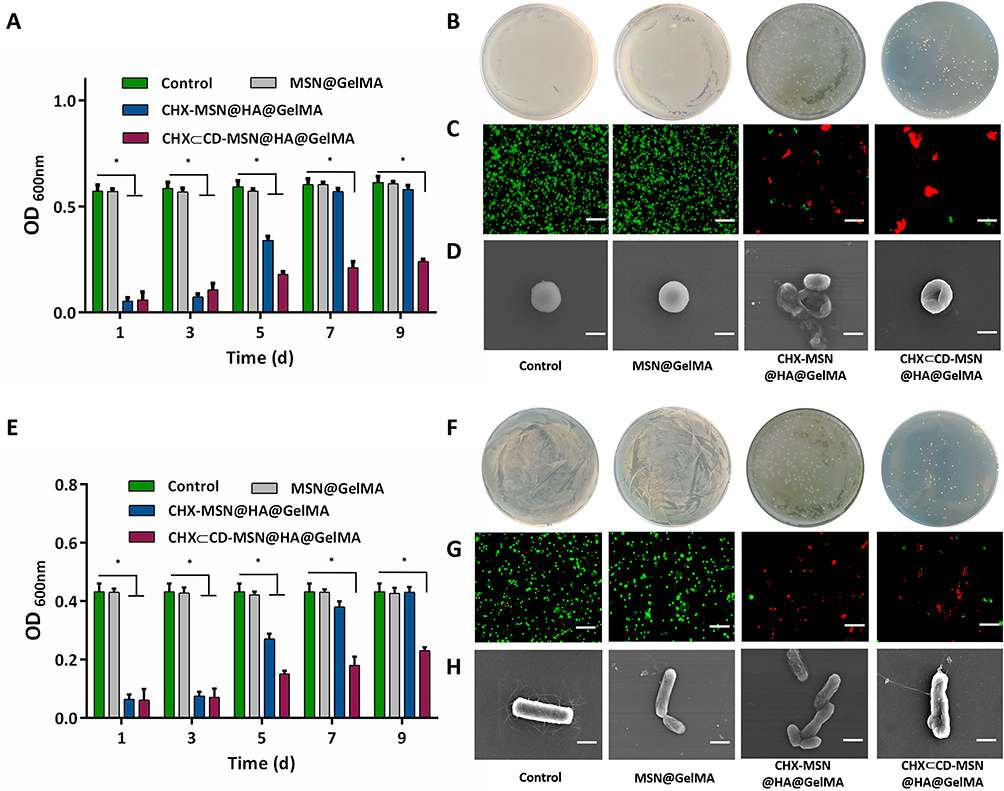 |
Figure 5 The antibacterial effects of CHX⊂CD-MSN@HA@GelMA hydrogels. (A) The antibacterial effects of CHX⊂CD-MSN@HA@GelMA hydrogels at 1, 3, 5, 7, and 9 days against S. aureus. (B) The antibacterial effects of CHX⊂CD-MSN@HA@GelMA hydrogels at 9 days against S. aureus were evaluated by a standard plate count method. (C) Live/dead fluorescence images of S. aureus samples. Viable cells are green fluorescent, and dead cells are red fluorescent. Scale bars=10 µm. (D) SEM morphologic observation of S. aureus. Scale bars=0.5 µm (E) The antibacterial effects of CHX⊂CD-MSN@HA@GelMA hydrogels at 1, 3, 5, 7, and 9 days against E. coli. (F) The antibacterial effects of CHX⊂CD-MSN@HA@GelMA hydrogels at 9 days against E. coli were evaluated by a standard plate count method. (G) Live/dead fluorescence images of E. coli samples. Scale bars = 10 µm. (H) SEM morphologic observation of E. coli. Scale bars = 1 µm with n = 3 independent samples. Data are expressed as mean ± SD. A significant difference was detected by the multiple t-test comparisons test. *P<0.05. |
In vivo Wound Healing Efficiency
We evaluated the antibacterial and wound healing capabilities of created hydrogel dressings in vivo using an S. aureus-infected cutaneous wound model in mice. The wounds were photographed on days 1, 5, 10, and 15. Figure 6A and B show that the control group developed an infection with an intense and severe bacterium and showed the smallest wound size decrease after 5 days of therapy. By comparison, wounds treated with MSN@GelMA, CHX-MSN@HA@GelMA, and CHX⊂CD-MSN@HA@GelMA hydrogels had no visible bacterium. This indicates that these groups had fewer bacterial infections and had a noticeable size reduction after 5 days of treatment, particularly the CHX-MSN@HA@GelMA and CHX⊂CD-MSN@HA@GelMA hydrogels. Additionally, the CHX⊂CD-MSN@HA@GelMA hydrogels showed superior debridement and antibacterial properties relative to the other groups, and the wounds healed significantly and almost completely after 15 days of treatment. Additionally, the wounds of the other groups did not heal effectively (Figure 6A). Here, we utilized the agar plate method to detect the antibacterial effect (Figure 6C). Like the in vitro results, there were many bacteria in the control and MSN groups, whereas few bacteria were detected in the CHX⊂CD-MSN@HA@GelMA hydrogels group. Additionally, using H&E analysis to evaluate the newly formed skin on day 15, it was observed that the scar width (Figure 6D and E) was the smallest in the CHX⊂CD-MSN@HA@GelMA hydrogels (0.35 mm) when compared with the control group (1.45 mm), MSN@GelMA hydrogels (1.09 mm), and CHX-MSN@HA@GelMA hydrogels (0.72 mm). When MSN@GelMA hydrogels, CHX-MSN@HA@GelMA hydrogels, and CHX⊂CD-MSN@HA@GelMA hydrogels were applied to wounds, it is assumed that the hydrogels all performed a debridement function and prevented bacterium from growing fast because of their outstanding hydrophilicity.
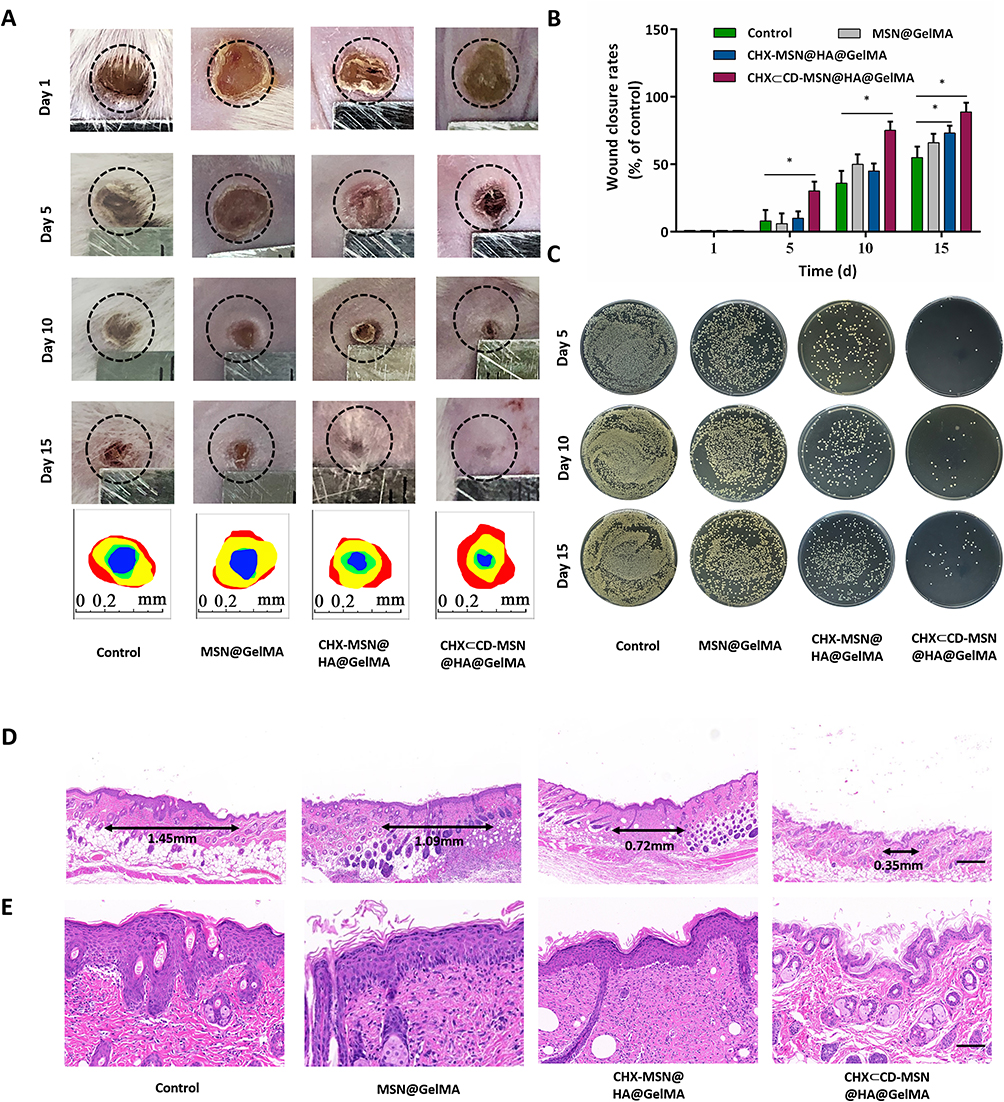 |
Figure 6 Evaluation of skin wound repair in vivo. (A) Representative photographs of rat wounds infected with S. aureus in four treatment groups (Control and MSN@GelMA hydrogels, CHX-MSN@HA@GelMA hydrogels, and CHX⊂CD-MSN@HA@GelMA hydrogels) within 15 days. (circle diameter: 5 mm). Traces of wound area during 15 days (red: day 1; yellow: day 5; green: day 10; blue: day 15). (B) Wound closure rates in all four groups. (C) Images of bacterial colonies on agar plates from the wounds treated with different experimental conditions. (D) H&E staining images of full-thickness wounds on day 15. Scale bar = 400 μm. (E) Partially enlarged H&E images. Scale bar = 100 μm. n = 3 independent samples. Data are expressed as mean ± SD. A significant difference was detected by the multiple t-test comparisons test. *P<0.05. |
Compared with MSN@GelMA hydrogels, CHX-MSN@HA@GelMA hydrogels and CHX⊂CD-MSN@HA@GelMA hydrogels containing CHX had higher therapeutic effectiveness and a similar initial healing time. Considering the varying quantities and times of drug release, CHX⊂CD-MSN@HA@GelMA, with a sustained drug release profile, has a somewhat shorter healing time (Figure 3F). The quantitative hemolysis ratios of CHX⊂CD-MSN@HA@GelMA hydrogel groups showed almost no apparent variation with PBS groups (Figure 7A and B), indicating the prominent hemocompatibility of the CHX⊂CD-MSN@HA@GelMA hydrogels. In addition, after the administration of CHX⊂CD-MSN@HA@GelMA hydrogels to essential organs of lab rats, an H&E staining experiment was performed to examine tissue morphology. The results in Figure 7C showed that all the tissues in the CHX⊂CD-MSN@HA@GelMA hydrogels had the same morphology as the control group, indicating that CHX⊂CD-MSN@HA@GelMA hydrogels had no systemic harmful effect on the main organs.
 |
Figure 7 Biocompatibility of CHX⊂CD-MSN@HA@GelMA hydrogels. (A and B) Hemolysis evaluation of the CHX⊂CD-MSN@HA@GelMA hydrogels after incubation with rat erythrocytes. (C) H&E staining assay of vital organs. Scale bar = 100 μm with n = 3 independent samples. Data are expressed as mean ± SD. A significant difference was detected by multiple t-test comparisons test. *P<0.05. |
Conclusion
In summary, we constructed pH-sensitive CHX⊂CD-MSN@HA@GelMA hydrogels that enable ultra-long-acting drug release and sustained antibacterial properties. Furthermore, in vitro drug release experiments indicated that the drug release from the CHX⊂CD-MSN@HA@GelMA hydrogels was sensitive to pH values. Compared with the CHX-MSN@HA@GelMA hydrogels, CHX⊂CD-MSN@HA@GelMA hydrogels have a relatively prolonged release period of CHX and are therefore preferred in clinical applications. Importantly, in vitro analysis demonstrated that the CHX⊂CD-MSN@HA@GelMA hydrogels provide superior therapeutic efficacy and possess an antibacterial efficacy of 59.71% and 47.56% against S. aureus and E. coli over 9 days, respectively. Meanwhile, in vivo experiments revealed that the CHX⊂CD-MSN@HA@GelMA hydrogels safely promote skin wound healing and enhance therapeutic efficacy. All the results indicated that hydrogels with long-term antibacterial properties had great potential applications in wound healing.
Acknowledgments
This research was supported by Zhejiang Provincial Science and Technology Project for Public Welfare (Grant No. LGF20H140003, LGF21H140006, LTGY23H140008); the Public Welfare Science and Technology Plan of Wenzhou (Y20190101, Y20170242, Y20210115); Zhejiang Provincial Medical and Health Science and Technology Plan (2019KY444).
Disclosure
All authors have approved the final version of this manuscript. The authors report no conflicts of interest in this work.
References
1. Jin L, Guo X, Gao D, et al. An NIR photothermal-responsive hybrid hydrogel for enhanced wound healing. Bioact Mater. 2022;16:162–172. doi:10.1016/j.bioactmat.2022.03.006
2. Hussain M, Suo H, Xie Y, et al. Dopamine-substituted multidomain peptide hydrogel with inherent antimicrobial activity and antioxidant capability for infected wound healing. ACS Appl Mater Interfaces. 2021;13(25):29380–29391. doi:10.1021/acsami.1c07656
3. Liu H, Li Z, Zhao Y, et al. Novel diabetic foot wound dressing based on multifunctional hydrogels with extensive temperature-tolerant, durable, adhesive, and intrinsic antibacterial properties. ACS Appl Mater Interfaces. 2021;13(23):26770–26781. doi:10.1021/acsami.1c05514
4. Wang S, Zheng H, Zhou L, Cheng F, Zhang Q. Injectable redox and light responsive MnO2 hybrid hydrogel for simultaneous melanoma therapy and multidrug-resistant bacteria-infected wound healing. Biomaterials. 2020;260:120314. doi:10.1016/j.biomaterials.2020.120314
5. Liu W, Ou-Yang W, Zhang C, et al. Staphylococcus aureus-synthetic polymeric antibacterial hydrogel for methicillin-resistant infected wound healing: nano antimicrobial self-assembly, drug- and cytokine-free strategy. ACS nano. 2020;14(10):12905–12917. doi:10.1021/acsnano.0c03855
6. Zhou C, Yang Z, Xun X, et al. De novo strategy with engineering a multifunctional bacterial cellulose-based dressing for rapid healing of infected wounds. Bioact Mater. 2022;13:212–222. doi:10.1016/j.bioactmat.2021.10.043
7. Yuan M, Liu K, Jiang T, et al. GelMA/PEGDA microneedles patch loaded with HUVECs-derived exosomes and Tazarotene promote diabetic wound healing. J Nanobiotechnology. 2022;20(1):147. doi:10.1186/s12951-022-01354-4
8. Zhou W, Duan Z, Zhao J, Fu R, Zhu C, Fan D. Fan, Glucose and MMP-9 dual-responsive hydrogel with temperature sensitive self-adaptive shape and controlled drug release accelerates diabetic wound healing. Bioact Mater. 2022;17:1–17. doi:10.1016/j.bioactmat.2022.01.004
9. Zou C, Lei X, Hu J, et al. Multi-crosslinking hydrogels with robust bio-adhesion and pro-coagulant activity for first-aid hemostasis and infected wound healing. Bioact Mater. 2022;16:388–402. doi:10.1016/j.bioactmat.2022.02.034
10. Peng H, Rossetto D, Mansy S, Jordan M, Roos K, Chen I. Treatment of wound infections in a mouse model using zn-releasing phage bound to gold nanorods. ACS Nano. 2022;16(3):4756–4774. doi:10.1021/acsnano.2c00048
11. Liang Y, Li M, Yang Y, Qiao L, Xu H, Guo B. pH/Glucose dual responsive metformin release hydrogel dressings with adhesion and self-healing via dual-dynamic bonding for athletic diabetic foot wound healing. ACS nano. 2022;16(2):3194–3207. doi:10.1021/acsnano.1c11040
12. Yang X, Zhang C, Deng D, Gu Y, Wang H, Zhong Q. Multiple stimuli-responsive mxene-based hydrogel as intelligent drug delivery carriers for deep chronic wound healing. Small. 2022;18(5):e2104368. doi:10.1002/smll.202104368
13. He Y, Leng J, Li K, et al. A multifunctional hydrogel coating to direct fibroblast activation and infected wound healing via simultaneously controllable photobiomodulation and photodynamic therapies. Biomaterials. 2021;278:121164. doi:10.1016/j.biomaterials.2021.121164
14. Zhao P, Chen W, Feng Z, et al. Electrospun nanofibers for periodontal treatment: a recent progress. Int J Nanomedicine. 2022;4137–4162. doi:10.2147/IJN.S370340
15. Huang C, Xu X, Fu J, Yu D, Liu Y. Recent progress in electrospun polyacrylonitrile nanofiber-based wound dressing. Polymers. 2022;14(16):3266–3299. doi:10.3390/polym14163266
16. Wu H, Li F, Wang S, et al. Ceria nanocrystals decorated mesoporous silica nanoparticle based ROS-scavenging tissue adhesive for highly efficient regenerative wound healing. Biomaterials. 2018;151:66–77. doi:10.1016/j.biomaterials.2017.10.018
17. Wang H, Zhang Y, Xu X, Wang A. An injectable mesoporous silica-based analgesic delivery system prolongs the duration of sciatic nerve block in mice with minimal toxicity. Acta Biomater. 2021;135:638–649. doi:10.1016/j.actbio.2021.09.008
18. Sun Y, Han Y, Dou Y, et al. Polymeric coating lubricates nanocontainers to escape macrophage uptake for bioreceptor recognition. Bioact Mater. 2022;8:14.
19. Sun Y, Zhang H, Wang Y, Wang Y. Charged polymer brushes-coated mesoporous silica nanoparticles for osteoarthritis therapy: a combination between hydration lubrication and drug delivery. J Control Release. 2017;259:e45–e46. doi:10.1016/j.jconrel.2017.03.114
20. Yan Y, Fu J, Wang T, Lu X. Controlled release of silyl ether camptothecin from thiol-ene click chemistry-functionalized mesoporous silica nanoparticles. Acta Biomater. 2017;51:471–478. doi:10.1016/j.actbio.2017.01.062
21. Poostforooshan J, Belbekhouche S, Shaban M, et al. Aerosol-assisted synthesis of tailor-made hollow mesoporous silica microspheres for controlled release of antibacterial and anticancer agents. ACS Appl Mater Interfaces. 2020;12(6):6885–6898. doi:10.1021/acsami.9b20510
22. Dening T, Zemlyanov D, Taylor L. Application of an adsorption isotherm to explain incomplete drug release from ordered mesoporous silica materials under supersaturating conditions. J Control Release. 2019;307:186–199. doi:10.1016/j.jconrel.2019.06.028
23. Sun T, Sun Y, Zhang H. Phospholipid-coated mesoporous silica nanoparticles acting as lubricating drug nanocarriers. Polymers. 2018;10(5):513–523. doi:10.3390/polym10050513
24. Zhu H, Zheng K, Boccaccini A. Multi-functional silica-based mesoporous materials for simultaneous delivery of biologically active ions and therapeutic biomolecules. Acta Biomater. 2021;129:1–17. doi:10.1016/j.actbio.2021.05.007
25. Bindini E, Ramirez M, Rios X, et al. In vivo tracking of the degradation of mesoporous silica through zr radio-labeled core-shell nanoparticles. Small. 2021;17(30):e2101519. doi:10.1002/smll.202101519
26. Bindini E, Chehadi Z, Faustini M, et al. Following in situ the degradation of mesoporous silica in biorelevant conditions: at last, a good comprehension of the structure influence. ACS Appl Mater Interfaces. 2020;12(12):13598–13612. doi:10.1021/acsami.9b19956
27. García-Fernández A, Sancenón F, Martínez-Máñez R. Mesoporous silica nanoparticles for pulmonary drug delivery. Adv Drug Deliv Rev. 2021;177:113953. doi:10.1016/j.addr.2021.113953
28. Akram Z, Aati S, Ngo H, Fawzy A. pH-dependent delivery of chlorhexidine from PGA grafted mesoporous silica nanoparticles at resin-dentin interface. J Nanobiotechnology. 2021;19(1):43. doi:10.1186/s12951-021-00788-6
29. Chen C, Tang W, Jiang D, et al. Hyaluronic acid conjugated polydopamine functionalized mesoporous silica nanoparticles for synergistic targeted chemo-photothermal therapy. Nanoscale. 2019;11(22):11012–11024. doi:10.1039/C9NR01385G
30. Yilmaz M. Layer-by-layer hyaluronic acid/chitosan polyelectrolyte coated mesoporous silica nanoparticles as pH-responsive nanocontainers for optical bleaching of cellulose fabrics. Carbohydr Polym. 2016;146:174–180. doi:10.1016/j.carbpol.2016.03.037
31. Tabary N, Chai F, Blanchemain N, et al. A chlorhexidine-loaded biodegradable cellulosic device for periodontal pockets treatment. Acta Biomater. 2014;10(1):318–329. doi:10.1016/j.actbio.2013.09.032
32. Yue I, Poff J, Cortés M, et al. A novel polymeric chlorhexidine delivery device for the treatment of periodontal disease. Biomaterials. 2004;25(17):3743–3750. doi:10.1016/j.biomaterials.2003.09.113
33. Hemine K, Skwierawska A, Kleist C, et al. Effect of chemical structure on complexation efficiency of aromatic drugs with cyclodextrins: the example of dibenzazepine derivatives. Carbohydr Polym. 2020;250:116957. doi:10.1016/j.carbpol.2020.116957
34. Xue F, Wang Y, Zhang Q, et al. Self-assembly of affinity-controlled nanoparticles via host-guest interactions for drug delivery. Nanoscale. 2018;10(26):12364–12377. doi:10.1039/C8NR01518J
35. Sévery L, Szczerbiński J, Taskin M, et al. Immobilization of molecular catalysts on electrode surfaces using host-guest interactions. Nat Chem. 2021;13(6):523–529. doi:10.1038/s41557-021-00652-y
36. Kettel M, Heine E, Schaefer K, Moeller M. Chlorhexidine loaded cyclodextrin containing PMMA nanogels as antimicrobial coating and delivery systems. Macromol Biosci. 2017;17(2):182–197. doi:10.1002/mabi.201600230
37. Sun Y, Zhou Y, Li Q, Yang Y. Enzyme-responsive supramolecular nanovalves crafted by mesoporous silica nanoparticles and choline-sulfonatocalix[4]arene [2]pseudorotaxanes for controlled cargo release. Chem Commun. 2013;49(79):9033–9035. doi:10.1039/c3cc45216f
38. Sun Y, Yang Y, Chen D, et al. Mechanized silica nanoparticles based on pillararenes for on-command cargo release. Small. 2013;9(19):3224–3229. doi:10.1002/smll.201300445
39. Lavoine N, Tabary N, Desloges I, Martel B, Bras J. Controlled release of chlorhexidine digluconate using β-cyclodextrin and microfibrillated cellulose, Colloids. Surf B Biointerfaces. 2014;121:196–205. doi:10.1016/j.colsurfb.2014.06.021
40. Inoue B, Streit S, Dos Santos Schneider AL, Meier MM. Bioactive bacterial cellulose membrane with prolonged release of chlorhexidine for dental medical application. Int J Biol Macromol. 2020;148:1098–1108. doi:10.1016/j.ijbiomac.2020.01.036
41. Denadai A, Teixeira K, Santoro M, Pimenta A, Cortés M, Sinisterra R. Supramolecular self-assembly of beta-cyclodextrin: an effective carrier of the antimicrobial agent chlorhexidine. Carbohydr Res. 2007;342(15):2286–2296. doi:10.1016/j.carres.2007.05.002
42. Szente L, Singhal A, Domokos A, Song B. Cyclodextrins: assessing the impact of cavity size, occupancy, and substitutions on cytotoxicity and cholesterol homeostasis. Molecules. 2018;23(5):1228. doi:10.3390/molecules23051228
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.