")
Back to Journals » Journal of Inflammation Research » Volume 13
Human Secretary Phospholipase A2 Mutations and Their Clinical Implications
Authors Khan MI, Hariprasad G
Received 26 June 2020
Accepted for publication 13 August 2020
Published 16 September 2020 Volume 2020:13 Pages 551—561
DOI https://doi.org/10.2147/JIR.S269557
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ning Quan
Mohd Imran Khan, Gururao Hariprasad
Department of Biophysics, All India Institute of Medical Sciences, New Delhi 110029, India
Correspondence: Gururao Hariprasad Tel +91-11-26594240
Fax +91-11-26588663
. Email [email protected]
Abstract: Phospholipases A2 (PLA2s) belong to a superfamily of enzymes responsible for hydrolysis of the sn-2 fatty acids of membrane phospholipids to release arachidonic acid. PLA2s are the rate limiting enzyme for the downstream synthesis of prostaglandins and leukotrienes that are the main mediators of inflammation. The extracellular forms of this enzyme are also called the secretary phospholipase A2 (sPLA2) and are distributed extensively in most of the tissues in the human body. Their integral role in inflammatory pathways has been the primary reason for the extensive research on this molecule. The catalytic mechanism of sPLA2 is initiated by a histidine/aspartic acid/calcium complex within the active site. Though they are known to have certain housekeeping functions, certain mutations of sPLA2 are known to be implicated in causation of certain pathologies leading to diseases such as atherosclerosis, cardiovascular diseases, benign fleck retina, neurodegeneration, and asthma. We present an overview of human sPLA2 and a comprehensive compilation of the mutations that result in various disease phenotypes. The study not only helps to have a holistic understanding of human sPLA2 mutations and their clinical implications, but is also a useful platform to initiate research pertaining to structure–function relationship of the mutations to develop effective therapies for management of these diseases.
Keywords: secretary phospholipase A2, sPLA2, mutations, clinical implications, structure–function relationship
Phospholipase A2
Phospholipase hydrolyzes phospholipids into fatty acids and other lipophilic substances. The phospholipases are divided into groups depending on which glycerol ester bond they are capable of cleaving (these bonds are marked in Figure 1). The phospholipases are thus called phospholipase A, B, C and D. For phospholipase A, a subscript 1 or 2 is added depending on whether the cleaved bond involved is at the sn-1 or sn-2, position of the phospholipid substrate.1 1 Phospholipase B cleaves at both sn-1 and sn-2 positions. This is demonstrated by a diagrammatic illustration in Figure 1.
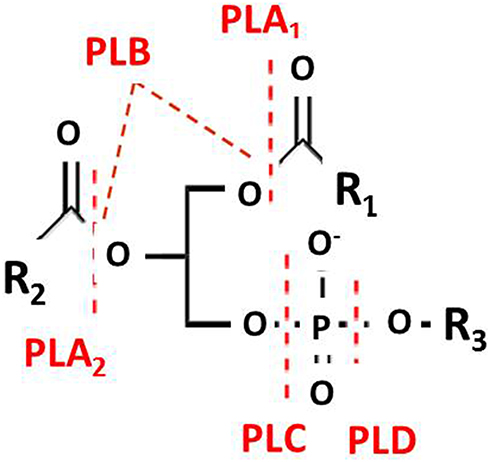 |
Figure 1 Diagrammatic representation showing the chemical bonds on the phospholipid substrate that are cleaved by the enzymes in the phospholipase family. |
The discovery of PLA2 was based on the observation that pancreatic juice and cobra venom were able to hydrolyze phosphatidylcholine (PC).2 PLA2 are major constituents in mammalian tissues as well as in insect and snake venoms. They are widespread in living organisms both as intracellular and extracellular forms and are the most extensively studied among all phospholipases. Phospholipases A2 (PLA2s, EC 3.1.1.4) are upstream regulators of many inflammatory processes which hydrolyze phospholipids at the sn-2 position of the glycerol backbone, releasing lysophospholipids and fatty acids.1 They hydrolyze various naturally occurring phospholipids such as phosphatidylcholine, phosphatidylethanolamine, phosphatidylserine, phosphatidylinositol, phosphatidylglycerol, plasmalogen, plasmenylcholine and 1-alkylether phosphatidylcholine and release the arachidonic acid. Upon downstream modification by cyclooxygenases, arachidonic acid is modified into active compounds called eicosanoids, which include prostaglandins, thromboxanes and leukotrienes which are categorized as inflammatory mediators which lead to pharmacological interest in this reaction.1 Mammalian PLA2 enzymes play an important role in the maintenance of the cellular phospholipid pools and membrane repair through deacylation/reacylation pathways and biosynthesis of prostaglandins and leukotrienes.3
PLA2s are classified into 15 groups due to the advent of genomics which has seen an increase in the number of PLA2 subgroups, leading to the characterization of exciting new PLA2s.4,5 Additional forms of secreted PLA2s (sPLA2s) utilizing a catalytic histidine have been discovered in recent years, which are clearly related to the GI, GII, and GIII PLA2, but do not easily fit into these groups. This led to the establishment of groups V, IX, X, XI, XII, XIII, and XIV. PLA2s using serine as catalytic residue, cytosolic PLA2s (cPLA2s) were classified into GIV and Ca2+ independent PLA2s (iPLA2) formed GVI. PLA2s using the serine/histidine/aspartate triad in catalysis, platelet-activating factor acetylhydrolases (PAF-AH) forms GVII and GVIII, and the recently discovered lysosomal PLA2s forms GXV. This review highlights the salient features of sPLA2s and the effect of their mutations on clinical phenotypes.
Structure of Secretary PLA2
Secreted PLA2 (sPLA2) family bags more than one third of the isoforms. The family contains 11 calcium dependent isoforms (IB, IIA, IIC, IID, IIE, IIF, III, V, X, XIIA and XIIB) in mammals.6 The secretory PLA2s are characterized by relatively low molecular weights (typically <20 kDa) proteins. Groups III and XII, are structurally distinct from rest of the members of the group based on the protein sequence identity.7 sPLA2s have some common structural elements that include a conserved CCXHDXC motif with His-Asp catalytic dyad at the active site, a highly conserved calcium binding domain and extensively conserved disulfide-stabilized tertiary structure (Figure 2). The core conformation of sPLA2 includes three α-helices, two anti-parallel β-strands, calcium binding loop, and a substrate binding hydrophobic channel. Few of the sPLA2s, like in the human group III, isoform have a C-terminal extension.7 Some of the characteristic structural features of sPLA2 is highlighted in the following sections.
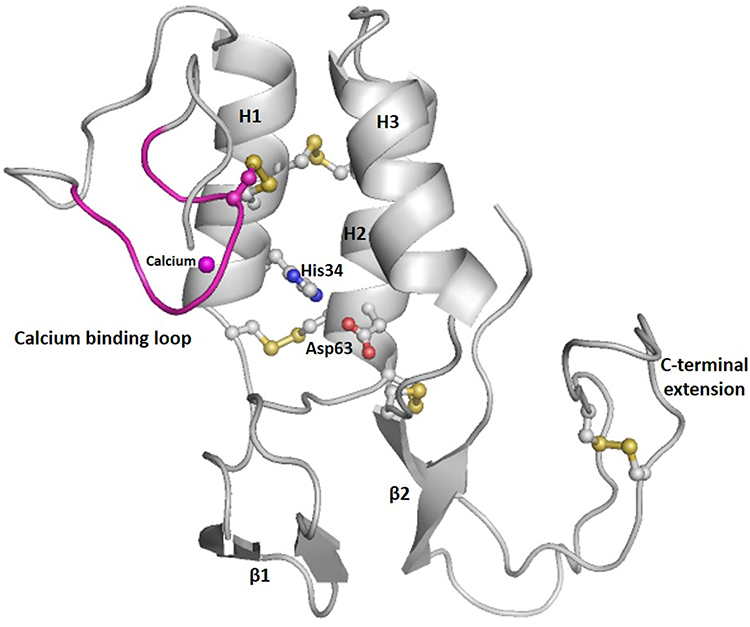 |
Figure 2 Ribbon diagram showing the overall structure of human group III PLA2, a prototype human sPLA2. Structure comprises of three helices, indicated as H1, H2, and H3; two β-wings, indicated as β1 and β2; calcium binding loop (pink) with the calcium ion (sphere in magenta); active site with residues histidine 34 and aspartic acid 63; C-terminal extension; and stabilized by five disulfide bonds (yellow). The structure was done using homology modeling on human group III PLA2 sequence (Q9NZ20), and viewed on Pymol. |
sPLA2 GI/II coordinate the essential Ca2+ through a conserved and conformationally flexible loop of residues with the consensus sequence Y25-G-C-Y/F-C-G-X-G-G33 which contributes three closely spaced backbone carbonyl oxygens (O28, O30 and O32).8 These carbonyl oxygen atoms, along with oxygen atoms donated by the carboxylate of Asp49 and two water molecules, form a tight pentagonal bipyramidal coordinate cage for the calcium ion. An analogous arrangement is found for the human GIII PLA2 enzyme, where Trp9, Gly11 and Gly13 contribute the backbone carbonyls and the carboxylate group from Asp36 form the coordinate cage around the calcium ion.7 Calcium coordination stabilizes the loop which otherwise has conformation that is flexible, to form the left-superior wall of the hydrophobic channel and orders the local protein structure in a manner that appears to optimize substrate interactions.
sPLA2s contain three spatially conserved α-helices. Two long anti-parallel helices are riveted together by twin disulfide bridges to create a rigid backbone brace that forms the back wall of the substrate-binding pocket. Disulfide bridges found in sPLA2s incorporate half-cysteines derived from this substructure. Conserved side-chains arising from the anti-parallel helices assist in the coordination of Ca2+ form the deeper contours of the hydrophobic channel and creates a catalytic network comprising of histidine, aspartic acid, and tyrosine, which ensures fixed active site geometry.
In GI PLA2s there is a distinctive loop of surface exposed residues arising from the distal tip of the first antiparallel helix. This loop is absent in GII enzymes, moderately developed among the GI elapids and most prominent among the GI enzymes from exocrine pancreas. There is little three-dimensional homology among sPLA2s in the region despite the presence of a conserved disulfide bridge (Cys61–Cys91). Deletion of residues 62–66 from the porcine pancreatic sPLA2 improves activity on micellar zwitterionic lecithins by up to 16-fold.9 This effect is likely to reflect the formation of a more favourable surface for interfacial adsorption.
All sPLA2 structures contain either one (GI/II) or two (GIII) well-developed β-wing(s). This substructure consists of a single loop of anti-parallel β-sheet that extends outwards from the molecule into the bulk solvent. The distal portions of these substructures are poorly anchored and may adapt a variety of orientations with respect to the enzyme proper. In some enzyme species the composition of the β-wing may confer ancillary pharmacological properties such as anti-coagulation.10
The C-terminal extension of GII enzymes forms a semicircular barrier around the Ca2+-binding loop. It is secured proximally (Cys126– Cys27) and distally (Cys134– Cys50) by disulfide bridges. The seven- or eight-residue loop is rich in proline and charged residues. In the human GIII sPLA2, despite the fact that all the functional motifs of the enzyme are within the first hundred residues, there is an additional 42-amino acid that adapts a long loop conformation. This last part of the loop is in close proximity to the third helix and the overall compactness of the enzyme is thereby maintained.11 This substructure is remote from the residues implicated in interfacial adsorption, substrate binding, and catalysis.11 This is in contrast to heterodimeric structural variants of group III sPLA2 from scorpions, wherein a single enzyme transcript codes for three distinct products that include a large enzymatic subunit, a pentameric peptide and a small non-enzymatic subunit.12,13 The enzymatic subunit comprises of three helices, the calcium binding loop and a substrate binding hydrophobic channel, and the non-enzymatic subunit comprises of extensive hydrophobic residues with a conformation of an anti-parallel β-sheets making it ideal for tissue specific targeting.12
This stereochemical mechanism of sPLA2 is reminiscent of the catalytic system of the serine proteases.14 A conserved water molecule acts as the attacking nucleophile attacks the sn-2 bond. A conserved histidine in the active site of PLA2 abstracts a proton from the water molecule at the Nδ1 position. The positive charge acquired by the histidine is stabilized through an extended hydrogen-bonded network that includes the carboxylate group of aspartic acid and the phenolic group tyrosine residues. It may be noted that the invariant aspartate and tyrosines at the active site are from nonanalogous backbone positions in different groups of the sPLA2 enzymes. The histidine, aspartate and the single/dual tyrosine residues are rightly called as catalytic residues of the sPLA2. It is noteworthy that this pattern of catalytic machinery is consistent in most groups of the sPLA2 ranging from group I, II IV, V, X, and XII.15–18 In the human group III sPLA2 (human), there is a phenylalanine at the active site where its aromatic ring fails to make any hydrogen bonded interactions with either the histidine or the aspartate which are in its vicinity. It is therefore established that tyrosine is not an essential requisite for the stabilization of the aspartic acid residue at the active site of PLA2 for its functionality.7 Interestingly, sPLA2 from the liver fluke parasite show classical features of histidine-aspartic acid-tyrosine in hydrogen bond formation at the active site.19 This difference at the active site between the target enzyme and the housekeeping human isoform is, therefore, an important structural parameter that can be exploited to design-specific drug molecules against the parasite Clonorchis sinensis.20
Unlike several sPLA2s in reptiles and scorpions that exist in solution as stable multichain complexes in the form of homodimers, homotrimers and heterodimers, native human sPLA2s do not have quaternary conformations.21,22 The human GIII sPLA2 has a C-terminal extension that is reminiscent of the non-enzymatic subunit of a sPLA2 from a scorpion that is associated with main enzymatic subunit by a disulfide bond.12,13 However, ligand associated mutimerization is known wherein bisindole compounds and anionic molecules induce dimers in certain sPLA2s.23,24
Physiological Functions of sPLA2
The functions of PLA2s go beyond their role in membrane homeostasis and they also function in such diverse roles such as digestion of nutrients to the formation of bioactive molecules involved in cell regulation. There are indications that a few phospholipases may carry out a biological function independent of their catalytic activity by binding to a regulatory membrane receptor. Phospholipase-like proteins with toxic properties, yet which lack a functional catalytic site, are found in venoms. It is of interest that most, but not all, phospholipases studied in detail thus far are soluble proteins. The soluble nature of many phospholipases suggests that their interaction with cellular membranes is one of the regulatory mechanisms that exist to prevent membrane degradation or to precisely control the formation of phospholipid-derived signalling molecules. In addition to the well-established functions of one of the sPLA2 enzymes in digestion of dietary phospholipids and another in host defense against bacterial infections, accumulating evidence shows that some of these sPLA2s are involved in arachidonic acid release from cellular phospholipids for the biosynthesis of eicosanoids, especially during inflammation.4,25 sPLA2s have also been involved in physiological and pathophysiological conditions in skin.26
PLA2s as Mediators of Inflammation
PLA2s participates in the inflammatory reaction in several different ways. PLA2s liberates free fatty acids and lysophospholipids by their hydrolytic action on phospholipids that are found in membranes. In inflammation, AA is the key fatty acid liberated from phospholipids by PLA2s. This reaction regulates the availability of AA which, in turn, is the rate limiting precursor for the formation of prostaglandins.25 AA is also the precursor of leukotrienes which are formed via the lipoxygenase pathway. The turnover of lysophospholipids in some conditions results in the formation of the platelet activating factor (PAF) which is another potent mediator of inflammation.27 Platelet activating factor-acetylhydrolases (PAF-AH) is an enzyme that is essential for this reaction. There are four members of this family, of which three are intracellular, and the fourth member, the plasma PAF-AH, is secreted extracellularly. The structure of plasma PAF-AH enzyme is a member of the serine-dependent class of sPLA2s. This enzyme has therefore been considered for discussion here.
Around 70–80% of circulating PAF-AH (Lp-PLA2) is bound to low-density lipoprotein (LDL) and the remainder is linked to high density lipoprotein (HDL) and some very low-density lipoproteins. The two α helices help the enzyme associate with these lipoproteins. Low density lipid has affinity in the region between residues 114 and 126 while high density lipid is associated with the residues 362 to 369 of the two α helices. The enzyme possess a classic lipase α/β- serine hydrolase fold and a catalytic triad consisting of Ser273, Asp296, and His351. Ser273 is located on the N-terminus of an alpha helix and on the conserved motif GXSXG classical to other lipases and serine esterases. Ser273 is a nucleophilic residue activated for catalysis through other two residues—Asp296 and His351. Residues Leu153 and Phe274 serve as oxyanion hole and stabilize the negative charge of tetrahedral intermediate through their amide nitrogens. The catalytic triad is oriented within a hydrophobic pocket and positioned towards its lipid substrate. PAF is a phospholipid-signaling molecule that binds its specific receptor leading to a cascade of proinflammatory signals; thus, PAF is a prominent pro-inflammatory mediator.28
Certain sPLA2s regulate a variety of biological functions through certain receptors called as sPLA2 receptor.29 These receptors are mannose type transmembrane glycoproteins that are related to the C-type animal lectin family. Some of the important receptor mediated functions are: (1) group X sPLA2-receptor interaction for arachidonic acid release from spleen cells;30 (2) group IB sPLA2-receptor interaction for cell proliferation and lipid mediator production;31 (3) group V sPLA2-receptor interaction for proangiogenic and anti-angiogenic factors by human neutrophils;32 (4) group I-receptor interaction for prostaglandin E2 production;33 (5) group IV-receptor interaction for cytokine release.34 In addition to physiological functions, the sPLA2-receptor interactions are also implicated in causing certain diseases including cancer.35,36
Human sPLA2 Mutations and Their Clinical Implications
sPLA2 are expressed in different tissues of the human body.37 In addition to its role in inflammation, they also known have a housekeeping role in some of these tissues.38 Mutations on these sPLA2 have an impact on the structure–function relationship of these enzymes that have various clinical implications. The various mutations on human sPLA2 and their clinical fallouts have been given in Table 1.
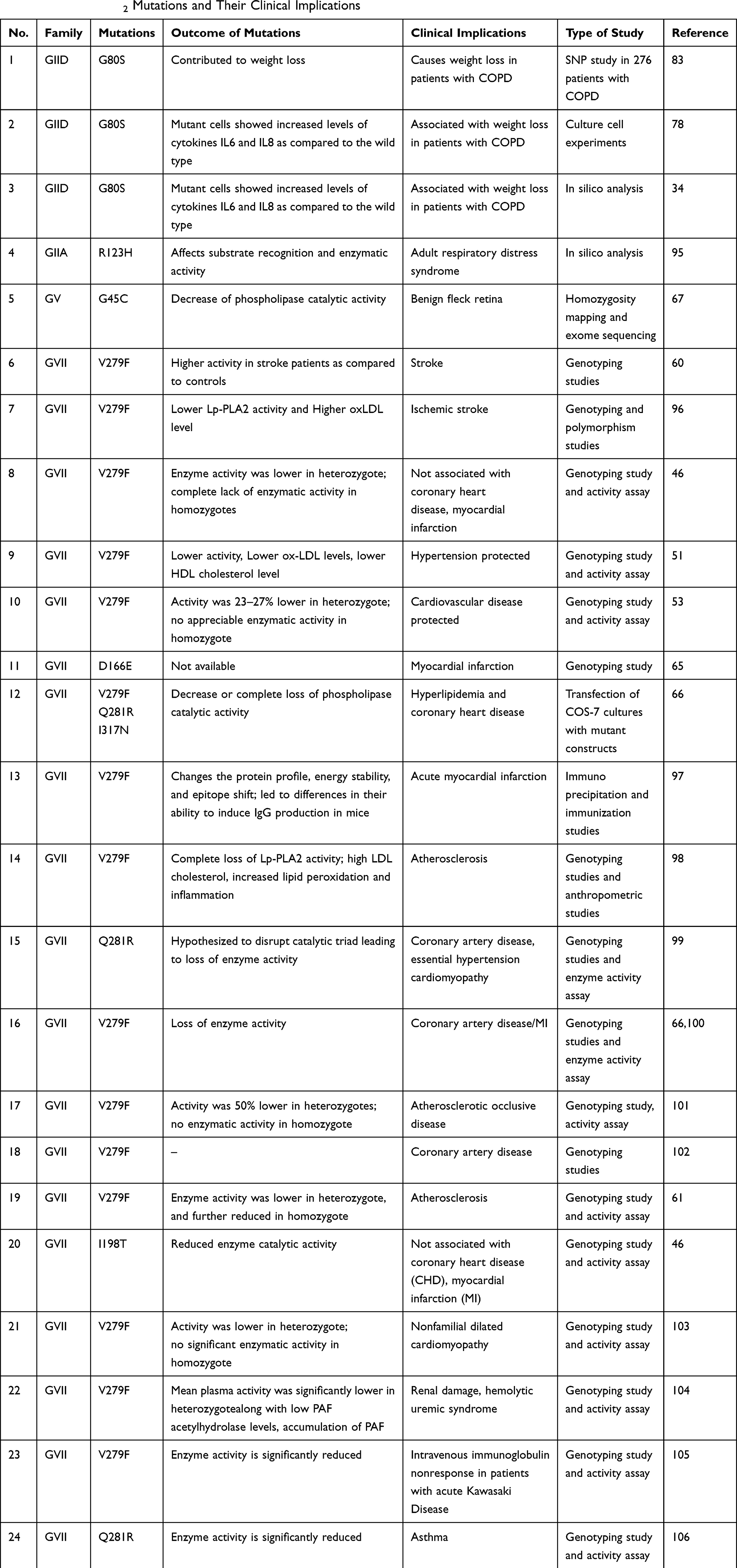 |
Table 1 Human sPLA2 Mutations and Their Clinical Implications |
sPLA2 Mutations and Coronary Artery Diseases
Platelet-activating factor acetylhydrolase (PAF-AH) is a calcium independent secreted enzyme that is classified as sPLA2, and associates both with LDL and HDL in human plasma.5 It acts on the sn-2 position of the glycerol backbone of the biologically active lipid messenger PAF molecule causing its deacetylation to an inactive lyso-PAF, a product that is no longer recognized by the PAF receptor.39 The PAF-AH activity may be considered anti-inflammatory and anti-atherogenic.40 Apart from PAF, the enzyme due to its broad substrate specificity also hydrolyzes phosphatidylethanolamine (PE) and phosphatidylcholines (PC) with short chain sn-2 moeties to generate oxidized fatty acid and lyso-phosphatidylcholine, which are pro-inflammatory, procalcifying and proapoptotic lipid mediators.41–44 These mediators play an important role in atherosclerotic plaque inflammation and development of atherosclerotic necrotic cores.45 The products of these two separate enzymatic reactions have varying effects on plaque formation in coronary arteries thereby leading to pro-atherosclerotic and anti-atherosclerotic clinical phenotypic outcomes.40,46-50 The varying clinical effects seen by effect of PAF-AH enzyme is probably due to the following reasons: (1) varying expression of the substrates; (2) varying affinity of the enzyme towards these substrates; and (3) varying enzyme kinetics leading to differential product concentrations that dictate the molecular pathogenesis.
It is clear from the above discussion that mutations on the enzyme PAF-AH can result in two clinical outcomes. While few studies show the mutations like V279F, R92H, A379V, D166E to be offering a favourable cardiovascular protective role.51–57 Few other studies show V279F, R92H, A379V, D166E to be having a frank cardiovascular disease (CVD) risk.54,58-65
sPLA2 Mutations and Benign Fleck Retina
GV sPLA2 enzyme is located at the short arm of the chromosome 1 between p36 and p34; in humans it is expressed in the eyes and has the ability to hydrolyze glycerophospholipids, releasing bioactive lipids and free fatty acids.66,67 This lipofuscin-like aggregates which is primarily composed of cross-linked lipid and protein at the retinal pigment epithelium (RPE) layer that gives rise to the focal thickening is referred to as 'fleck'.68 Lipofuscin is a marker of membrane, mitochondria and lysosomes damage.68,69 Usually lipofuscin is regarded as aggregates of undigested cell materials that accumulates over a lifetime, occupying major portions of the RPE cell in elderly individuals and is a hallmark of aging.69–72 Benign familial fleck retina (BFFR) is a congenital abnormality characterized by multifocal small, round, distinctive diffuse yellow-white fleck-like lesions of varying size involving the postequatorial retina without the involvement of the central macula.73 It is asymptomatic ocular condition with normal visual acuities in both eyes and the anterior segments of both eyes remains normal.73 This trait is genetically inherited as an autosomal recessive disease related to a G45C mutation in GV sPLA2.66,68 GV sPLA2 is known to contain 12 cysteines that form six disulfide bonds to stabilize its 3D structure.74 It is not clear as to how the mutation affects the structure–function of the enzyme. However, with another cysteine in 46th position on a flexible loop, it is likely that a wrong disulfide bond is formed that affects its conformation while folding, thereby leading to a loss of function.
sPLA2 Mutations and Weight Loss in Patients with Chronic Obstructive Pulmonary Disease (COPD)
COPD is airway diseases characterized by impaired airflow in the respiratory tract, chronic airway inflammation, as well as symptoms such as coughing, dyspnea, and wheezing.75 The disease is not just confined to problems of airflow obstruction, but also has a major impact on cardiac function and air exchange, thereby resulting in systemic manifestations such as cardiovascular dysfunction, anemia, gastroesophageal reflux, depression, anxiety, osteoporosis, and weight loss in patients.76 The cachexia that occurs in COPD is a result of muscle wasting and adipose tissue depletion and is linked to the systemic inflammation.77–83 It assumes clinical importance because it limits patient’s physical performance, compromises their quality of life and is also related to disease prognosis.84,85 The weight loss is related to elevated levels of pro-inflammatory mediators such as tumor necrosis factor and interleukins.78 However, in patients with COPD, the extent of weight loss varied and there was a wide distribution of pro-inflammatory mediators in the serum.79,83,86,87 The sPLA2 GIID protein consists of 125 amino acids and is constitutively expressed in the immune tissues humans and is upregulated by systemic pro-inflammatory stimuli in certain tissue tissues including the lung, suggesting its functional role in the progression of the inflammatory process.6,88 G80S is a missense mutation that lies on a loop that forms the interfacial binding surface (IBS). Our team has shown that the mutant enzyme adopts an open conformation which increases interfacial binding surface area, thereby binding more potently to the M-type receptor compared to the wild sPLA2.34 Hence G80S mutation on human sPLA2 GIID leads to enhanced expression of the cytokines that are responsible for the weight loss.34,78
Others Diseases Caused by sPLA2 Mutations
A159T mutation in sPLA2GVI is the causative for the neurological condition exhibiting familial cortical myoclonic tremor with epilepsy (FCMTE), an autosomal dominant epileptic syndrome, characterized by adult onset cortical myoclonic tremors of the extremities, epileptic seizures.89,90 I198T and V379A are two mutations on PAF-AH that decreases substrate affinity and thus increased PAF concentration causing prolonged B cell survival that consequently leading to higher IgE levels leading to atopy and asthma.91–93 R92H mutation in PAF-AH enzyme is reported to contribute to ischemic stroke susceptibility in the eastern Chinese Han population studied.94
Conclusions
sPLA2s are among the smallest enzymes which perform various vital physiological functions. Despite their small size, sPLA2 enzymes are complex puzzles with respect to their structure–function relationships. Certain sPLA2s are expressed in different body tissues and have regulated function and housekeeping properties. While their upregulation is responsible for various disease states including cancer, there are also mutations that cause pathologies leading to a number of clinical scenarios. This review pertains to a detailed understanding of these mutations; their role on the structure–function relationship of the enzyme and the plausible mechanisms that lead to disease is fascinating. This knowledge is useful to comprehensively understand the role of these enzymes in human disease, and lays the platform for the designing of appropriate therapeutics for patient care.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Van Deenen L, Haas G. The substrate specificity of phospholipase A. Biochimica Et Biophysica Acta (BBA) Spec Section Lipids Rel Sub. 1963;70:538–553.
2. Wittcoff H. The phosphatides. The Phosphatides. 1951.
3. Dennis EA. Diversity of group types, regulation, and function of phospholipase A2. J Biol Chem. 1994;269(18):13057–13060.
4. Lambeau G, Gelb MH. Biochemistry and physiology of mammalian secreted phospholipases A2. Annu Rev Biochem. 2008;77:495–520.
5. Schaloske RH, Dennis EA. The phospholipase A2 superfamily and its group numbering system. Biochimica Et Biophysica Acta (BBA)-Molecular and Cell Biology of Lipids. 2006;1761(11):1246–1259.
6. Murakami M, Yoshihara K, Shimbara S, et al. Group IID heparin‐binding secretory phospholipase A2 is expressed in human colon carcinoma cells and human mast cells and up‐regulated in mouse inflammatory tissues. Eur J Biochem. 2002;269(11):2698–2707.
7. Hariprasad G, Kumar M, Kaur P, Singh TP, Kumar RP. Human group III PLA2 as a drug target: structural analysis and inhibitor binding studies. Int J Biol Macromol. 2010;47(4):496–501.
8. Bekkers AC, Franken PA, Toxopeus E, Verheij HM, de Haas GH. The importance of glycine-30 for enzymatic activity of phospholipase A2. Biochimica Et Biophysica Acta (BBA) Protein Struct Mol Enzymol. 1991;1076(3):374–378.
9. Kuipers OP, Thunnissen M, De Geus P, et al. Enhanced activity and altered specificity of phospholipase A2 by deletion of a surface loop. Science. 1989;244(4900):82–85.
10. Kini RM, Evans HJ. Structure-function relationships of phospholipases. The anticoagulant region of phospholipases A2. J Biol Chem. 1987;262(30):14402–14407.
11. Nicolas JP, Lin Y, Lambeau G, Ghomashchi F, Lazdunski M, Gelb MH. Localization of structural elements of bee venom phospholipase A2 involved in N-type receptor binding and neurotoxicity. J Biol Chem. 1997;272(11):7173–7181.
12. Hariprasad G, Kumar M, Srinivasan A, Kaur P, Singh TP, Jithesh O. Structural analysis of a group III Glu62-phospholipase A2 from the scorpion, Mesobuthustamulus: targeting and reversible inhibition by native peptides. Int J Biol Macromol. 2011;48(3):423–431.
13. Hariprasad G, Hariprasad G, Singh B, et al. Cloning, sequence analysis and homology modeling of a novel phospholipase A2 from Heterometrusfulvipes (Indian black scorpion) Full Length Research Paper. DNA Seq. 2007;18(3):242–246.
14. Annand RR, Kontoyianni M, Penzotti JE, Dudler T, Lybrand TP, Gelb MH. Active site of bee venom phospholipase A2: the role of histidine-34, aspartate-64 and tyrosine-87. Biochemistry. 1996;35(14):4591–4601.
15. Jabeen T, Singh N, Singh RK, et al. Crystal structure of a novel phospholipase A2 from Najanajasagittifera with a strong anticoagulant activity. Toxicon. 2005;46(8):865–875.
16. Jasti J, Paramasivam M, Srinivasan A, Singh TP. Structure of an acidic phospholipase A2 from Indian saw-scaled viper (Echiscarinatus) at 2.6 A resolution reveals a novel intermolecular interaction. Acta Crystallogr D Biol Crystallogr. 2004;60(Pt 1):66–72.
17. Pan YH, Yu BZ, Singer AG, et al. Crystal structure of human group X secreted phospholipase A2. Electrostatically neutral interfacial surface targets zwitterionic membranes. J Biol Chem. 2002;277(32):29086–29093.
18. Six DA, Barbayianni E, Loukas Vet al,. Structure− activity relationship of 2-oxoamide inhibition of group IVA cytosolic phospholipase A2 and group V secreted phospholipase A2. J Med Chem. 2007;50(17):4222–4235.
19. Hariprasad G, Kaur P, Srinivasan A, Singh TP, Kumar M. Structural analysis of secretory phospholipase A 2 from Clonorchis sinensis: therapeutic implications for hepatic fibrosis. J Mol Model. 2012;18(7):3139–3145.
20. Hariprasad G, Kota D, Singh SB, Srinivasan A, Adhikary S. Delineation of the structural elements of oriental liver fluke PLA 2 isoforms for potent drug designing. Indian J Clin Biochem. 2014;29(4):430–441.
21. Fremont DH, Anderson DH, Wilson IA, Dennis EA, Xuong N-H. Crystal structure of phospholipase A2 from Indian cobra reveals a trimeric association. Proc Natl Acad Sci. 90(1):342–346.
22. Gu L, Wang Z, Song S, Shu Y, Lin Z. Crystal structures of an acidic phospholipase A2 from the venom of Najakaouthia. Toxicon. 2002;40(7):917–922.
23. Bahnson BJ. Structure, function and interfacial allosterism in phospholipase A2: insight from the anion-assisted dimer. Arch BiochemBiophys. 2005;433(1):96–106.
24. Zhou L, Fang C, Wei P, Liu S, Liu Y, Lai L. Chemically induced dimerization of human nonpancreatic secretory phospholipase A2 by bis-indole derivatives. J Med Chem. 2008;51(12):3360–3366.
25. Schaefers HJ, Haselmann J, Goppelt-Struebe M. Regulation of prostaglandin synthesis in Madin Darby canine kidney cells: role of prostaglandin G/H synthase and secreted phospholipase A2. Biochimica Et Biophysica Acta (BBA) Lipids Lipid Metabol. 1996;1300(3):197–202.
26. Scott GA, Jacobs SE, Pentland AP. sPLA2-X stimulates cutaneous melanocyte dendricity and pigmentation through a lysophosphatidylcholine-dependent mechanism. J Invest Dermatol. 2006;126(4):855–861.
27. Benveniste J, Chignard M, Le Couedic J, Vargaftig B. Biosynthesis of platelet-activating factor (PAF-acether) II. Involvement of phospholipase A2 in the formation of PAF-acether and lyso-PAF-acether from rabbit platelets. Thromb Res. 1982;25(5):375–385.
28. Prescott SM, Fitzpatrick F. Cyclooxygenase-2 and carcinogenesis. Biochim Biophys Acta. 2000;1470(2):M69–78.
29. Hanasaki K, Arita HJP. mediators ol. Phospholipase A2 receptor: a regulator of biological functions of secretory phospholipase A2. Prostaglandins Other Lipid Mediators. 2002;68:71–82.
30. Morioka Y, Saiga A, Yokota Y, et al. Mouse group X secretory phospholipase A2 induces a potent release of arachidonic acid from spleen cells and acts as a ligand for the phospholipase A2 receptor. Arch Biochem Biophys. 2000;381(1):31–42.
31. Hanasaki K. Mammalian phospholipase A2: phospholipase A2 receptor. J Biol Pharm Bull. 2004;27(8):1165–1167.
32. Loffredo S, Borriello F, Iannone R, et al. Group V secreted phospholipase A2 induces the release of proangiogenic and antiangiogenic factors by human neutrophils. Front Immunol. 2017;8:443.
33. Kishino J, Kawamoto K, Ishizaki J, Verheij HM, Ohara O, Arita HJ. Pancreatic-type phospholipase A2 activates prostaglandin E2 production in rat mesangial cells by receptor binding reaction. J Biochem. 1995;117(2):420–424.
34. Khan MI, Gupta AK, Kumar DR, Kumar M, Ethayathulla AS, Hariprasad G. Molecular modeling of Gly80 and Ser80 variants of human group IID phospholipase A2 and their receptor complexes: potential basis for weight loss in chronic obstructive pulmonary disease. J Mol Model. 2016;22(9):232.
35. Friedemann M, Nacke B, Hagelgans A, et al. Diverse effects of phospholipase A2 receptor expression on LNCaP and PC-3 prostate cancer cell growth in vitro and in vivo. Oncotarget. 2018;9(89):35983.
36. Sukocheva O, Menschikowski M, Hagelgans A, et al. Current insights into functions of phospholipase A2 receptor in normal and cancer cells: more questions than answers. Paper presented at: Seminars in cancer biology 2019.
37. Valentin E, Ghomashchi F, Gelb MH, Lazdunski M, Lambeau G. Novel human secreted phospholipase A(2) with homology to the group III bee venom enzyme. J Biol Chem. 2000;275(11):7492–7496.
38. Masuda S, Yamamoto K, Hirabayashi T, et al. Human group III secreted phospholipase A2 promotes neuronal outgrowth and survival. Biochem J. 2008;409(2):429–438.
39. Tjoelker LW, Wilder C, Eberhardt C, et al. Anti-inflammatory properties of a platelet-activating factor acetylhydrolase. Nature. 1995;374(6522):549–553.
40. Karasawa K. Clinical aspects of plasma platelet-activating factor-acetylhydrolase. Biochimica Et Biophysica Acta (BBA)-Molecular and Cell Biology of Lipids. 2006;1761(11):1359–1372.
41. Macphee CH, Nelson JJ, Zalewski A. Lipoprotein-associated phospholipase A2 as a target of therapy. Curr Opin Lipidol. 2005;16(4):442–446.
42. Dennis EA, Cao J, Hsu Y-H, Magrioti V, Phospholipase KG. A2 enzymes: physical structure, biological function, disease implication, chemical inhibition, and therapeutic intervention. Chem Rev. 2011;111(10):6130–6185.
43. Stremler K, Stafforini D, Prescott S, McIntyre T. Human plasma platelet-activating factor acetylhydrolase. Oxidatively fragmented phospholipids as substrates. J Biol Chem. 1991;266(17):11095–11103.
44. Murakami M, Taketomi Y, Miki Y, Sato H, Hirabayashi T, Yamamoto K. Recent progress in phospholipase A2 research: from cells to animals to humans. Prog Lipid Res. 2011;50(2):152–192.
45. Santoso A, Maulana R, Alzahra F, Maghfirah I, Putrinarita AD, Heriansyah T. Associations between four types of single-nucleotide polymorphisms in PLA2G7 gene and clinical atherosclerosis: a meta-analysis. Am J Cardiovasc Dis. 2017;7(6):122.
46. Hou L, Chen S, Yu H, et al. Associations of PLA2G7 gene polymorphisms with plasma lipoprotein-associated phospholipase A2 activity and coronary heart disease in a Chinese Han population: the Beijing atherosclerosis study. Hum Genet. 2009;125(1):11–20.
47. Sabatine MS, Morrow DA, O’Donoghue M, et al. Prognostic utility of lipoprotein-associated phospholipase A2 for cardiovascular outcomes in patients with stable coronary artery disease. Arterioscler Thromb Vasc Biol. 2007;27(11):2463–2469.
48. Koenig W, Twardella D, Brenner H, Rothenbacher D. Lipoprotein-associated phospholipase A2 predicts future cardiovascular events in patients with coronary heart disease independently of traditional risk factors, markers of inflammation, renal function, and hemodynamic stress. Arterioscler Thromb Vasc Biol. 2006;26(7):1586–1593.
49. Lavi S, Herrmann J, Lavi R, McConnell JP, Lerman LO, Lerman A. Role of lipoprotein-associated phospholipase A 2 in atherosclerosis. Curr Atheroscler Rep. 2008;10(3):230.
50. Lerman A, McConnell JP. Lipoprotein-associated phospholipase A2: a risk marker or a risk factor? Am J Cardiol. 2008;101(12):S11–S22.
51. Kim M, Kim M, Yoo HJ, Jang HY, Lee S-H, Lee JH. Effects of overweight and the PLA2G7 V279F polymorphism on the association of age with systolic blood pressure. PLoS One. 2017;12(3):e0173611.
52. LI JH, Wang LM, Li YC, Zhang M, Wang LH. Prevalence of major cardiovascular risk factors and cardiovascular disease in women in China: surveillance efforts. Biomed Environ Sci. 2016;29(3):205–211.
53. Jang Y, Kim OY, Koh SJ, et al. The Val279Phe variant of the lipoprotein-associated phospholipase A2 gene is associated with catalytic activities and cardiovascular disease in Korean men. J Clin Endocrinol Metab. 2006;91(9):3521–3527.
54. Xu L, Zhou J, Huang S, et al. An association study between genetic polymorphisms related to lipoprotein-associated phospholipase A2 and coronary heart disease. Exp Ther Med. 2013;5(3):742–750.
55. Ninio E, Tregouet D, Carrier J-L, et al. Platelet-activating factor-acetylhydrolase and PAF-receptor gene haplotypes in relation to future cardiovascular event in patients with coronary artery disease. Hum Mol Genet. 2004;13(13):1341–1351.
56. Abuzeid A, Hawe E, Humphries S, Talmud PJ, Group HS. Association between the Ala379Val variant of the lipoprotein associated phospholipase A2 and risk of myocardial infarction in the north and south of Europe. Atherosclerosis. 2003;168(2):283–288.
57. Jang Y, Waterworth D, Lee J-E, et al. Carriage of the V279F null allele within the gene encoding Lp-PLA 2 is protective from coronary artery disease in South Korean males. PLoS One. 2011;6(4):e18208.
58. Packard CJ, O’Reilly DS, Caslake MJ, et al. Lipoprotein-associated phospholipase A2 as an independent predictor of coronary heart disease. N Engl J Med. 2000;343(16):1148–1155.
59. Caslake MJ, Packard CJ, Suckling KE, Holmes SD, Chamberlain P, Macphee CH. Lipoprotein-associated phospholipase A2, platelet-activating factor acetylhydrolase: a potential new risk factor for coronary artery disease. Atherosclerosis. 2000;150(2):413–419.
60. Hiramoto M, Yoshida H, Imaizumi T, Yoshimizu N, Satoh K. A mutation in plasma platelet-activating factor acetylhydrolase (Val279→ Phe) is a genetic risk factor for stroke. Stroke. 1997;28(12):2417–2420.
61. Yamamoto I, Fujitsu J, Nohnen S, et al. Association of plasma PAF acetylhydrolase gene polymorphism with IMT of carotid arteries in Japanese type 2 diabetic patients. Diabetes Res Clin Pract. 2003;59(3):219–224.
62. Yamada Y, Ichihara S, Fujimura T, Yokota M. Identification of the G994→ T missense mutation in exon 9 of the plasma platelet-activating factor acetylhydrolase gene as an independent risk factor for coronary artery disease in Japanese men. Metabolism. 1998;47(2):177–181.
63. Sutton BS, Crosslin DR, Shah SH, et al. Comprehensive genetic analysis of the platelet activating factor acetylhydrolase (PLA2G7) gene and cardiovascular disease in case–control and family datasets. Hum Mol Genet. 2008;17(9):1318–1328.
64. Hong M, Zhang M, Lu X. Nonsynonymous polymorphisms in PLA2G7 gene are associated with the risk of coronary heart disease in a southern Chinese population. Mammalian Genome. 2015;26(3–4):191–199.
65. Zhang M, Zhang C, Yang C, Zhao P, Li Y. The association between the D166E polymorphism of the lipoprotein associated phospholipase A2 and risk of myocardial infarction. Eur Rev Med Pharmacol Sci. 2019;23(9):3960–3966.
66. Sergouniotis PI, Davidson AE, Mackay DS, et al. Biallelic mutations in PLA2G5, encoding group V phospholipase A2, cause benign fleck retina. Am J Human Genetics. 2011;89(6):782–791.
67. Six DA, Dennis EA. The expanding superfamily of phospholipase A2 enzymes: classification and characterization. Biochimica Et Biophysica Acta (BBA)-Molecular and Cell Biology of Lipids. 2000;1488(1–2):1–19.
68. Bin NJ, Heng HM, Poh R, Noor SM, Phospholipase SV. A2 group v in benign familial fleck retina in a set of triplets. Retina. 2015;35(6):1266–1272.
69. Brunk UT, Terman A. Lipofuscin: mechanisms of age-related accumulation and influence on cell function. Free Radic Biol Med. 2002;33(5):611–619.
70. Ach T, Huisingh C, McGwin G, et al. Quantitative autofluorescence and cell density maps of the human retinal pigment epithelium. Invest Ophthalmol Vis Sci. 2014;55(8):4832–4841.
71. Ach T, Tolstik E, Messinger JD, Zarubina AV, Heintzmann R, Curcio CA. Lipofuscin redistribution and loss accompanied by cytoskeletal stress in retinal pigment epithelium of eyes with age-related macular degeneration. Invest Ophthalmol Vis Sci. 2015;56(5):3242–3252.
72. Feeney-Burns L, Hilderbrand E, Eldridge S. Aging human RPE: morphometric analysis of macular, equatorial, and peripheral cells. Invest Ophthalmol Vis Sci. 1984;25(2):195–200.
73. Aish SS, Dajani B. Benign familial fleck retina. Br J Ophthalmol. 1980;64(9):652–659.
74. Chen J, Engle SJ, Seilhamer JJ, Tischfield JA. Cloning and recombinant expression of a novel human low molecular weight Ca (2+)-dependent phospholipase A2. J Biol Chem. 1994;269(4):2365–2368.
75. Pniewska E, Pawliczak R. The involvement of phospholipases A2 in asthma and chronic obstructive pulmonary disease. Mediators Inflamm. 2013;2013.
76. Barnes P, Celli B. Systemic manifestations and comorbidities of COPD. Eur Respir J. 2009;33(5):1165–1185.
77. Kotler DP. Cachexia. Ann Intern Med. 2000;133(8):622–634.
78. Igarashi A, Shibata Y, Yamauchi K, et al. Gly80Ser polymorphism of phospholipase A2-IID is associated with cytokine inducibility in A549 cells. Respiration. 2009;78(3):312–321.
79. Oudijk ED, Lammers JJ, Koenderman L. Systemic inflammation in chronic obstructive pulmonary disease. Eur Respir J. 2003;22(46 suppl):5s–13s.
80. Andreassen H, Vestbo J. Chronic obstructive pulmonary disease as a systemic disease: an epidemiological perspective. Eur Respir J Suppl. 2003;46:2s–4s.
81. Wouters EF, Creutzberg EC, Schols AM. Systemic effects in COPD. Chest. 2002;121(5):127S–130S.
82. Agusti A, Noguera A, Sauleda J, Sala E, Pons J, Busquets X. Systemic effects of chronic obstructive pulmonary disease. Eur Respir J. 2003;21(2):347–360.
83. Takabatake N, Nakamura H, MINAMIHABA O, et al. A novel pathophysiologic phenomenon in cachexic patients with chronic obstructive pulmonary disease: the relationship between the circadian rhythm of circulating leptin and the very low-frequency component of heart rate variability. Am J Respir Crit Care Med. 2001;163(6):1314–1319.
84. Schols AM, Slangen J, Volovics L, Wouters EF. Weight loss is a reversible factor in the prognosis of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 1998;157(6):1791–1797.
85. Landbo C, Prescott E, Lange P, Vestbo J, Almdal TP. Prognostic value of nutritional status in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 1999;160(6):1856–1861.
86. Gan WQ, Man S, Senthilselvan A, Sin D. Association between chronic obstructive pulmonary disease and systemic inflammation: a systematic review and a meta-analysis. Thorax. 2004;59(7):574–580.
87. Takabatake N, Nakamura H, Abe S, et al. The relationship between chronic hypoxemia and activation of the tumor necrosis factor-α system in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2000;161(4):1179–1184.
88. Ishizaki J, Suzuki N, Higashino K-I, et al. Cloning and characterization of novel mouse and human secretory phospholipase A2s. J Biol Chem. 1999;274(35):24973–24979.
89. Mikami M, Yasuda T, Terao A, et al. Localization of a gene for benign adult familial myoclonic epilepsy to chromosome 8q23. 3-q24. 1. Am J Human Genetics. 1999;65(3):745–751.
90. Gao L, Li L, Ye J, et al. Identification of a novel mutation in PLA2G6 gene in a Chinese pedigree with familial cortical myoclonic tremor with epilepsy. Seizure. 2016;41:81–85.
91. Toledano BJ, Bastien Y, Noya F, Mazer B. Characterization of B lymphocytes rescued from apoptosis by platelet-activating factor. Cell Immunol. 1999;191(1):60–68.
92. Hsieh K-H, Ng C-K. Increased plasma platelet-activating factor in children with acute asthmatic attacks and decreased in vivo and in vitro production of platelet-activating factor after immunotherapy. J Allergy Clin Immunol. 1993;91(2):650–657.
93. Kruse S, Mao X-Q, Heinzmann A, et al. The Ile198Thr and Ala379Val variants of plasmatic PAF-acetylhydrolase impair catalytical activities and are associated with atopy and asthma. Am J Human Genetics. 2000;66(5):1522–1530.
94. Ma Y. Associations of platelet-activating factor acetylhydrolase gene polymorphisms with risk of ischemic stroke. Biomed Rep. 2016;4(2):246–250.
95. Righino B, Minucci A, Pirolli D, et al. In silico investigation of the molecular effects caused by R123H variant in secretory phospholipase A2-IIA associated with ARDS. J Mol Graph Model. 2018;81:68–76.
96. Ni J, Gu H, Hu W, Zhou F, Zhu X, Wang K. Association of Lp‐PLA2 G994T gene polymorphism with risk of ischemic stroke in Chinese population. J Biochem Mol Toxicol. 2017;31(12):e21999.
97. Widodo W, Ramadhani AN, Nofitasari A, et al. The V279F polymorphism might change protein character and immunogenicity in Lp-PLA2 protein. Egypt J Med Human Genetics. 2018;19(2):107–112.
98. Paik JK, Chae JS, Jang Y, et al. Effects of V279F in the Lp-PLA2 gene on markers of oxidative stress and inflammation in Koreans. Clinica Chimica Acta. 2010;411(7–8):486–493.
99. Yamada Y, Yokota M. Loss of activity of plasma platelet-activating factor acetylhydrolase due to a novel Gln281→ Arg mutation. Biochem Biophys Res Commun. 1997;236(3):772–775.
100. Samanta U, Bahnson BJ. Crystal structure of human plasma platelet-activating factor acetylhydrolase: structural implication to lipoprotein binding and catalysis. J Biol Chem. 2008;283(46):31617–31624.
101. Unno N, Nakamura T, Kaneko H, et al. Plasma platelet-activating factor acetylhydrolase deficiency is associated with atherosclerotic occlusive disease in Japan. J Vasc Surg. 2000;32(2):263–267.
102. Li L, Qi L, Lv N, et al. Association between lipoprotein‐associated phospholipase A2 gene polymorphism and coronary artery disease in the Chinese Han population. Ann Hum Genet. 2011;75(5):605–611.
103. Ichihara S, Yamada Y, Yokota M. Association of a G994→ T missense mutation in the plasma platelet-activating factor acetylhydrolase gene with genetic susceptibility to nonfamilial dilated cardiomyopathy in Japanese. Circulation. 1998;98(18):1881–1885.
104. Xu H, Iijima K, Shirakawa T, et al. Platelet-activating factor acetylhydrolase gene mutation in Japanese children with Escherichia coli O157–associated hemolytic uremic syndrome. Am J Kidney Dis. 2000;36(1):42–46.
105. Minami T, Suzuki H, Takeuchi T, Uemura S, Sugatani J, Yoshikawa N. A polymorphism in plasma platelet-activating factor acetylhydrolase is involved in resistance to immunoglobulin treatment in Kawasaki disease. J Pediatr. 2005;147(1):78–83.
106. Stafforini DM, Numao T, Tsodikov A, et al. Deficiency of platelet-activating factor acetylhydrolase is a severity factor for asthma. J Clin Invest. 1999;103(7):989–997.
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.