")
Back to Journals » Journal of Inflammation Research » Volume 13
Human Lung Macrophages Challenged to Oxidants ex vivo: Lysosomal Membrane Sensitization is Associated with Inflammation and Chronic Airflow Limitation
Authors Persson HL , Sioutas A, Jacobson P, Vainikka LK
Received 4 September 2020
Accepted for publication 20 October 2020
Published 16 November 2020 Volume 2020:13 Pages 925—932
DOI https://doi.org/10.2147/JIR.S280419
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Ning Quan
Hans Lennart Persson,1,2 Apostolos Sioutas,1,2 Petra Jacobson,1,2 Linda K Vainikka3,4
1Department of Respiratory Medicine in Linköping, Linköping University, Linköping, Sweden; 2Department of Health, Medicine and Caring Sciences, Linköping University, Linköping, Sweden; 3Department of Experimental Pathology, Linköping University, Linköping, Sweden; 4Department of Biomedical and Clinical Sciences, Linköping University, Linköping, Sweden
Correspondence: Hans Lennart Persson
Department of Respiratory Medicine in Linköping, Linköping University, Linköping SE-581 85, Sweden
Tel +46 010 1033621
Email [email protected]
Background: The lung macrophage (LM) is involved in most inflammatory processes of the human lung by clearance of dying cells and by wound repair. Upon cellular stress by oxidant challenge in vivo lysosomes may rupture in LMs and leakage of cellular content and cell debris may trigger airway inflammation and fibrosis, which may lead to chronic airflow limitation (CAL).
Objective: The aim of this study was to determine whether lysosomal membrane permeabilization (LMP) in LMs challenged to oxidants ex vivo is associated with airway inflammation and CAL, the latter assessed as the reduced forced expiratory volume in one second (FEV1) expressed as % of predicted.
Materials and Methods: Twenty-eight subjects were investigated; 13 lung-healthy subjects and 15 subjects with a variety of inflammatory disorders, demonstrating CAL on dynamic spirometry (defined as an FEV1/FVC ratio < 0.70). LMs were harvested by broncho-alveolar lavage (BAL) and challenged ex vivo by oxidants. LMP in oxidant-exposed LMs was assessed as the emitted acridine orange (AO) green fluorescence from oxidant-exposed LMs (using macrophage-like murine J774 cells as positive controls). Inflammatory cells in BAL were counted and lung volumes were recorded.
Results: Oxidant-induced LMP in LMs was significantly greater among subjects with CAL and particularly among those with ongoing inflammation. Previous tobacco history did not influence LMP. Among subjects with CAL, oxidant-induced LMP correlated negatively with FEV1% of predicted.
Conclusion: Lysosomes of LMs harvested from patients with CAL demonstrate an increased sensitivity to oxidants, which may trigger mechanisms behind CAL, eg, chronic airway inflammation and fibrotic re-modelling. The study suggests a mechanistic role for LMP in LMs on airway inflammation, suggesting an anti-inflammatory effect by drugs that prevent increased LMP.
Keywords: acridine orange, lung macrophages, BAL, COPD, LMP, pulmonary fibrosis
Introduction
Lately, lysosomal pathology behind the inflammation and fibrosis observed in the airways of humans with chronic obstructive pulmonary disease (COPD) has attracted increasing attention.1,2 Hallmarks of COPD are typical symptoms from the respiratory tract and a spirometry showing chronic airflow limitation (CAL), defined as an FEV1/FVC ratio <0.70 after bronchodilatation. Recently, a proteomic analysis of proteins from BAL cells retrieved from smokers with or without COPD demonstrated decreased levels of proteins of the lysosomal pathway in female smoker vs never-smokers, which were further decreased in female smoking COPD subjects.3,4 This observation may suggest that a dysregulation of the lysosomal pathway may represent early signs of disease development in individuals particularly susceptible to tobacco-smoke exposure.3,4 Another study of fibroblasts, harvested from the airways of subjects with COPD and healthy control subjects, showed that the endoplasmic reticulum, Golgi, and lysosomes were permanently disorganized in fibroblasts from COPD patients, and, moreover, in culture, these fibroblasts demonstrated increased vulnerability to cellular stress.5
Oxidants probably play an important role for COPD development due to damage on single cells and tissues.6 Such damage is possibly potentiated by a disturbed metabolism of iron.7 Reparative mechanisms such as autophagy, executed by lysosomes, seems to be involved as well.8 Therefore, we have for some time studied how lung macrophages (LMs), retrieved from the airways of patients with inflammatory lung diseases, respond to oxidative stress ex vivo, focusing upon the leakage of lysosomal content, ie, lysosomal membrane permeabilization (LMP), from disrupted lysosomes.9–12 We have previously demonstrated that lysosomes in LMs harvested from subjects with inflammatory and/or fibrotic lung disease are much more vulnerable to an oxidant challenge ex vivo, thus, experimentally mimicking the exposure to oxidants occurring in vivo.10–12 We have also shown that autophagy of iron-rich material (mainly oxidatively damaged ferritin and mitochondria) plays a major role behind the observed susceptibility of lysosomes to oxidative stress.10–13 Indeed, in cultured human respiratory epithelial cells a vicious circle of oxidative cell damage, enhanced autophagy, increased LMP and further cellular stress and autophagy triggered epithelial–mesenchymal transition (EMT).13 EMT and autophagy are mechanisms supposed to be implicated in the fibrosis development in the airways of COPD patients,14–16 resulting in CAL by fibrotic re-modelling.
In the present study, the aims were to investigate whether LMP during oxidant challenge ex vivo, in LMs harvested from subjects with inflammatory lung disease and CAL, exhibited abnormalities, and, if so, whether an abnormal response of LMP to oxidative stress was associated with signs of an ongoing airway inflammation and measure of CAL, in the present study assessed by the forced expiratory volume in one second (FEV1) and expressed as % of predicted post-dilatation. We hypothesized that an inflammatory milieu in vivo, with an ongoing mild exposure to oxidants, would make the lysosomes more sensitive to a greater oxidant challenge ex vivo, possibly being a reflection of the burst of oxidants that may occur in vivo during inflammatory exacerbations.
Materials and Methods
Ethical Considerations
The study protocol was approved by the local Ethical Committee of Linköping, Sweden (Dnr: M32-07) according to the guidelines of the Declaration of Helsinki.
Study Population
Following informed and written consent broncho-alveolar lavage (BAL) was performed during a fibreoptic bronchoscopy with sterile 0.9% (w/v) saline solution. Bronchoscopy was performed for medical reasons only, but the subjects included gave their consent to include BAL fluid (BALF) for study purpose. The BALF retrieved (without blood stain) were prepared as previously described.11,12 Healthy lungs were confirmed among all 13 control subjects (4 ex-smokers and 9 never-smokers) by a negative history of respiratory discomfort and a negative lung examination and were further confirmed by negative findings on bronchoscopy (including cultures), chest X-ray, and lung function tests. The 15 patients included with CAL (7 ex-smokers and 8 never-smokers) were 5 subjects with sarcoidosis, 4 subjects with COPD, 3 subjects with asthma and 3 subjects with fibrosis due to either cryptogenic organizing pneumonia (COP), rheumatoid arthritis (RA) or idiopathic pulmonary fibrosis (IPF). All these subjects fulfilled the criteria for CAL on dynamic spirometry post-dilatation, that is an FEV1/FVC ratio <0.7, and demonstrated stable disease without clinical signs of ongoing exacerbation or infection. In addition, cultures on BALF were negative. A cell count of each BALF was also performed. Lung volumes post-dilatation (>15 min after inhalation of 0.6 mg salbutamol) was measured with a spirometer using Hedenström as a reference. Smoking habits were recorded as exposure to tobacco smoke oxidatively stress cells and lysosomes. Use of medications and supplements, including those with lysosomotropic properties,17 was also recorded. Particularly, attention was paid to ongoing use of proton-pump inhibitors (PPIs), since these drugs may theoretically interfere on LMP in this experimental model.
Assessment of LMP ex vivo in Human LMs
LMs were prepared for experiments as previously described in more detail.11,12 LMs were seeded at 75–95% confluence in a black 96-well clear-bottom plate. After 4 h, cells were rinsed with phosphate-buffered saline (PBS), and the culture medium (DMEM containing 10% FBS, 100 IU/mL penicillin, 100 µg/mL streptomycin and 0.25 μg/mL amphotericin B (all were from GIBCO, Paisley, UK)) was changed. The next day, 2 µg/mL acridine orange (AO; Gurr, Poole, UK) was added to the cells (diluted in medium) and cells were incubated at 37°C for 15 min, followed by 3 x washing with DMEM. Immediately after, glucose oxidase (GO; Sigma-Aldrich Inc.) was added at a concentration of 18 µg/mL. Control cells received only medium. To assess the dynamics of LMP, cells were assessed using real-time LMP. Real-time LMP was analyzed by measuring the green fluorescence in an incubated SPARK 10M plate reader (Tecan). Green (Ex 485 nm/Em 535 nm) fluorescence was measured every two minutes for 2 hours at 37°C. In parallel experiments, murine macrophage-like J774 cells were exposed and analyzed in the same way and results were used as a positive control for further standardization. The variation of AO-green fluorescence emitted from the J774 cells between separate experiments was <10%. By real-time LMP, a peak of AO-green fluorescence was determined at 60 min of GO-exposure (Figure 1). The peak of AO-green fluorescence derived from oxidant-exposed LMs was then standardized to the corresponding peak of AO-green fluorescence derived from oxidant-exposed J774 cells. For this purpose, following assessment of the peak of green fluorescence for both cell types, an index was constructed by dividing the AO-green peak value for oxidant-exposed LMs with the corresponding AO-green peak value for J774 cells, hereafter termed “the AO-green peak fluorescence Index”. For further analysis, LMP was analyzed at 60 min of GO exposure.
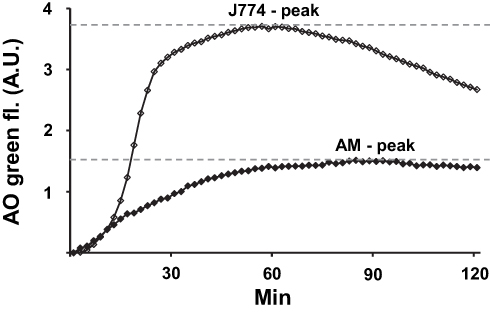 |
Figure 1 In parallel experiments murine macrophage-like J774 cells and LMs, harvested from a patient with CAL or not, were subjected to the same oxidant challenge ex vivo. Increase of cytosolic and nuclear AO-induced green fluorescence, reflecting increased LMP (leakage of AO from lysosomes to the cytosol), was assessed every 2 min and expressed as AU. AO-induced green peak fluorescence of J774 cells and LMs are indicated in the figure image. An index was calculated by dividing the green peak fluorescence value of the LMs with the green peak fluorescence value of the J774 cells at 60 min of GO-exposure. The index value was used to quantify LMP in LMs exposed ex vivo to oxidants. The positive control, oxidant-exposed J774 cells, varied < 10% over time. For further details, see Material and Method section. |
Statistical Analysis
The results are reported as the means (± 1 S.D.) for continuous variables. For comparison between independent groups, the Student’s T-test was used for normally distributed continuous variables and the Mann Whitney U-test for non-normally distributed continuous variables. Differences in categorical data were analyzed by the Chi-square test and Fisher’s exact test. For correlations, Pearson’s correlation coefficient was calculated for normally distributed continuous variables and Spearman correlation coefficient for non-normally distributed variables. As exposure to tobacco smoke, along with an inflammation in the airway mucosa, are the most important contributors of oxidative cell damage, subjects presenting with CAL were statistically analyzed and compared regarding smoking habits and evidence of ongoing airway inflammation. P-values <0.05 were considered statistically significant.
Results
Main Characteristics of Subjects with CAL and Healthy Controls
Fifteen subjects with CAL associated with various chronic inflammatory lung disorders (sarcoidosis, COPD, asthma or lung fibrosis due to COP or RA) and IPF, and 13 lung healthy subjects (hereafter referred to as “control group/subjects”) completed the study. The characteristics of the study population are presented in Table 1.
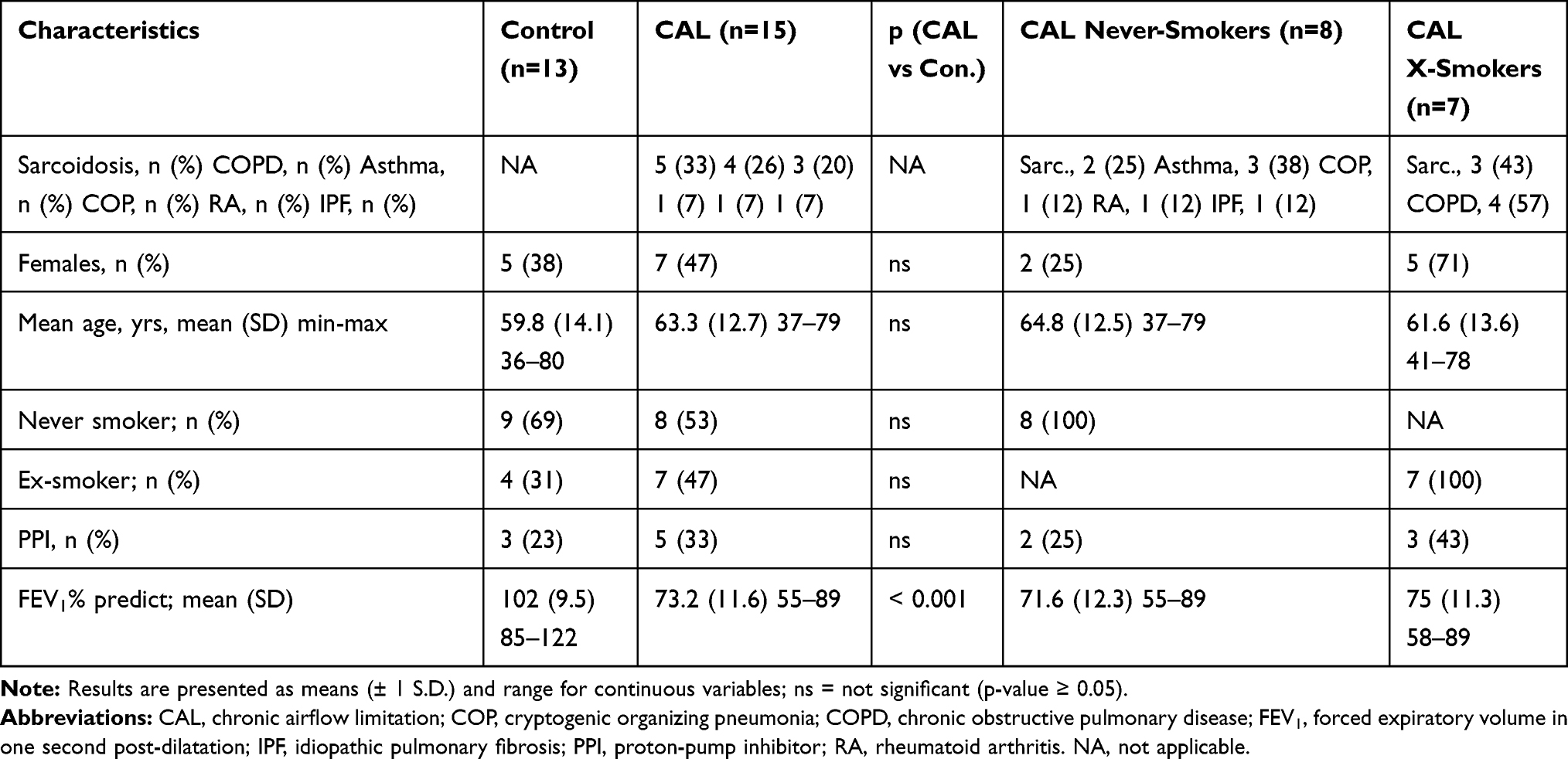 |
Table 1 Characteristics of the Study Sample Controls versus Subjects with CAL |
Gender, age and smoking history did not differ significantly between control subjects and subjects with CAL (Table 1). Seven of 15 subjects with lung disease had a history of previous tobacco use, but all 7 subjects had stopped smoking ≥12 yrs prior to bronchoscopy (Table 1). In the control group, 4 of 13 subjects had a previous history of tobacco smoking, but all had stopped smoking ≥10 yrs prior to bronchoscopy. Particular attention was paid to ongoing use of PPIs, since these drugs may theoretically interfere on LMP in this experimental model due to their lysosomotropic properties.17 The frequency of medication with PPIs did not differ significantly between subjects with CAL and the control group (Table 1). None of the subjects were on any other drug interfering with LMP. Subjects with COPD or asthma medicated with inhaled steroids, but none of the subjects included were on medication with potent immunosuppressants. Compared to the control group, FEV1% of predicted post-dilatation was significantly lower in the CAL group (Table 1). Compared to never-smokers with CAL, significantly more patients with CAL were females in the X-smoker group (Table 1). Otherwise, no significant difference was observed.
LMs from CAL Subjects Exhibit Increased LMP Upon Oxidative Stress
Ex vivo oxidant-induced LMP in LMs, expressed as the AO-green peak fluorescence index, was significantly greater among subjects with CAL compared to control subjects (Figure 2). A history of previous tobacco use did not influence significantly on LMP (Figure 2), nor did gender or PPI medication (results not shown).
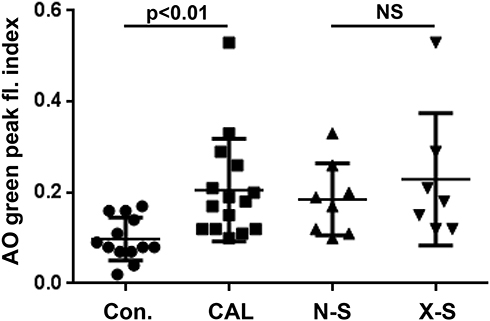 |
Figure 2 AO green peak fluorescence index, as measure of increased LMP, in oxidatively stressed LMs harvested from control subjects (Con; n = 13) and subjects with CAL (CAL; n = 15). In the group with CAL subjects X-smokers (X-S) did not differ significantly, when compared with never-smokers (N-S). Means ± 1 S.D. are indicated. NS = no significance. |
Increase of LMP is Associated with Ongoing Inflammation
Using BALF cell count, subjects with CAL were divided into two groups; those who displayed evidence of ongoing inflammation and those who did not. BALF cell count of control subjects and subjects with CAL, the latter grouped by type of inflammation, is shown in Table 2. Inflammation was defined by a cellular pattern dominated by neutrophils (>3%), eosinophils (>1%) or lymphocytes (>15%).18 Significant differences between subjects with CAL and ongoing inflammation and subjects with CAL and no ongoing inflammation are presented in Table 3.
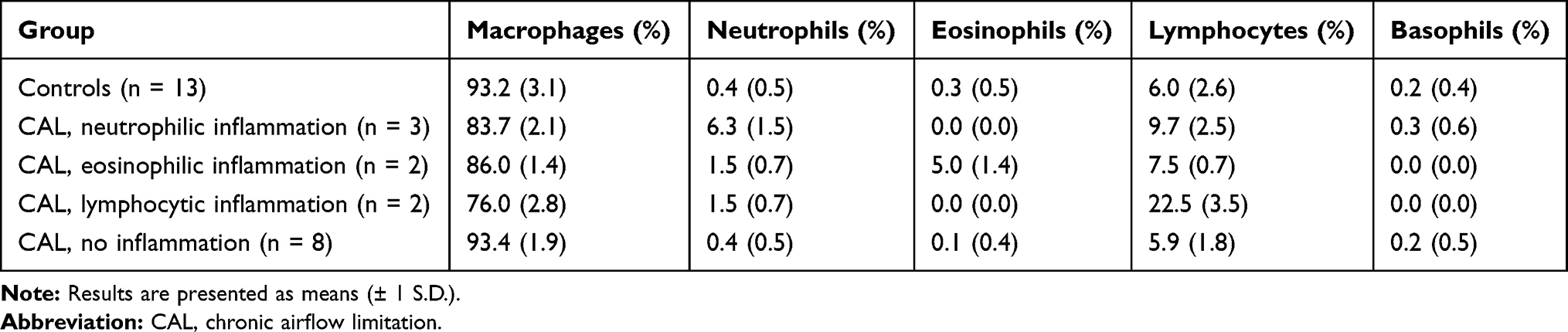 |
Table 2 BALF Cell Count of Controls and Subjects with CAL, Presented by Type of Inflammation (or Not) |
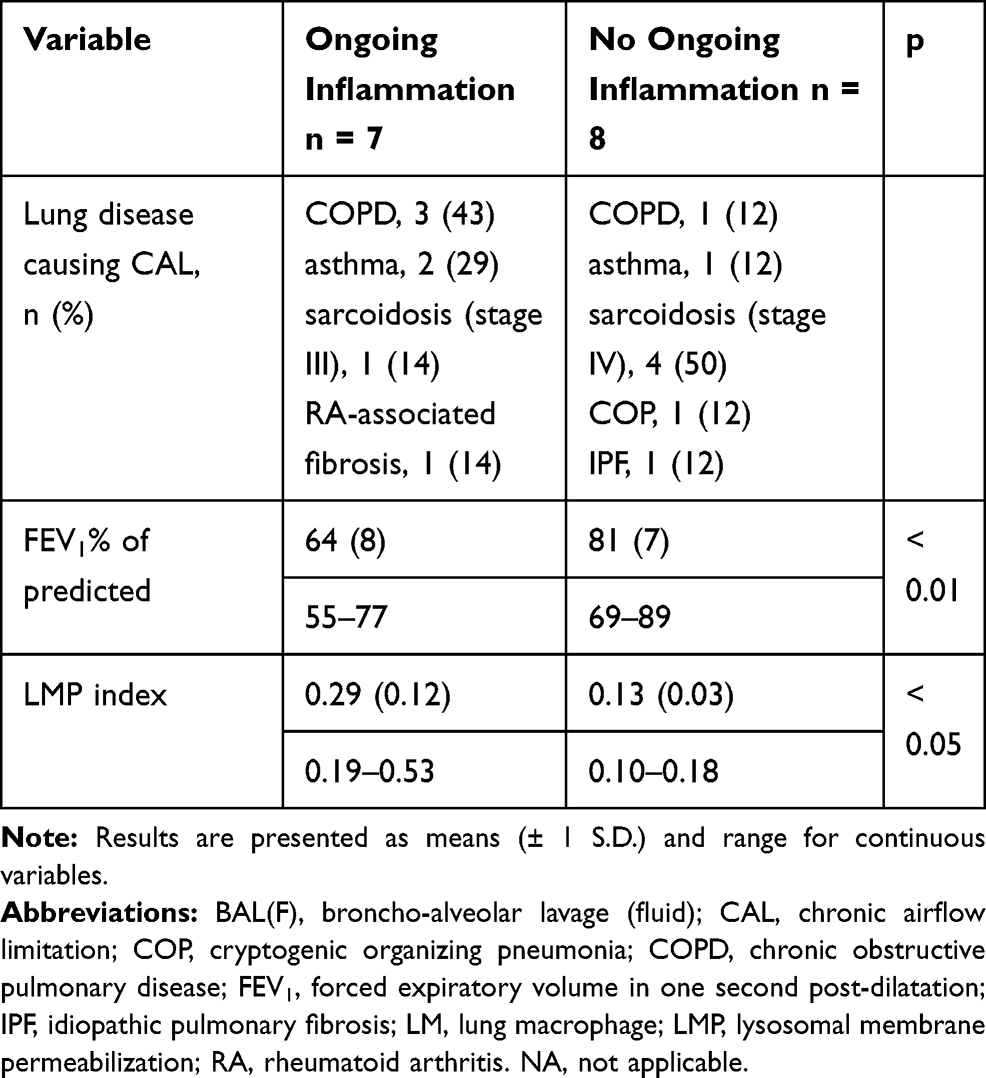 |
Table 3 Subjects with CAL Were Divided in Two Groups That Were Compared; Subjects Demonstrating Ongoing Inflammation (Judged by BALF Cell Count) and Subjects Without Signs of Inflammation. Infections Were Excluded in All Subjects by Negative History, Blood Analyses and Cultures on BALF |
Subjects with CAL showing ongoing inflammation on BALF cell count consisted of COPD subjects with neutrophilic inflammation, asthma subjects with eosinophilic inflammation, and subjects with stage III sarcoidosis or RA-associated lung fibrosis with lymphocytic inflammation (Table 3). Subjects with CAL displaying no inflammation were mainly subjects with stage IV sarcoidosis (Table 3).
Compared to CAL subjects with no inflammation present in BALF, CAL subjects with ongoing inflammation exhibited significantly lower FEV1% of predicted and significantly higher LMP index (Table 3). Likewise, compared to control subjects, CAL subjects with no inflammation present in BALF demonstrated significantly lower FEV1% of predicted (p < 0.001) and significantly higher LMP (p < 0.05).
Increase of LMP Correlates to Obstructive Lung Volume Impairment
Ex vivo oxidant-induced LMP in AMs, expressed as the AO-green peak fluorescence index, was significantly and negatively correlated to FEV1% of predicted (Figure 3). No other significant correlation was found.
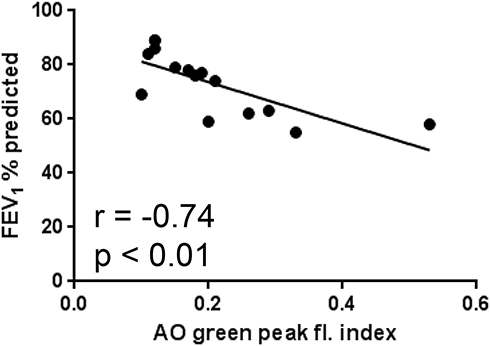 |
Figure 3 Correlation between LMP, expressed as the AO-green peak fluorescence index and CAL (n = 15), expressed as FEV1% of predicted. |
Discussion
Human LMs were in the focus of the present study. This cell type is the most abundant one in BAL from the healthy human lung and makes up 95% of all retrieved cells.19 LMs are supposed to play a pivotal role in chronic inflammatory processes in the airways, regardless of the origin and the characteristics of the inflammation.19,20 This idea is based on the role that LMs execute as professional phagocytes of cell debris, resulting from excessive regulated (apoptosis) and unregulated cell death, in conditions of chronic inflammation.19,20 If the clearance of apoptotic cells, a process called efferocytosis, and cell debris is hampered by some mechanism interfering with the phagocytic capacity of LMs, this will lead to an uncontrolled activation of the inflammatory system.19,20 Indeed, data from patients with neutrophilic or eosinophilic airway inflammation and non-inflammatory conditions, represented by subjects with COPD or asthma and IPF, respectively, suggests that disturbed clearance in lung diseases may not be specific for individual diagnoses, but rather is a general hallmark of chronic inflammation.19,20 Moreover, LMs are also key-player in tissue-repair, leading to fibrotic airway re-modelling and CAL.20–22 Consequently, in the present study LMs, derived from humans with a variety of inflammatory and/or fibrotic conditions presenting with CAL, were investigated regarding the impact of their lysosomes on CAL.
One obvious reason for defective phagocytic activity of LMs is oxidative stress in vivo. Such oxidant challenge may increase LMP, which in turn may lead to impaired functions of LMs due to the cellular stress executed by increased LMP, and, if pronounced, the increase of LMP may even kill the LMs.11,12,23,24 Unregulated death of LMs by necroptosis is associated with tobacco-smoke induced airway damage and COPD.24 This scenario may be prevented by azithromycin,25 a macrolide antibiotic used in the clinic to prevent COPD deterioration due to frequent exacerbations. In a series of studies of a variety of inflammatory and fibrotic lung diseases, we have previously presented observations suggesting that oxidant-induced LMP, mainly driven by a disturbed metabolism of iron and an accumulation of this metal inside lysosomes,26 may be an important up-stream event in the cascade of events that trigger inflammation and/or fibrosis,11,12,27,28 ultimately leading to CAL. We have previously shown that azithromycin may protect human LMs against oxidant-induced LMP and ensuing cell death ex vivo due to its lysosomotropic and iron-chelating properties.11 Thus, azithromycin, being a weak base, accumulates thousand-fold in the acidic lysosomes and prevents intra-lysosomal harmful oxidative reactions by binding iron in an unreactive state.11 These previous studies served to us as a logic back-ground for a study on the role that increased LMP in oxidant-challenged LMs may play for development of airway inflammation and CAL.
The present study is the first we know of that demonstrates a relationship between an increase of LMP in human LMs, oxidatively stressed ex vivo, and different types of airway inflammation (Figure 2 and Table 3). LMP was assessed as the increase of AO-related green fluorescence, being a much more sensitive marker of lysosomal rupture than the decline in AO-related red fluorescence.29 This observation suggests that oxidant-induced LMP in human LMs may be a cornerstone in the development of most types of inflammation. We have previously demonstrated that LMP and ensuing cell death both are increased in cultures of oxidant-challenged LMs, harvested from lung transplants and human fibrotic lungs.11,12 Importantly, the present study is the first to show a relationship between an increase of LMP and CAL (Figure 3). Thus, a mechanistic role for an increase of LMP in oxidatively stressed LMs and airway obstruction is suggested. CAL, here expressed as FEV1% of predicted, was more pronounced in the CAL group demonstrating ongoing inflammation (Table 3), suggesting that both inflamed mucosa and fibrotic airway re-modelling may have served as causes behind the observed airway obstruction.
The disease mechanisms behind inflammatory and fibrotic conditions of the lungs are known to involve a polarization of LMs to a continuum with subpopulations in between the phenotypes M1 and M2, expressing varying levels of M1 and M2 markers and activities.19–23 The action of M1 LMs is mainly proinflammatory and cytotoxic, while M2 LMs works in an anti-inflammatory way and execute the repair of the wounded airway epithelium.19–23 The M2 LMs produce ornithine, which promote cell proliferation and local production of collagen.19–23 It is thought that a prolonged or exaggerated response of M1 LMs leads to chronic airway inflammation, while hyper-responsive subpopulations of M2 LMs cause airway fibrosis.19–23 It has also been suggested that profibrotic M2 LMs are largely derived from proinflammatory M1 LMs.19–23 In the healthy human lung, the ratio of M1/M2 LMs is highly regulated.19–23 The ratio increases during the inflammation process, whereas in fibrosis M2 LMs will dominate.19–23
In the present study, M phenotypes were not quantified. As M1 and M2 LMs exhibit differences regarding their metabolism of iron, including their lysosomal turn-over of the metal,30 additional information regarding the M1/M2 ratio would be of some value. We cannot rule out that the M1/M2 ratio of the subjects studied may play a role for the degree of LMP displayed in the performed experiments. This lack of information about the M1/M2 ratio we regard as the main weakness of the present study. On the other hand, the purpose of the present study was to investigate the collective lysosomal response of all LMs to see whether increased LMP in LMs related or not to ongoing airway inflammation and measure of CAL. Other limitations of the present study needed to address is that it is observational and shows correlative data derived from rather few subjects with different types of inflammation as cause to CAL. Clearly, larger studies are needed in the future to clarify a potential mechanistic role for increased oxidant-induced LMP behind the development of CAL.
In summary, the present study provides further observations indicating that cellular stress, here reflected by increased LMP on oxidant challenge, may play a role for the development of different types of airway inflammation, which in turn may lead to fibrotic airway remodelling and ultimately CAL.
Conclusion
Lysosomes of LMs harvested from patients with CAL demonstrate an increased sensitivity to oxidants, suggesting that an increase of LMP may trigger chronic inflammation of various types, fibrotic airway re-modelling and CAL.
Abbreviations
AO, acridine orange; A.U, arbitrary unit; BAL(F), broncho-alveolar lavage (fluid); CAL, chronic airflow limitation; COP, cryptogenic organizing pneumonia; COPD, chronic obstructive pulmonary disease; DMEM, Dulbecco’s Modified Eagle Medium; EMT, epithelial–mesenchymal transition; FBS, fetal bovine serum; FEV1, forced expiratory volume in one second post-dilatation; FEV1/FVC, the ratio of FEV1 and FVC post-dilatation; FVC, forced vital capacity post-dilatation; GO, glucose oxidase; IPF, idiopathic pulmonary fibrosis; LM, lung macrophage; LMP, lysosomal membrane permeabilization; PBS, phosphate-buffered saline solution; PPI, proton-pump inhibitor; RA, rheumatoid arthritis.
Data Sharing Statement
The data upon which this analysis was based are available from the corresponding author in anonymized form, upon receipt of a reasonable request.
Acknowledgments
The authors greatly appreciate the contribution from patients and personnel at the Department of Pulmonary Medicine at the University Hospital of Linköping, which made this study possible to perform. This study was financially supported by grants to H.L.P. from Region Östergötland (ALF; LIO-534741, LIO-432791, LIO-355681 and LIO-275911) and the Medical Research Council of Southeast Sweden (FORSS-474691, FORSS-386881, FORSS-222801 and FORSS-82031). They had no role in study design, data collection, analysis and interpretation; in the writing of the manuscript; nor in the decision to submit the manuscript for publication.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
The authors have no financial or non-financial conflicts to disclose in relation to the present study.
References
1. Hikichi M, Mizumura K, Maruoka S, et al. Pathogenesis of chronic obstructive pulmonary disease (COPD) induced by cigarette smoke. J Thorac Dis. 2019;11(S17):S2129–S2140. doi:10.21037/jtd.2019.10.43
2. Brown R, Nath S, Lora A, et al. Cathepsin S: investigating an old player in lung disease pathogenesis, comorbidities, and potential therapeutics. Respir Res. 2020;21:111. doi:10.1186/s12931-020-01381-5
3. Yang M, Kohler M, Heyder T, et al. Long-term smoking alters abundance of over half of the proteome in bronchoalveolar lavage cell in smokers with normal spirometry, with effects on molecular pathways associated with COPD. Respir Res. 2018;19(1):40. doi:10.1186/s12931-017-0695-6
4. Yang M, Kohler M, Heyder T, et al. Proteomic profiling of lung immune cells reveals dysregulation of phagocytotic pathways in female-dominated molecular COPD phenotype. Respir Res. 2018;19(1):39. doi:10.1186/s12931-017-0699-2
5. Weidner J, Jarenbäck L, Åberg I, et al. Endoplasmic reticulum, Golgi, and lysosomes are disorganized in lung fibroblasts from chronic obstructive pulmonary disease patients. Physiol Rep. 2018;6(5):e13584. doi:10.14814/phy2.13584
6. Aja M, Sapey E. Oxidative Stress in COPD: sources, Markers, and Potential Mechanisms. J Clin Med. 2017;6:21. doi:10.3390/jcm6020021
7. Zhang WZ, Butler JJ, Cloonan SM. Smoking-induced Iron Dysregulation in the Lung. Free Radic Biol Med. 2019;133:238–247. doi:10.1016/j.freeradbiomed.2018.07.024
8. Liao S-X, Sun P-P, Gu Y, et al. Autophagy and pulmonary disease. Ther Adv Respir Dis. 2019;13:1753466619890538. doi:10.1177/1753466619890538
9. Persson HL, Richardson DR. Iron-binding drugs targeted to lysosomes: a potential strategy to treat inflammatory lung disorders. Expert Opin Investig Drugs. 2005;14(8):997–1008. doi:10.1517/13543784.14.8.997
10. Persson HL. Iron-dependent lysosomal destabilization initiates silica-induced apoptosis in murine macrophages. Toxicol Lett. 2005;159(2):124–133. doi:10.1016/j.toxlet.2005.05.002
11. Persson HL, Vainikka LK, Sege M, et al. Leaky lysosomes in lung transplant macrophages: azithromycin prevents oxidative damage. Respir Res. 2012;13(1):83. doi:10.1186/1465-9921-13-83
12. Persson HL, Vainikka LK. Increased lysosomal membrane permeabilization in oxidant-exposed macrophages of human fibrotic lungs. J Cell Death. 2013;6:69–74. doi:10.4137/JCD.S13271
13. Sioutas A, Vainikka LK, Kentson M, et al. Oxidant-induced autophagy and ferritin degradation contribute to epithelial-mesenchymal transition through lysosomal iron. J Inflamm Res. 2017;10:29–39. doi:10.2147/JIR.S128292
14. Milara J, Peiró T, Serrano A, et al. Epithelial to mesenchymal transition is increased in patients with COPD and induced by cigarette smoke. Thorax. 2013;68(5):410–420. doi:10.1136/thoraxjnl-2012-201761
15. Mahmood MQ, Sohal SS, Shukla SD, et al. Epithelial mesenchymal transition in smokers: large versus small airways and relation to airflow obstruction. Int J Chron Obstruct Pulmon Dis. 2015;10:1515–1524. doi:10.2147/COPD.S81032
16. Kota A, Deshpande DA, Haghi M, et al. Autophagy and airway fibrosis: is there a link? Res. 2017;6:409. doi:10.12688/f1000research.11236.2
17. Mattsson JP, Väänänen K, Wallmark B, et al. Omeprazole and bafilomycin, two proton pump inhibitors: differentiation of their effects on gastric, kidney and bone H(+)-translocating ATPases. Biochim Biophys Acta. 1991;1065(2):261–268. doi:10.1016/0005-2736(91)90238-4
18. Meyer KC, Raghu G, Baughman RP, et al. on behalf of the American Thoracic Society Committee on BAL in Interstitial Lung Disease. An Official American Thoracic Society Clinical Practice Guideline: the Clinical Utility of Bronchoalveolar Lavage Cellular Analysis in Interstitial Lung Disease. Am J Respir Crit Care Med. 2012;185:1004–1014. doi:10.1164/rccm.201202-0320ST
19. Grabiec AM, Hussell T. The role of airway macrophages in apoptotic cell clearance following acute and chronic lung inflammation. Semin Immunopathol. 2016;38(4):409–423. doi:10.1007/s00281-016-0555-3
20. Allard B, Panariti A, Martin JG. Alveolar Macrophages in the Resolution of Inflammation, Tissue Repair, and Tolerance to Infection. Front Immunol. 2018;9:1777. doi:10.3389/fimmu.2018.01777
21. Arora S, Dev K, Agarwal B, et al. Macrophages: their role, activation and polarization in pulmonary diseases. Immunobiology. 2018;223(4–5):383–396. doi:10.1016/j.imbio.2017.11.001
22. Laskin DL, Malaviya R, Laskin JD. Role of Macrophages in Acute Lung Injury and Chronic Fibrosis Induced by Pulmonary Toxicants. Toxicol Sci. 2019;168(2):287–301. doi:10.1093/toxsci/kfy309
23. Fan EKY, Fan J. Regulation of alveolar macrophage death in acute lung inflammation. Respir Res. 2018;19:50. doi:10.1186/s12931-018-0756-5
24. Wang Y, Wang X-K, Wu -P-P, et al. Necroptosis Mediates Cigarette Smoke-Induced Inflammatory Responses in Macrophages. Int J Chron Obstruct Pulmon Dis. 2020;15:1093–1101. doi:10.2147/COPD.S233506
25. Hodge S, Tran HB, Hamon R, et al. Nonantibiotic macrolides restore airway macrophage phagocytic function with potential anti-inflammatory effects in chronic lung diseases. Am J Physiol Lung Cell Mol Physiol. 2017;312(5):L678–L687. doi:10.1152/ajplung.00518.2016
26. Yu Z, Persson HL, Eaton JW, et al. Intralysosomal iron: a major determinant of oxidant-induced cell death. Free Radic Biol Med. 2003;34(10):1243–1252. doi:10.1016/S0891-5849(03)00109-6
27. Persson HL, Vainikka LK. Lysosomal Iron in Pulmonary Alveolar Proteinosis: A Case Report. Eur Respir J. 2009;33(3):673–679. doi:10.1183/09031936.00044108
28. Persson HL, Vainikka LK, Eriksson HB, et al. Lane-Hamilton Syndrome: ferritin Protects Lung Macrophages Against Iron and Oxidation. Chest. 2011;139(2):361–367. doi:10.1378/chest.10-0818
29. Antunes F, Cadenas E, Brunk UT. Apoptosis induced by exposure to a low steady-state concentration of H2O2 is a consequence of lysosomal rupture. Biochem J. 2001;356(Pt2):549–555. doi:10.1042/0264-6021:3560549
30. Brüne B, Dehne N, Grossmann N, et al. Redox Control of Inflammation in Macrophages. Antioxid Redox Signal. 2013;19(6):595–637. doi:10.1089/ars.2012.4785
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.