")
Back to Journals » International Journal of Nanomedicine » Volume 14
Hyaluronic acid-decorated redox-sensitive chitosan micelles for tumor-specific intracellular delivery of gambogic acid
Authors Xu W , Wang H, Dong L, Zhang P , Mu Y, Cui X, Zhou J, Huo M, Yin T
Received 10 January 2019
Accepted for publication 25 April 2019
Published 27 June 2019 Volume 2019:14 Pages 4649—4666
DOI https://doi.org/10.2147/IJN.S201110
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Lei Yang
Wei Xu,1,2 Honglan Wang,3 Lihui Dong,3 Pan Zhang,3 Yan Mu,2 Xueyan Cui,2 Jianping Zhou,3 Meirong Huo,3 Tingjie Yin3
1Department of Pharmacy, Shandong Provincial Qian Foshan Hospital, Shandong University, Jinan 250014, People’s Republic of China; 2Qianfoshan Hospital, The First Hospital Affiliation with Shandong First Medical University, Jinan 250012, People’s Republic of China; 3State Key Laboratory of Natural Medicines, Department of Pharmaceutics, China Pharmaceutical University, Nanjing 210009, People’s Republic of China
Introduction: Herein, a hyaluronic acid (HA)-coated redox-sensitive chitosan-based nanoparticle, HA(HECS-ss-OA)/GA, was successfully developed for tumor-specific intracellular rapid delivery of gambogic acid (GA).
Materials and methods: The redox-sensitive polymer, HECS-ss-OA, was prepared through a well-controlled synthesis procedure with a satisfactory reproducibility and stable resulted surface properties of the assembled cationic micelles. GA was solubilized into the inner core of HECS-ss-OA micelles, while HA was employed to coat outside HECS-ss-OA/GA for CD44-mediated active targeting along with protection from cation-associated in vivo defects. The desirable redox-sensitivity of HA(HECS-ss-OA)/GA was demonstrated by morphology and particle size changes alongside in vitro drug release of nanoparticles in different simulated reducing environments.
Results: The results of flow cytometry and confocal microscopy confirmed the HA-receptor mediated cellular uptake and burst drug release in highly reducing cytosol of HA(HECS-ss-OA)/GA. Consequently, HA(HECS-ss-OA)/GA showed the highest apoptosis induction and cytotoxicity over the non-sensitive (HA(HECS-cc-OA)/GA) and HA un-coated (HECS-ss-OA/GA) controls against A549 NSCLC model both in vitro and in vivo. Furthermore, a diminished systemic cytotoxicity was observed in HA(HECS-ss-OA)/GA treated mice compared with those treated by HA un-coated cationic ones and GA solution.
Keywords: gambogic acid, nanoparticles, redox-sensitive, intracellular drug delivery, tumor
Introduction
Lung cancer, of which non-small-cell lung cancer (NSCLC) is the most common form, remains the leading cause of cancer death around the world.1–3 Medicinal chemotherapy is currently one of the three main methods in the clinical treatment. Specifically, as the main active compound of gamboge resin, gambogic acid (GA) has recently been established as a potent anticancer agent that shows significant inhibitive activities against a variety of cancer cells including NSCLC.4–6 Notably, GA treatment for lung cancer has been approved by the Chinese Food and Drug Administration.7 In a Phase II study of GA injection in China demonstrated that GA has a promising control rate of advanced malignant tumors.8 As a promising anticancer agent with multiple targets, GA mediates various effects including induction of cell apoptosis, inhibition of cell proliferation, prevention of tumor angiogenesis, and interception of tumor invasion and metastasis, which shows great advantages over many current used anticancer agents.9–11
However, like many other chemotherapeutic drugs, great limitations such as poor water solubility (∼10 μg/mL), rapid system clearance (t1/2=15.7 mins), and non-selective cytotoxicity against cancer cells confuse its clinical application.12 Consequently, the nanocarrier-based anti-tumor therapeutic strategy, especially the self-assembled polymeric micelle, attracted lots of attention due to its efficient solubilizing ability for hydrophobic drugs and passive targeting-mediated site-specific delivery.13,14 Nevertheless, the conventional polymeric micellar delivery system was usually faced with a poor cellular uptake into cancer cells owing to the steric shielding from its highly hydrophilic outer shell. Worse still, the gradual degradation of micellar structures led to a sustained drug release over a long period of time.15 Resultantly, a deficient cellular internalization along with a mild drug liberation simultaneously contributed to a suboptimal drug potency. The design of polymeric micelles to achieve active targeting as well as stimuli-triggered burst drug release at tumor microenvironment for a sufficient drug treatment concentration is of a high demand.
Currently, physiological signals (redox potential,16 pH,17 enzymes18) and external stimuli signals (light,19 temperature,20 magnetic field,21 ultrasound22) have enabled drug delivery systemsto achieve triggered payloads liberation inside tumor cells.23,24 Our group consistently focuses on the redox potential-based technology. To date, we have successfully developed redox-sensitive poly(ethylene glycol) (PEG)-based prodrug [OCT(Phe)-PEG-ss-PTX],25 chitosan (CS)-based micellar system (HECS-ss-OA)26 or polyethyleneimine (PEI)-based amphiphilic micelles [HA-ss-(OA-g-bPEI)]27 containing disulfide bond as redox-responsive linkages for effective solubilization and intracellular controlled-release of model anticancer drug, paclitaxel (PTX). The existing data validated a notable improvement on PTX potency ascribed to the raised tumor accumulation and the tumor cytoplasm-specific burst drug release by these designed vehicles. However, the present redox-sensitive micelle based on O,N-hydroxyethyl CS−octylamine conjugates (HECS-ss-OA) by our group still exhibited limitations.26 The primary one is the chemical modification-associated un-controllability. In detail, the synthesis of HECS-ss-OA started with conjugating the terminal carboxyl groups of 3,3'-dithiodipropionic acid (DPA), a bio-reducible linkage, to the free amino groups of hydroxyethyl CS (HECS) to get HECS-DPA intermediates. In the following, the free carboxyl in HECS-DPA was conjugated with n-octylamine (OA) to obtain HECS-ss-OA. The couple rate between HECS-DPA and OA relied on the remained amounts of free carboxyl groups in conjugated DPA. However, both the two terminal carboxyl groups of DPA might be coupled to HECS, and the remained amounts of carboxyl groups in HECS-DPA were uncertain. Therefore, the HECS-ss-OA polymer was confronted with an un-accurate OA grafting ratio and a varying amount of surface-free carboxyl groups, which would eventually affect the microscopic structure, surface properties and thus in vivo distribution of the micelles.
In light of aforementioned limitations of HECS-ss-OA, we herein further improve the accuracy, controllability, and reproducibility of the polymer synthesis procedure. The n-octylamine (OA) is first conjugated with 3, 3'-dithiodipropionic anhydride (DPAH) to form OA-ss-COOH intermediates which possess disulfide bond and one terminal carboxyl group. Thus, HECS can be accurately modified by OA-ss-COOH with a well-controlled grafting ratio and a satisfactory reproducibility. The obtained redox-sensitive HECS-ss-OA conjugates stably assemble in water to achieve definitely positive charged micelles. Nevertheless, the cationic HECS-ss-OA micelles are not only lacking of active tumor targeting, but also encountered with serum protein-mediated aggregation and elimination by the reticular epithelial system.27,28 Accordingly, hyaluronic acid (HA) was explored to shield the cationic CS-based micelles owing to its natural electronegativity, low immunogenicity, biocompatibility, and specific targeting ability to receptors like cluster determinant 44 (CD44) overexpressed in many tumors.15,29,30
As shown in Scheme 1, the HA-coated redox-sensitive nanoparticles, HA(HECS-ss-OA), were developed for the purpose of tumor cytoplasm-specific rapid delivery of GA to achieve a highly efficient treatment of NSCLC. The GA loaded HA(HECS-ss-OA) nanoparticles, HA(HECS-ss-OA)/GA, are expected to stably transport in blood circulation, selectively accumulate within tumor sites, efficiently internalize into tumor cells via CD44-mediated endocytosis and complete GSH triggered GA release in cytoplasm. In this study, we investigated the basic physicochemical characteristics of HA(HECS-ss-OA), including its self-assembly behaviors as well as the redox-sensitivity. Subsequently, the CD44 receptor-mediated tumor active targeting and the redox-responsive rapid drug release of HA(HECS-ss-OA)/GA were collectively confirmed by an enhanced cytotoxicity and apoptosis-inducing ability of GA against A549 cell lines both in vitro and in vivo.
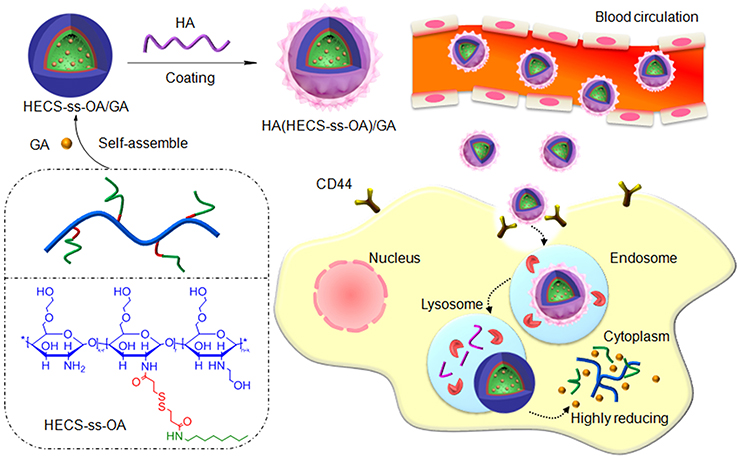 |
Scheme 1 Schematic design of HECS-ss-OA and the illustration of tumor cytoplasm-selective rapid GA delivery by the HA-coated redox-sensitive HA(HECS-ss-OA)/GA nanoparticles. Abbreviations: HECS-ss-OA, redox-sensitive O, N-hydroxyethyl chitosan−octylamine conjugates; GA, gambogic acid; HA, hyaluronic acid. |
Materials and methods
Materials
CS (average molecular weight 87 kDa, 90% deacetylated) was purchased from Yuhuan Biochemical Co. Ltd. (Zhejiang, People's Republic of China). Sodium HA (molecular weights 50 kDa) was purchased from Freda Biochem Co., Ltd. (Shangdong, People's Republic of China). DL-Dithiothreitol (DTT), 3, 3'-dithiodipropionic acid (DPA), succinic anhydride (SA), Octylamine (OA), 1-ethyl-3-(3-(dimethylamino) propyl) carbodiimide hydrochloric acid salt (EDC·HCl), N-hydroxysuccinimide (NHS), and 4-dimethylaminopyridine (DMAP) were purchased from Aladdin Reagent Co. Ltd. (Shanghai, People's Republic of China). MTT was purchased from Beyotime Biotechnology (Shanghai, People's Republic of China). The near-infrared dye DiR was obtained from Beijing Fanbo Science and Technology Co., Ltd. (Beijing, People's Republic of China). Coumarin 6 (C6) and Nile red (NR) were purchased from Sigma-Aldrich (St Louis, MO, USA). All the chemicals and reagents unless mentioned above were analytical grade without further purification, obtained commercially.
Cell culture
A549 cells were provided by Origin Biosciences Inc. (Nanjing, People's Republic of China), and were grown in RPMI-1640 media containing FBS (10%, v/v) at 37°C in 5% CO2 atmosphere. The cells were sub-cultivated every 3 days at a split ratio of 1:3.
Animals and tumor xenograft models
BALB/c nude mice (age, 5–6 weeks; weight, 18−22 g) were obtained from Qing long shan animal farms (Nanjing, People's Republic of China). All animal care and experiments were conducted in compliance with the National Institute of Health Guide for the Care and Use of Laboratory Animals. The animals were pathogen free and allowed to access food and water freely. All experiments were conducted in strict accordance with the relevant laws and followed the institutional guidelines of China Pharmaceutical University. The China Pharmaceutical University institutional committee approved the experiments. The nude mice were subcutaneously inoculated in the armpit region with A549 cells (1×107 cells/mouse). The tumor volume (V) was monitored by caliper measurements and calculated as V = a × b2/2, where a was the largest diameter and b the smallest.
Synthesis of HECS-ss-OA and HECS-cc-OA conjugates
The HECS-ss-OA and HECS-cc-OA conjugates were synthesized following the steps presented in Scheme 2.
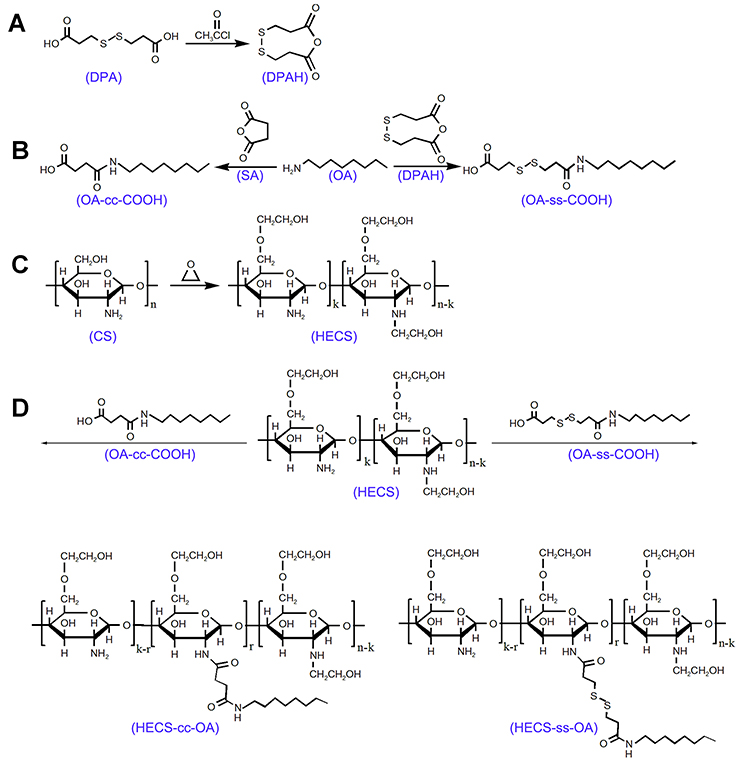 |
Scheme 2 Synthetic scheme of DPAH (A), OA-cc-COOH and OA-ss-COOH (B), HECS conjugate (C), HECS-cc-OA and HECS-ss-OA conjugates (D). Abbreviations: DPA, 3, 3'-dithiodipropionic acid; DPAH, 3, 3'-dithiodipropionic acid anhydride; OA, octylamine; SA, succinic anhydrid; HECS, hydroxyethyl chitosan; HECS-cc-OA, redox-insensitive O,N-hydroxyethyl chitosan−octylamine conjugates; HECS-ss-OA, redox-sensitive O,N-hydroxyethyl chitosan−octylamine conjugates. |
Synthesis and structural characterization of HECS conjugate
HECS was synthesized by conjugating glycol groups to CS as we previously reported (yield = 95%).31 The chemical structure of the HECS conjugate was confirmed by 1H NMR at 300 MHz (AVACE, Bruker) dissolved in D2O. The degree of substitution (DS) of hydroxyethyl groups was calculated by comparing the C and N molar ratio obtained from elemental analysis via a Vario EL III analyzer (Elementar, Hanau, Germany), using the following equation:32
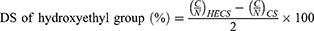
Synthesis and structural characterization of OA-ss-COOH and OA-cc-COOH conjugates
DPAH was prepared according to the general methods as reported.33 2 g of DPA was refluxing in 10 mL of acetyl chloride at 65°C for 2 hrs. Then, most of the acetyl chloride in the solution was evaporated under vacuum. An excess ice-cold ethyl ether was used to remove the remaining acetyl chloride, while precipitating and washing the product. Finally, the obtained DPAH was vacuum dried with a yield of 75.8%.
To synthesize OA-ss-COOH, 610.2 mg of OA was added into 20 mL of dichloromethane (CH2Cl2) which dissolved 1.0 g of DPAH, 473.0 mg of triethylamine and 29.0 mg of N, N-dimethyl-4-aminopyridine (DMAP). The solution reacted at 25°C for 24 hrs, followed by several washes with weak acid water solution and saturated sodium chloride (NaCl) solution. The mixture was further dried with anhydrous sodium sulfate (Na2SO4) before evaporation. Afterward, the crude product was purified by silica gel column chromatography with CH2Cl2/CH3OH (v/v, 100:1) to obtain the pure intermediate, OA-ss-COOH (yield = 48.5%).
To synthesize OA-cc-COOH, 990.0 mg of succinic anhydride, 1.2 mg of OA, 900.0 mg of triethylamine, and 55.0 mg of DMAP were dissolved in 20 mL of CH2Cl2. The reaction proceeded at 25°C for 24 hrs, and then the solution was washed with weak acid water solution and saturated NaCl solution for several times. After dried with anhydrous Na2SO4, the solvent was evaporated to achieve the pure intermediate, OA-cc-COOH (yield = 82.4%).
The chemical structure of OA-ss-COOH and OA-cc-COOH was confirmed by 1H NMR at 300 MHz (AVACE, Bruker), and the intermediates were dissolved in d6-DMSO. In addition, the mass spectra of OA-ss-COOH and OA-cc-COOH were scanned using High-Resolution Mass Spectrometer (HR-MS, JMS-800D).
Synthesis and structural characterization of HECS-ss-OA and HECS-cc-OA conjugates
HECS-ss-OA and HECS-cc-OA conjugates were obtained by conjugating OA-ss-COOH and OA-cc-COOH to HECS, respectively. In detail, OA-ss-COOH (32.0 mg, 0.1 mmol) or OA-cc-COOH (23.0 mg, 0.1 mmol) was dissolved in 10 mL of DMSO containing EDC (57.5 mg, 0.3 mmol) and NHS (34.5 mg, 0.3 mmol). The reaction proceeded under stirring for 30 mins at 0°C to form active esters. Then, the activated OA-ss-COOH or OA-cc-COOH was added dropwise to 10 mL of DMSO which contained 205.0 mg of HECS, and the mixture was stirred at 25°C for 24 hrs. Finally, the solution was diluted by 200 mL of distilled water, dialyzed against distilled water and lyophilized.
The chemical structures of HECS-ss-OA and HECS-cc-OA conjugates were confirmed by 1H NMR at 300 MHz (AVACE, Bruker) with HECS, OA-ss-COOH, and OA-cc-COOH as controls. In this experiment, HECS, HECS-ss-OA, and HECS-cc-OA conjugates were dissolved in D2O. The DS of OA in HECS-ss-OA and HECS-cc-OA conjugates were estimated according to data from 1H NMR.34
Preparation and characterization of nanoparticles
Preparation and characterization of GA-loaded micelles
The GA-loaded polymeric micelles, HECS-ss-OA/GA and HECS-cc-OA/GA, were prepared by dialysis technique. Briefly, 18 mg of HECS-ss-OA or HECS-cc-OA powders was dispersed in 3 mL of distilled water under continuous stirring. 5 mg of GA dissolved in 200 µL ethanol were added dropwise to the blank micelle solutions (6 mg/mL) while stirring. The mixture was stirred for 0.5 hr at room temperature followed by being sonicated using a probe-type ultrasonicator (JY92-2D; Ningbo Scientz Biotechnology Co., Ltd, Ningbo, People's Republic of China) at 100 W for 0.5 hr in an ice bath. The resulting solution was dialyzed against distilled water overnight to remove ethanol. The unloaded GA was removed by filtration with a 0.45 μm filter and then lyophilized.
The dispersion state of GA in HECS-ss-OA micelles was evaluated by different scanning calorimetry (DSC) analysis using a NETZSCH DSC 204 equipment with the temperature and heating rate of 40–300°C and 10°C/min, separately. Herein, the samples including GA, blank HECS-ss-OA, and the physical mixture of GA and HECS-ss-OA were used as controls.
Preparation and characterization of HA(HECS-ss-OA)/GA
HA was coated outside GA-loaded micelles via electrostatic interactions. Briefly, the GA-loaded micelles (1 mg/mL) were added slowly into the HA solution (1 mg/mL) under vigorous stirring at different weight ratios. Then, the mixture was kept at 37°C for 30 mins. The optimal weight ratio for HA and GA-loaded micelles was selected according to the measured particle size and zeta potential of HA(HECS-ss-OA)/GA and HA(HECS-cc-OA)/GA.
The particle size and zeta potential of the prepared nanoparticles were measured by dynamic light scattering (DLS) using a Malvern Zetasizer nano-ZS90 (Malvern Instruments, UK) with HA-uncoated micelles (HECS-ss-OA/GA, HECS-cc-OA/GA) as controls. The morphology was observed by transmission electron microscopy (TEM, H-600, Hitachi, Japan). Samples for TEM analysis were negatively stained by phosphotungstic acid (1%, v/v). The drug-loading (DL) content and entrapment efficiency (EE) were calculated by the following formulas:

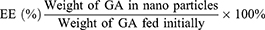
GA concentrations were measured by high-performance liquid chromatography (LC-2010C, Shimadzu, Japan) equipped with a Lichrospher C18 column (4.6 mm × 250 mm, 5 μm) at 30°C. The mobile phase was the mixed solution of methanol and water (93:7, v/v) containing 0.1% acetic acid, while the detection wavelength, flow rate, and the sample injected volume were set at 360 nm, 1 mL/min, and 20 μL, respectively.
In vitro response to the reducing environment
First, the disassembly of redox-sensitive HA(HECS-ss-OA)/GA nanoparticle in response to reducing environment was monitored by TEM to observe the morphology change. Briefly, HA(HECS-ss-OA)/GA nanoparticles in pH 7.4 HEPES buffer solution were incubated with 10 mM DTT for 4 hrs. Then, the morphology of nanoparticles was observed using TEM after stained by phosphotungstic acid (1%, v/v). The morphology of non-sensitive HA(HECS-cc-OA)/GA nanoparticles incubated with 10 mM DTT was also observed as comparison.
In order to further verify the reducing environment triggered drug release of HA(HECS-ss-OA)/GA, the fluorescent probe NR was loaded into the nanoparticles instead of the model drug, GA. The NR-loaded nanoparticles, HA(HECS-ss-OA)/NR and HA(HECS-cc-OA)/NR, were treated in pH 7.4 HEPES buffer containing 10 mM DTT. The fluorescence emission spectra of NR were monitored at 0, 6, 12, 24, and 36 hrs with an excitation wavelength of 326 nm. Moreover, the change of particle size of these two nanoparticles in response to different reductive conditions was also measured by DLS. 1 mg/mL of HA(HECS-ss-OA)/NR and HA(HECS-cc-OA)/NR nanoparticles in pH 7.4 HEPES were treated with various concentrations of DTT (0 mM, 5 mM, 10 mM). Samples were placed in shaking bed at 37°C with a rotation speed of 100 rmp. The samples were detected at 0, 1, 2, 4, 8, and 12 hrs by DLS.
In vitro cellular studies
Cellular uptake and intracellular release behaviour
The cellular uptake effects of nanoparticles in A549 cells were monitored by confocal laser scanning microscopy (CLSM) and flow cytometry. Firstly, coumarin 6 (C6) was loaded into the HECS-ss-OA and HECS-cc-OA micelles as a fluorescent marker according to the protocol for the encapsulation of GA, followed a coating with HA. For CLSM analyses, A549 cells (5×104 cells/mL) were seeded in glass bottom dishes and incubated in complete RPMI-1640 for 24 hrs. Then, cells were incubated with fresh media containing HA(HECS-ss-OA)/C6 or HA(HECS-cc-OA)/C6 nanoparticles at C6 concentration of 100 ng/mL for 1, 2, or 4 hrs, followed by three washes with cold PBS. In addition, to demonstrate the CD44 receptor-mediated specific internalization of HA(HECS-ss-OA)/C6, another group of cells were incubated with 10 mg/mL of free HA for 2 hrs before being incubated with the nanoparticles for 4 hrs. Subsequently, the cell nuclei were stained by Hoechst 33342 (10 µg/mL) for 10 mins and the samples were observed under CLSM. For the flow cytometric analyses, the above-mentioned cell groups were washed by cold PBS for three times and re-suspended in PBS. The fluorescent intensity of C6 in cell samples was detected by a flow cytometer (BD FACS Calibur, USA).
To visualize the intracellular release behavior of HA-coated nanoparticles, fluorescent probe NR was loaded into HECS-ss-OA and HECS-cc-OA micelles as described above. A549 cells (5×104 cells/mL) were planted in glass bottom dishes and incubated in complete RPMI-1640 for 24 hrs. After an incubation with HA(HECS-ss-OA)/NR and HA(HECS-cc-OA)/NR at a NR concentration of 100 ng/mL for 4 h or 24 h, the nuclei of A549 cells were stained by Hoechst 33342 (10 µg/mL) for 10 min and the cells were observed using CLSM.
Cytotoxicity assay
The cytotoxicity of blank or drug-loaded nanoparticles was evaluated by MTT assay on A549 cells. To the biomaterial MTT assay, A549 cells seeded in 96-well plates were treated with HA(HECS-ss-OA), HA(HECS-cc-OA), HECS-ss-OA or HECS-cc-OA for 48 hrs with the conjugate (HECS-ss-OA or HECS-cc-OA) concentration ranging from 0.05 to 100 µg/mL. In the case of the MTT assay for drug-loaded nanoparticles, A549 cells were cultured with HA(HECS-ss-OA)/GA, HA(HECS-cc-OA)/GA and free GA with different concentrations of GA (0.05–10 µg/mL) for 48 h. After specified durations, 20 µL MTT (5 mg/mL) was added to each well for 4 hrs, followed by an addition of 100 µL DMSO to dissolve the formazan crystals. The OD was measured by a microplate reader (Multiskan Mk3, Thermo Lab systems, Beverly, MA, USA) at the wavelength of 570 nm. Cell sample treated with blank medium was served as control. Cell viability was calculated using the following formulas: cell viability = ODsample/ODcontrol × 100%. The toxicity of different formulations was expressed as the inhibitory concentration at which 50% of cell growth was inhibited (IC50).
Apoptosis assay
Apoptosis of A549 cells induced by GA-loaded nanoparticles was further assessed using Annexin V-FITC and propidium iodide (PI) double staining assay. A549 cells seeded in 6-well plates were treated with HA(HECS-ss-OA)/GA or HA(HECS-cc-OA)/GA micelles at a GA concentration of 0.4 µg/mL, In the receptor competitive inhibition experiment, cells were incubated with 10 mg/mL of free HA for 2 hrs before adding HA(HECS-ss-OA)/GA. The cell sample treated with blank medium was set as control. After incubated for 48 hrs, the cells were washed with cold PBS for three times, followed by re-suspended in PBS. Subsequently, Annexin V-FITC and PI were added according to the manufacturer’s protocol before detected by flow cytometry.
In vivo bio-distribution
BALB/c nude mice bearing A549 tumors around 200 mm3 were intravenously injected with DiR and GA co-loaded nanoparticles, HA(HECS-ss-OA)/GA+DiR and HA(HECS-cc-OA)/GA + DiR, via the tail vein at a DiR dose of 0.5 mg/kg. In the competition group, the mice were injected with 50 mg/kg of free HA 30 mins before the treatment with HA(HECS-ss-OA)/GA+DiR. NIR fluorescent imaging experiments were performed using an in vivo imaging system (FX PRO, Kodak, USA) at pre-scheduled time intervals. Afterward, the mice were sacrificed and the major organs including heart, liver, spleen, lung, and kidney, as well as tumor, were excised for ex vivo imaging using the same system described above.
In vivo antitumor efficacy
Tumor-bearing BALB/c nude mice were randomly divided into five groups (n = 6) when the tumor volume grew to approximately 100 mm3, and injected with saline, the free GA, HECS-ss-OA/GA, HA(HECS-ss-OA)/GA or HA(HECS-cc-OA)/GA via tail vein at a constant GA dose of 8.0 mg/kg. In order to administrate via tail vein injection, GA was dissolved in 20% trimethylene glycol aqueous solution due to its poor water solubility and HECS-ss-OA/GA, HA(HECS-ss-OA)/GA or HA(HECS-cc-OA)/GA solutions were prepared from rehydration of the lyophilized formulations. These preparations were accomplished immediately before drug administration. The injections were performed once 2 days for fivetimes. The body weight and the tumor volumes of mice were recorded. Afterward, the mice were sacrificed and tumors were extracted and weighted. The tumor weight inhibition ratio (IR) was calculated by the following equation: IR = (TWcontrol – TWtest)/TWcontrol, where TWcontrol and TWtest represented the mean tumor weight of saline group and treated groups, respectively.
In addition, tumors were tested for histological evaluation by H&E staining and TUNEL assays. To evaluate the safety of the formulations in vivo, the body weight of the experiment mice, the levels of hepatic function markers (alanine aminotransferase (ALT), aspartate transaminase (AST)), and renal function markers (serum creatinine [Scr], blood urea nitrogen [BUN]) in serum from the treated mice were also detected. In addition, normal organs including heart, liver, spleen, lung, and kidney were separated from the sacrificed mice for histological evaluation by H&E staining.
Statistics
All quantitative data were expressed as mean ± SD unless otherwise noted. Statistical evaluation was performed through one-way or two-way ANOVA, followed by the Tukey–Kramer test. A value of p < 0.05 was considered statistically significant.
Results and discussion
Synthesis and characterization of HECS-ss-OA and HECS-cc-OA conjugates
A redox-sensitive CS-based amphiphilic conjugate, HECS-ss-OA, alongside its non-sensitive control, HECS-cc-OA, were synthesized following the procedures shown in Scheme 2. Preliminarily, the hydroxyethyl groups were incorporated onto CS backbone to improve water solubility. As mentioned in the “Introduction” section, we herein improved the synthetic procedure of these two conjugates published in our previous paper.26 To ensure the accuracy, controllability and reproducibility of HECS-ss-OA and HECS-cc-OA, the redox-sensitive and non-sensitive intermediates (OA-ss-COOH and OA-cc-COOH) were firstly synthesized. The structures of the pure OA-ss-COOH and OA-cc-COOH were confirmed by 1H NMR and HR-MS. Data in Figure S1 demonstrated 1H NMR characteristic peaks of DPA or SA with octyl. Additionally, [M-H]− for OA-ss-COOH and OA-cc-COOH was 320.2 and 228.2, respectively, which was consistent with the theory number that DPAH or SA covalently linked to OA at a molar ratio of 1:1. Thereafter, EDC activated OA-ss-COOH and OA-cc-COOH intermediates were conjugated with free amine of HECS by amide coupling to obtain HECS-ss-OA and HECS-cc-OA conjugates. The chemical structures of HECS-ss-OA and HECS-cc-OA were characterized using 1H NMR. As shown in Figure 1, the peaks in the range of 3.5−4.0 ppm of HECS were attributed to CS-based backbone (H3, H4, H5, H6) and protons (−OCH2CH2OH) from glycol group. Compared with the spectrum of HECS, the successful introduction of OA-ss-COOH or OA-cc-COOH was demonstrated by the well-observed −CH3 protons (0.8 ppm) belong to octyl.
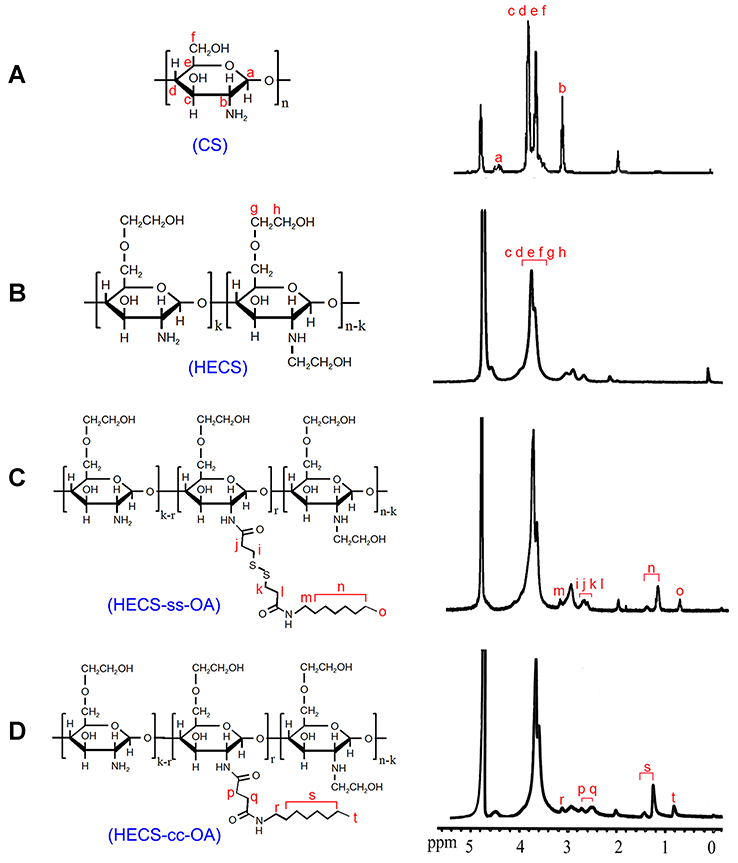 |
Figure 1 Structure and 1H NMR spectrum of CS (A), HECS (B), HECA-ss-OA (C) and HECS-cc-OA (D). a, b, c, etc indicated the attribution of spectral peaks to protons from the structural units of conjugates.Abbreviations: CS, chitosan; HECS, hydroxyethyl chitosan; HECS-cc-OA, redox-insensitive O, N-hydroxyethyl chitosan−octylamine conjugates; HECS-ss-OA, redox-sensitive O, N-hydroxyethyl chitosan−octylamine conjugates; NMR, nuclear magnetic resonance. |
The DS of glycol groups in HECS-ss-OA and HECS-OA conjugates were determined to be 110.1% and 107.3%, respectively, using elemental analysis. The amounts of OA in HECS-ss-OA and HECS-cc-OA were estimated to be 8.6% and 9.2%, respectively, according to the ratio of integration values of the methyl protons in OA (0.8 ppm, 3H, −CH3) to values ascribed to CS protons in (3.5−4.0 ppm, 8H, H3, H4, H5, H6 and −OCH2CH2OH).
Preparation and characterization of HA(HECS-ss-OA)/GA nanoparticles
As described above, GA was incorporated into the hydrophobic inner core of HECS-ss-OA micelles by dialysis technique. Due to the well controllability of conjugate synthesis procedure used here, HECS-ss-OA/GA and HECS-cc-OA/GA micelles obtained a stably reproduced zeta potential and particle size of around +20 mV and 210 nm, respectively, and a high DL of ~18% (Table 1). Besides, DSC was utilized to investigate the existing state of GA encapsulated in the micelles. As exhibited in Figure 2A, free GA crystalline state was found to have one endothermic peak at 66.0°C and two exothermic peaks at 214.1°C and 287.2°C, while the lyophilized HECS-ss-OA had an endothermic peak at 57.9°C and an exothermic peak at 229.9°C. In addition, the characteristic peaks of GA were exhibited in the physical mixture of GA+HECS-ss-OA, but almost absent in HECS-ss-OA/GA micelles. This result illustrated a significantly reduced crystalline state of GA entrapped in HECS-ss-OA micelles.35
 |
Table 1 The characteristics of HECS-ss-OA, HECS-cc-OA, HECS-ss-OA/GA, HECS-cc-OA/GA, HA(HECS-ss-OA)/GA and HA(HECS-cc-OA)/GA nanoparticles |
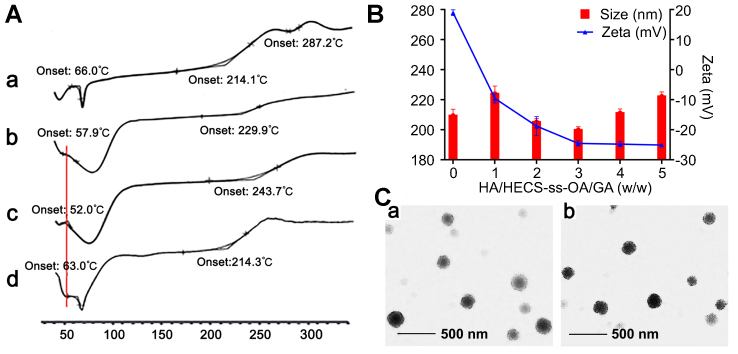 |
Figure 2 (A) DSC thermograms of GA (a), HECS-ss-OA (b), HECS-ss-OA/GA (c), and the physical mixture of GA + HESC-ss-OA (d). (B) Particle size and zeta potential of HA(HECS-ss-OA)/GA at different weight ratio of HA and HECS-ss-OA/GA. (C) TEM images of HA(HECS-ss-OA)/GA (a) and HA(HECS-cc-OA)/GA (b). Scale bar is 500 nm. Abbreviations: DSC, differential scanning calorimetry; GA, gambogic acid; HECS, hydroxyethyl chitosan; OA, octylamine; HA, hyaluronic acid; TEM, transmission electron microscopy. |
Next, the HA(HECS-ss-OA)/GA nanoparticle was prepared through electrostatic attraction between HA and HECS-ss-OA/GA micelles. After a screening of the weight ratio between HA and HECS-ss-OA/GA (Figure 2B), HA(HECS-ss-OA)/GA nanoparticles achieved a smallest particle size of 200.0 nm and a highly negative zeta potential of −24.53 mV when the weight ratio was fixed at 3/1 (Table 1). The shrinkage in particle size after the HA coating was attributed to an ionic compression effect between HA and the cationic CS-based micelles. This surface charge inversion assigned by the HA coating facilitated the stable in vivo transport and reduced immunogenicity of the positive CS-based micelles. Resultantly, the weight ratio of 3/1 was selected into the final preparation process for HA(HECS-ss-OA)/GA and its comparison, HA(HECS-cc-OA)/GA. As shown in Table 1, these two nanoparticles possessed similar characteristics including a uniform size distribution around 200 nm, a negative surface charge around −24 mV, a DL of ~5% and EE of ~80%. The HA coating did not affect the drug loading capacity of the HECS-based micelles. Finally, TEM images in Figure 2C demonstrated the core/shell spherical morphologies and the uniform size distributions (~180 nm) of HA(HECS-ss-OA)/GA and HA(HECS-cc-OA)/GA nanoparticles. The sizes estimated from TEM were smaller than that from DLS due to micellar collapse during the drying process of TEM sampling.
In vitro response to the reducing environment
To verify the sharp responsibility of the objective nanoparticles to the bio-reducing environment, the morphology change of HA(HECS-ss-OA)/GA and HA(HECS-cc-OA)/GA when treated by 10 mM DTT at pH 7.4 was firstly observed using TEM. As exhibited in Figure 3A, HA(HECS-ss-OA)/GA was notably destructed by DTT to be irregular and loose particles. In contrast, no significant change of morphology was observed in HA(HECS-cc-OA)/GA group after the same treatment, which demonstrated the attribution of disulfide bond to the redox-sensitivity of HA(HECS-ss-OA)/GA nanoparticles. This redox-sensitivity was further confirmed by its size change in response to different DTT concentrations using DLS detection. Results in Figure 3C showed that HA(HECS-ss-OA)/GA nanoparticle displayed a DTT concentration- and time-dependent size change. For instance, the particle size of HA(HECS-ss-OA)/GA kept ~200 nm without DTT in 24 hrs, but increased to ~300 nm with 5 mM DTT and ~550 nm with 10 mM DTT in 24 hrs. However, only negligible size changes were discerned in HA(HECS-cc-OA)/GA group even with 10 mM DTT in 24 hrs. All results here confirmed that HA(HECS-ss-OA)/GA nanoparticle would remain structural integrity in normal conditions but quickly loosened upon within intracellular bio-reducing environment.15
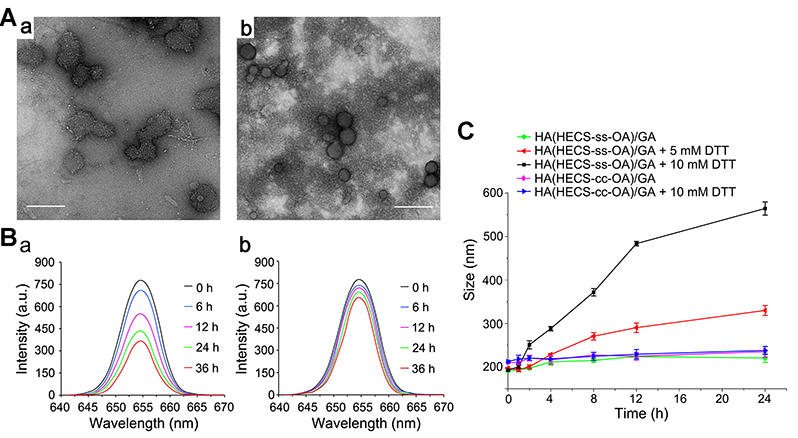 |
Figure 3 (A) TEM images of HA(HECS-ss-OA)/GA (a) and HA(HECS-cc-OA)/GA (b) nanoparticles after 4-hr incubation with 10 mM DTT in pH 7.4 HEPES. Scar bar is 500 nm. (B) Fluorescence spectra of HA(HECS-ss-OA)/NR (a) and HA(HECS-cc-OA)/NR (b) nanoparticles in pH 7.4 HEPES solution supplemented with 10 mM DTT at the excitation wavelength of 326 nm for different times. (C) Size change of HA(HECS-ss-OA)/GA (a) and HA(HECS-cc-OA)/GA (b) nanoparticles in response to DTT determined by DLS measurement. Abbreviations: TEM, transmission electron microscopy; HA, hyaluronic acid; HECS, hydroxyethyl chitosan; OA, octylamine; GA, gambogic acid; DTT, dithiothreitol; HEPES, 2-[4-(2-Hydroxyethyl)-1-piperazinyl] ethanesulfonic acid; NR, Nile red; DLS, dynamic light scattering. |
Subsequently, the fluorescent probe NR was encapsulated into the designed nanoparticles instead of GA to conveniently investigate their redox-triggered drug release behavior. It was observed in Figure 3B that the emission intensity of HA(HECS-ss-OA)/NR decreased notably with time extending when incubated in 10 mM DTT supplemented HEPES solution. Nevertheless, such a drug release-mediated significant increase in NR fluorescence was absent in HA(HECS-ss-OA)/NR group due to the deficiency of redox-sensitivity. Accordingly, a cytoplasm-specific rapid GA delivery would be realized by using HA(HECS-ss-OA)/GA nano-system.
In vitro cellular studies
Cellular uptake and intracellular release behaviour
The HA-mediated active cellular uptake and redox-triggered drug liberation was intuitively confirmed using CLSM. C6 was used as a substitution of GA to label the two nanoparticles (HA(HECS-ss-OA)/C6 and HA(HECS-cc-OA)/C6). As illustrated in Figure 4A and B, C6 was efficiently delivered into A549 cells by these two nanoparticles over time, indicating a time-dependent cellular internalization. Additionally, the diminished intracellular fluorescence intensity of C6 at 4 h time point when cells pre-treated with free HA demonstrated the role of HA in mediating specific cellular uptake of nanoparticles. This HA-mediated effective intracellular C6 delivery was also validated through a quantitative evaluation of C6 fluorescence intensity using the flow cytometry (Figure 4C). Consistent with CLSM results, A549 cells incubated with HA(HECS-ss-OA)/C6 had a comparable fluorescence intensity to those treated with HA(HECS-cc-OA)/C6 for 4 hrs, but exhibited a remarkably decreased one when cells were pre-cultured with molecular HA.
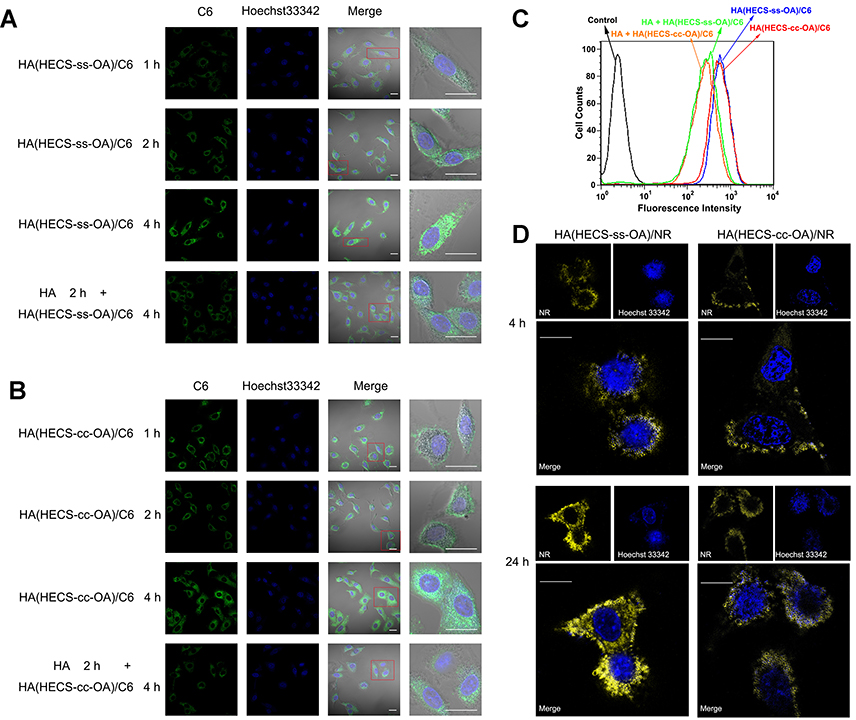 |
Figure 4 CLSM images of the intercellular uptake of HA(HECS-ss-OA)/C6 (A) and HA(HECS-cc-OA)/C6 (B) in A549 cells at 37°C after incubation for 1, 2, and 4 hrs with the absence or presence of free HA. Scale bars are 20 μm. (C) Flow cytometric curves of intracellular uptake of HA(HECS-ss-OA)/C6 and HA(HECS-cc-OA)/C6 in A549 cells after 4 hrs of incubation with the absence or presence of free HA. (D) CLSM images of A549 cells incubated with HA(HECS-ss-OA)/NR and HA(HECS-cc-OA)/NR nanoparticles for 4 and 24 hrs. Scale bars are 5 μm. Abbreviations: CLSM, confocal laser scanning microscopy; HA, hyaluronic acid; HECS, hydroxyethyl chitosan; OA, octylamine; C6, coumarin 6; NR, Nile red. |
NR is an outstanding vital stain when viewed as yellow-gold fluorescence for the observation of intracellular lipid droplets.13 Resultantly, NR was loaded into nanoparticles (HA(HECS-ss-OA)/NR and HA(HECS-cc-OA)/NR) as fluorescence probe to visually display the redox-triggered drug release of this sensitive vehicle within cytoplasm. As time elapsed from 4 to 24 hrs, a greater number of stained intracellular lipid droplets was seen in redox-sensitive HA(HECS-ss-OA)/NR treated cells over that in non-sensitive HA(HECS-cc-OA)/NR treated ones (Figure 4D). A greater number of yellow lipid droplets corresponded to a more efficient NR release from nanoparticles within the reducing cytoplasm. Consequently, this data intuitively confirmed the sharp redox responsibility of HA(HECS-ss-OA) nanoparticles to achieve burst release of cargoes within tumor cytoplasm.
Cytotoxicity and apoptosis assay
To investigate whether GA-loaded redox-sensitive nanoparticles could achieve a much higher inhibition in cell proliferation, MTT assay was performed against A549 cells. Results in Figure 5A indicated that bare HECS-ss-OA and HECS-cc-OA micellar conjugates exhibited negligible toxicities toward A549 cells only when the concentration was below 10 µg/mL. However, the introduction of HA coating increased the safe concentration of blank materials to 100 µg/mL, demonstrating an effective shielding of surface positive charges of the two CS-based micelles and thus an increase of their biocompatibility. In the following MTT assay for free GA and GA-loaded nanoparticles, the highest tested concentration of GA was limited to 10 µg/mL with a highest material concentration <100 µg/mL. As shown in Figure 5B, the free GA was the lowest among all text groups due to its passive diffusion pathway into the cells. Although free GA is potent in vitro, it lacks focus targeting and can cause serious side effects. HA(HECS-ss-OA)/GA exhibited an IC50 value of 1045.0 μg/mL, 2.15-fold lower compared with that of non-sensitive HA(HECS-cc-OA)/GA (2246.8 μg/mL). The enhancement shown here was attributed to the accelerated GA release in highly reducing tumor cytoplasm.
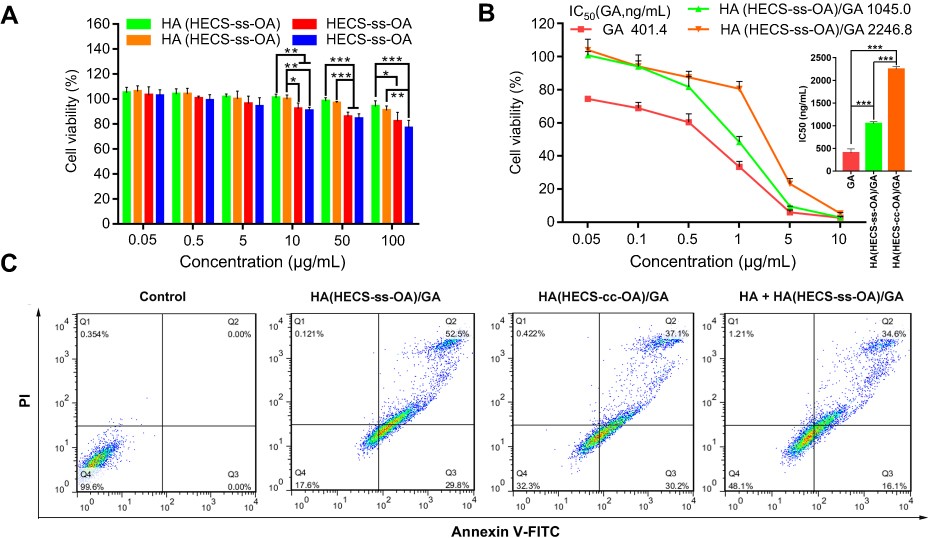 |
Figure 5 (A) Viability of A549 cells when incubated with various concentrations of HA(HECS-ss-OA), HA(HECS-cc-OA), HECS-ss-OA and HECS-cc-OA for 48 h. Data are presented as the mean ± SD (n = 5). ***P < 0.001; **P < 0.01; *P < 0.05. (B) Viability of A549 cells after an incubation with GA, HA(HECS-ss-OA)/GA and HA(HECS-cc-OA)/GA at different GA concentrations for 48 h. Data are presented as the mean ± SD (n = 5). ***p < 0.001. (C) Flow cytometric analysis of A549 cell apoptosis induced by different formulations at a GA concentration of 0.4 µg/mL for 48 hrs by using Annexin V-FITC/PI staining. Q4, Q3, and Q2 quadrant represent normal cells, early apoptotic cells, apoptotic plus necrotic cells, respectively. Abbreviations: HA, hyaluronic acid; HECS, hydroxyethyl chitosan; OA, octylamine; GA, gambogic acid; FITC, fluorescein isothiocyanate; PI, propidium iodide. |
Next, the burst drug release-resulted enhancement in drug potency of HA(HECS-ss-OA)/GA was further verified through an Annexin V-FITC/PI detection assay (Figure 5C). It was observed that HA(HECS-ss-OA)/GA induced the highest total apoptotic ratio (AR, AR = Q2 + Q3) of 82.3% compared with the other groups. Nevertheless, the apoptotic ratio induced by HA(HECS-ss-OA)/GA was notably decreased to 50.7% when cells pre-treated with HA. Collectively, HA(HECS-ss-OA)/GA nanoparticles enhanced GA potency through HA receptor-mediated active targeting and tumor cytoplasm-selective burst drug release.
In vivo distribution
To investigate the in vivo biodistribution of our designed nano-system, the DiR co-loaded nanoparticles (HA(HECS-ss-OA)/GA+DiR, HA(HECS-cc-OA)/GA+DiR) were intravenously administrated into A549 tumor-bearing nude mice and detected using non-invasive near-infrared optical imaging.36 As illustrated in Figure 6A, a similar biodistribution was achieved between HA(HECS-ss-OA)/GA+DiR and HA(HECS-cc-OA)/GA+DiR owning to their accordant dimensional and superficial characteristics. At 2 hrs after administration, a strong signal was observed at liver. As time extended, a preferential accumulation of DiR signal was found in tumor site after 24 hrs, demonstrating a successful tumor targeting of the both HA-coated nanoparticles. Moreover, to visualize the ability of HA in active targeting through specific receptor-mediated endocytosis, the mice were pre-injected intravenously with a large dose of free HA before the HA(HECS-ss-OA)/GA+DiR injection. As shown, a weakened DiR signal was obtained in tumor site, demonstrating the role of HA-mediated in vivo targeting. The mice were sacrificed 24 hrs post-injection, and the major normal organs as well as tumors were excised for ex vivo imaging (Figure 6B). Consistent with results from in vivo imaging, the DiR signal in tumor of HA(HECS-ss-OA)/GA+DiR group was comparable to that of HA(HECS-cc-OA)/GA+DiR, but much higher than that of HA pre-injected HA(HECS-ss-OA)/GA+DiR and the corresponding normal tissues. Overall, these results demonstrated the excellent in vivo tumor targeting of HA(HECS-ss-OA) nanoparticles based on a combination of passive and active targeting mechanisms.
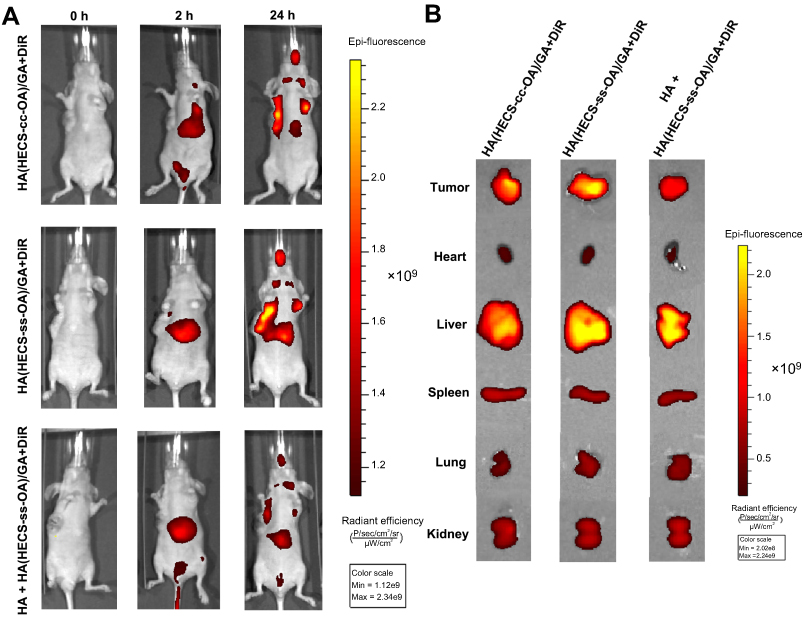 |
Figure 6 (A) In vivo fluorescence imaging of A549 tumor-bearing mice administrated with GA and DiR co-loaded nanoparticles with or without pre-injection of free HA at 2 and 24 hrs. (B) Ex vivo fluorescence imaging of tissues including tumor, heart, liver, spleen, lung, and kidney excised from mice after administrated with GA and DiR co-loaded micelles for 24 hrs. Abbreviations: HA, hyaluronic acid; HECS, hydroxyethyl chitosan; OA, octylamine; GA, gambogic acid. |
In vivo antitumor efficacy
The in vivo antitumor efficacy of HA(HECS-ss-OA)/GA was further investigated on A549 tumor-bearing BALB/c nude mice. All GA formulations were effective in suppressing tumor growth compared with saline (Figure 7A and B). Based on the results of tumor volume and tumor weight measurement, HA(HECS-ss-OA)/GA achieved the most effective tumor growth inhibition compared with other GA formulations including free GA solution, non-sensitive HA(HECS-cc-OA)/GA and HA un-coated HECS-ss-OA/GA. It was ascribed to an effective tumor targeting via HA-mediated specific endocytosis and triggered burst release of GA by intracellular reducing microenvironment.37,38 The tumor weight inhibition ratio (IR) was calculated based on the tumor weight results (Table 2). IR values of HA(HECS-ss-OA)/GA was 89.24%, 1.13-fold, 1.31-fold and 1.58-fold higher than that of HA(HECS-cc-OA)/GA, HECS-ss-OA/GA and free GA, respectively, which further suggested the most effective tumor suppression effect of HA(HECS-ss-OA)/GA. Moreover, the notable cell remission observed in HE-stained tumor sectioning (Figure 7C(a)) and the greatest degree of tumor apoptosis exhibited in terminal deoxynucleotidyl transferase dUTP nick end labeling assay (Figure 7C(b)) also confirmed the prominent anti-tumor potential of HA(HECS-ss-OA)/GA.
 |
Table 2 Tumor weight and growth inhibition of A549 tumor-bearing mice by multiple injections of different GA formulations |
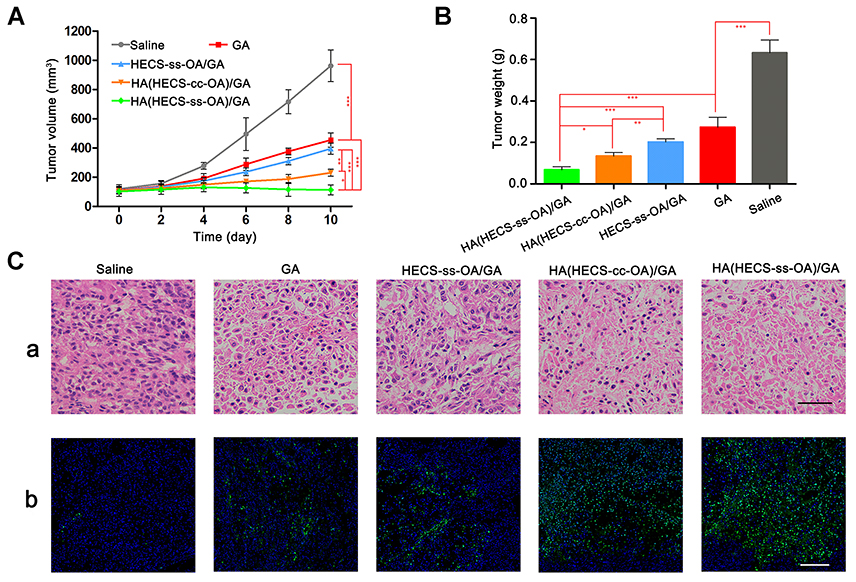 |
Figure 7 Tumor growth (A) and tumor weight (B) of A549 tumor-bearing mice administrated with saline, GA, HECS-ss-OA/GA, HA(HECS-ss-OA)/GA and HA(HECS-cc-OA)/GA at a GA dose of 8 mg/kg. Data are represented as the mean ± SD (n = 5). ***P < 0.001; **P < 0.01; *P <0.05. (C) Images of H&E-stained (a) and TUNEL-stained (b) tumor sections, scale bar is 200 μm. Abbreviations: HA, hyaluronic acid; HECS, hydroxyethyl chitosan; OA, octylamine; GA, gambogic acid. |
Apart from the anti-tumor efficacy, the systemic safety evaluation is essential for systemic drug delivery nanoparticles. As shown in Figure 8A, no significant weight loss was observed in GA-loaded nanoparticle treated groups, while the free GA treated mice showed a significant weight loss at the same dose level due to a nonspecific biodistribution. In addition, the levels of hepatic and renal function markers (ALT, AST, Scr, BUN) in serum from these treated groups exhibited no significant difference compared with those from saline-treated mice with the exception of GA group (Figure 8B and C). Furthermore, the results in Figure 8D from histological analysis of organs through HE staining indicated the severe organ damage after free GA treatment including cardiomyocyte separation, alveolar capillaries congestion, and renal tubular lumen red staining (indicated by the black arrows). It was worth to be noted that the alveolar capillaries congestion was also observed in HECS-ss-OA/GA group, which was ascribed to its cation-associated lung damage. In contrast, no pathological change was displayed compared with the saline group when mice were injected with HA(HECS-ss-OA)/GA. This marked systemic safety of HA-coated nanoparticles over free GA and HECS-ss-OA/GA was attributed to a charge inversion assigned by HA coating and a selective tumor accumulation through a combination of passive and active targeting mechanism. Additionally, the selective drug release rate of HA(HECS-ss-OA)/GA in response to the much highly reducing tumor cytoplasmic environment compared with normal cells, would certainly improve tumor specificity without undesired drug leakage and toxicity to normal tissues.
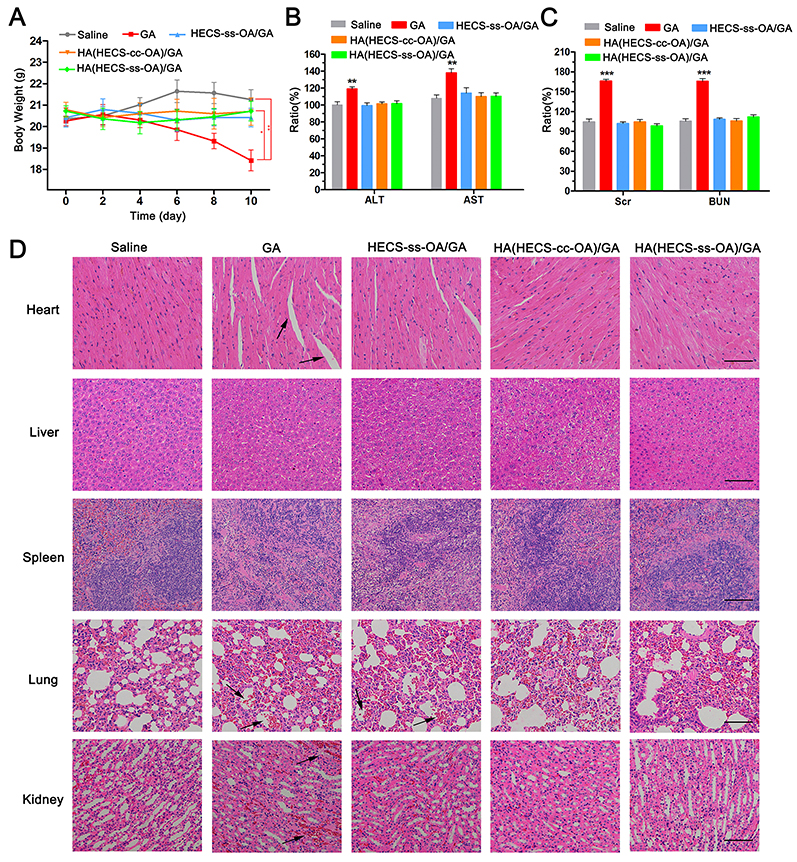 |
Figure 8 (A) The weight of mice during the treatment period. Data are represented as the mean ± SD (n = 5). **P < 0.01; *P < 0.05. The changes of serum ALT, AST (B), Scr and BUN (C) levels from A549 tumor-bearing mice after administration of different formulations. Data are represented as the mean ± SD (n = 3). (**p < 0.01 vs Saline). (D) Images of H&E-stained heart, liver, spleen, lung and kidney tissues from A549 tumor-bearing mice after intravenous injection of different formulations. Scar bar is 100 μm. The black arrows indicated the severe organ damages. Abbreviations: HA, hyaluronic acid; HECS, hydroxyethyl chitosan; OA, octylamine; GA, gambogic acid; ALT, alanine transaminase; AST, aspartate transaminase; Scr, serum creatinine; BUN, blood urea nitrogen. |
Conclusion
In this study, we further improved the accuracy, controllability, and reproducibility of the preparation procedure for HECS-ss-OA and developed an HA-coated redox-sensitive system for tumor cytoplasm-selective rapid delivery of the natural anti-cancer drug, GA. HA(HECS-ss-OA)/GA nanoparticles exhibited excellent drug loading for GA. The results of morphology and particle size change, in vitro drug release in response to simulated reducing environments well demonstrated the desirable redox-sensitivity of HA(HECS-ss-OA)/GA. In vitro and in vivo experiments proved the strongest antitumor efficacy of HA(HECS-ss-OA)/GA nanoparticles compared with the non-sensitive and the HA un-coated controls. In a word, this designed HA(HECS-ss-OA) nanoparticle offers a promising new avenue for tumor-specific intracellular controlled delivery of natural antitumor drugs for efficient tumor therapy.
Acknowledgments
This work was supported by the project of the National Natural Science Foundation of China (Nos. 81872424, 81703442, 81503261, 81573613), the Natural Science Foundation of Shandong Province (ZR2013HQ011), the Natural Science Foundation of Jiangsu Province (BK20130655), Primary Research & Development Plan of Shandong Province (2017GSF218016), Qing Lan Project of Jiangsu Province (No. 02432009), the Project Program of State Key Laboratory of Natural Medicines, China Pharmaceutical University (No. JKPZ2013004), and the Open Fund of State Key Laboratory of Natural Medicines (SKLNMKF201612), Double First-rate construction project of China Pharmaceuticla University: Advanced Technology in New Drug Discovery and its Inversion and Application (CPU2018GY26).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Chen W, Zheng R, Baade PD, et al. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66(2):115–132. doi:10.3322/caac.21338
2. Zhou C, Wu Y-L, Chen G, et al. Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): a multicentre, open-label, randomised, phase 3 study. Lancet Oncol. 2011;12(8):735–742. doi:10.1016/S1470-2045(11)70184-X
3. Molina JR, Yang P, Cassivi SD, Schild SE, Adjei AA. Non-small cell lung cancer: epidemiology, risk factors, treatment, and survivorship. Mayo Clin Proc. 2008;83(5):584–594. doi:10.4065/83.5.584
4. Li C, Qi Q, Lu N, et al. Gambogic acid promotes apoptosis and resistance to metastatic potential in MDA-MB-231 human breast carcinoma cells. Biochem Cell Biol. 2012;90(6):718–730. doi:10.1139/o2012-030
5. Zhu X, Zhang H, Lin Y, et al. Mechanisms of gambogic acid-induced apoptosis in non-small cell lung cancer cells in relation to transferrin receptors. J Chemother. 2009;21(6):666–672. doi:10.1179/joc.2009.21.6.666
6. Wang X, Deng R, Lu Y, et al. Gambogic acid as a non-competitive inhibitor of ATP-binding cassette transporter B1 reverses the multidrug resistance of human epithelial cancers by promoting ATP-binding cassette transporter B1 protein degradation. Basic Clin Pharmacol Toxicol. 2013;113(1):25–33. doi:10.1111/bcpt.12058
7. Banik K, Harsha C, Bordoloi D, et al. Therapeutic potential of gambogic acid, a caged xanthone, to target cancer. Cancer Lett. 2018;416:75–86. doi:10.1016/j.canlet.2017.12.014
8. Chi Y, Zhang XK, Yu H, et al. An open-labeled, randomized, multicenter phase IIa study of gambogic acid injection for advanced malignant tumors. Chin Med J (Engl). 2013;126(9):1642–1646.
9. Wang X, Chen W. Gambogic acid is a novel anti-cancer agent that inhibits cell proliferation, angiogenesis and metastasis. Anticancer Agents Med Chem. 2012;12(8):994–1000.
10. Ishaq M, Khan MA, Sharma K, Sharma G, Dutta RK, Majumdar S. Gambogic acid induced oxidative stress dependent caspase activation regulates both apoptosis and autophagy by targeting various key molecules (NF-kappaB, Beclin-1, p62 and NBR1) in human bladder cancer cells. Biochim Biophys Acta. 2014;1840(12):3374–3384. doi:10.1016/j.bbagen.2014.08.019
11. Wang S, Wang L, Chen M, Wang Y. Gambogic acid sensitizes resistant breast cancer cells to doxorubicin through inhibiting P-glycoprotein and suppressing survivin expression. Chem Biol Interact. 2015;235:76–84. doi:10.1016/j.cbi.2015.03.017
12. Xu Y, Wang S, Chan HF, et al. Triphenylphosphonium-modified poly(ethylene glycol)-poly(epsilon-caprolactone) micelles for mitochondria- targeted gambogic acid delivery. Int J Pharm. 2017;522(1–2):21–33. doi:10.1016/j.ijpharm.2017.01.064
13. Yin T, Wang L, Yin L, Zhou J, Huo M. Co-delivery of hydrophobic paclitaxel and hydrophilic AURKA specific siRNA by redox-sensitive micelles for effective treatment of breast cancer. Biomaterials. 2015;61:10–25. doi:10.1016/j.biomaterials.2015.05.022
14. Shi S, Shi K, Tan L, et al. The use of cationic MPEG-PCL-g-PEI micelles for co-delivery of Msurvivin T34A gene and doxorubicin. Biomaterials. 2014;35(15):4536–4547. doi:10.1016/j.biomaterials.2014.02.010
15. Li J, Huo M, Wang J, et al. Redox-sensitive micelles self-assembled from amphiphilic hyaluronic acid-deoxycholic acid conjugates for targeted intracellular delivery of paclitaxel. Biomaterials. 2012;33(7):2310–2320. doi:10.1016/j.biomaterials.2011.11.022
16. Yang X, Cai X, Yu A, Xi Y, Zhai G. Redox-sensitive self-assembled nanoparticles based on alpha-tocopherol succinate-modified heparin for intracellular delivery of paclitaxel. J Colloid Interface Sci. 2017;496:311–326. doi:10.1016/j.jcis.2017.02.033
17. Raja MA, Arif M, Feng C, Zeenat S, Liu CG. Synthesis and evaluation of pH-sensitive, self-assembled chitosan-based nanoparticles as efficient doxorubicin carriers. J Biomater Appl. 2017;31(8):1182–1195. doi:10.1177/0885328216681184
18. Li N, Cai H, Jiang L, et al. Enzyme-sensitive and amphiphilic PEGylated dendrimer-paclitaxel prodrug-based nanoparticles for enhanced stability and anticancer efficacy. ACS Appl Mater Interfaces. 2017;9(8):6865–6877. doi:10.1021/acsami.6b15505
19. Huu VA, Luo J, Zhu J, et al. Light-responsive nanoparticle depot to control release of a small molecule angiogenesis inhibitor in the posterior segment of the eye. J Control Release. 2015;200:71–77. doi:10.1016/j.jconrel.2015.01.001
20. Yamazaki N, Sugimoto T, Fukushima M, et al. Dual-stimuli responsive liposomes using pH- and temperature-sensitive polymers for controlled transdermal delivery. Polym Chem. 2017;8(9):1507–1518. doi:10.1039/C6PY01754A
21. Tabatabaei SN, Girouard H, Carret AS, Martel S. Remote control of the permeability of the blood-brain barrier by magnetic heating of nanoparticles: a proof of concept for brain drug delivery. J Control Release. 2015;206:49–57. doi:10.1016/j.jconrel.2015.02.027
22. Bae YJ, Yoon YI, Yoon TJ, Lee HJ. Ultrasound-guided delivery of siRNA and a chemotherapeutic drug by using microbubble complexes: in vitro and in vivo evaluations in a prostate cancer model. Korean J Radiol. 2016;17(4):497–508. doi:10.3348/kjr.2016.17.4.497
23. Sabu C, Rejo C, Kotta S, Pramod K. Bioinspired and biomimetic systems for advanced drug and gene delivery. J Control Release. 2018;287:142–155. doi:10.1016/j.jconrel.2018.08.033
24. Ramasamy T, Ruttala HB, Gupta B, et al. Smart chemistry-based nanosized drug delivery systems for systemic applications: a comprehensive review. J Control Release. 2017;258:226–253. doi:10.1016/j.jconrel.2017.04.043
25. Yin T, Wu Q, Wang L, Yin L, Zhou J, Huo M. Well-defined redox-sensitive polyethene glycol-paclitaxel prodrug conjugate for tumor-specific delivery of paclitaxel using octreotide for tumor targeting. Mol Pharm. 2015;12(8):3020–3031. doi:10.1021/acs.molpharmaceut.5b00280
26. Huo M, Liu Y, Wang L, et al. Redox-sensitive micelles based on O,N-hydroxyethyl chitosan-octylamine conjugates for triggered intracellular delivery of paclitaxel. Mol Pharm. 2016;13(6):1750–1762. doi:10.1021/acs.molpharmaceut.5b00696
27. Yin T, Liu J, Zhao Z, et al. Smart nanoparticles with a detachable outer shell for maximized synergistic antitumor efficacy of therapeutics with varying physicochemical properties. J Control Release. 2016;243:54–68. doi:10.1016/j.jconrel.2016.09.036
28. Ito T, Koyama Y, Otsuka M. Analysis of the surface structure of DNA/polycation/hyaluronic acid ternary complex by Raman microscopy. J Pharm Biomed Anal. 2010;51(1):268–272. doi:10.1016/j.jpba.2009.07.024
29. Zhong Y, Zhang J, Cheng R, et al. Reversibly crosslinked hyaluronic acid nanoparticles for active targeting and intelligent delivery of doxorubicin to drug resistant CD44+ human breast tumor xenografts. J Control Release. 2015;205:144–154. doi:10.1016/j.jconrel.2015.01.012
30. Dosio F, Arpicco S, Stella B, Fattal E. Hyaluronic acid for anticancer drug and nucleic acid delivery. Adv Drug Deliv Rev. 2016;97:204–236. doi:10.1016/j.addr.2015.11.011
31. Huo M, Zou A, Yao C, et al. Somatostatin receptor-mediated tumor-targeting drug delivery using octreotide-PEG-deoxycholic acid conjugate-modified N-deoxycholic acid-O, N-hydroxyethylation chitosan micelles. Biomaterials. 2012;33(27):6393–6407. doi:10.1016/j.biomaterials.2012.05.052
32. Li H, Huo M, Zhou J, et al. Enhanced oral absorption of paclitaxel in N-deoxycholic acid-N, O-hydroxyethyl chitosan micellar system. J Pharm Sci. 2010;99(11):4543–4553. doi:10.1002/jps.22159
33. Moyuan C, Haixia J, Weijuan Y, Peng L, Liqun W, Hongliang J. A convenient scheme for synthesizing reduction-sensitive chitosan-based amphiphilic copolymers for drug delivery. J Appl Polym Sci. 2012;123(5):3137–3144.
34. Luo Y, Prestwich GD. Synthesis and selective cytotoxicity of a hyaluronic acid-antitumor bioconjugate. Bioconjugate Chem. 1999;10(5):755−63. doi:10.1021/bc9900338
35. Li Z, Liu R, Mai B, et al. Temperature-induced and crystallization-driven self-assembly of polyethylene-b-poly(ethylene oxide) in solution. Polymer. 2013;54(6):1663–1670. doi:10.1016/j.polymer.2013.01.044
36. Hou L, Yao J, Zhou J, Zhang Q. Pharmacokinetics of a paclitaxel-loaded low molecular weight heparin-all-trans-retinoid acid conjugate ternary nanoparticulate drug delivery system. Biomaterials. 2012;33(21):5431–5440. doi:10.1016/j.biomaterials.2012.03.070
37. Wang X, Ouyang X, Chen J, Hu Y, Sun X, Yu Z. Nanoparticulate photosensitizer decorated with hyaluronic acid for photodynamic/photothermal cancer targeting therapy. Nanomedicine. 2019;14(2):151–167. doi:10.2217/nnm-2018-0204
38. Ramasamy T, Tran TH, Choi J, et al. Layer-by-layer coated lipid–polymer hybrid nanoparticles designedfor use in anticancer drug delivery. Carbohydr Polym. 2014;102:653–661. doi:10.1016/j.carbpol.2013.10.048
Supplementary material
The 1H NMR and HR-MS spectrum of OA-ss-COOH and OA-cc-COOH.
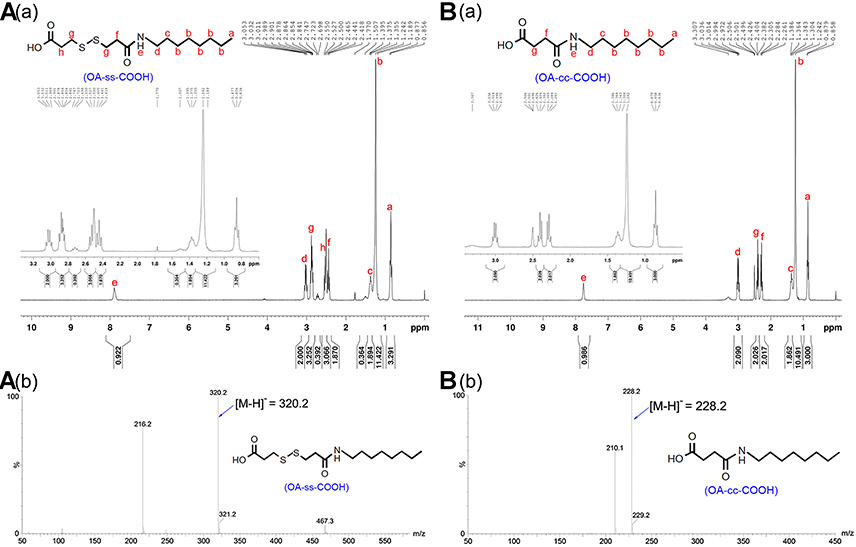 |
|
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.