")
Back to Journals » Drug Design, Development and Therapy » Volume 11
Humanin rescues cultured rat cortical neurons from NMDA-induced toxicity through the alleviation of mitochondrial dysfunction
Authors Cui A, Zhang Y , Li J, Song T , Liu X, Wang H, Zhang C, Ma G, Zhang H, Li K
Received 23 January 2017
Accepted for publication 15 March 2017
Published 18 April 2017 Volume 2017:11 Pages 1243—1253
DOI https://doi.org/10.2147/DDDT.S133042
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Sukesh Voruganti
Ai-Ling Cui,1 Ying-Hua Zhang,2 Jian-Zhong Li,3 Tianbin Song,4 Xue-Min Liu,1 Hui Wang,2 Ce Zhang,5 Guo-Lin Ma,6 Hui Zhang,7 Kefeng Li8
1Anatomy Department, Changzhi Medical College, Changzhi, Shanxi, 2Key Laboratory of Tissue Regeneration of Henan Province, Xinxiang Medical University, Xinxiang, Henan, 3Clinical Laboratory of Heji Hospital Affiliated to Changzhi Medical College, Changzhi, Shanxi, 4Department of Nuclear Medicine, Xuanwu Hospital, Capital Medical University, Beijing, 5Department of Physiology, Shanxi Medical University, Taiyuan, Shanxi, 6Department of Radiology, China-Japan Friendship Hospital, Beijing, 7Department of Radiology, First Clinical Medical College, Shanxi Medical University, Taiyuan, Shanxi, People’s Republic of China; 8School of Medicine, University of California – San Diego, San Diego, CA, USA
Abstract: N-methyl-D-aspartate (NDMA) receptor-mediated excitotoxicity has been implicated in a variety of pathological situations such as Alzheimer’s disease (AD) and Parkinson’s disease. However, no effective treatments for the same have been developed so far. Humanin (HN) is a 24-amino acid peptide originally cloned from the brain of patients with AD and it prevents stress-induced cell death in many cells/tissues. In our previous study, HN was found to effectively rescue rat cortical neurons. It is still not clear whether HN protects the neurons through the attenuation of mitochondrial dysfunction. In this study, excitatory toxicity was induced by NMDA, which binds the NMDA receptor in primarily cultured rat cortical neurons. We found that NMDA (100 µmol/L) dramatically induced the decrease of cell viability and caused mitochondrial dysfunction. Pretreatment of the neurons with HN (1 µmol/L) led to significant increases of mitochondrial succinate dehydrogenase (SDH) activity and membrane potential. In addition, HN pretreatment significantly reduced the excessive production of both reactive oxygen species (ROS) and nitric oxide (NO). Thus, HN could attenuate the excitotoxicity caused by the overactivation of the NMDA receptor through the alleviation of mitochondrial dysfunction.
Keywords: NMDA-induced toxicity, mitochondrial dysfunction, humanin, alleviation
Introduction
Glutamate is an essential excitatory neurotransmitter in the central nervous system (CNS). Its accumulation in the synaptic cleft, however, triggers the excessive activation of the N-methyl-D-aspartate (NMDA) receptor and, consequently, the influx of extracellular calcium.1–3 The fine spatial and temporal organization of intracellular calcium signals is fundamental to the functions of the CNS, perhaps more than for any other tissues.4 Calcium signals are conveyed throughout the CNS by local changes in the calcium concentration ([Ca2+]c). The rapid increase of ([Ca2+]c) in the cytoplasm activates Ca2+ channels on the mitochondrial membrane and triggers calcium flux into the mitochondrial matrix. In the matrix, calcium upregulates the activity of Ca2+-sensitive dehydrogenases of the Krebs cycle and the F1F0-ATP synthase and thereby controls the rate of ATP production.5,6 A pathological aspect of this process is linked with the mitochondrial Ca2+ overload, which might trigger the activation of the mitochondrial permeability transmission pore, which in turn releases apoptotic and necrotic signal factors, leading to cell death.7,8 Neuronal energy supplies are, meanwhile, entirely based on mitochondrial oxidative phosphorylation, making them especially vulnerable to mitochondrial dysfunction.4
Because the overactivation of the NMDA receptor is the pivotal factor in the damage process, NMDA receptor antagonists have been explored for many years as therapeutic agents for the treatment of excitatory neurotoxicity-oriented neurological disorders, such as stroke, epilepsy, pain, and Parkinson’s disease. However, it has been discovered that many of these compounds can cause adverse behavioral effects and produce neurotoxicity.9,10
Humanin (HN) is a 24-amino acid peptide located in the mitochondria and cloned in 2001 from the occipital lobe of patients with Alzheimer’s disease (AD) during autopsy.11,12 It was considered an AD-selective neuroprotective peptide at the very beginning.13 Later, however, several HN homologs have been discovered in other species of animals, including rat, mouse, monkey, and nematode11,14 and identified in other tissues apart from the brain, including testes, colon, skeletal muscles, and human vascular walls.14–17 HN’s widespread distribution implies that its role is versatile, not restricted to just attenuating AD-related insults.
In our previous study, HN was used to attenuate NMDA-induced neurotoxicity.18 The results showed that HN could effectively rescue cultured rat cortical neurons. However, HN did not block the intracellular Ca2+ overload triggered by NMDA. It might induce the reverting of the high Ca2+ concentration quickly.18 It is still not clear whether HN protects the neurons through attenuation of NMDA-induced mitochondrial dysfunction.
Materials and methods
Primary cerebral cortical neuronal culture
The study protocol was reviewed and approved by the institutional review board (IRB) of the Changzhi Medical College, People’s Republic of China (IRB number 2015-021453) in accordance with the guidance for the care and use of laboratory animals, issued by the Ministry of Science and Technology of China. The primarily cultured cortical neurons were prepared using neonatal Wistar rats (P1–3). Briefly, the pups were decapitated and the cerebral cortices were isolated and immersed into ice-cold D-Hanks buffer containing NaCl 136.7 mM, KCl 5.4 mM, NaHCO3 4.2 mM, KH2PO4 0.4 mM, NaH2PO4 0.6 mM, and glucose 5.6 mM, with pH =7.4. To dissociate the cortices into single cells, they were minced mechanically into grains of ~1 mm3. The minced cortices were then digested with trypsin (0.03%, pH 7.4, 37°C, Sigma) for 1 min, centrifuged at 2,500 rpm for 5 min, and then resuspended in Dulbecco’s Modified Eagle’s Medium (DMEM) (Gibco) containing fetal calf serum (FCS, Sigma) 20% (v/v) and penicillin–streptomycin 100 U/mL. Cortical cells were plated on poly-D-lysine-coated 6-well (3 mL) or 96-well (100 μL) plates (Costar) at 5×105 cells/mL. Cells grown on 6-well plates with coverslips were used for the visualization experiments under a confocal microscope (reactive oxygen species [ROS] and mitochondrial membrane potential). Cells grown on 6-well plates without coverslips were used for the quantification of ROS by flow cytometry, and the medium was collected for the quantitative analysis of nitric oxide (NO) and lactate dehydrogenase (LDH) release. Cells grown on 96-well plates were used for the measurement of succinate dehydrogenase (SDH) activity. Cytosine arabinoside (Ara-C) (Sigma) (10 μmol/L) was added 24 h after cell plating to limit the proliferation of nonneuronal cells. The culture media were half-refreshed every 2 days. All experiments were performed on 9th day in vitro (DIV9).
Experimental groups and chemical treatments
In this study, the cultured neurons were grouped as follows: 1) control group, containing cultured neurons only; 2) HN group, containing cultured neurons incubated with HN (1 μmol/L; Shanghai Shenggong Co. Ltd., Shanghai, People’s Republic of China); 3) NMDA group, containing cultured neurons incubated with NMDA (100 μmol/L; Sigma Aldrich Co., St Louis, MO, USA) and glycine (10 μmol/L; Sigma Aldrich Co.) for 2.5 h; 4) NMDA + MK-801 group, wherein MK-801 (10 μmol/L; Sigma Aldrich Co.) was added to the NMDA treatment; 5) NMDA + HN group, in which HN (1 μmol/L) was added to NMDA treatment; 6) NMDA + S7A-HN (inactive form of HN; 1 μmol/L; Beijing SBS Genetech, Beijing, People’s Republic of China), in which S7A-HN was added along with NMDA; 7) NMDA + AGA-HNG (100× more active form; 1 μmol/L; Beijing SBS Genetech), with AGA-HNG being added to NMDA-treated cells. Cortical neurons isolated from the pups were cultured for 8 days before chemical treatment. HN was administered 16 h before NMDA treatment. The cultured neurons were preincubated with or without HN, and neurotoxicity was induced by treatment with NMDA, as described previously.19 Acute neurotoxic exposure (NMDA challenge) was achieved by removing the culture medium from the cultured neurons. The collected medium was filter-sterilized and stored for future analysis. To remove traces of growth medium, cells were washed 3 times with prewarmed Locke’s buffer containing (in millimoles per liter) NaCl 154, KCl 5.6, NaHCO3 3.6, CaCl2 2.3, MgCl2 1.2, glucose 5.6, and 4-(2-hydroxyethyl)-1-piperazine ethanesulfonic acid (HEPES) 5.0, with pH 7.4. The cells were then incubated for 2.5 h with either drug-free or neurotoxin-containing Mg2+-free Locke’s buffer. NMDA and MK-801/HN were added at the concentrations mentioned earlier. The incubation was terminated by the removal of the drug-containing buffer, followed by washing with prewarmed drug-free Locke’s buffer (1 mmol/L Mg2+). Cells were harvested for analysis after 24 h of incubation.
Quantification of LDH release
Cell damage was estimated by measuring the activity of LDH in the culture medium after release from the damaged cells. In the experiment, the culture medium was collected at 24 h after NMDA exposure. The activity of LDH was measured as described by Koh and Choi.20 The values were expressed as the percentage compared to the control group, which was set as 100%. The data presented were mean ± standard deviation (SD) for 5 independent experiments using different batches of cells.
Dimethylthiazolyldiphenyl tetrazolium bromide (MTT) assay for cell viability
The cell viability was evaluated using a cell proliferation kit (MTT assay; Sigma-Aldrich, St Louis, MO, USA) according to the manufacturer’s protocol. Briefly, the cells were incubated with MTT for 4 h at 37°C. After incubation, dimethyl sulfoxide was added to lyse the cells and dissolve the formazan crystals. The absorbance was recorded at 490 nm using a microplate reader (iMark, Bio-Rad). Cell viability was expressed as the percentage of absorption relative to the control group, which was exposed to drug-free buffer (set as 100%).
SDH activity measurement
The SDH activity was measured using an SDH activity colorimetric assay kit (Biovision, Milpitas, CA, USA), which was performed 24 h after NMDA treatment, as described earlier.21 The absorbance was recorded at 600 nm in kinetic mode for 10–30 min in a microplate reader (Benchmark, Bio-Rad). The activity of SDH was expressed as the percentage of the absorption in the control group, which was exposed to drug-free buffer (and set as 100%).
Measurement of intracellular Ca2+ concentration
On DIV8, the cultured neurons on coverslips were treated with HN. After 16 h, all groups were washed 3 times in Locke’s buffer. They were then treated with 5 μmol/L of Fluo 3-AM (calcium inhibitor) (Biotium, Fremont, CA, USA) in the dark and incubated at 37°C for 40 min. The neurons were then washed 3 times with D-Hanks buffer and visualized under a confocal microscope (TCS SP5 II; Leica). Ca2+-dependent fluorescence intensity was measured at an excitation wavelength of 488 nm and an emission wavelength of 530 nm. Dynamic changes in the levels of cytoplasmic Ca2+ were captured and recorded every 660 ms for total 300 successive measurements (~198 s) for each neuron. Further, 5–10 neurons were measured in each group, and the average intensity represented the intracellular instantaneous Ca2+ concentration of each group.
Detection of cellular mitochondrial membrane potential (ΔΨm)
The mitochondrial membrane potential (ΔΨm) was measured by staining the cells with JC-1 (5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolylcarbocyanine iodide) fluorescent probe. JC-1 was added into the serum-free DMEM (final concentration: 5 μg/mL) and incubated at 37°C for 30 min in the dark. After washing 3 times with phosphate-buffered saline (PBS) (0.01 mol/L, pH 7.4), the neurons with the coverslips were fixed in freshly prepared paraformaldehyde at 4°C for 30 min. The JC-1 polymer was then washed 3 times with PBS (0.01 mol/L, pH 7.4) again, and the red fluorescence was visualized under a confocal laser scanning microscope. The neurons on plates without coverslips were digested with trypsin–EDTA (trypsin 0.027%, EDTA 0.025%; pH 7.4) and collected in 500 μL PBS at the cell concentration of >1×106/mL. The cell fluorescence intensity was measured using a fluorescence-activated cell sorting (FACS) flow cytometer (Becton Dickinson, Franklin Lakes, NJ, USA). The maximum excitation wavelength of JC-1 polymer (J-aggregates) was 530 nm, and the maximum emission wavelength was 590 nm.
Measurement of intracellular ROS
To measure the intracellular accumulation of ROS, a fluorometric assay using intracellular oxidation of dichlorodihydrofluorescein diacetate (DCFH-DA) was performed. Briefly, the cultured cortical neurons were washed twice with serum-free DMEM. DCFH-DA was added to the medium (final concentration: 10 μmol/L) and incubated with the neurons in the dark for 40 min. The neurons were then washed 3 times with Locke’s buffer and treated with NMDA to induce cytotoxicity as described. After washing twice with PBS (0.01 M, pH 7.4), the neurons on the coverslips were fixed in freshly prepared paraformaldehyde at 4°C for 30 min. The fluorescent dichlorofluorescein (DCF) was visualized by a confocal microscope. The neurons on the plates without coverslips were digested with trypsin–EDTA (trypsin 0.027%, EDTA 0.025%; pH 7.4) and collected in 500 μL of PBS at a cell concentration of 1×106/mL for testing by flow cytometry. The cells’ fluorescence intensity was measured using the flow cytometer (Becton Dickinson, USA) at 490 nm excitation and 530 nm emission.
NO determination
NO was determined by the colorimetric method using a commercial kit (Nanjing Jiangcheng Biological Engineering Company, People’s Republic of China). Briefly, after NMDA treatment, the cultured cortical neurons were collected, and NO concentration was measured according to the manufacturer’s protocol.
Protein extraction and Western blot analysis
Mitochondrial-enriched proteins were isolated with a Mitochondrial/Cytosolic Fractionation kit (Abcam, Cambridge, MA, USA) according to the manufacturer’s protocol. Briefly, the cells were harvested and lysed with cytosol extraction buffer mix. The homogenized cells were then centrifuged at 16,000× g for 10 min at 4°C. The pellet contained the intact mitochondria. The pellet was then resuspended in mitochondrial extraction buffer mix containing dithiothreitol (DTT) and protease inhibitors. Protein concentration was determined using Pierce bicinchoninic acid protein assay (Pierce Chemicals). Twenty micrograms of protein was treated with 1× reducing agent (Life Technologies) in 1× LDS buffer (Life Technologies), heated for 10 min at 95°C, loaded onto a 10% bis-Tris polyacrylamide gel, and electrophoresed at 150 V for 1.5 h. Proteins were transferred to nitrocellulose, followed by blocking with 3% milk for 1 h. The filter was washed with 1× Tris-buffered saline containing Tween-20 (TBST) and probed with the primary antibody anti-mitochondrial succinate dehydrogenase subunit B (SDHB) (Abcam, 1:1,000 dilution) or anti-inducible nitric oxide synthase (iNOS) (Abcam, 1:2,000 dilution) overnight. A monoclonal antibody raised against the structural protein beta-actin (1:1,000 dilution, Sigma) was used as the control. Horseradish peroxidase-conjugated anti-rabbit/mouse secondary antibody (GE Healthcare, Wauwatosa, WI, USA, 1:15,000) and an ECL plus kit (Pierce, Waltham, MA, USA) were used to detect the protein. The intensity of the bands was analyzed using ImageJ 1.3.8. Fold change (experimental group/control) was calculated and plotted.
Statistical analysis
All data were expressed as mean ± SD (n=5). The significance of the difference between groups was tested using 1-way analysis of variance (ANOVA), followed by Tukey’s multiple-comparisons test; P-values <0.05 were considered statistically significant.
Results
NMDA induced leakage of LDH from cultured rat cortical neurons in a dose-dependent manner
As shown in Figure 1, NMDA treatment resulted in the leakage of LDH from the neurons in a dose-dependent manner. NMDA is an excitotoxin that induces the overactivation of the NMDA receptor and causes excitotoxicity. Compared with the control group, the LDH activity was significantly increased by 39.3% and 70.7%, respectively, in the 2 NMDA treatment groups (50 and 100 μmol/L), and the neurons were dramatically damaged. MK-801 is an uncompetitive channel blocker of the NMDA receptor. MK-801 inhibited the increase of LDH release, suggesting that the increase of LDH release was triggered by NMDA.
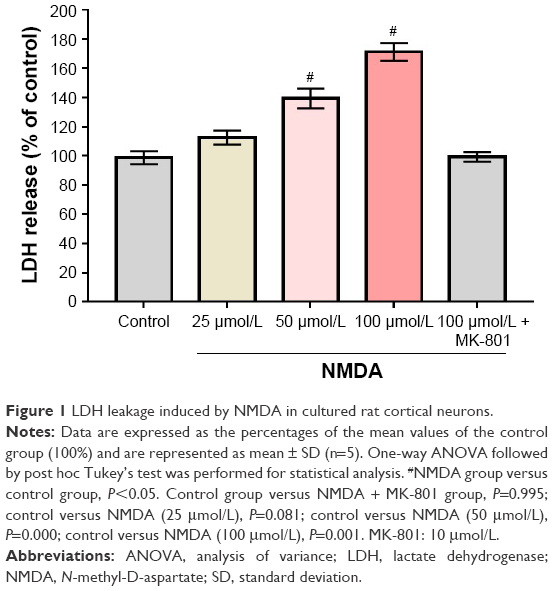 | Figure 1 LDH leakage induced by NMDA in cultured rat cortical neurons. |
HN caused dose-dependent restoration of the NMDA-triggered cell viability reduction in cultured rat cortical neurons
NMDA exposure (100 μmol/L) induced the damage of neuron cells and caused a decrease of cell viability to levels ~53.2% of the controls, as shown in Figure 2. The cell viability was significantly increased by the addition of HN in a dose-dependent manner. When the concentration of HN reached 0.1μmol/L, the decrease of cell viability began to be attenuated in the NMDA treatment group. When 10 μmol/L of HN was added into the medium, the viability of the neurons was ~98.3% of the control group (Figure 2). Furthermore, NMDA exposure did not induce any obvious reduction in cell viability when 100 μmol/L of HN was present in the medium.
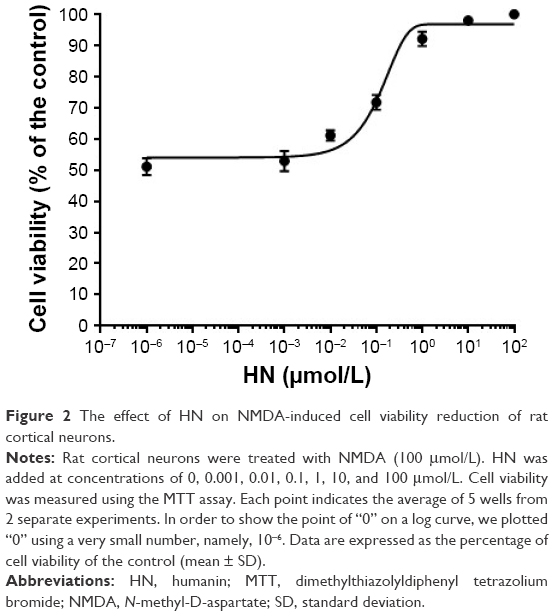 | Figure 2 The effect of HN on NMDA-induced cell viability reduction of rat cortical neurons. |
HN did not block NMDA-induced intracellular increase of calcium ions
NMDA (100 μmol/L) exposure induced a quick increase of cytoplasmic Ca2+ concentration, which reached its peak value within 50 s (Figure 3, green curve). The intracellular Ca2+ remained on the plateau during the whole observation (Figure 3, green curve). The treatment with MK-801, a blocker of the NMDA receptor, completely inhibited the increase of cytoplasmic Ca2+ triggered by NMDA (Figure 3, red curve). In contrast, 0.1 μmol/L of HN treatment did not show the inhibitory effect of NMDA-induced increase of cytoplasmic Ca2+ concentration (Figure 3, pink curve). HN (1 μmol/L) also did not block the initial sharp increase of cytoplasmic Ca2+ even though a gradual decrease of Ca2+ concentration was observed after 100 s (Figure 3, purple curve).
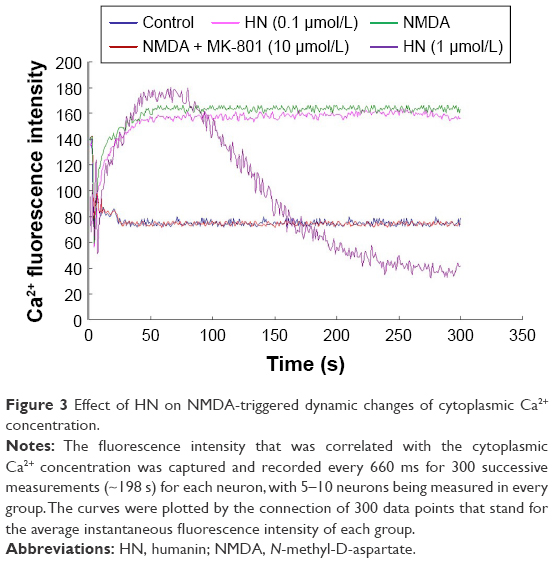 | Figure 3 Effect of HN on NMDA-triggered dynamic changes of cytoplasmic Ca2+ concentration. |
HN attenuated the decrease of SDH activity caused by NMDA exposure
NMDA treatment resulted in a decrease in SDH activity by 47% compared with the control group (Figure 4). As an uncompetitive channel blocker of the NMDA receptor, MK-801 completely inhibited the decrease of SDH activity triggered by NMDA (control group against NMDA + MK-801 group, P=0.766), suggesting that the decrease of SDH activity was triggered by NMDA exposure. HN (1 μmol/L) did not change the activity of SDH in the control neurons. However, HN (1 μmol/L) significantly attenuated the decrease of SDH activity triggered by NMDA (P<0.05 compared with the NMDA group) (Figure 4). S7A-HN is the inactive form of HN. The SDH activity in the NMDA + S7A-HN group was significantly lower than that in the control cells (P=0.032, control group versus NMDA + S7A-HN group), suggesting that the inactive form of HN did not protect against NMDA toxicity. AGA-HNG is 100× more active than the wild-type HN. The presence of AGA-HNG significantly increased SDH activity in NMDA-treated cells (P=0.001, NMDA versus NMDA + AGA-HNG) (Figure 4). Western blot analysis showed that NMDA treatment significantly inhibited the expression of mitochondrial SDH subunit B (SDHB) (P<0.05, Figure S1A). The expression of SDHB was normalized by pretreatment with HN for 16 h prior to the addition of NMDA. HN itself did not alter SDHB expression.
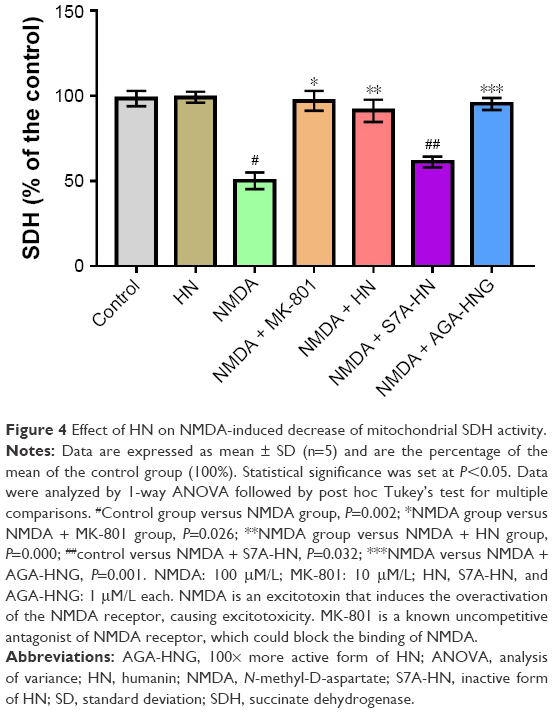 | Figure 4 Effect of HN on NMDA-induced decrease of mitochondrial SDH activity. |
HN inhibited the decrease of mitochondrial membrane potential (ΔΨm) induced by NMDA exposure
Compared to the control group, the NMDA group was darker and the average fluorescence intensity was significantly lower (Figure 5A and B). This indicated that NDMA administration resulted in significant reduction of the membrane potential. Similarly, quantification of the membrane potential showed a 25% decrease in the NMDA-treated group compared with the control (P=0.002). As an uncompetitive channel blocker of the NMDA receptor, MK-801 inhibited the reduction of the membrane potential triggered by NMDA (P=0.026, NMDA versus NMDA + MK-801). The fluorescence intensity of the NMDA + HN group was similar to that of the NMDA + MK-801 group (Figure 5B). The addition of HN (1 μmol/L) could significantly attenuate the decrease of mitochondrial membrane potential triggered by NMDA (P=0.000, NMDA versus NMDA + HN).
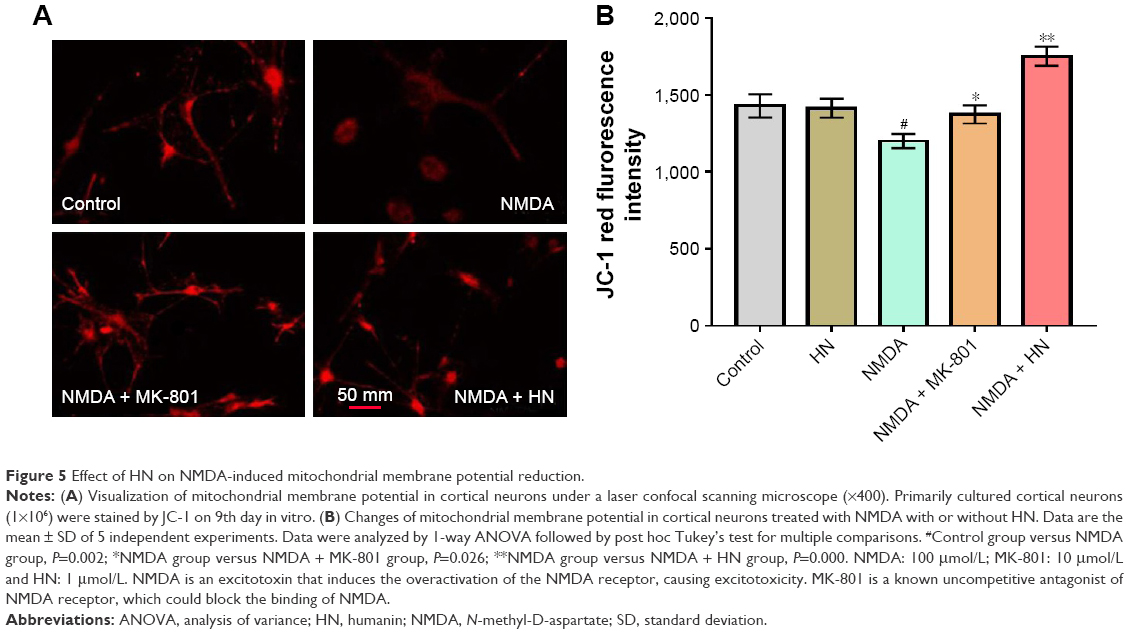 | Figure 5 Effect of HN on NMDA-induced mitochondrial membrane potential reduction. |
HN reduced the production of ROS triggered by NMDA
Although cells could be observed in a bright field, the fluorescence was weak in the control group (Figure 6A). On the contrary, the image was brighter and the cells were very clear in the NMDA group, suggesting that much more ROS had been generated compared with the control group (Figure 6A). ROS quantification revealed that the amount of ROS in the NMDA group was ~20% more than that in the control group (P=0.000, control versus NMDA). As an uncompetitive channel blocker of the NMDA receptor, MK-801 inhibited ROS production triggered by NMDA (P=0.000, NMDA versus NMDA + MK-801) (Figure 6B). These results indicated that the increase of ROS production in the experiment was triggered by NMDA. The ROS level in the NMDA + HN group was similar to that of the NMDA + MK-801 group and significantly lower than that in the NMDA group (P=0.001, NMDA versus NMDA + HN), suggesting that HN (1 μmol/L) could attenuate NMDA-triggered ROS production. Similarly, we found that the inactive form of HN, S7A-HN did not lower ROS production in NMDA-treated cells, while AGA-HNG did (P=0.022, NMDA versus NMDA + AGA-HNG) (Figure 6B).
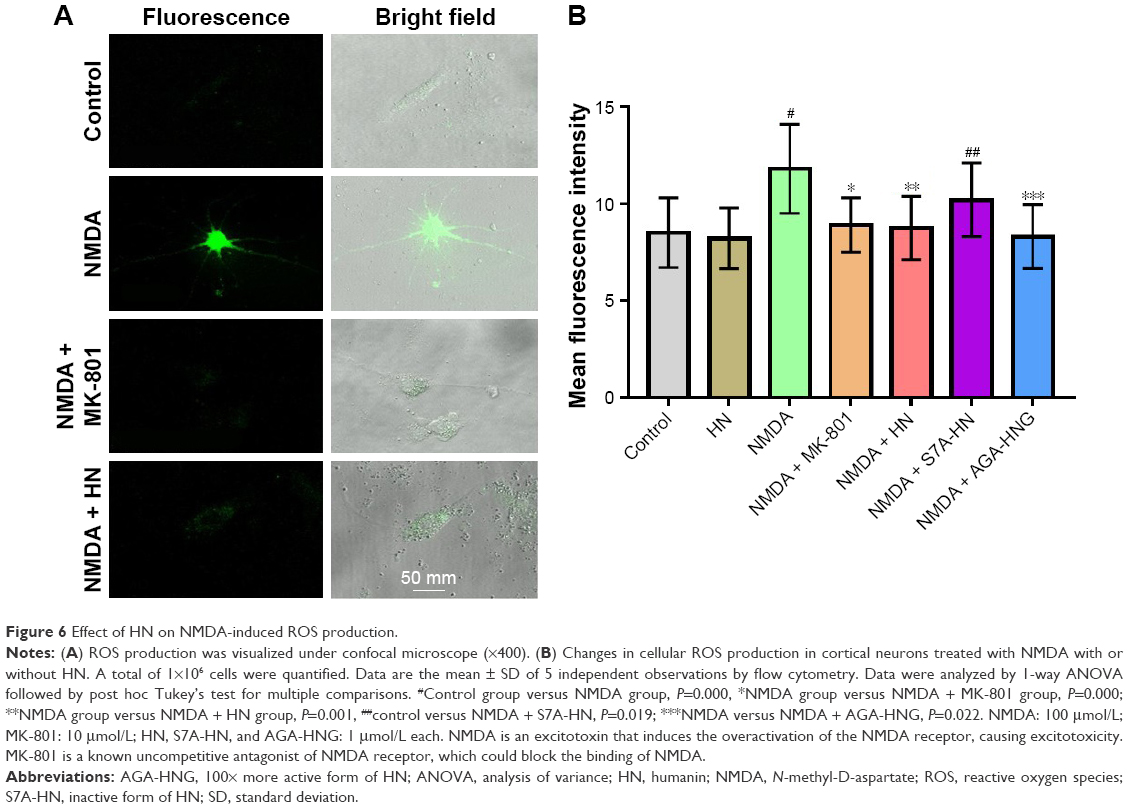 | Figure 6 Effect of HN on NMDA-induced ROS production. |
HN inhibited NMDA-triggered NO production
As shown in Figure 7, the production of NO increased significantly in the NMDA group compared with the control group (P=0.000). As an uncompetitive channel blocker of the NMDA receptor, MK-801 treatment dramatically inhibited NO overproduction triggered by NMDA (P=0.003, NMDA versus NMDA + MK-801), suggesting that the increase of NO was triggered by NMDA. The production of NO in the NMDA + HN group was similar to that in the NMDA + MK-801 group and significantly lower than that in the NMDA group (P=0.002, NMDA versus NMDA + HN), suggesting that HN (1 μmol/L) could attenuate NMDA-triggered NO production effectively when the cells were treated 16 h in advance. NO production in the NMDA + S7A-HN group was significantly higher than that in the control group (P=0.004, control versus NMDA + S7A-HN). Preincubation with AGA-HNG significantly decreased NO production in NMDA-treated cells (P=0.005, NMDA versus NMDA + AGA-HNG) (Figure 7). Western blot analysis of inducible nitric oxide synthase (iNOS) expression showed that iNOS was dramatically upregulated by NMDA treatment (P<0.05). There was no difference in the iNOS expression levels between the control and the HN-treated cells (Figure S1B).
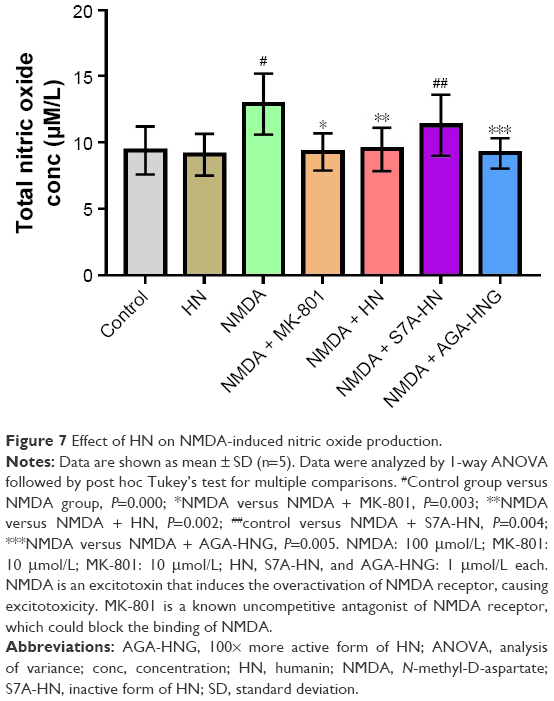 | Figure 7 Effect of HN on NMDA-induced nitric oxide production. |
Discussion
NMDA receptor-mediated Ca2+ overloading is a pivotal characteristic of glutamate toxicity and is implicated in a variety of pathogeneses, such as stroke, traumatic brain injury, schizophrenia, Parkinson’s disease, cerebral ischemia, and neurodegenerative disorders.22,23 NMDA receptor antagonists have been explored for many years as therapeutic agents for the treatment of Ca2+ overloading. However, these antagonists often cause severe adverse effects.9,10 Not only is HN a potential versatile neuroprotective agent, but it is also encoded, transcribed, synthesized, and secreted from the occipital lobe of the human brain. Therefore, HN could be a promising candidate for the therapy of excitatory neurotoxicity. In our previous study, we found that HN effectively rescued cultured rat cortical neurons.18 Its mode of action was not due to the blocking of intracellular Ca2+ overloading triggered by NMDA.
Mitochondria and the endoplasmic reticulum can actively sequester Ca2+ in response to high Ca2+ concentration in the cytoplasm, which might be the explanation for the reverting of the high Ca2+ concentration in the cytoplasm. The mitochondrial energy-transducing capacity is essential for the maintenance of neuronal function. NMDA exposure triggered the activation of Ca2+ influx. The mitochondria then actively sequestered Ca2+ to prevent pathologic increases in cytosolic [Ca2+]. Intramitochondrial [Ca2+] increase is a potent signal for the activation of mitochondrial respiration, leading to increased respiration and excessive ROS generation.24 Cytochrome C (Cytc) and other apoptogenic proteins are tethered to the inner mitochondrial membrane by cardiolipin. Excess ROS production in the mitochondria then induced peroxidation and subsequently oxidation of cardiolipin, thereby releasing Cytc and other apoptogenic proteins into the intermitochondrial space. Upon pore formation or alterations in the permeability of the outer mitochondrial membrane, free Cytc and other apoptogenic proteins are released into the cytosol and these subsequently promote the formation of the apoptosome (a complex containing Cytc, caspase 9, and Apaf-1), which activates caspase-related apoptosis.25,26 In other words, mitochondrial Ca2+ overload is a critical sensitizing signal in the proapoptotic transition of mitochondria and plays a key role in the regulation of cell death.
To assess the mitochondrial functional situation, 4 functional indexes were tested. The first was mitochondrial SDH activity, an important enzyme in the tricarboxylic acid (TCA) cycle. Neurons are exclusively dependent on mitochondrial ATP generation and have almost no capacity to upregulate energy supply through glycolysis. Therefore, the activity of SDH is the biomarker for the mitochondrial situation. The second is the mitochondrial membrane potential, the foundation of ATP production. On the one hand, the transient mitochondrial membrane hyperpolarization triggered by mitochondrial Ca2+ overload could induce pathological ROS generation and subsequent potential insults. On the other hand, the following depolarization collapse is the pivotal hallmark of irreversible mitochondrial injury and a critical step in the pathway to cell death. The third, namely, the level of ROS, which is one of the by-products of ATP production, is critical. Under normal conditions, >90% of oxygen is fully reduced to H2O by Cytc oxidase (CcO). Only a small number of electrons “leak” and thereby lead to the partial reduction of O2 to superoxide. Pathophysiologic levels of ROS are triggered by stressful stimuli, such as ischemia, and are produced during the hyperpolarization of ΔΨm. ROS initiates oxidative damage to cellular proteins, lipids, nucleic acids, and polysaccharides throughout the cell. The fourth is the NO level. Electrons escape from the electron transport chain and react with O2 to form superoxide (O2–), a potent ROS, or interact with NO to form equally cytotoxic reactive nitrogen species. Among the 4 indexes, SDH can reflect the ability of mitochondria for ATP generation. The mitochondrial membrane potential can reflect the integrity of the ATP machinery. ROS and NO reflect the degree of damage to the ATP machinery and the potential insult to the neurons. ROS and NO both are superoxides, but NO is the major ROS species involved in NMDA receptor-mediated toxicity.4,27 The analysis of NO is crucial for the assessment of mitochondrial function.
HN has been found to prevent stress-induced apoptosis in many cells/tissues. The putative underlying mechanisms include HN binding to an interleukin (IL)-12-like trimeric membrane receptor, BAX, or IGFBP-3 (a proapoptotic factor).28–30 In this research, the HN concentration used to inhibit NMDA-induced toxicity was much lower than that resulting in HN’s protection of the death systems (1–10 μmol/L), as mentioned earlier. This difference suggested that the HN receptor that mediates the HN-induced protection of rat cortical neurons against NMDA toxicity might be different from the receptor that has been reported in earlier studies.30,31
The results that HN could attenuate NMDA-induced mitochondrial dysfunction effectively in cultured rat cortical neurons were coincident with the report by Sreekumar et al.32 In their research,32 Sreekumar et al found that HN protects human retinal pigment epithelial (hRPE) cells from tert-butyl hydroperoxide (tBH)-induced oxidative stress, senescence, and mitochondrial dysfunction.32 Exogenous HN was taken up by the RPE cells and colocalized with the mitochondria, wherein it regulated ROS production,32,33 which gives us a clue about the mechanism underlying HN’s attenuation of NMDA-induced mitochondrial dysfunction. HN might be directly taken up by the neurons and subsequently translocated into the mitochondria, thereby inhibiting pathological ROS production.32 HN derivatives were reported to inhibit necrotic cell death in neurons by mitigating necrosis-associated decrease in ATP levels. Further, the peptide’s direct enhancement of the activity of ATP synthase has also been reported.34 Several laboratories have tested the protective effect of HN on various types of stresses or diseases. Although the specific molecular mechanism of cytotoxicity is unclear for some of these models, eg, Aβ and 3xTg-AD, many have oxidative stress as a common mechanistic denominator, suggesting a specific oxidative insult-related cytoprotective role of HN.35 ATP synthase and oxidative insult are closely related to mitochondrial function, which gives a good explanation for HN’s versatile cytoprotection, from the broad cell types to different death models.
Conclusion
We found that NMDA exposure triggered a decrease in cell viability and caused LDH leakage in rat cortical neurons, which could be restored by HN treatment. We further showed that HN (1 μmol/L) could attenuate NMDA-induced mitochondrial dysfunctions effectively by restoring SDH activity, blocking the decrease in mitochondrial potential, and neutralizing the excessive ROS and NO produced. The possible underlying mechanism is that HN is directly absorbed by the neurons and subsequently translocated to the mitochondria; thereafter, it inhibits pathological ROS production.
Acknowledgments
This study was supported by grants from The National Key Research and Development Program of China (no 2016YFC0100105), the National Science Foundation of China (nos 81628008, 81571641, and 81471652), and an internal grant from the China-Japan Friendship Hospital, Beijing, People’s Republic of China (no 2014-3-MS-18).
Disclosure
The authors report no conflicts of interest in this work.
References
Choi DW. Excitotoxic cell death. J Neurobiol. 1992;23(9):1261–1276. | ||
Papouin T, Ladépêche L, Ruel J, et al. Synaptic and extrasynaptic NMDA receptors are gated by different endogenous coagonists. Cell. 2012;150(3):633–646. | ||
Zhou X, Hollern D, Liao J, Andrechek E, Wang H. NMDA receptor-mediated excitotoxicity depends on the coactivation of synaptic and extrasynaptic receptors. Cell Death Dis. 2013;4:e560. | ||
Duchen MR. Mitochondria, calcium-dependent neuronal death and neurodegenerative disease. Pflugers Arch. 2012;464(1):111–121. | ||
Glancy B, Balaban RS. Role of mitochondrial Ca2+ in the regulation of cellular energetics. Biochemistry. 2012;51(14):2959–2973. | ||
Palty R, Hershfinkel M, Sekler I. Molecular identity and functional properties of the mitochondrial Na+/Ca2+ exchanger. J Biol Chem. 2012;287(38):31650–31657. | ||
Rueda CB, Llorente-Folch I, Traba J, et al. Glutamate excitotoxicity and Ca2+ regulation of respiration: Role of the Ca2+ activated mitochondrial transporters (CaMCs). Biochim Biophys Acta. 2016;1857:1158–1166. | ||
Fraysse B, Nagi SM, Boher B, et al. Ca2+ overload and mitochondrial permeability transition pore activation in living delta-sarcoglycan-deficient cardiomyocytes. Am J Physiol Cell Physiol. 2010;299(3): C706–C713. | ||
Wu YN, Johnson SW. Memantine selectively blocks extrasynaptic NMDA receptors in rat substantia nigra dopamine neurons. Brain Res. 2015;1603:1–7. | ||
Wrighton DC, Baker EJ, Chen PE, Wyllie DJ. Mg2+ and memantine block of rat recombinant NMDA receptors containing chimeric NR2A/2D subunits expressed in Xenopus laevis oocytes. J Physiol. 2008;586(1):211–225. | ||
Hashimoto Y, Niikura T, Ito Y, et al. Detailed characterization of neuroprotection by a rescue factor humanin against various Alzheimer’s disease-relevant insults. J Neurosci. 2001;21(23):9235–9245. | ||
Hashimoto Y, Ito Y, Niikura T, et al. Mechanisms of neuroprotection by a novel rescue factor humanin from Swedish mutant amyloid precursor protein. Biochem Biophys Res Commun. 2001;283(2):460–468. | ||
Tajima H, Niikura T, Hashimoto Y, et al. Evidence for in vivo production of humanin peptide, a neuroprotective factor against Alzheimer’s disease-related insults. Neurosci Lett. 2002;324(3):227–231. | ||
Tajima H, Kawasumi M, Chiba T, et al. A humanin derivative, S14G-HN, prevents amyloid-beta-induced memory impairment in mice. J Neurosci Res. 2005;79(5):714–723. | ||
Mamiya T, Ukai M. [Gly(14)]-humanin improved the learning and memory impairment induced by scopolamine in vivo. Br J Pharmacol. 2001;134(8):1597–1599. | ||
Krejcova G, Patocka J, Slaninova J. Effect of humanin analogues on experimentally induced impairment of spatial memory in rats. J Pept Sci. 2004;10(10):636–639. | ||
Caricasole A, Bruno V, Cappuccio I, Melchiorri D, Copani A, Nicoletti F. A novel rat gene encoding a humanin-like peptide endowed with broad neuroprotective activity. FASEB J. 2002;16(10): 1331–1333. | ||
Cui AL, Li JZ, Feng ZB, et al. Humanin rescues cultured rat cortical neurons from NMDA-induced toxicity not by NMDA receptor. ScientificWorldJournal. 2014;2014:341529. | ||
Cebere A, Liljequist S. Ethanol differentially inhibits homoquinolinic acid- and NMDA-induced neurotoxicity in primary cultures of cerebellar granule cells. Neurochem Res. 2003;28(8): 1193–1199. | ||
Koh JY, Choi DW. Vulnerability of cultured cortical neurons to damage by excitotoxins: differential susceptibility of neurons containing NADPH-diaphorase. J Neurosci. 1988;8(6):2153–2163. | ||
Li R, Guo M, Zhang G, Xu X, Li Q. Neuroprotection of nicotiflorin in permanent focal cerebral ischemia and in neuronal cultures. Biol Pharm Bull. 2006;29(9):1868–1872. | ||
Chen Z, Zhou Q, Zhang M, Wang H, Yun W, Zhou X. Co-activation of synaptic and extrasynaptic NMDA receptors by neuronal insults determines cell death in acute brain slice. Neurochem Int. 2014;78:28–34. | ||
Toglia P, Cheung KH, Mak DD, Ullah G. Impaired mitochondrial function due to familial Alzheimer’s disease-causing presenilins mutants via Ca(2+) disruptions. Cell Calcium. 2016;59(5):240–250. | ||
Takeuchi A, Kim B, Matsuoka S. The destiny of Ca(2+) released by mitochondria. J Physiol Sci. 2015;65(1):11–24. | ||
Sanderson TH, Reynolds CA, Kumar R, Przyklenk K, Hüttemann M. Molecular mechanisms of ischemia-reperfusion injury in brain: pivotal role of the mitochondrial membrane potential in reactive oxygen species generation. Mol Neurobiol. 2013;47(1):9–23. | ||
Corona JC, Duchen MR. Impaired mitochondrial homeostasis and neurodegeneration: towards new therapeutic targets. J Bioenerg Biomembr. 2015;47(1–2):89–99. | ||
Drechsel DA, Estévez AG, Barbeito L, Beckman JS. Nitric oxide-mediated oxidative damage and the progressive demise of motor neurons in ALS. Neurotox Res. 2012;22(4):251–264. | ||
Guo B, Zhai D, Cabezas E, et al. Humanin peptide suppresses apoptosis by interfering with Bax activation. Nature. 2003;423(6938):456–461. | ||
Ikonen M, Liu B, Hashimoto Y, et al. Interaction between the Alzheimer’s survival peptide humanin and insulin-like growth factor-binding protein 3 regulates cell survival and apoptosis. Proc Natl Acad Sci U S A. 2003;100(22):13042–13047. | ||
Ying G, Iribarren P, Zhou Y, et al. Humanin, a newly identified neuroprotective factor, uses the G protein-coupled formylpeptide receptor-like-1 as a functional receptor. J Immunol. 2004;172(11): 7078–7085. | ||
Hashimoto Y, Kurita M, Aiso S, Nishimoto I, Matsuoka M. Humanin inhibits neuronal cell death by interacting with a cytokine receptor complex or complexes involving CNTF receptor alpha/WSX-1/gp130. Mol Biol Cell. 2009;20(12):2864–2873. | ||
Sreekumar PG, Ishikawa K, Spee C, et al. The mitochondrial-derived peptide humanin protects RPE cells from oxidative stress, senescence, and mitochondrial dysfunction. Invest Ophthalmol Vis Sci. 2016;57(3):1238–1253. | ||
Paharkova V, Alvarez G, Nakamura H, Cohen P, Lee KW. Rat humanin is encoded and translated in mitochondria and is localized to the mitochondrial compartment where it regulates ROS production. Mol Cell Endocrinol. 2015;413:96–100. | ||
Cohen A, Lerner-Yardeni J, Meridor D, Kasher R, Nathan I, Parola AH. Humanin derivatives inhibit necrotic cell death in neurons. Mol Med. 2015;21:505–514. | ||
Lee C, Yen K, Cohen P. Humanin: a harbinger of mitochondrial-derived peptides. Trends Endocrinol Metab. 2013;24(5):222–228. |
Supplementary material
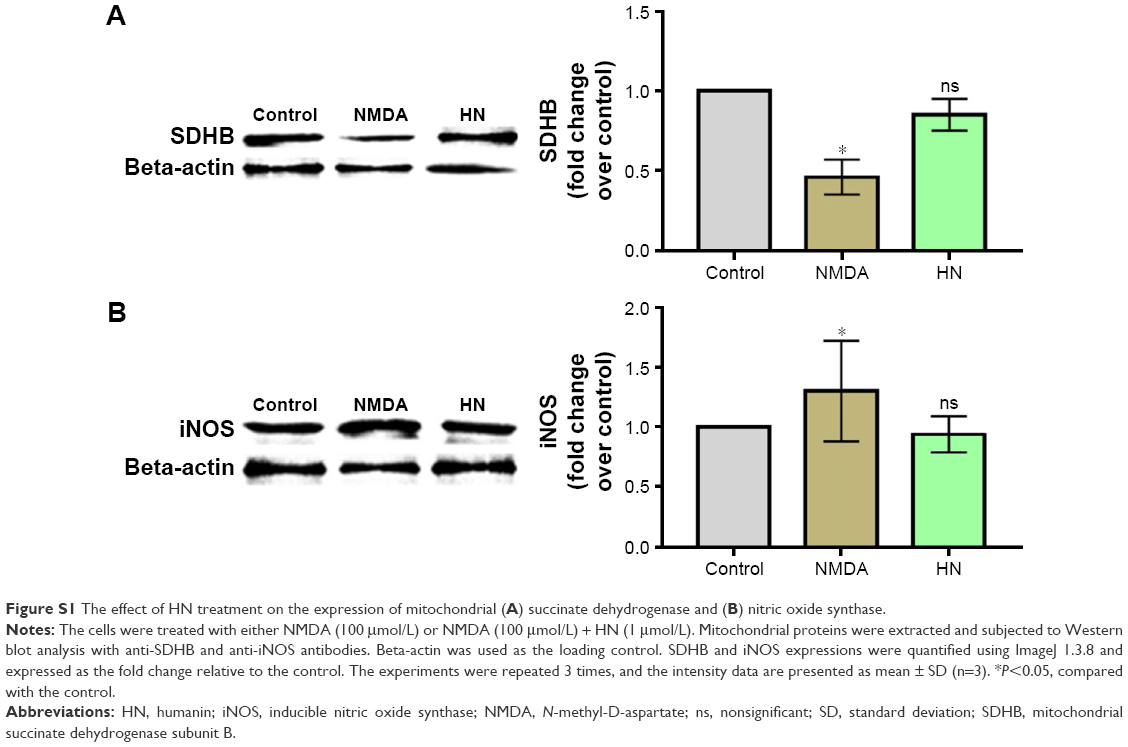 | Figure S1 The effect of HN treatment on the expression of mitochondrial (A) succinate dehydrogenase and (B) nitric oxide synthase. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.