")
Back to Journals » Degenerative Neurological and Neuromuscular Disease » Volume 6
Human immune globulin infusion in the management of multifocal motor neuropathy
Authors Jovanovich E, Karam C
Received 12 September 2015
Accepted for publication 19 November 2015
Published 22 December 2015 Volume 2016:6 Pages 1—12
DOI https://doi.org/10.2147/DNND.S96258
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Thomas Müller
Elizabeth Jovanovich,1 Chafic Karam2
1Department of Physical Medicine and Rehabilitation, 2Department of Neurology, The University of North Carolina, Chapel Hill, NC, USA
Abstract: Multifocal motor neuropathy (MMN) is a debilitating and rare disease causing profound weakness with minimal to no sensory symptoms. Conduction block is frequently seen on electrodiagnostic testing. An immune-mediated pathology is suspected though the exact underlying pathophysiology has yet to be elucidated. The presence of anti-GM1 ganglioside IgM antibodies coupled with favorable response to intravenous and subcutaneous immunoglobulins supports a complement-mediated mechanism which leads to destruction of nerve tissue with probable predilection to the nodes of Ranvier. High-dose immunoglobulin currently is the only treatment with proven efficacy for MMN patients. Unfortunately, many patients experience decreased responsiveness to immunoglobulins over time, requiring higher and more frequent dosing. In this review, we will focus on the pharmacology, efficacy, safety, and tolerability of intravenous and subcutaneous immune globulin infusion for treatment of MMN.
Keywords: intravenous immunoglobulin, subcutaneous immunoglobulin, multifocal motor neuropathy, immunosuppressive therapies, anti-GM1 ganglioside IgM antibodies, conduction block
Introduction
Multifocal motor neuropathy (MMN) is an immune-mediated peripheral neuropathy affecting only motor nerves. MMN is rare; one study in the Netherlands estimated the prevalence to be approximately 0.6 per 100,000 individuals. The disease is more common in men than women by a ratio of 2.7:1.1 Though there have been case reports of MMN in patients as young as 6 years of age,2 onset typically occurs between 20 and 70 years of age with approximately 80% of patients reporting onset between 20 and 50 years of age.3 Classic presenting symptoms include progressive painless, distal, asymmetric weakness in the upper extremities.4–6 The ulnar, radial, median, and tibial nerves frequently are affected.4 Though exceedingly rare, involvement of cranial nerves has been reported.7 Autonomic dysfunction is absent.3 Muscle cramps, fasciculations, and exacerbation of weakness in cold weather may occur.4,8 In early stages of disease, there is weakness without significant muscle atrophy since the disease is caused by conduction block. With disease progression, however, muscle atrophy can occur.3,9
Prominent sensory symptoms are not typical and this may help differentiate MMN from compression neuropathies.4,5 Electrophysiological studies demonstrating conduction block in motor nerves not exposed to compression or entrapment and sparing of sensory nerves are characteristics of MMN.4,10 Notably, some patients with MMN will not have detectable conduction block.11–13 Routine nerve conduction studies may miss conduction block if present proximally.14 Activity-dependent conduction block, defined as a temporary conduction block induced by exercise, can also go undetected on routine nerve conduction studies.12,13 Conduction block may also be difficult to demonstrate in cases of advanced disease with severe, confluent denervation.11 Though serum anti-GM1 ganglioside IgM antibodies may be increased in MMN, these antibodies are not sensitive markers for the disease since they may be detected in as few as 25% of patients.15
MMN occasionally may be misdiagnosed as motor neuron disease, progressive muscular atrophy, or lower motor neuron predominant amyotrophic lateral sclerosis (ALS).11 However, clinical findings as summarized in Table 1 and electrophysiology can help differentiate these.9,16 It is important to distinguish among these diseases as each has a dramatically different course. Most forms of ALS are rapidly progressive and the disease is incurable with little more than supportive treatments available currently. In contrast, MMN frequently responds well to treatment with IVIG, giving patients the potential for a normal lifespan.16 IVIG may also be used in a diagnostic trial11 for confirmation if a diagnosis of MMN cannot be made on the basis of physical examination and electrodiagnostic studies.11
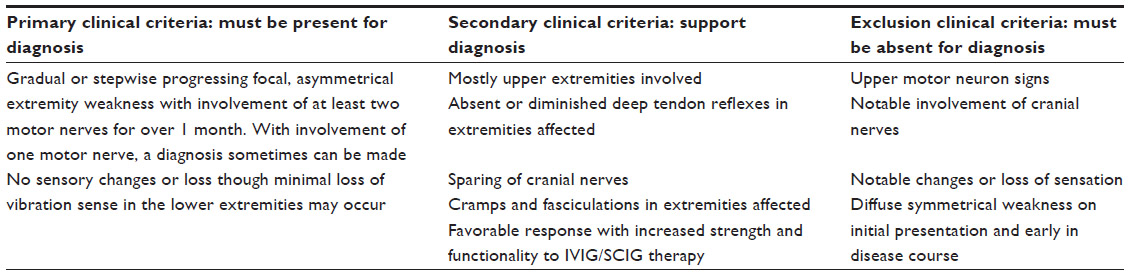 | Table 1 Diagnostic clinical criteria for multifocal motor neuropathy |
MMN typically follows a chronic progressive course. However, some patients may present with an acute form of the disease. The goal of treatment is to improve motor deficits by reducing conduction block, slowing axonal degeneration, and promoting reinnervation. Most patients require treatment for many years though some do achieve prolonged remission.16 The mainstay of treatment is intravenous immunoglobulin (IVIG) or subcutaneous immunoglobulin (SCIG). Other treatments which have been investigated include corticosteroids and plasma exchange. These currently are not recommended due to potential to worsen weakness. Cyclophosphamide, mycophenolate mofetil (MMF), rituximab, and eculizumab have been explored as well though all have worse side effect profiles and are not as effective as IVIG or SCIG which remain the gold standard treatments for MMN.6 In some patients, IVIG may lose its efficacy. In those patients, IV cyclophosphamide may reset the immune system and provide prolonged recovery.11 In this review, we will focus on the pharmacology, efficacy, safety, and tolerability of IVIG and SCIG infusion for treatment of MMN.
Overview of IVIG pharmacology in MMN
The pathophysiology behind MMN is not entirely clear. The few available studies investigating tissue pathology of motor nerves have shown differing findings,11,17,18 though demyelination typically is absent as MMN likely is not a demyelinating neuropathy.17 The presence of anti-GM1 ganglioside IgM antibodies, efficacy of IVIG treatment, and similar clinical features to the pure motor axonal variant of Guillain–Barre syndrome (acute motor axonal neuropathy) all are suggestive of an autoimmune pathophysiology.16,17,19
A proposed mechanism of injury from anti-GM1 ganglioside IgM antibodies in MMN involves complement-mediated damage to the sodium channels in the nodes of Ranvier11 which has been observed in the rabbit model of acute motor axonal neuropathy. This model demonstrates how IgM GM1 activates the complement cascade, causing production of a membrane attack complex which weakens membrane integrity by disrupting sodium channels and facilitating antibody binding to the axolemma.20 In this model, complement inhibitors prevent further antibody-mediated damage, supporting a complement-mediated mechanism of injury.20 By extension, a 2015 case-control study demonstrated that increased activity of the classical complement pathway and anti-GM1 ganglioside IgM antibodies determined disease severity in MMN patients, providing further evidence in support of a complement-mediated disease mechanism.21 Since not all patients with MMN have detectable anti-GM1 ganglioside IgM antibodies, T-cells and cytokines are thought to play a significant role as well.22
As with MMN, the exact mechanism by which IVIG affects immunomodulation in the treatment of MMN and other inflammatory neuropathies is not fully understood, though several mechanisms have been proposed. IVIG contains antibodies against components of the classical complement pathway to prevent membrane attack complex formation leading to tissue degradation at the axolemma.23,24 The presence of anti-GM1 ganglioside IgM antibodies and possible complement-mediated mechanism described suggests the effect of IVIG on the complement pathway may play a significant role in its efficacy for treating MMN as shown in Figure 1.
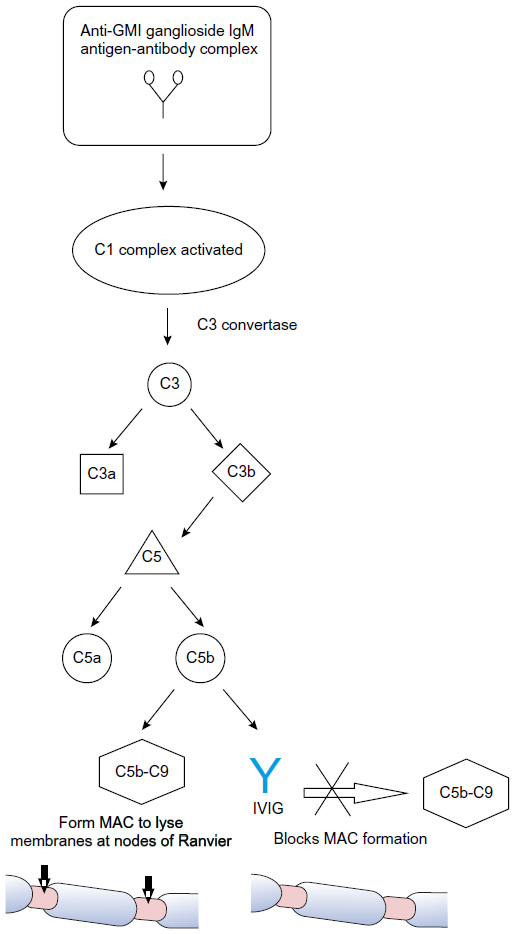 | Figure 1 Proposed mechanism of IVIG within the classical complement pathway for treatment of multifocal motor neuropathy. |
Other mechanisms may be at work. IVIG affects B-cells and antibodies. The anti-idiotype antibodies to different gangliosides present in IVIG may prevent binding of pathogenic autoantibodies to target epitopes in MMN, helping to regulate immune function.25 IVIG also has been shown to inhibit antibody production26 and B-cell differentiation27 and to downregulate certain autoreactive B-cells28,29 all of which may have an effect. Another proposed mechanism of IVIG involves T-cell regulation. In 2008, several regulatory T-cell epitopes (Tregitopes) were discovered in the heavy and light chains of IgG, and administration of these Tregitopes in vivo for several animal models of autoimmune disease suppressed immune responses to antigen.30 Similarly, in humans IVIG treatment selectively activated Tregitopes, enhancing suppressive function and potentially explaining how IVIG restores balanced immune function.22,31 Other proposed mechanisms include IVIG’s effect on cytokines,32 mediation of Fc receptor blockade on macrophages,33 and cell migration.33,34 None of these proposed mechanisms has been proven as the dominant pathway and several mechanisms may work synergistically in treatment of MMN and other autoimmune diseases for which IVIG is effective.22
MMN typically responds well to treatment with IVIG; however, the degree of recovery is variable, and optimal dosing and treatment intervals for IVIG maintenance therapy have not conclusively been established.35 In our experience, patients treated early in their disease, ie, prior to significant muscle atrophy, tend to do better than patients treated later when atrophy clearly is present. Increased IgG concentration (ΔIgG) after IVIG infusion recently was identified as a major factor in determining outcomes of IVIG therapy for GBS36 and some posit ΔIgG could be a useful indicator for optimizing outcomes of IVIG therapy and dosing for treatment of MMN.37 A recent small study37 examined the variability of IVIG pharmacokinetics among MMN patients relative to treatment response. ΔIgG was greater in patients who responded favorably to IVIG treatment. Researchers also examined if variability in pharmacokinetics was associated with genetic polymorphisms in the endothelial IgG receptor (FcRn), a determinant of IgG half-life. IVIG likely saturates these receptors, thereby accelerating the degradation of endogenous IgG and potentially balancing immune function.38 Researchers found no association of ΔIgG levels and responsiveness to treatment with genetic variation in the FcRn gene. However, there was a high degree of variability in ΔIgG levels among patients who received identical dosing protocols as the underlying mechanisms behind variable IVIG efficacy and metabolism have yet to be elucidated. This poses a challenge for establishing standardized treatment dosages and interval protocols.
Efficacy studies
IVIG remains the first-line treatment for patients with MMN and has been investigated thoroughly with studies evaluating efficacy, dosing, and routes of administration.6,39,40 Meta-analysis of multiple randomized, placebo-controlled, double-blind trials41,42 showed that nearly 80% of all subjects had a significant improvement in strength short term after treatment with IVIG as compared to less than 5% after treatment with placebo.43
A 2008 retrospective study (Study A, Table 2) examined dosing in 40 patients with MMN, 22 of whom were IVIG naïve.44 A cumulative dose of 2 g/kg IVIG was effective in 70% of patients. Though not statistically significant, researchers found that positive predictors for favorable response to IVIG were female and lower Medical Research Council (MRC) muscle strength scores. No correlation was found between electrophysiological findings of conduction block and clinical improvements in this study, though correlation has been found in other trials. A recent randomized, double-blind, controlled trial45 from 2013 (Study B, Table 2) assessed efficacy, safety, and tolerability of 10% liquid IVIG. Forty-four patients were randomized to 12 weeks of treatment with IVIG followed by 12 weeks of placebo or vice versa. To ensure stability, all patients received IVIG for 12 weeks at the beginning and end of the study. To prevent carry-over effects, IVIG also was given between the two double-blind 12-week periods. While on IVIG, patients’ mean maximal grip strength increased by nearly 4% and while on placebo it declined by approximately 31% (P=0.005). Using Guy’s Neurological Disability Scale to assess disability related to upper limb function, researchers found a significant number of subjects worsened while on placebo with over two-thirds of participants needing to stop placebo prematurely and resume IVIG therapy. These findings supported researchers’ conclusions that IVIG significantly improves muscle strength and function in MMN patients.
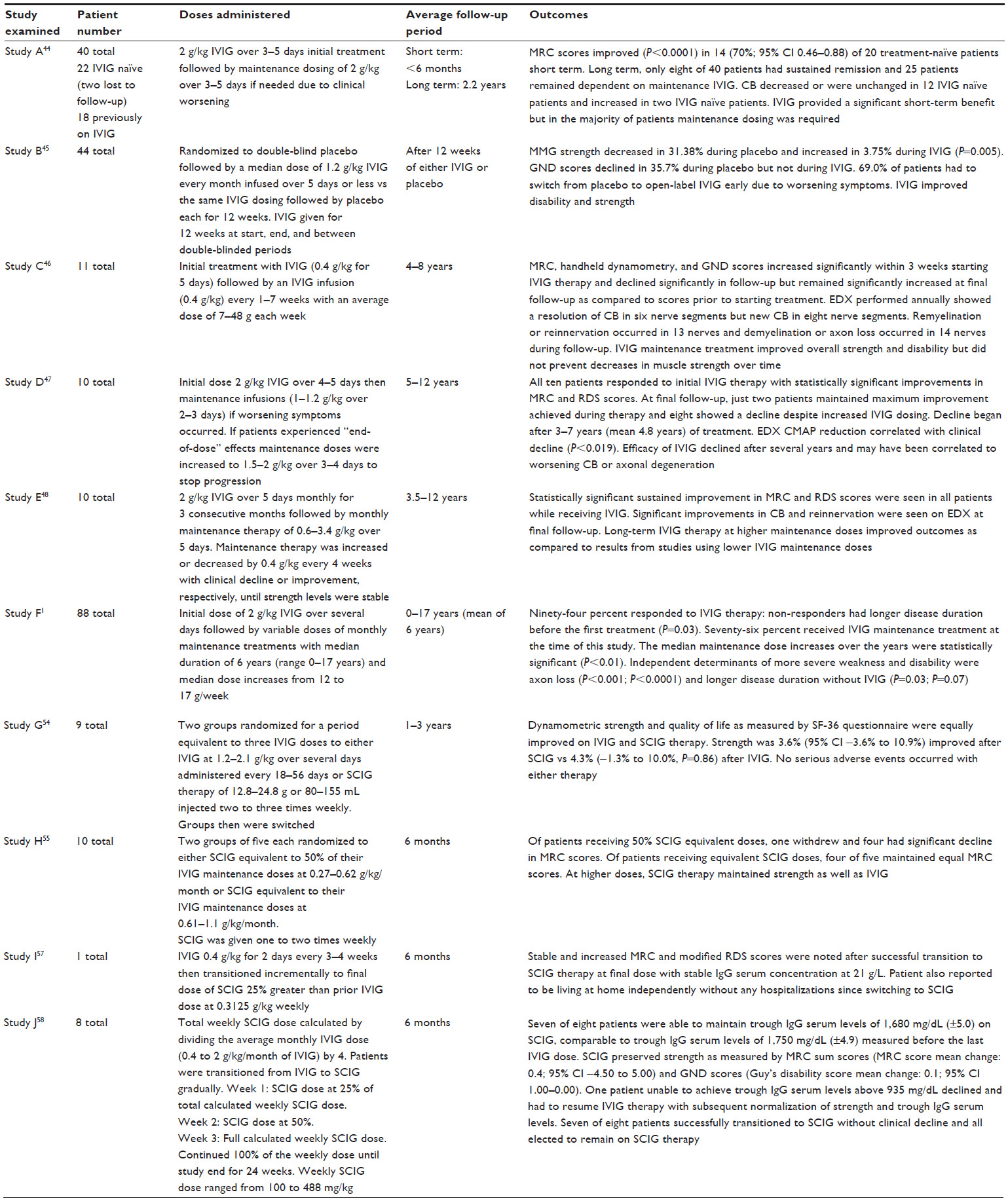 | Table 2 Summary of studies evaluating efficacy of IVIG/SCIG for treatment of MMN |
Though IVIG therapy is helpful for nearly all MMN patients to some degree, the treatment does not always prevent loss of muscle strength and function, and patients often require increased doses and frequency of treatments over time. MMN follows a chronic progressive course and most patients require maintenance therapy with IVIG for years. Prolonged remission is achieved for a minority, some remain stable, and many experience a gradual decline in strength despite being on maintenance therapy. Long-term therapy has been investigated in several studies. A 2002 study46 (Study C, Table 2), over a period of 4–8 years, followed eleven patients with MMN initially treated with one full course of IVIG (2 g/kg) followed by 0.4 g/kg every week and then with maintenance therapy of one infusion every 1–7 weeks. Patients’ strength was evaluated using the MRC score summated for 20 muscle groups, handheld dynamometry, electrodiagnostic studies, and Guy’s Neurological Disability Scale. Muscle strength was reevaluated within 3 weeks after starting IVIG and significant gains were noted for all patients as compared to baseline evaluations performed prior to starting treatment. Slight but significant decreases in strength were noted during subsequent follow-ups over the 4–8-year period. Electrodiagnostic evaluations showed improvement in 13 nerves and decline in 14 nerves. Researchers concluded that maintenance treatment with IVIG improved overall muscle strength and function over time but did not prevent minimal but statistically significant decreases in muscle strength. These findings correlated with electrophysiological data showing a link between IVIG treatments and diminished ongoing axon loss. A 2004 study47 (Study D, Table 2) of ten MMN patients revealed similar findings clinically and electrophysiologically with ongoing maintenance therapy. Patients responded well to initial treatment with IVIG, but by the time of the final follow-up, after anywhere from 5 to 12 years of maintenance treatments, only two maintained the maximum gains, while eight showed a decline after 3–7 years despite ongoing treatments. Decreased responsiveness to IVIG leading to clinical decline correlated with electrodiagnostic findings showing reduced compound motor action potential amplitudes. Diminished efficacy of IVIG with decreasing compound motor action potential supports the theory that loss of efficacy of IVIG over time occurs due to worsening axonal loss with more advanced stages of disease.46,48,49 As compared to the previous two studies, another study from 200450 (Study E, Table 2) examined MMN patients on significantly higher IVIG dosing regimens of 2 g/kg over 5 days monthly for 3 consecutive months, followed by monthly maintenance therapy over 3.5–12 years. Patients on higher doses had improved outcomes, with ongoing statistically significant gains in strength and function and lower disability scores based on the modified Rankin Disability Scale.50 Clinical improvements for these patients correlated with improvements on electrodiagnostic testing including resolution of conduction blocks, decreased evidence of axonal degeneration, and ongoing reinnervation. A 2008 retrospective study44 (Study A, Table 2) of 40 patients with MMN found that only eight participants from the original cohort remained in remission, defined as clinical improvement without further treatment lasting for at least 6 months. Twenty-five participants required IVIG maintenance therapy to preserve motor function. Of these, eight were selected at various times to try adjunct immunosuppressive agents but with limited success. A retrospective study from 20101 (Study F, Table 2) examined increases in strength with IVIG therapy as well as dosage increases required for maintenance therapy over time. Of 88 patients, 94% responded favorably to IVIG. The 6% who did not respond had been diagnosed much later in their disease course and had far more advanced MMN symptoms. Delayed initiation of treatment with IVIG was a statistically significant independent determinant for greater weakness and disability. Seventy-six percent of patients evaluated were given IVIG maintenance therapy with a median duration of 6 years. Median dosage increased from 12 to 17 g/week for these patients. Thirty-five patients tried immunosuppressive agents other than immunoglobulin without improvement. In light of these findings, guidelines established in 2010 by the European Federation of Neurological Societies/Peripheral Nerve Society6 recommends initial dosing of 2 g/kg infused over 2–5 consecutive days. If clinical improvement occurs but is not sustained, then maintenance therapy of 1 g/kg infused every 2–4 weeks or 2 g/kg every 1–2 months is recommended. Some clinicians recommend holding initiation of maintenance therapy until symptoms plateau or begin to decline.51 Other clinicians advocate starting a specific regimen every 14 days and making adjustments gradually based upon clinical response.52 Typical maintenance therapy dosages are 0.4 g/kg weekly or 1–2 g/kg every 2–6 weeks depending on responsiveness and ability to tolerate therapy.9,53 Adjunctive or alternative immunosuppressive treatments can be considered if IVIG alone is not sufficient.6
While effective, IVIG is not without associated risks, which include potentially life threatening complications such as thromboembolic events and anaphylaxis. Intravenous administration is also expensive and burdensome, requiring patients to be monitored closely in a hospital or clinic setting while undergoing infusion. Because of these risks, expenses, and logistical burdens, a popular alternative is subcutaneous administration of immunoglobulins which can be self-administered at home.
A 2009 randomized, controlled, single-blinded study54 (Study G, Table 2) of nine IVIG responsive patients with MMN evaluated dynamometric strength of muscles weakened by disease and quality of life as measured by the SF-36 quality of life questionnaire. Patients treated with IVIG and SCIG saw statistically equivalent mean improvements in muscle strength and no significant difference was noted for SF-36 scores between the two groups. One study participant had irritation, erythema, and swelling at injection sites for a few weeks but all other adverse reactions to SCIG were mild and short-lived. A majority, five of nine participants, decided to continue with SCIG upon completion of the study. After 2 years, researchers followed up with the participants who continued maintenance therapy with SCIG. Dosages varied between 12.8 and 24.8 g or 80 and 155 mL infused two to three times per week. There were no serious adverse side effects reported and any reactions occurring at injection sites were mild and short-lived. Strength and SF-36 scores were stable over the 2-year period. Another 2009 trial55 (Study H, Table 2) examined SCIG dosing. Ten patients were randomized to SCIG dosing equivalent to 50% of prior IVIG maintenance dosing or SCIG dosing equivalent to 100% of prior IVIG maintenance dosing. Of patients receiving SCIG dosing at 50% of prior IVIG dosing, one withdrew and four had a significant decline in MRC scores. Of patients receiving equivalent SCIG doses, four of five maintained equal MRC scores. This study demonstrated SCIG therapy to be safe and as effective as IVIG in maintaining motor function when used at equivalent but not lesser doses to treat MMN. To date, weekly dosing of SCIG has been shown to have a similar efficacy to IVIG.16
SCIG may even reduce fluctuations in strength related to troughs in IgG serum concentration between IVIG dosage intervals known as “end of dose” weakness.56 Though a direct link between serum IgG levels and degree of strength and functionality has yet to be established conclusively, two small studies examining SCIG and IVIG dosing show results supporting the hypothesis that strength is related directly to serum IgG concentration. One study57 (Study I, Table 2) examined a patient with MMN who suffered cyclical fluctuations in strength while on IVIG therapy dosed every 3–4 weeks with trough serum IgG serum levels at 1,500 mg/dL. The patient showed improvement in strength with fewer fluctuations after switching to weekly SCIG infusions with total monthly dosing increased by 25% resulting in a steady-state concentration of IgG serum levels at 2,100 mg/dL. A small, open-label, multicenter, Phase II study58 (Study J, Table 2) examined transitioning from equivalent monthly IVIG to weekly SCIG in eight MMN patients and showed similar dose-dependent effects. Seven of eight patients were able to maintain trough IgG serum levels of 1,680 mg/dL (±5.0) on SCIG, comparable to trough IgG serum levels of 1,750 mg/dL (±4.9) measured before the last IVIG dose. SCIG preserved strength as measured by MRC sum scores for over 6 months. One patient, despite receiving equivalent doses of SCIG and subsequent dosage increases by 25%, had deterioration in strength and correspondingly low IgG serum trough at 935 mg/dL. This was significantly lower than the IgG serum trough of 1,890 mg/dL while on IVIG. Once this patient resumed IVIG therapy, IgG serum troughs rose to previous levels and strength improved to baseline. These studies support the idea that higher steady-state concentrations of serum IgG are beneficial for maintaining strength. Since all studies to date have been small, larger trials are needed comparing IVIG and SCIG to determine if one form of delivery is more effective than the other. Since a minority of patients with MMN are non-responders to IVIG and SCIG and since a majority of patients who do respond favorably to immunoglobulins require progressively more aggressive regimens to remain in remission, alternate or adjunctive immunosuppressive treatments are necessary. Unfortunately, no other immunosuppressive therapies used in place of or in addition to IVIG or SCIG conclusively have demonstrated benefit in clinical trials. Corticosteroids and plasma exchanges can lead to worsening of motor function and are not recommended for treatment of MMN.6 A randomized controlled trial59 of 28 patients at a single center examined MMF as an adjunctive treatment for MMN. Researchers used 1 g MMF twice daily in addition to pre-established IVIG maintenance therapy over the course of a year. Patients did not have significant increases in muscle strength, functional scores, or a reduction in maintenance IVIG dosing. Since no adverse side effects occurred, researchers determined that adjunctive treatment with MMF was safe but ineffective.59,60
Eculizumab, a monoclonal antibody successfully used to treat complement-mediated disorders, has shown some promise as an adjunctive therapy. Eculizumab binds to and inactivates complement factor C5, thereby blocking terminal complement activation and subsequent lysis of membranes via membrane attack complexes in a manner similar to the complement-mediated mechanism of IVIG outlined in Figure 1. A single small clinical trial61 of 14 patients with aggressive neuromyelitis optica, a complement-mediated disease, demonstrated favorable responses including reduced frequency of attacks and disability scores following treatment with eculizumab. Further studies including a Phase III randomized, double-blind, open-label clinical trial have yet to be completed.62 Similarly, studies of eculizumab’s efficacy in treating myasthenia gravis (MG), another complement-mediated disease, are limited. A single pilot Phase II trial63 of eculizumab in 14 patients with severe MG refractory to other treatments demonstrated significant clinical benefits of increased strength and functionality for patients receiving eculizumab as compared to those receiving placebo. As discussed previously, a proposed mechanism of MMN involves anti-GM1 ganglioside IgM antibodies activating complement-mediated destruction of axolemmal membranes. Inhibition of the complement cascade could protect motor nerves.16 A 2011 open-label clinical trial was conducted testing eculizumab over a period of 14 weeks on 13 MMN patients. Of the patients included in the study, ten were concurrently receiving maintenance therapy with IVIG.64 Improvements were noted in patient-rated subjective scores, myometric measurements of muscle strength, and prevalence of nerves with conduction block on electrodiagnostic testing. Researchers noted a small treatment effect in patients with higher baseline motor function whether eculizumab was used in conjunction with or independent of IVIG. Given limited available studies of eculizumab for treatment of MMN, MG, and neuromyelitis optica, it is difficult to compare efficacy for the drug among these disorders though the preliminary data suggest that eculizumab may be a promising treatment for all of these conditions.
Treatment with rituximab, a monoclonal antibody against CD20 surface antigen for B-cell apoptosis, has also demonstrated some clinical improvement in patients with diminishing or insufficient responsiveness to IVIG. However, reports of efficacy are inconsistent and based on a small number of patients. Established dosing recommendations call for 375 mg/m2 weekly for 2–4 weeks for B-cell depletion with single booster doses of 375 mg/m2 given for maintenance if patients develop worsening weakness. The largest study65 with 14 patients showed strength improved by 13% in patients taking rituximab as compared to 3% in controls over 1 year. Strength improved 23% over 2 years. However, the next largest study66 with six IVIG responsive patients showed no significant improvement in strength and patients could not have their IVIG dosage reduced while on adjuvant therapy.
Though cyclophosphamide carries a much greater risk of toxicity compared to rituximab, there is evidence of its efficacy for treating MMN even in patients with no response to IVIG.9 High-dose but not low-dose IV cyclophosphamide has been shown to be effective in up to half of patients in small uncontrolled trials.67,68 Unfortunately, use of high-dose IV cyclophosphamide is limited due to its toxicity, which include bone marrow suppression, hemorrhagic cystitis, bladder cancer, risk of infection, teratogenic effects, and infertility. In the studies listed earlier, several patients involved experienced severe side effects.
Safety/tolerability of IVIG
IVIG carries risks of adverse side effects with initial dosing and maintenance therapy. There is always potential for anaphylaxis, though risks are increased for patients with anti-IgA antibodies and selective IgA deficiency.69,70 More commonly, patients can develop flu-like symptoms9 and should be offered symptomatic therapy.
One of the most common and serious adverse side effects is thromboembolic events including deep venous thrombosis, stroke, pulmonary embolism, and arterial ischemia leading to myocardial infarction,71 with average incidence reported anywhere from 3% to 13%.72 Thrombotic events are more likely to occur in patients receiving higher doses of IVIG.73 Males and patients over 60 years of age are at higher risk for IVIG-induced thrombosis, as are those with diabetes, hyperlipidemia, coronary and peripheral vascular disease, renal insufficiency, hypertension, immobility, and coronary disease.70 Patients predisposed to form blood clots due to atrial fibrillation, pregnancy, and prolonged immobility as well as those with a family history of thromboembolic disease are also at increased risk. A retrospective review demonstrated that arterial ischemic events such as stroke and myocardial infarction are more likely to occur within 12 hours following infusion, with over half occurring within this time frame, whereas venous thromboses are far more likely to occur later, with three-quarters occurring over 24 hours after infusion. No correlation between total number of infusions and arterial or venous thrombotic events was seen.74 Assessment of patient risk factors and careful monitoring for thrombosis through serial physical examinations and, if indicated, Doppler ultrasound could be helpful in preventing adverse events.70
Mechanisms underlying IVIG-associated thrombosis have yet to be elucidated. Theories proposed include a hypercoaguable state due to increased blood viscosity75 and the passive transfer of anticardiolipin antibodies76 or high-molecular-weight proteins77 via IVIG. Passive transfer of factor XIa and other clotting factors could also occur due to insufficient anticoagulation of donated blood/plasma due to neglecting safety protocols or altering established manufacturing processes.70
Other less common serious side effects include renal tubular necrosis, hemolytic anemia, and aseptic meningitis. Patients with pre-existing renal dysfunction are at greater risk of developing renal tubular necrosis on IVIG and must have renal function monitored regularly and receive proper hydration prior to infusion.9 Hemolytic anemia typically occurs only with high-dose IVIG (over 100 g of IVIG in 2–4 days) with a 5.8% incidence rate.78 Low-dose IgG replacement therapy rarely causes hemolysis with only a few cases reported.79,80 Females with blood group type A, B, or AB are at greater risk for IVIG-induced hemolysis. Basic lab work including complete blood counts before starting therapy with IVIG coupled with close follow-up is helpful for early detection of hemolytic anemia.9 Aseptic meningitis is a rare complication and is usually self-limited, though systemic steroids sometimes are needed for severe cases. Infusing at a slower rate coupled with proper hydration prior to infusion and antihistamines when indicated can be helpful for prevention.9,81
Most patients do not develop severe complications on IVIG. Subcutaneous formulations of immunoglobulins may further reduce the risk of serious systemic complications as compared to intravenous formulations. This could be due to limitations of patients tolerating large volume doses of immunoglobulin subcutaneously. A 2009 study54 of SCIG as compared to IVIG showed good results for SCIG without severe adverse events and reported only minimal and transient side effects, primarily local irritation at injection sites. Review of long-term treatment with SCIG also showed good outcomes without severe complications.82
Patient-focused perspectives
MMN profoundly affects patients’ ability to function and definitive diagnosis can be difficult. Due to focal distribution, patients frequently are misdiagnosed with entrapment mononeuropathies despite lack of sensory symptoms and may undergo unnecessary surgeries before being correctly diagnosed.5 Progressive weakness coupled with fasciculations and cramping can lead to misdiagnosis of ALS. Many patients labor under an ALS diagnosis for years prior to receiving the correct determination of MMN.83 Median time from symptom onset to diagnosis is approximately 4 years.5 Though IVIG is highly effective in reversing symptoms, the longer the treatment is delayed, typically, the less effective IVIG will be.1 Univariate analysis from a 2010 study1 suggested a correlation with greater severity of disability and greater length of time untreated; thus, earlier diagnosis is crucial and could prevent patients from acquiring debilitating impairments. Nearly one-fifth of patients in this study were found to have severe disability on the Overall Disability Sum Score due to profound upper extremity weakness. Another study84 from 2010 evaluated how patients’ weakness impacted their daily functioning. This cross-sectional study examined 47 patients with MMN and found that, in addition to muscle weakness, overall functionality was affected by multiple factors including fatigue, impairments in dexterity, and impaired ambulation. Fatigue, a factor often overlooked, had a significant impact. Some electrodiagnostic studies have demonstrated activity-dependent conduction block12 which may relate to fatigue.9 Though fatigue was an ever-present issue, use of gait and hand aids significantly improved scores for autonomy and 94% of these patients were employed.
MMN clearly impacts strength and functionality, but there continues to be no consensus on how to evaluate degree of disability and quality of life for patients. Progress has been made however and in 2013 at the 196th ENMC International workshop, a panel of experts recommended that the primary outcome of patient activity and participation levels be measured by the disease-specific Rasch-built Overall Disability Scale (R-ODS).85 These researchers anticipate creating a Rasch-transformed quality of life scale based on findings of future studies to help standardize assessments of quality of life, a parameter which is essential but difficult to quantify.9,85 Using these standardized measurements hopefully will provide a more reliable method with which to assess patients’ functioning, thereby making it easier to tailor interventions such as walking and hand aids, physical and occupational therapies, and immunoglobulin dosing protocols to better meet their needs.
Conclusion/place in therapy
MMN is a rare but treatable neuropathy. Early diagnosis and treatment is a crucial factor for preserving strength and functionality long term. It is critical to recognize MMN’s unique symptoms and to differentiate MMN from mimicking conditions such as compression neuropathies and ALS for which immunomodulatory therapies are not effective. The only proven treatments available at present are IVIG and SCIG. Further studies are needed to determine optimum steady-state IgG serum concentrations for therapy as well as the best individual doses, dosing intervals, and routes of administration to achieve these concentrations.
Though effective, IVIG and SCIG are costly to produce and administer. Unfortunately, patients still typically experience progressive motor decline due to diminished responsiveness to treatment over time. Patients often require higher and more frequent doses, thereby increasing the risk of adverse and potentially fatal outcomes including myocardial infarction and stroke. Research to elucidate the underlying pathophysiology behind MMN is ongoing. Hopefully, a more complete understanding of disease mechanisms will lead to development of more targeted and economical treatments with lasting effectiveness. For now, IVIG and SCIG remain the first-line agents for treatment of MMN.
Disclosure
The authors report no conflicts of interest in this work.
References
Cats EA, van der Pol WL, Piepers S, et al. Correlates of outcome and response to IVIg in 88 patients with multifocal motor neuropathy. Neurology. 2010;75(9):818–825. | |
Moroni I, Bugiani M, Ciano C, Bono R, Pareyson D. Childhood-onset multifocal motor neuropathy with conduction blocks. Neurology. 2006;66(6):922–924. | |
Nguyen TP, Chaudhry V. Multifocal motor neuropathy. Neurol India. 2011;59(5):700–706. | |
Vlam L, van der Pol WL, Cats EA, et al. Multifocal motor neuropathy: diagnosis, pathogenesis and treatment strategies. Nat Rev Neurol. November 22, 2011;8(1):48–58. | |
Taylor BV, Wright RA, Harper CM, Dyck PJ. Natural history of 46 patients with multifocal motor neuropathy with conduction block. Muscle Nerve. 2000;23(6):900–908. | |
Joint Task Force of the EFNS and the PNS. European federation of neurological societies/peripheral nerve society guideline on management of multifocal motor neuropathy. Report of a joint task force of the european federation of neurological societies and the peripheral nerve society – first revision. J Peripher Nerv Syst. 2010;15(4):295–301. | |
Pringle CE, Belden J, Veitch JE, Brown WF. Multifocal motor neuropathy presenting as ophthalmoplegia. Muscle Nerve. 1997;20(3):347–351. | |
Straver DC, van Asseldonk JT, Notermans NC, Wokke JH, van den Berg LH, Franssen H. Cold paresis in multifocal motor neuropathy. J Neurol. 2011;258(2):212–217. | |
Lawson VH, Arnold WD. Multifocal motor neuropathy: a review of pathogenesis, diagnosis, and treatment. Neuropsychiatr Dis Treat. 2014;10:567–576. | |
Galassi G, Girolami F, Ariatti A, Monelli M, Sola P. Fulminant multifocal motor neuropathy: a report of two cases. Int J Neurosci. 2012;122(7):395–400. | |
Karam C, Dyck PJ, Engelstad JK, Macgowan DJ. Clinical reasoning: a 34-year-old man with recurrent limb weakness. Neurology. 2011; 77(12):e68–e72. | |
Kaji R, Bostock H, Kohara N, Murase N, Kimura J, Shibasaki H. Activity-dependent conduction block in multifocal motor neuropathy. Brain. 2000;123(Pt 8):1602–1611. | |
Straver DC, van den Berg LH, van den Berg-Vos RM, Franssen H. Activity-dependent conduction block in multifocal motor neuropathy. Muscle Nerve. 2011;43(1):31–36. | |
Delmont E, Azulay JP, Giorgi R, et al. Multifocal motor neuropathy with and without conduction block: a single entity? Neurology. 2006; 67(4):592–596. | |
Slee M, Selvan A, Donaghy M. Multifocal motor neuropathy: the diagnostic spectrum and response to treatment. Neurology. 2007;69(17):1680–1687. | |
Leger JM, Guimaraes-Costa R, Iancu Ferfoglia R. The pathogenesis of multifocal motor neuropathy and an update on current management options. Ther Adv Neurol Disord. 2015;8(3):109–122. | |
Taylor BV, Dyck PJ, Engelstad J, Gruener G, Grant I, Dyck PJ. Multifocal motor neuropathy: pathologic alterations at the site of conduction block. J Neuropathol Exp Neurol. 2004;63(2):129–137. | |
Auer RN, Bell RB, Lee MA. Neuropathy with onion bulb formations and pure motor manifestations. Can J Neurol Sci. 1989;16(2):194–197. | |
Takigawa T, Yasuda H, Terada M, et al. Increases in K+ conductance and Ca2+ influx under high glucose with suppressed Na+/K+-pump activity in rat myelinated nerve fibers. Neuroreport. 2000;11(11):2547–2551. | |
O’Hanlon GM, Humphreys PD, Goldman RS, et al. Calpain inhibitors protect against axonal degeneration in a model of anti-ganglioside antibody-mediated motor nerve terminal injury. Brain. 2003;126(Pt 11):2497–2509. | |
Vlam L, Cats EA, Harschnitz O, et al. Complement activity is associated with disease severity in multifocal motor neuropathy. Neurol Neuroimmunol Neuroinflamm. 2015;2(4):e119. | |
Jacob S, Rajabally YA. Current proposed mechanisms of action of intravenous immunoglobulins in inflammatory neuropathies. Curr Neuropharmacol. 2009;7(4):337–342. | |
Halstead SK, Zitman FM, Humphreys PD, et al. Eculizumab prevents anti-ganglioside antibody-mediated neuropathy in a murine model. Brain. 2008;131(Pt 5):1197–1208. | |
Yuki N, Watanabe H, Nakajima T, Spath PJ. IVIG blocks complement deposition mediated by anti-GM1 antibodies in multifocal motor neuropathy. J Neurol Neurosurg Psychiatry. 2011;82(1):87–91. | |
Malik U, Oleksowicz L, Latov N, Cardo LJ. Intravenous gamma-globulin inhibits binding of anti-GM1 to its target antigen. Ann Neurol. 1996;39(1):136–139. | |
Kondo N, Kasahara K, Kameyama T, et al. Intravenous immunoglobulins suppress immunoglobulin productions by suppressing Ca(2+)-dependent signal transduction through fc gamma receptors in B lymphocytes. Scand J Immunol. 1994;40(1):37–42. | |
Stohl W, Elliot JE. In vitro inhibition by intravenous immunoglobulin of human T cell-dependent B cell differentiation induced by staphylococcal superantigens. Clin Immunol Immunopathol. 1996;79(2):122–133. | |
Vassilev T, Yamamoto M, Aissaoui A, et al. Normal human immunoglobulin suppresses experimental myasthenia gravis in SCID mice. Eur J Immunol. 1999;29(8):2436–2442. | |
Korporal-Kuhnke M, Haas J, Schwarz A, Jarius S, Wildemann B. Plasmacytosis is a common immune signature in patients with MMN and CIDP and responds to treatment with IVIg. J Neuroimmunol. 2015;278:60–68. | |
De Groot AS, Cousens L, Mingozzi F, Martin W. Tregitope peptides: the active pharmaceutical ingredient of IVIG? Clin Dev Immunol. 2013;2013:493138. | |
Tjon AS, Tha-In T, Metselaar HJ, et al. Patients treated with high-dose intravenous immunoglobulin show selective activation of regulatory T cells. Clin Exp Immunol. 2013;173(2):259–267. | |
Crow AR, Song S, Semple JW, Freedman J, Lazarus AH. A role for IL-1 receptor antagonist or other cytokines in the acute therapeutic effects of IVIg? Blood. 2007;109(1):155–158. | |
Creange A, Gregson NA, Hughes RA. Intravenous immunoglobulin modulates lymphocyte CD54 and monocyte FcgammaRII expression in patients with chronic inflammatory neuropathies. J Neuroimmunol. 2003;135(1–2):91–95. | |
Vassilev TL, Kazatchkine MD, Duong Van Huyen JP, et al. Inhibition of cell adhesion by antibodies to arg-gly-asp (RGD) in normal immunoglobulin for therapeutic use (intravenous immunoglobulin, IVIg). Blood. 1999;93(11):3624–3631. | |
Kerr J, Quinti I, Eibl M, et al. Is dosing of therapeutic immunoglobulins optimal? A review of a three-decade long debate in Europe. Front Immunol. 2014;5:629. | |
Kuitwaard K, de Gelder J, Tio-Gillen AP, et al. Pharmacokinetics of intravenous immunoglobulin and outcome in Guillain-Barre syndrome. Ann Neurol. 2009;66(5):597–603. | |
Vlam L, Cats EA, Willemse E, et al. Pharmacokinetics of intravenous immunoglobulin in multifocal motor neuropathy. J Neurol Neurosurg Psychiatry. 2014;85(10):1145–1148. | |
Yu Z, Lennon VA. Mechanism of intravenous immune globulin therapy in antibody-mediated autoimmune diseases. N Engl J Med. 1999;340(3):227–228. | |
Leger JM, Viala K, Maisonobe T, Bouche P. Multifocal motor neuropathy: a retrospective study of the response to high-dose intravenous immunoglobulin (IVIg) and current perspectives for diagnosis and treatment. Bull Acad Natl Med. 2007;191(7):1395–1407; discussion 1407-9. | |
Donofrio PD, Berger A, Brannagan TH 3rd, et al. Consensus statement: the use of intravenous immunoglobulin in the treatment of neuromuscular conditions report of the AANEM ad hoc committee. Muscle Nerve. 2009;40(5):890–900. | |
Leger JM, Chassande B, Musset L, Meininger V, Bouche P, Baumann N. Intravenous immunoglobulin therapy in multifocal motor neuropathy: a double-blind, placebo-controlled study. Brain. 2001;124(Pt 1):145–153. | |
Federico P, Zochodne DW, Hahn AF, Brown WF, Feasby TE. Multifocal motor neuropathy improved by IVIg: randomized, double-blind, placebo-controlled study. Neurology. 2000;55(9):1256–1262. | |
van Schaik IN, van den Berg LH, de Haan R, Vermeulen M. Intravenous immunoglobulin for multifocal motor neuropathy. Cochrane Database Syst Rev. 2005;(2)(2):CD004429. | |
Leger JM, Viala K, Cancalon F, et al. Intravenous immunoglobulin as short- and long-term therapy of multifocal motor neuropathy: a retrospective study of response to IVIg and of its predictive criteria in 40 patients. J Neurol Neurosurg Psychiatry. 2008;79(1):93–96. | |
Hahn AF, Beydoun SR, Lawson V, et al. A controlled trial of intravenous immunoglobulin in multifocal motor neuropathy. J Peripher Nerv Syst. 2013;18(4):321–330. | |
Van den Berg-Vos RM, Franssen H, Wokke JH, Van den Berg LH. Multifocal motor neuropathy: long-term clinical and electrophysiological assessment of intravenous immunoglobulin maintenance treatment. Brain. 2002;125(Pt 8):1875–1886. | |
Terenghi F, Cappellari A, Bersano A, Carpo M, Barbieri S, Nobile-Orazio E. How long is IVIg effective in multifocal motor neuropathy? Neurology. 2004;62(4):666–668. | |
Van Asseldonk JT, Van den Berg LH, Kalmijn S, et al. Axon loss is an important determinant of weakness in multifocal motor neuropathy. J Neurol Neurosurg Psychiatry. 2006;77(6):743–747. | |
Baumann A, Hess CW, Sturzenegger M. IVIg dose increase in multifocal motor neuropathy: a prospective six month follow-up. J Neurol. 2009;256(4):608–614. | |
Vucic S, Black KR, Chong PS, Cros D. Multifocal motor neuropathy: decrease in conduction blocks and reinnervation with long-term IVIg. Neurology. 2004;63(7):1264–1269. | |
Vlam L, van den Berg LH, Cats EA, Piepers S, van der Pol WL. Immune pathogenesis and treatment of multifocal motor neuropathy. J Clin Immunol. 2013;33(Suppl 1):S38–S42. | |
Nowacek DG, Teener JW. Multifocal motor neuropathy. Semin Neurol. 2012;32(5):500–505. | |
Nobile-Orazio E, Gallia F. Multifocal motor neuropathy: current therapies and novel strategies. Drugs. 2013;73(5):397–406. | |
Harbo T, Andersen H, Hess A, Hansen K, Sindrup SH, Jakobsen J. Subcutaneous versus intravenous immunoglobulin in multifocal motor neuropathy: a randomized, single-blinded cross-over trial. Eur J Neurol. 2009;16(5):631–638. | |
Eftimov F, Vermeulen M, de Haan RJ, van den Berg LH, van Schaik IN. Subcutaneous immunoglobulin therapy for multifocal motor neuropathy. J Peripher Nerv Syst. 2009;14(2):93–100. | |
Dimberg EL. Treatment of multifocal motor neuropathy with immunoglobulin: does route of administration matter? Eur J Neurol. 2009; 16(5):553–554. | |
Dacci P, Riva N, Scarlato M, et al. Subcutaneous immunoglobulin therapy for the treatment of multifocal motor neuropathy: a case report. Neurol Sci. 2010;31(6):829–831. | |
Misbah SA, Baumann A, Fazio R, et al. A smooth transition protocol for patients with multifocal motor neuropathy going from intravenous to subcutaneous immunoglobulin therapy: an open-label proof-of-concept study. J Peripher Nerv Syst. 2011;16(2):92–97. | |
Piepers S, Van den Berg-Vos R, Van der Pol WL, Franssen H, Wokke J, Van den Berg L. Mycophenolate mofetil as adjunctive therapy for MMN patients: a randomized, controlled trial. Brain. 2007;130(Pt 8):2004–2010. | |
Umapathi T, Hughes RA, Nobile-Orazio E, Leger JM. Immunosuppressant and immunomodulatory treatments for multifocal motor neuropathy. Cochrane Database Syst Rev. 2015;3:CD003217. | |
Pittock SJ, Lennon VA, McKeon A, et al. Eculizumab in AQP4-IgG-positive relapsing neuromyelitis optica spectrum disorders: an open-label pilot study. Lancet Neurol. 2013;12(6):554–562. | |
Sherman E, Han MH. Acute and chronic management of neuromyelitis optica spectrum disorder. Curr Treat Options Neurol. 2015;17(11):48,015-0378-x. | |
Howard JF Jr, Barohn RJ, Cutter GR, et al. A randomized, double-blind, placebo-controlled phase II study of eculizumab in patients with refractory generalized myasthenia gravis. Muscle Nerve. 2013; 48(1):76–84. | |
Fitzpatrick AM, Mann CA, Barry S, Brennan K, Overell JR, Willison HJ. An open label clinical trial of complement inhibition in multifocal motor neuropathy. J Peripher Nerv Syst. 2011;16(2):84–91. | |
Pestronk A, Florence J, Miller T, Choksi R, Al-Lozi MT, Levine TD. Treatment of IgM antibody associated polyneuropathies using rituximab. J Neurol Neurosurg Psychiatry. 2003;74(4):485–489. | |
Chaudhry V, Cornblath DR. An open-label trial of rituximab (rituxan(R)) in multifocal motor neuropathy. J Peripher Nerv Syst. 2010;15(3):196–201. | |
Feldman EL, Bromberg MB, Albers JW, Pestronk A. Immunosuppressive treatment in multifocal motor neuropathy. Ann Neurol. 1991;30(3):397–401. | |
Pestronk A, Cornblath DR, Ilyas AA, et al. A treatable multifocal motor neuropathy with antibodies to GM1 ganglioside. Ann Neurol. 1988;24(1):73–78. | |
Rachid R, Bonilla FA. The role of anti-IgA antibodies in causing adverse reactions to gamma globulin infusion in immunodeficient patients: a comprehensive review of the literature. J Allergy Clin Immunol. 2012;129(3):628–634. | |
Spath PJ, Granata G, La Marra F, Kuijpers TW, Quinti I. On the dark side of therapies with immunoglobulin concentrates: the adverse events. Front Immunol. 2015;6:11. | |
Orbach H, Katz U, Sherer Y, Shoenfeld Y. Intravenous immunoglobulin: adverse effects and safe administration. Clin Rev Allergy Immunol. 2005;29(3):173–184. | |
Rajabally YA, Kearney DA. Thromboembolic complications of intravenous immunoglobulin therapy in patients with neuropathy: a two-year study. J Neurol Sci. 2011;308(1–2):124–127. | |
Ramirez E, Romero-Garrido JA, Lopez-Granados E, et al. Symptomatic thromboembolic events in patients treated with intravenous-immunoglobulins: results from a retrospective cohort study. Thromb Res. 2014;133(6):1045–1051. | |
Marie I, Maurey G, Herve F, Hellot MF, Levesque H. Intravenous immunoglobulin-associated arterial and venous thrombosis: report of a series and review of the literature. Br J Dermatol. 2006; 155(4):714–721. | |
Dalakas MC. High-dose intravenous immunoglobulin and serum viscosity: risk of precipitating thromboembolic events. Neurology. 1994;44(2):223–226. | |
Sakem B, Matozan K, Nydegger UE, Weigel G, Griesmacher A, Risch L. Anti-red blood cell antibodies, free light chains, and antiphospholipid antibodies in intravenous immunoglobulin preparations. Isr Med Assoc J. 2013;15(10):617–621. | |
Grosse-Wilde H, Blasczyk R, Westhoff U. Soluble HLA class I and class II concentrations in commercial immunoglobulin preparations. Tissue Antigens. 1992;39(2):74–77. | |
Kahwaji J, Barker E, Pepkowitz S, et al. Acute hemolysis after high-dose intravenous immunoglobulin therapy in highly HLA sensitized patients. Clin J Am Soc Nephrol. 2009;4(12):1993–1997. | |
Quinti I, Pulvirenti F, Milito C, et al. Hemolysis in patients with antibody deficiencies on immunoglobulin replacement treatment. Transfusion. 2015;55(5):1067–1074. | |
Wilson JR, Bhoopalam H, Fisher M. Hemolytic anemia associated with intravenous immunoglobulin. Muscle Nerve. 1997;20(9):1142–1145. | |
Jain RS, Kumar S, Aggarwal R, Kookna JC. Acute aseptic meningitis due to intravenous immunoglobulin therapy in guillain-barre syndrome. Oxf Med Case Reports. 2014;2014(7):132–134. | |
Harbo T, Andersen H, Jakobsen J. Long-term therapy with high doses of subcutaneous immunoglobulin in multifocal motor neuropathy. Neurology. 2010;75(15):1377–1380. | |
Dimachkie MM, Barohn RJ, Katz J. Multifocal motor neuropathy, multifocal acquired demyelinating sensory and motor neuropathy, and other chronic acquired demyelinating polyneuropathy variants. Neurol Clin. 2013;31(2):533–555. | |
Erdmann PG, Lindeman E, Cats EA, van den Berg LH. Functioning of patients with multifocal motor neuropathy. J Peripher Nerv Syst. 2010; 15(2):113–119. | |
Vanhoutte EK, Faber CG, Merkies IS, PeriNomS Study Group. 196th ENMC international workshop: outcome measures in inflammatory peripheral neuropathies, 8–10 February 2013, Naarden, the Netherlands. Neuromuscul Disord. 2013;23(11):924–933. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.