")
Back to Journals » Journal of Inflammation Research » Volume 14
Human Amniotic Mesenchymal Stem Cells Inhibit aGVHD by Regulating Balance of Treg and T Effector Cells
Authors Gao Y, Li W, Bu X, Xu Y, Cai S, Zhong JM, Du M, Sun H, Huang L, He Y, Hu X , Liu Q, Jin H , Wang Q, Ping B
Received 10 June 2021
Accepted for publication 27 July 2021
Published 16 August 2021 Volume 2021:14 Pages 3985—3999
DOI https://doi.org/10.2147/JIR.S323054
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ning Quan
Ya Gao,1,* Weiru Li,1,* Xiaoyin Bu,1,* Ying Xu,1 Shengchun Cai,1 Jinman Zhong,1 Meixue Du,1 Haitao Sun,2 Liping Huang,3 Yongjian He,4 Xiumei Hu,4 Qifa Liu,1 Hua Jin,1 Qian Wang,2,4 Baohong Ping5
1Department of Hematology, Nanfang Hospital, Southern Medical University, Guangzhou, 510515, People’s Republic of China; 2Department of Laboratory Medicine, Zhujiang Hospital, Southern Medical University, Guangzhou, 510280, People’s Republic of China; 3Department of Obstetrics and Gynecology, Nanfang Hospital, Southern Medical University, Guangzhou, 510515, People’s Republic of China; 4Department of Laboratory Medicine, Nanfang Hospital, Southern Medical University, Guangzhou, 510515, People’s Republic of China; 5Department of Hematology, Huiqiao Medical Center, Nanfang Hospital, Southern Medical University, Guangzhou, 510515, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Baohong Ping
Department of Hematology, Huiqiao Medical Center, Nanfang Hospital, Southern Medical University, Guangzhou, 510515, People’s Republic of China
Tel +8615625042109
Fax +862061642231
Email [email protected]
Qian Wang
Department of Laboratory Medicine, Nanfang Hospital, Southern Medical University, Guangzhou, 510515, People’s Republic of China
Tel +8613822251298
Fax +862061643018
Email [email protected]
Background: Acute graft versus host disease (aGVHD) remains a leading cause of transplant-related mortality following allogeneic haematopoietic cell transplantation (allo-HCT). Human amniotic mesenchymal stem cells (hAMSCs) are a novel mesenchymal stem cells (MSCs), which have stronger proliferation and immunomodulatory ability compared with bone marrow mesenchymal stem cells (BM-MSCs). Besides, as the amniotic membrane is often treated as medical waste after delivery, hAMSCs can be obtained conveniently and noninvasively. The aim of this study was to explore the therapeutic efficacy and underlying mechanisms of hAMSCs transplantation for the humanized aGVHD mouse model.
Methods: We established a humanized aGVHD mouse model by transplanting human peripheral blood mononuclear cells (PBMCs) into NOD-PrkdcscidIL2rγnull (NPG) mice, human amniotic membrane collected from discarded placenta of healthy pregnant women after delivery and hAMSCs were extracted from amniotic membrane and expanded in vitro. Mice were divided into untreated group (Control), aGVHD group (aGVHD), and hAMSCs treatment group (aGVHD+hAMSCs), the hAMSCs labeled with GFP were administered to aGVHD mice to explore the homing ability of hAMSCs. T effector and regulatory T cells (Tregs) levels and cytokines of each group in target organs were detected by flow cytometry and cytometric bead array (CBA), respectively.
Results: We successfully established a humanized aGVHD mouse model using NPG mice. The hAMSCs have the ability to inhibit aGVHD in this mouse model through reduced villous blunting and lymphocyte infiltration of the gut while reducing inflammatory edema, tissue destruction and lymphocyte infiltration into the parenchyma of the liver and lung. hAMSCs suppressed CD3+CD4+ T and CD3+CD8+ T cell expression and increased the proportion of Tregs, and besides, hAMSCs can reduce the levels of IL-17A, INF-γ, and TNF in aGVHD target organs.
Conclusion: The NPG murine environment was capable of activating human T cells to produce aGVHD pathology to mimic aGVHD as in humans. The hAMSCs controlled aGVHD by decreasing inflammatory cytokine secretion within target organs by modulating the balance of Tregs and T effector cells in humanized mice.
Keywords: acute graft versus host disease, humanized mouse model, NPG mice, amniotic mesenchymal stem cells, immunomodulatory
Introduction
Graft-versus-host disease (GVHD) is reported as a syndrome in which donor immunocompetent cells recognize and attack host tissues in immunocompromised allogeneic recipients after hematopoietic stem cell transplantation (HSCT).1,2 Steroids are the standard first line treatment for this disease with the response rate of 50~80%.3,4 However, 35~50% of patients become refractory to systemic steroid therapy, with higher mortality risk.5 Likewise, there are many treatment-related complications such as increased risk of infections, osteopenia and aseptic necrosis.6 Besides, around half of patients develop aGVHD, even when preventive measures are adequately used.7 Unmet need for second-line/third-line treatment including anti-thymocyte globulin, ruxolitinib and infliximab to steroid-refractory aGVHD remains.5 Thus, there is an urgent need to develop more potent immunosuppressive treatment strategies for patients suffering from acute steroid refractory aGVHD while maintaining the graft versus tumor effect to avoid a potential rise in relapse-related mortality.
Mesenchymal stem cells (MSCs) are non-hematopoietic multipotent progenitor cells that can be isolated from embryonic and adult tissues, such as bone marrow, adipose tissue, dental pulp, placenta, and skin. MSCs have potent immunomodulatory effects on both innate and adaptive immune cells which have been demonstrated in preclinical and clinical studies concerning the variety of inflammatory and autoimmune diseases.8–11 Currently, the most commonly used sources of MSCs is bone marrow, however, bone marrow derived MSCs (BM-MSCs) have their own limitations including necessity to accompany invasive procedure to procure and decreasing ability and number of cells to differentiate with increasing donor’s age.12,13 Besides, multiple infusions of MSCs may be requiring multiple harvesting of different tissue samples.14,15 Human amniotic mesenchymal stem cells (hAMSCs) are a type of MSCs that can be easily, massively and safely isolated from the amniotic membrane of placenta from pregnant women when the placenta is delivered and usually discarded.16 The merits of using hAMSCs lie in that they are free from ethical concern in obtain and that a relatively enormous of hAMSCs can be readily gained. Thus, hAMSCs have been recognized as one of the most promising stem cells in the field of regenerative medicine.17 The hAMSCs possess immunomodulatory properties that are thought to enable damaged tissues to form a balanced inflammatory and regenerative micro-environment in the presence of vigorous inflammation.18 hAMSCs have low immunogenicity and high histocompatibility, due to their low expression levels of MHC class I antigens (HLA-A, HLA-B and HLA-C) but almost no MHC class II antigens (HLA-DR) or co-stimulatory molecules.19 Meanwhile, the amniotic membrane is an avascular structure only composed of epithelium and mesenchyme, which makes it easy to remove epithelial layer by Dispase II and thus obtain hAMSCs without contamination of endothelial cells and hematopoietic cells.20 Our previous study found that the immunomodulatory and proliferation activity of hAMSCs is higher than that of human bone marrow MSCs in vitro.21,22 Yamahara et al confirmed that human amnion-derived MSCs had immunomodulatory activity equal to or higher than that of human bone marrow MSCs.23 Lee et al also found that placenta-derived stem cells may have additional advantage over the bone marrow derived MSCs (BM-MSCs) and adipose tissue derived MSCs (AD-MSCs) MSCs in terms of immunomodulation.13 hAMSCs have already been applied in miscellaneous researches for tissue regeneration, including corneal tissue, spinal cord injury, and brain infarction. However, relatively research in GVHD is few.24 Pianta et al found that, in vitro, hAMSCs could inhibit the proliferation of CD4+T cells and CD8+T cells, down regulate the proportion of Th1/Th17 cells, and induce the proliferation and differentiation of regulatory T cells (Tregs).25 Our previous study demonstrated that hAMSCs induced Th1 cells into Th2 cells in vitro.22 Yamahara et al showed that hAMSCs could reduce the degree of weight loss and improve the pathological situation of aGVHD in aGVHD mice, but the mechanism of hAMSCs regulating aGVHD inflammation was scarcely discussed.23 A Phase I/II clinical trial also found that steroid-refractory aGVHD infusion of hAMSCs was safe and effective.26 According to our knowledge, there are few reports about the mechanisms by which hAMSCs exert their therapeutic effects in vivo.
Due to ethical constraints, relevant research cannot be conducted on humans and GVHD pathogenesis has been mostly studied in mouse models of transplantation. Until recently, C57/Bl6 (H2b) donors in BALB/c (H2d) recipients have been widely used in the establishment of aGVHD model mice. However, mouse to mouse aGVHD models have some limitations. To generate a system whereby human T cell-mediated aGVHD can be studied and manipulated in vivo, Immunodeficiency mice, which can be humanized, have been rapidly developed.27 Currently, the most popular immune deficient mouse strains, NOD/SCID-IL2Rγ−/-, NOD.Cg-Prkdcscid IL2rγtm1Wjl/SzJ (NSG), and NOD.Cg-Prkdcscid-IL2rγtm1Sug/Jic (NOG), play an important role in the study of the immunological pathological mechanisms of GVHD and the improvement of therapy. Those mice lack T, B, and NK cells, and also have reduced macrophage and dendritic cell function that allow efficient human peripheral blood mononuclear cells (PBMCs) engraftment. Transplantation of PBMCs can cause aGVHD syndrome because human APCs process mouse antigens and present them in the presence of class II MHC.28 Compared to mouse models of aGVHD, the humanized model has many advantages such as the use of human cells to induce and control xenogeneic (xeno)-aGVHD, the possibility of using donors with high genetic diversity, and the possibility of using older donors previously exposed to various pathogens.29 Here, we used NOD-PrkdcscidIL2rγnull (NPG) mice to establish a xenogeneic aGVHD humanized model and investigated the therapeutic potential of hAMSCs in preventing aGVHD in vivo. We demonstrated that hAMSCs ameliorated aGVHD by modulating the balance of Tregs and T effector cells and reducing inflammatory cytokine secretion in target organs. Therefore, hAMSCs are potential candidate for aGVHD treatment and have certain clinical transformation value.
Materials and Methods
Animals
NPG mice 8–10 weeks of age and weighed 25–30g were obtained from Beijing Vitalstar Biotechnology Co., Ltd. (laboratory animal production license no. SCXK2019-0002). Animals were housed in a specific pathogen-free facility in microisolator cages, given autoclaved food, and maintained on acidified autoclaved water and a solution of gentamicin. All animal experiments were approved by the laboratory animal ethics review committee of Southern Medical University. All animal procedures were in accordance with the National Institute regarding laboratory animal care and use.
Human PBMCs Collection
Human PBMCs were collected from healthy volunteers. Each donor provided written informed consent. Peripheral blood was collected in sodium citrate and PBMCs were isolated from peripheral blood by Ficoll-Hypaque (Solarbio, catalog number: P8900) density centrifugation, washed in PBS, suspended in red blood cell lysis buffer (Solarbio, catalog number: R1010) at 4°C for 15 min, washed again in PBS, and suspended in PBS for tail vein injection into NPG mice.
Induction of aGVHD in NPG Mice
Mice were divided into groups by randomization according to weight. In aGVHD group (n=6), mice were given 200 cGy irradiation 3–4h before cell injection followed by tail vein injection of 3×106 PBMCs/mouse suspended in 500μL PBS while mice of control group (n=3) were just injected 500μL PBS from teil vein. During all experiments, each mouse was graded according to the aGVHD clinical scoring system from the one originally described by Cooke.30 The symptoms of aGVHD included weight loss, hunched posture, ruffled fur, reduced mobility, and diarrhea. Mice survived to the 28 day endpoint and those that suffered from severe GVHD (weight loss of 25%, severe hunched posture, severe ruffled fur, less or no mobility, or hematochezia) were euthanized.
Histopathological Analyses
Tissues were collected at the time of necropsy, fixed in 10% buffered formalin, and embedded in paraffin. For routine histology, sections were stained with hematoxylin and eosin. According to the degree of tissues structure destruction and lymphocyte infiltration, the sections of each organ were graded as normal, mild, moderate and severe, with a histological score of 0, 1, 2 and 3, respectively. For immunohistochemistry, 2μm sections of tissues were immunostained with anti-human CD45 antibody (Abcam, catalog number: ab40763) at 37°C for 1 hour, then rinse 5 times with PBS, and incubated with goat anti-rabbit IgG antibody-HRP secondary antibody (Genetech, catalog number: GK600711-B) at 37°C for 30 min. Develop with DAB for 2min after washing with PBS. For immunofluorescence, target organs were fixed with 4% paraformaldehyde for 24 h and dehydrated with 30% sucrose. After more than 48 h, the tissues were embedded in a frozen slicer at −25°C to generate slices. Mount the slides with anti-fluorescence attenuation mounting tablets (including DAPI). The huCD45 positive area were analyzed by ImageJ 1.52a.
Isolation and Culture of hAMSCs
Human placentas were obtained from caesarean sections from healthy women with informed consent. The amnion layer was separated from the chorion layer and washed several times with phosphate-buffered saline (PBS), cut into 0.5–1.0 cm2 pieces, and digested with 0.25% trypsin at 37°C for 30 min. Amniotic epithelial cells were removed gently and the amnion was digested again under the same conditions. Finally, the amnion was washed with PBS and cut into 1 mm2 pieces and cultured in α-MEM (1×; Gibco, catalog number: C12571) supplemented with 10% fetal bovine serum (FBS; Gibco, catalog number: A31608-02), 0.01 mM/mL sodium pyruvate (Corning, catalog number: 15719005), 100 U/mL penicillin streptomycin (Corning, catalog number: 15719005), and 0.01 mM/mL nonessential amino acids (Solarbio, catalog number: N1250) at 37°C and 5% CO2. Cells were recovered using 0.25% Trypsin and passaged when the cell density reached 80–90% between 5 to 7 days. Passaged cells were cultured in 25 cm2 culture flasks at a density of 1 × 105/cm2. hAMSCs used in experiments were third- to fifth-passage.
Identification of hAMSCs
Biomarkers of hAMSCs. For identification, at a concentration of 1 × 106cells/mL in PBS and stained with Mouse IgG1 kappa Isotype Control (P3.6.2.8.1), PE-Cyanine7 (eBioscience, catalog number: 25-4714-80), Mouse IgG1 kappa Isotype Control (P3.6.2.8.1), PE (eBioscience, catalog number: 12-4714-42), mouse anti-human CD90-PE antibody (BD Pharmingen, catalog number: 561970), mouse anti-human CD105-PE antibody (eBioscience, catalog number: 12-1057-42), mouse anti-human CD73-PE antibody (BD Pharmingen, catalog number: 561014), mouse anti-human CD45-PE antibody (BD Pharmingen, catalog number: 560975), mouse anti-human CD11b-PE-Cy™7 antibody (BD Pharmingen, catalog number: 561098), mouse anti-human CD34-PE antibody (BD Pharmingen, catalog number: 560941), and mouse anti-human HLA-DR-PE antibody (BD Pharmingen, catalog number: 560943) at 4°C for 30 min respectively. Cells were washed with PBS, adjusted to 300 μL, and analyzed by flow cytometry. Differentiation potential of hAMSCs, to analyze adipogenic and osteogenic differentiation, passage three hAMSCs were seeded at a density of 1.5×105 cells/well in a six-well plate. When the cells reached 85–90% confluence, cells were incubated in human MSCs adipogenic differentiation medium (Pythonbio, catalog number: 20191008), for 21 days; the adipogenic induction medium was replaced every 3 days. Oil red O (Solarbio, catalog number: G1260) staining was performed to assess the differentiation potential. For osteogenic differentiation, hAMSCs were cultured with human MSCs osteogenic differentiation medium (Pythonbio, catalog number: 20191009) for 21 days; the induction medium was replaced every 3 days. The differentiation potential for osteogenesis was assessed by Alizarin Red (pH 4.2, 40 mM) (Solarbio, catalog number: G1452) staining.
GFP Labeling Cells and in-vivo Tracing
For hAMSCs GFP labeling, cells were added to a 24-well plate at a concentration of 3×105/mL, 500 μL per well. The 24-well plate was incubated at 37 °C and 5% CO2. After 24 h, the culture media was disposed, and 250 μL of culture media and 1.6 μL of GFP-pseudovirion (Hanbio, catalog number: HH20191126GYQ-LP01) were added; the rest of the 250 μL culture media was added 4 h later and the plate was incubated. After 24 h, the culture media was disposed, 500 μL of culture media was added and the plate was incubated. For 293T (Procell, catalog number: CL-0005) GFP labeling, cells were added to a 24-well plate at a concentration of 3×105/mL, 500 μL per well. The 24-well plate was incubated at 37 °C and 5% CO2. After 24 h, the culture media was disposed, and 250 μL of culture media and 1.0 μL of GFP-pseudovirion were added; the rest of the 250 μL culture media was added 4 h later and the plate was incubated. After 24 h, the culture media was disposed, 500 μL of culture media was added and the plate was incubated. GFP-labeled hAMSCs and 293T were identified by fluorescence microscope after 2–3 days. The suspension of GFP-labeled hAMSCs was adjusted to 5×105 cells suspended in 500 μL PBS and injected into NPG mice via the tail vein. These mice were euthanized after 24 h and 72 h. Blood and tissues such as the liver, spleen, lung, and gut were collected. For immunofluorescence, target organs were fixed with 4% paraformaldehyde for 24 h and dehydrated with 30% sucrose. After more than 48 h, target organs were embedded with a frozen slicer at −25°C to generate slices. Recipient cells were distinguished by DAPI counterstaining.
Effect of hAMSCs in the Treatment of aGVHD in vivo
NPG mice of aGVHD group (n=27) and hAMSCs treatment group (n=5) were induced aGVHD according to the above protocol. Mice of aGVHD group were injected 500μL PBS from tail vein on the third day while those mice of hAMSCs treatment group were injected 5×105 hAMSCs suspended in 500μL PBS from tail vein. Mice of control group (n=3) were untreated. Each mouse was graded according the aGVHD clinical scoring system from the one originally described by Cooke.30 Mice survived to the 28 day endpoint and those that suffered from severe GVHD were euthanized.
Flow Cytometric Analysis in Lymphocytes of Mouse Target Organs
Mouse anti-human CD3-PE-Cyanine7 antibody (eBioscience, catalog number: 25-0038-42), mouse anti-human CD4-FITC antibody (BD Pharmingen, catalog number: 340133), mouse anti-human CD8-PE (BD Pharmingen, catalog number: 340046), mouse anti-human CD25-APC antibody (Biolegend, catalog number: 302610), and mouse anti-human Foxp3-PE (eBioscience, catalog number: 12-4777-42) Mouse IgG1 kappa Isotype Control (P3.6.2.8.1), PE (eBioscience, catalog number: 12-4714-42), Mouse IgG1 kappa Isotype Control (P3.6.2.8.1), FITC (eBioscience, catalog number: 11-4714-82), Mouse IgG1 kappa Isotype Control (P3.6.2.8.1), APC (eBioscience, catalog number: 17-4714-42) Mouse IgG1 kappa Isotype Control (P3.6.2.8.1), PE-Cyanine7 (eBioscience, catalog number: 25-4714-80) were used. Peripheral blood, livers, spleens, lungs and guts were collected at the time of necropsy and analyzed by flow cytometry. Single cell 1×106/mL suspensions were obtained by grinding the liver, spleen, lung, and gut, and blood samples were processed with red blood cell lysis buffer (Solarbio, catalog number: R1010) at 4°C according to the protocol. All samples were stained with antibodies or isotype matched control IgG for 30 min at 4°C in the dark and analyzed with a BD FACS CantoII flow cytometer with FlowJo 10.0.
Analysis of the Cytokines by Cytometric Bead Array (CBA)
The CBA human Th1/Th2/Th17 cytokine kit (BD, catalog number: 560484) was used. Peripheral blood and single cell suspensions of each target organ were centrifuged at 4°C, 3000 rpm/min for 20 min. Human Th1/Th2/Th17 cytokine capture beads were mixed and added to all assay tubes. Then, 50 µL of each sample was added to appropriately labeled sample tubes and 50 µL of the human Th1/Th2/Th17 PE detection reagent was added. The assay tubes were incubated for 3 h at room temperature in the dark. Then, 1 mL of wash buffer was added to each assay tube followed by centrifugation at 1500 rpm/min for 5 min. The supernatant from each assay tube was carefully aspirated and discarded, and 300 µL of wash buffer was added to resuspend the bead pellet. The samples were analyzed with flow cytometry (BD FACS CantoII) human Th1/Th2/Th17 cytokine data using FCAP Array software.
Statistical Analysis
Comparisons between two means were performed using the independent samples t-test. Comparisons of three or more means were performed using one-way ANOVA. Survival curves were generated with the Kaplan-Meier method. P < 0.05 was considered significant. All statistical analyses were performed with GraphPad Prism 8.0.
Results
aGVHD Animal Model Established by NPG Mice
aGVHD group developed symptoms of typical aGVHD, including weight loss, hunched posture, ruffed fur, reduced mobility, damaged skin, diarrhea, and hematochezia compared with control group (Figure 1A). The weight loss rate (aGVHD vs Control: day 14 P=0.008) and clinical score increase (aGVHD vs Control: day 14 P=0.029) of NPG mice in the aGVHD group are faster than those in Control group. aGVHD group died between 12 to 18 days after human PBMSCs transfer (P=0.009) (Figure 1B). Similar to humans, NPG mice with aGVHD showed severe inflammation, leukocyte infiltration, necrosis, and tissue damage in target organs such as the liver, spleen, lung and gut (aGVHD vs Control: Liver P=0.012; Spleen P=0.012; Lung P=0.012; Gut P=0.024) (Figure 1C). Brown lump-like human PBMCs infiltration was observed by immunohistochemistry in the liver, spleen, lung and gut. Compared with the control group, the target organ tissues of the aGVHD group showed obvious infiltration of PBMCSs (aGVHD vs Control: Liver P=0.035; Spleen P<0.001; Lung P=0.035; Gut P=0.448) (Figure 1D). These results indicated that the aGVHD NPG mice were mediated mainly by human T cells. PBMCs in the blood were detected by flow cytometry, which confirmed that PBMCs were successfully injected into the body (Figure 1E).
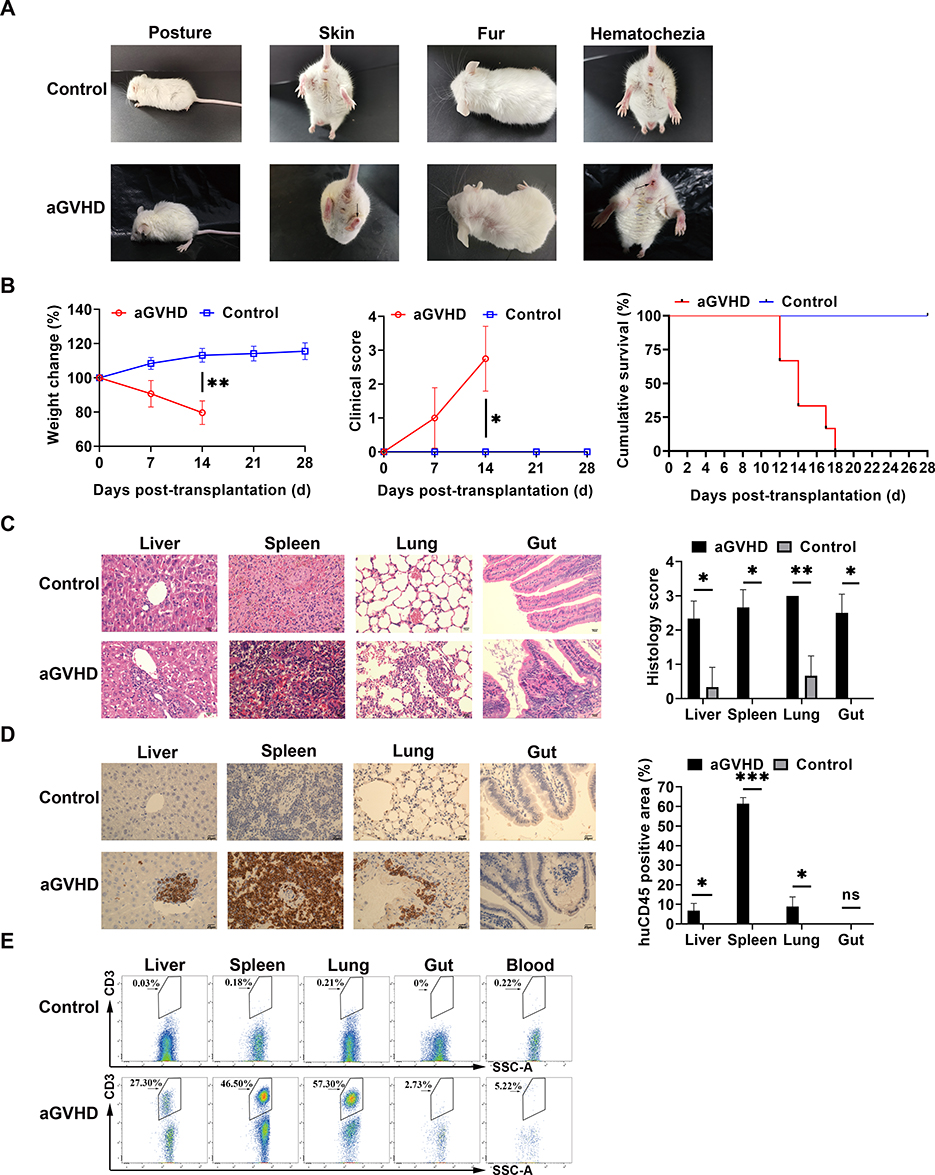 |
Figure 1 Induced aGVHD model in NPG mice. (A) Representative clinical manifestations including hunched posture, ruffed fur, damaged skin, and hematochezia in the aGVHD mouse model. (B) Measurement of weight change, clinical score, and survival time at different time points with those two groups. The weight loss rate in aGVHD group is faster than in the control group, the clinical score of aGVHD was higher than that of the control group, the survival time of the control group was also significantly longer than that of the aGVHD group. *p < 0.05 and **p < 0.01. (C) Representative histology of target organs, including the liver, spleen, lung and gut, showed inflammatory cell infiltration and tissue damage in aGVHD group. 400×. *p < 0.05 and **p < 0.01. (D) Representative immunohistochemistry of target organs. Human CD45+ cells were detected by immunohistochemistry in these target organs. 400×. *p < 0.05 and ***p < 0.001. (E) Human CD3+ T cells were not detected by flow cytometry in the control group. Human CD3+ T cells from each target organ were detected by flow cytometry in the aGVHD group. |
Identification of hAMSC and the Homing Ability of hAMSC to aGVHD Target Organs
Passage three hAMSCs appeared to be spindle- or polygon-shaped in morphology (Figure 2A). The hAMSCs possessed osteogenic and adipogenic differentiation potentials in vitro (Figure 2B and C). Flow cytometry analysis of the hAMSCs surface antigen phenotype revealed that CD90, CD105, and CD73 were expressed, while CD45, CD34, HLA-DR and CD11b were not (Figure 2D). As the GFP gene was present in the GFP-pseudovirion, the hAMSCs transduced with pseudovirion produced a high level of GFP (aGVHD+hAMSCs vs Control P<0.001); the efficiency of labeling was > 80% (84.33±1.73%) (n=6) (Figure 2E). GFP-hAMSCs were injected into NPG mice, after 24 and 72 h, mice were euthanized separately. GFP-labeled hAMSCs were detected in the liver, spleen, lung, and gut after 24 h. However, few GFP-labeled cells were observed in target organs except the lung after 72 h (Figure 3). The results demonstrated that after intravenous injection, hAMSCs migrated to damaged target organs.
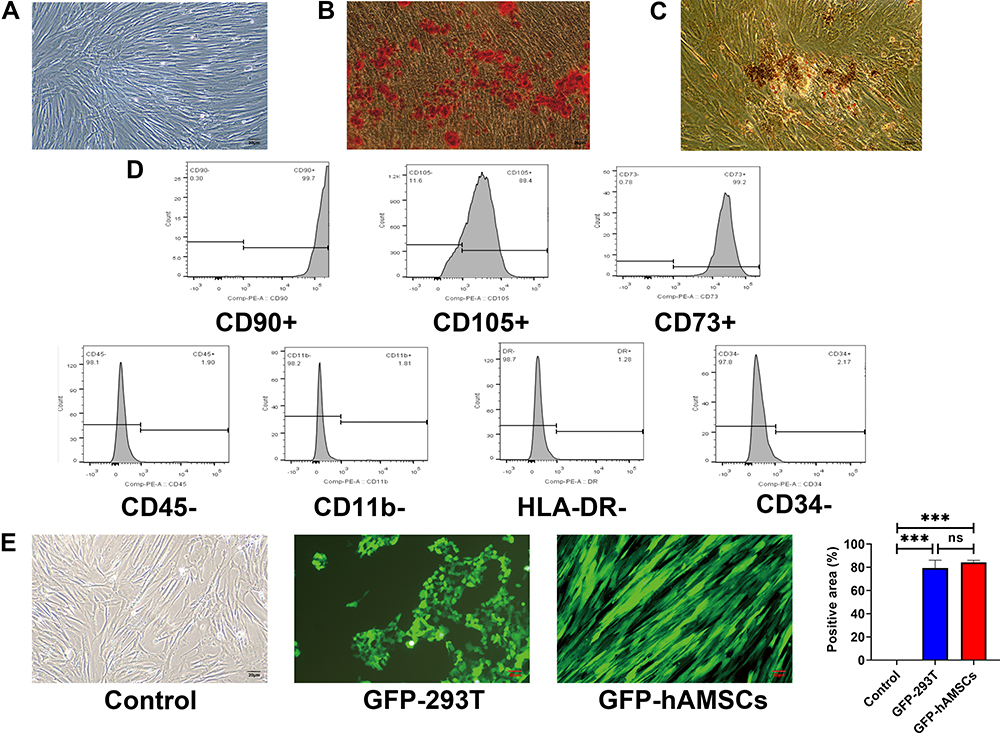 |
Figure 2 Identification of human amnion mesenchymal stem cells. (A) Characteristics of passage three hAMSCs. (B) Osteogenic differentiation of hAMSCs indicated by Alizarin Red S. 100×. (C) Adipogenic differentiation of hAMSCs indicated by Oil Red O staining. 100×. (D) Surface antigens of hAMSCs were detected by flow cytometry. Cells were positive for CD90, CD105, and CD73 and negative for CD45, CD11b, CD34, and HLA-DR. (E) GFP-labeled hAMSCs and 293T cells were detected by fluorescence microscopy at day 3 following transfection with GFP-pseudovirion. 100×. ***p < 0.001. |
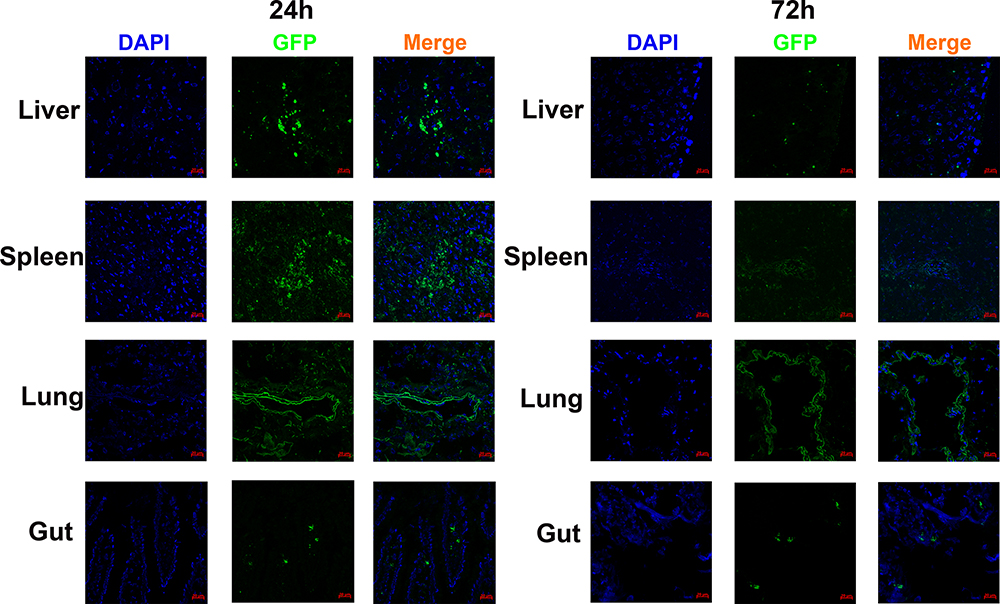 |
Figure 3 Human amnion mesenchymal stem cells migrated to target organs. GFP-labeled hAMSCs detected by confocal microscopy in target organs at 24 after injection in vivo. At 72 h, GFP fluorescence in the spleen, liver, and gut was weakened while increased in the lung. |
Therapeutic Effect of hAMSCs on aGVHD Mice
To verify the immunosuppressive ability of hAMSCs in vivo, we tested hAMSCs in the aGVHD model. As shown in Figure 4A, hAMSCs significantly ameliorated the severity of acute GVHD in terms of weight loss (aGVHD vs aGVHD+hAMSCs: day 7 P=0.043, day 14 P=0.037) and disease score (aGVHD vs aGVHD+hAMSCs: day 14 P=0.002). Moreover, aGVHD mice injected with hAMSCs survived for 26–28 days, while the aGVHD mice that received PBS died 6–18 days after transplantation (aGVHD vs aGVHD+hAMSCs P<0.001). The hAMSCs significantly ameliorated the severity of acute GVHD in terms of weight loss and disease score within 28 days of transplantation. Moreover, hAMSCs prevented leukocyte infiltration and reduced pathology in the liver, spleen, lung, and gut on day 6 after transplantation (aGVHD vs aGVHD+hAMSCs: Liver P=0.002; Spleen P=0.003; Lung P=0.002; Gut P=0.003) (Figure 4B). Immunohistochemistry experiments further confirmed this phenomenon (aGVHD vs Control: Liver P=0.002; Spleen P<0.001; Lung P=0.004; Gut P=0.711) (Figure 4C). These results demonstrated that hAMSCs control aGVHD clinical symptoms in the aGVHD mouse.
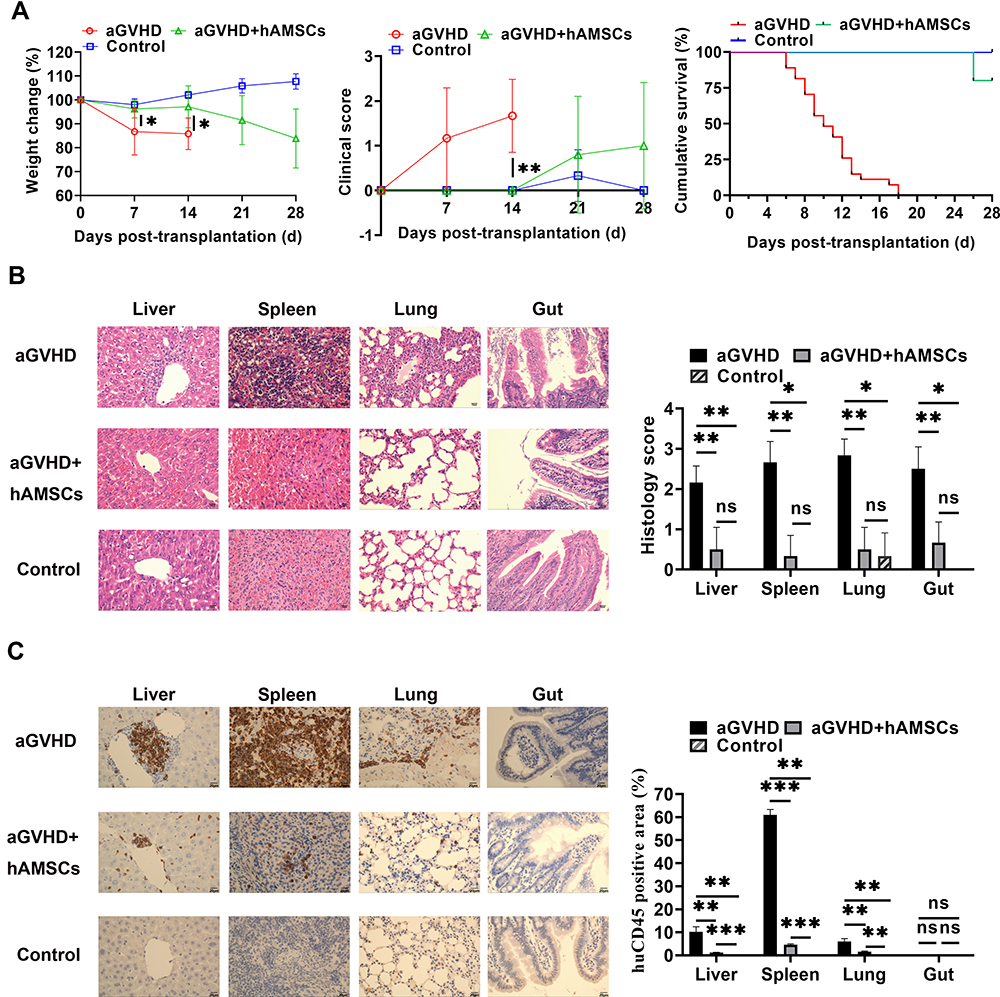 |
Figure 4 The capacity of hAMSCs inhibited the development of -aGVHD in vitro. (A) Measurement of weight change (aGVHD vs aGVHD+hAMSCs day 7 P=0.043, day 14 P=0.037), clinical score (aGVHD vs aGVHD+hAMSCs day 14 P=0.002), and survival in the different groups (aGVHD vs aGVHD+hAMSCs P<0.001). *p < 0.05 and **p < 0.01. (B) Representative histology of target organs, including the liver, spleen, lung, and gut. Compared with the hAMSCs treatment group, inflammatory cell infiltration and tissue damage in the aGVHD group were more severe. 400×. *p < 0.05 and **p < 0.01. (C) Representative immunohistochemistry of target organs, including the liver, spleen, lung, and gut. Infiltration and tissue damage in the aGVHD group were more severe than that of hAMSCs treatment group and Control group. 400×. **p < 0.01 and ***p < 0.001. |
Regulation of hAMSCs on T Cells in Target Organs of aGVHD Mice
To investigate the effect of hAMSCs on T effector and Tregs, single cell suspensions of target organs were analyzed by flow cytometry. The cell concentration was 1×106 cells/mL. We found that the proportion of CD3+CD4+ T cells in the liver (P=0.018), spleen (P=0.021) and lung (P=0.015) of mice in hAMSCs treatment group was significantly lower than that in aGVHD group, there was no significant statistically difference in gut (P=0.218) (Figure 5A). The proportion of CD3+CD8+ T cells in the liver (P=0.012), spleen (P=0.048), lung (P=0.0004) and gut (P=0.014) of mice in hAMSCs treatment group was lower compared with aGVHD group (Figure 5B). We further found that CD4+CD25+Foxp3+ Tregs from liver (P=0.037), spleen (P=0.013), and gut (P=0.011) in hAMSCs treatment group were increased, especially in the gut, there was no significant statistically difference in lung (P=0.072) (Figure 5C). Our data supported the finding that hAMSCs treatment significantly inhibited the proportion of donor CD3+CD4+ T and CD3+CD8+ T cells and increased the proportion of Tregs in target organs from day 3 to day 6 after transplantation.
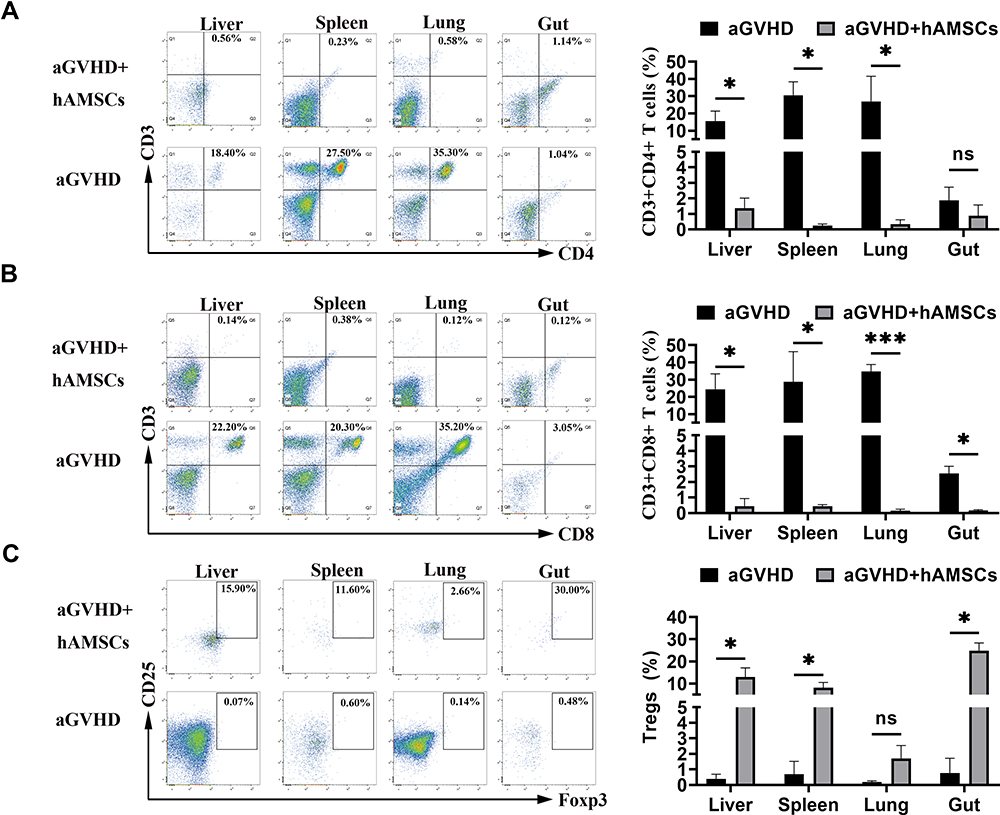 |
Figure 5 hAMSCs suppressed effector T cell and increased Tregs in target organs. (A) After hAMSCs treatment, the proportion of CD3+CD4+ T cells in liver, spleen and gut was significantly lower than that of aGVHD group. *p < 0.05. (B) After hAMSCs treatment, the proportion of CD3+CD8+ T in liver, spleen, lung and gut was significantly lower than that of aGVHD group. *p < 0.05 and ***p < 0.001. (C) After hAMSCs treatment, the proportion of CD4+CD25+Foxp3+ Tregs in liver, spleen, and gut was significantly increased compared with that of aGVHD group. *p < 0.05. |
Inflammatory Cytokines Secretion in vivo
The hAMSCs treatment significantly inhibited the secretion of human inflammatory cytokines, including IL-17A, IFN-γ, and TNF from the target organs. Level of IL-17A was significantly decreased in lung (P=0.017) and gut (P=0.008) in aGVHD+hAMSCs group than that in aGVHD group (n=3) (Figure 6A). Level of IFN-γ was significantly decreased in liver (P=0.032) and gut (P=0.046) in aGVHD+hAMSCs group than that in aGVHD group (n=3) (Figure 6B). Level of TNF was significantly decreased of the liver (P=0.033) and blood (P=0.004) in aGVHD+hAMSCs group than that in aGVHD group (n=3) (Figure 6C). Especially in the gut, an important aGVHD target organ, we found that both IFN-γ, and IL-17A were significantly suppressed by hAMSCs. These results demonstrated that hAMSCs ameliorated human allogeneic acute GVHD by reduction of inflammatory cytokine secretion in target organs.
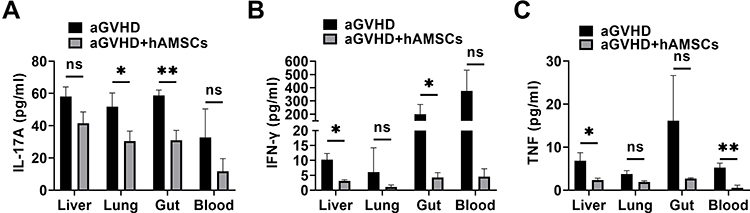 |
Figure 6 hAMSCs can inhibit the secretion of inflammatory cytokine in target organs. (A) IL-17A level in lung and gut were significantly decreased in aGVHD+hAMSCs group than that in aGVHD group. *p < 0.05 and **p < 0.01. (B) IFN-γ level in liver and gut were significantly decreased in aGVHD+hAMSCs group than that in aGVHD group. *p < 0.05. (C) TNF level in liver and blood were significantly decreased in aGVHD+hAMSCs group than that in aGVHD group. *p < 0.05 and **p < 0.01. |
Discussion
MSCs have been widely used in several clinical trials and numerous reports have shown that MSCs yielded a favorable therapeutic benefit for GVHD.17,31 Nevertheless, controversy about their safety and efficacy remains. Although bone marrow is the main source of MSCs, BM-MSCs have some deficiency that prevent wider application including invasive harvesting procedure, and the number of BM-MSCs declines with increasing cell aging, and the possibility of virus contamination.32,33 hAMSCs are readily available and easy to obtain from a waste product after delivery with abundant sources, no ethical and moral disputes, which have been reported to contain a population of multipotent stem cells exhibiting characteristics of MSCs. hAMSCs have no tumorigenicity, low immunogenicity and high histocompatibility.34 Furthermore, placenta is an organ that originates during the first period of embryological development and is fundamental for maintaining fetomaternal tolerance during pregnancy, which probably omen that hAMSCs might harbor similar characteristic as well.35 Therefore, amniotic membranes might be an alternative source of mesenchymal stem cells. The hAMSCs have intermediate levels of HLA MHC class I molecules, but do not have HLA class II antigens, FAS ligand, and the co-stimulatory molecules, and therefore, do not activate alloreactive T cells.36–38 The hAMSCs exhibited a fibroblast-shaped morphology and adherence to plastic, our data show that the cell-surface markers of the hAMSCs were positive for CD90, CD73 and CD105 while negative for CD45, CD11b, HLA-DR and CD34. These findings met the criteria for MSCs identification by International Society for Cell & Gene Therapy. Our observations further demonstrated GFP-labeled hAMSCs with a labeling rate of 84%. The hAMSCs labeled with GFP fluorescence could be reproducibly detected by immunofluorescence in the lung, liver, spleen, and gut at day one and day three, respectively, after cell infusion via the mouse tail vein. This phenomenon may be caused by secreted cytokines such as fibroblast growth factor, chemokine receptors, and stem cell homing factors39–41 secreted by hAMSCs and rich marginal blood flow in that region, which are beneficial for cell migration. We also found that from day one to day three, GFP labeled cells increased gradually and were observed in the lung but also can track to the liver, spleen, and gut. However, the number of hAMSCs gradually decreased over time in those organs. It has been reported that BM-MSCs were largely trapped in the lungs, liver, and spleen with abundant capillaries after intravenous transplantation.42 Our data were consistent with this research; we also found that hAMSCs reached target organs such as the small intestine and liver in early period. Therefore, hAMSCs exhibited homing ability to target damaged tissues, relieving the severity of inflammation and accelerating tissue repair. Homing of MSCs may also be associated with local microvascular changes, increased capillary permeability, hemostasis, and passive retention.
In the present study, we established an acute GVHD mouse model by adoptive transfer of hPBMCs into immunodeficient NOD-PrkdcscidIL2rγnull (NPG) mice,43,44 then analyzed the function of hAMSCs in the aGVHD mouse model. We revealed that hAMSCs therapy reduced villous blunting and lymphocyte infiltration of the gut while reducing inflammatory edema, tissue destruction and lymphocyte infiltration into the parenchyma of the liver and lung. In addition, hAMSCs suppressed of the CD3+CD4+ T and CD3+CD8+ T cell proportions and increased the expression of Tregs in target organs. The induction of immune tolerance involves a precise balance between activation and inhibition of T cell responses, which is important in the development of aGVHD.45 Tobin et al46 found that BM-MSCs blocked TNF-α secretion by dendritic cells via promotion of IL-10 and IL-4 secretion, which impeded T cell differentiation into Th1 cells, directing differentiation of these cells into Tregs and Th2 cells, respectively.47 A recent study showed that AMSCs reduced the activity of human CD8+T cells and TNF-α in the peripheral blood of GVHD mice. They also compared the immunomodulatory effects of AMSCs and BM-MSCs in vitro, and found that both AMSCs and BM-MSCs reduced the concentration of TNF-α and IFN-γ expressed by PBMCs.48 However, this study did not detect T cell and inflammatory cytokine levels in the target organs of aGVHD mice. We examined CD3+CD4+ T and CD3+CD8+ T cells in the main target organs of our aGVHD mice, and found a significant decrease in the proportion of CD3+CD4+ T and CD3+CD8+ T cells in these tissues of mice treated with hAMSCs compared to control group. MSCs further favor Tregs expansion in vitro indirectly by inhibiting dendritic cell maturation, and CD8+ T cell and NK cell expansion.49,50 Our observations demonstrated that the proportion of CD4+CD25+Foxp3+ Tregs cells was increased after hAMSCs treatment in the liver, spleen, and gut. Our data is consistent with the group that showed that murine CD4+CD25+Foxp3+ Tregs were induced during GVHD after allogeneic BMT, and the induction of these Tregs was positively correlated with the protection of GVHD in mice.51 GVHD involves a pathophysiology that includes host tissue damage, increased secretion of proinflammatory cytokines (TNF, IFN-γ, IL-1, IL-2, and IL-12), and activation of dendritic cells, macrophages, NK cells, and cytotoxic T cells.52 Inhibition of proinflammatory cytokines has been shown to be beneficial in resolution of the severity and incidence of GVHD.53,54 Although some in vitro studies have suggested that IL-10 or TGF-β may be involved in the suppression of MSCs,55,56 it remains unknown whether these molecules participate in the suppression mediated by hAMSCs in vivo. Here, we found that the amounts of IL-17A, IFN-γ, and TNF in target organs such as the liver, lung, and gut decreased after hAMSCs treatment. IL-17A was initially reported to be produced by T helper 17 (Th17) cells.57 In general, pro-inflammatory cytokines such as IFN-γ, TNF-α, IL-1α, or IL-1β have been extensively reported in MSCs activation in vitro.58 However, there are few studies on inflammatory cytokines by hAMSCs in vivo. Our data also showed that hAMSCs treatment inhibited IL-17A, INF-γ, and TNF, which are involved in the pathogenesis of GVHD target organs. Th17 cells and Th17-associated cytokines play a central role in the occurrence of aGVHD.59,60 IL-17 contributed to the development of aGVHD in recipient mice by recruiting or priming Th1 cells during the early stages of the disease, reflecting a shift from Th1 to Th17 cells in the physiopathology of aGVHD.59 A subset of Th17 cells in the gut has been described as having regulatory properties with high levels of IL-10 and low TNF and IL-2 production.61 We found that IL-17A, and IFN-γ were increased in the gut of the aGVHD model; after hAMSCs treatment, the levels of IL-17A, and IFN-γ were significantly decreased, which means hAMSCs can suppressed Th1 and Th17 cells in those aGVHD target organs. It has been well-established from both murine studies and immune reconstitution data in the clinic that the production of IFN-γ was an adverse effect of GVHD-associated cytokines.62–64 Aggarwal et al suggested that MSCs inhibited IFN-γ and increased IL-4 secretion, and may orchestrate a shift from the prominence of proinflammatory Th1 cells toward an increase in anti-inflammatory Th2 cells, beneficial for GVHD management.65 The hAMSCs possessed potent immunomodulatory properties capable of suppressing allogeneic T cell responses in vivo. The immune suppressive activity of hAMSCs in vivo was associated with a significant decrease in Th1 and Th17 cytokines, including IL-17A, IFN-γ, and TNF.
In summary, using humanized mice with a complete human immune system, we successfully established an aGVHD mouse model. Furthermore, our data also demonstrated that NPG mice could serve as a better model to study GVHD. Using this model, we demonstrated that hAMSCs could control acute GVHD by regulating the balance of Tregs and T effector cells. Our study provided a proof of concept of hAMSCs treatment to control GVHD, which providing a theoretical foundation for potentially preventing and treating GVHD clinical using hAMSCs alone or in combination with minimal conventional immunosuppression. Rigorous clinical research design needs to evaluate the safety and efficacy of hAMSCs in patients with steroid-refractory aGVHD after HSCT, which is thus a limitation of our study. Our follow-up research aimed at understanding the mechanisms underlying the immunomodulatory effects of hAMSCs on aGVHD afterallogeneic hematopoietic stem cell transplantation.
Conclusions
NPG mice are a valuable tool for studying GVHD induced by human immune cells. The hAMSCs could control acute GVHD by decreasing inflammatory cytokine secretion within target organs through modulating the balance of Tregs and T effector cells in aGVHD mice. Our study may provide a theoretical basis for the clinical application of hAMSCs.
Abbreviations
NPG, NOD-PrkdcscidIL2rγnull; SCID, severe combined immune deficiency; Prkdc, DNA-dependent protein kinase catalytic subunit; NOD-SCID, non-obese diabetic-SCID; NK, natural killer; IL2rγ, interleukin-2 receptor g common chain; NSG, NOD/SCID-IL2Rγnull; NOD, Cg-Prkdcscid IL2rγtm1Wjl/SzJ; NOG, NOD.Cg-Prkdcscid-IL2rγtm1Sug/Jic; MHC, major histocompatibility complex; HSCT, hematopoietic stem cell transplantation; xeno-GVHD, xenogeneic GVHD; GVHD, graft-versus-host disease; MSCs, mesenchymal stem cell; hAMSCs, human amniotic mesenchymal stem cells; BM-MSCs, bone marrow mesenchymal stem cells; PBMCs, peripheral blood mononuclear cells; Tregs, regulatory T cells.
Ethics Approval and Informed Consent
The study was approved by the Ethics Committee of Nanfang Hospital, Southern Medical University. Signed informed consent with permission to report individual patient data was obtained from all participants after a detailed description of the purposes of our study.
Acknowledgments
We would like to thank all staff at the Department of Laboratory Medicine of Nanfang Hospital of Southern Medical University for their kind assistance. We also thank all sample donors and their families who have kindly donated their sample.
Funding
The authors gratefully acknowledge the Natural Science Foundation of China (81701243, 81870145, 81772244, 82072334 and 81870144), the Science and Technology Planning Project of Guangzhou (201904010481), the Natural Science Fund of Guangdong Province (2020A1515010038), the Presidential Foundation of Nanfang Hospital of Southern Medical University (2017C030), and the Presidential Foundation of Zhujiang Hospital of Southern Medical University (yzjj2018rc03).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Ghimire S, Weber D, Mavin E, Wang XN, Dickinson AM, Holler E. Pathophysiology of GvHD and other HSCT-related major complications. Front Immunol. 2017;8:79. doi:10.3389/fimmu.2017.00079
2. Blazar BR, Murphy WJ, Abedi M. Advances in graft-versus-host disease biology and therapy. Nat Rev Immunol. 2012;12(6):443–458. doi:10.1038/nri3212
3. Wolf D, von Lilienfeld-toal M, Wolf AM, et al. Novel treatment concepts for graft-versus-host disease. Blood. 2012;119(1):16–25. doi:10.1182/blood-2011-08-339465
4. Wu X, Jiang J, Gu Z, Zhang J, Chen Y, Liu X. Mesenchymal stromal cell therapies: immunomodulatory properties and clinical progress. Stem Cell Res Ther. 2020;11(1):345. doi:10.1186/s13287-020-01855-9
5. Malard F, Huang XJ, Sim JPY. Treatment and unmet needs in steroid-refractory acute graft-versus-host disease. Leukemia. 2020;34(5):1229–1240. doi:10.1038/s41375-020-0804-2
6. Deeg HJ. How I treat refractory acute GVHD. Blood. 2007;109(10):4119–4126. doi:10.1182/blood-2006-12-041889
7. Tan Y, Xiao H, Wu D, et al. Combining therapeutic antibodies using basiliximab and etanercept for severe steroid-refractory acute graft-versus-host disease: a Multi-Center Prospective Study. Oncoimmunology. 2017;6(3):e1277307. doi:10.1080/2162402X.2016.1277307
8. Spaggiari GM, Capobianco A, Abdelrazik H, Becchetti F, Mingari MC, Moretta L. Mesenchymal stem cells inhibit natural killer-cell proliferation, cytotoxicity, and cytokine production: role of indoleamine 2,3-dioxygenase and prostaglandin E2. Blood. 2008;111(3):1327–1333. doi:10.1182/blood-2007-02-074997
9. Dazzi F, Lopes L, Weng L. Mesenchymal stromal cells: a key player in ‘innate tolerance? Immunology. 2012;137(3):206–213. doi:10.1111/j.1365-2567.2012.03621.x
10. Ben-Ami E, Berrih-Aknin S, Miller A. Mesenchymal stem cells as an immunomodulatory therapeutic strategy for autoimmune diseases. Autoimmun Rev. 2011;10(7):410–415. doi:10.1016/j.autrev.2011.01.005
11. Pittenger MF, Mackay AM, Beck SC, et al. Multilineage potential of adult human mesenchymal stem cells. Science. 1999;284(5411):143–147. doi:10.1126/science.284.5411.143
12. Rao MS, Mattson MP. Stem cells and aging: expanding the possibilities. Mech Ageing Dev. 2001;122(7):713–734. doi:10.1016/S0047-6374(01)00224-X
13. Lee JM, Jung J, Lee HJ, et al. Comparison of immunomodulatory effects of placenta mesenchymal stem cells with bone marrow and adipose mesenchymal stem cells. Int Immunopharmacol. 2012;13(2):219–224. doi:10.1016/j.intimp.2012.03.024
14. Kagia A, Tzetis M, Kanavakis E, et al. Therapeutic effects of mesenchymal stem cells derived from bone marrow, umbilical cord blood, and pluripotent stem cells in a mouse model of chemically induced inflammatory bowel disease. Inflammation. 2019;42(5):1730–1740. doi:10.1007/s10753-019-01033-x
15. Sharma RR, Pollock K, Hubel A, McKenna D. Mesenchymal stem or stromal cells: a review of clinical applications and manufacturing practices. Transfusion. 2014;54(5):1418–1437. doi:10.1111/trf.12421
16. Antoniadou E, David AL. Placental stem cells. Best Pract Res Clin Obstet Gynaecol. 2016;31:13–29.
17. Insausti CL, Blanquer M, García-Hernández AM, Castellanos G, Moraleda JM. Amniotic membrane-derived stem cells: immunomodulatory properties and potential clinical application. Stem Cells Cloning. 2014;7:53–63.
18. Ono M, Ohnishi S, Honda M, et al. Effects of human amnion-derived mesenchymal stromal cell transplantation in rats with radiation proctitis. Cytotherapy. 2015;17(11):1545–1559. doi:10.1016/j.jcyt.2015.07.003
19. Liu QW, Huang QM, Wu HY, et al. Characteristics and therapeutic potential of human amnion-derived stem cells. Int J Mol Sci. 2021;22(2):970. doi:10.3390/ijms22020970
20. Li J, Zhou Z, Wen J, Jiang F, Xia Y. Human amniotic mesenchymal stem cells promote endogenous bone regeneration. Front Endocrinol (Lausanne). 2020;11:543623. doi:10.3389/fendo.2020.543623
21. Hong JQ, Gao Y, Song J, Zhuo WB, Sun HT, Ping BH. Comparison of biological characteristics and immunosuppressive activity between human amniotic mesenchymal stem cells and human bone marrow mesenchymal stem cells. Zhongguo Shi Yan Xue Ye Xue Za Zhi. 2016;24(3):858–864.
22. Song J, Gao Y, Zhou WB, Yang CY, Xu Y, Ping BH. Immunomodulatory effects of human amniotic versus bone marrow-derived mesenchymal stem cells on peripheral blood T lymphocytes in vitro. Nan Fang Yi Ke Da Xue Xue Bao. 2017;37(6):780–785.
23. Yamahara K, Harada K, Ohshima M, et al. Comparison of angiogenic, cytoprotective, and immunosuppressive properties of human amnion- and chorion-derived mesenchymal stem cells. PLoS One. 2014;9(2):e88319. doi:10.1371/journal.pone.0088319
24. Sun H, Hou Z, Yang H, et al. Multiple systemic transplantations of human amniotic mesenchymal stem cells exert therapeutic effects in an ALS mouse model. Cell Tissue Res. 2014;357(3):571–582. doi:10.1007/s00441-014-1903-z
25. Pianta S, Bonassi Signoroni P, Muradore I, et al. Amniotic membrane mesenchymal cells-derived factors skew T cell polarization toward Treg and downregulate Th1 and Th17 cells subsets. Stem Cell Rev Rep. 2015;11(3):394–407. doi:10.1007/s12015-014-9558-4
26. Yamahara K, Hamada A, Soma T, et al. Safety and efficacy of amnion-derived mesenchymal stem cells (AM01) in patients with steroid-refractory acute graft-versus-host disease after allogeneic haematopoietic stem cell transplantation: a study protocol for a phase I/II Japanese trial. BMJ Open. 2019;9(7):e026403. doi:10.1136/bmjopen-2018-026403
27. Schroeder MA, DiPersio JF. Mouse models of graft-versus-host disease: advances and limitations. Dis Model Mech. 2011;4(3):318–333. doi:10.1242/dmm.006668
28. Lucas PJ, Shearer GM, Neudorf S, Gress RE. The human antimurine xenogeneic cytotoxic response. I. Dependence on responder antigen-presenting cells. J Immunol. 1990;144(12):4548–4554.
29. Ito R, Takahashi T, Katano I, Ito M. Current advances in humanized mouse models. Cell Mol Immunol. 2012;9(3):208–214. doi:10.1038/cmi.2012.2
30. Cooke KR, Kobzik L, Martin TR, et al. An experimental model of idiopathic pneumonia syndrome after bone marrow transplantation: I. The roles of minor H antigens and endotoxin. Blood. 1996;88(8):3230–3239. doi:10.1182/blood.V88.8.3230.bloodjournal8883230
31. Godoy JAP, Paiva RMA, Souza AM, Kondo AT, Kutner JM, Okamoto OK. Clinical translation of mesenchymal stromal cell therapy for graft versus host disease. Front Cell Dev Biol. 2019;7:255. doi:10.3389/fcell.2019.00255
32. Lim JE, Son Y. Endogenous stem cells in homeostasis and aging. Tissue Eng Regen Med. 2017;14(6):679–698. doi:10.1007/s13770-017-0097-3
33. Knuth CA, Kiernan CH, Palomares Cabeza V, Lehmann J, Witte-Bouma J, Ten Berge D. Isolating pediatric mesenchymal stem cells with enhanced expansion and differentiation capabilities. Tissue Eng Part C Methods. 2018;24(6):313–321. doi:10.1089/ten.tec.2018.0031
34. Umezawa A, Hasegawa A, Inoue M, et al. Amnion-derived cells as a reliable resource for next-generation regenerative medicine. Placenta. 2019;84:50–56. doi:10.1016/j.placenta.2019.06.381
35. Evangelista M, Soncini M, Parolini O. Placenta-derived stem cells: new hope for cell therapy? Cytotechnology. 2008;58(1):33–42. doi:10.1007/s10616-008-9162-z
36. Di Nicola M, Carlo-Stella C, Magni M, et al. Human bone marrow stromal cells suppress T-lymphocyte proliferation induced by cellular or nonspecific mitogenic stimuli. Blood. 2002;99(10):3838–3843. doi:10.1182/blood.V99.10.3838
37. Blanco B, Herrero-Sánchez MD, Rodríguez-Serrano C, et al. Immunomodulatory effects of bone marrow versus adipose tissue-derived mesenchymal stromal cells on NK cells: implications in the transplantation setting. Eur J Haematol. 2016;97(6):528–537. doi:10.1111/ejh.12765
38. Huang Y, Yin Y, Gu Y, et al. Characterization and immunogenicity of bone marrow-derived mesenchymal stem cells under osteoporotic conditions. Sci China Life Sci. 2020;63(3):429–442. doi:10.1007/s11427-019-1555-9
39. Keith MC, Bolli R. “String theory” of c-kit(pos) cardiac cells: a new paradigm regarding the nature of these cells that may reconcile apparently discrepant results. Circ Res. 2015;116(7):1216–1230. doi:10.1161/CIRCRESAHA.116.305557
40. Eseonu OI, De Bari C. Homing of mesenchymal stem cells: mechanistic or stochastic? Implications for targeted delivery in arthritis. Rheumatology (Oxford). 2015;54(2):210–218. doi:10.1093/rheumatology/keu377
41. Nitzsche F, Müller C, Lukomska B, Jolkkonen J, Deten A, Boltze J. Concise review: MSC adhesion cascade-insights into homing and transendothelial migration. Stem Cells. 2017;35(6):1446–1460. doi:10.1002/stem.2614
42. Thompson M, Mei SHJ, Wolfe D, et al. Cell therapy with intravascular administration of mesenchymal stromal cells continues to appear safe: an updated systematic review and meta-analysis. E Clin Med. 2020;19:100249.
43. King MA, Covassin L, Brehm MA, Racki W, Pearson T, Leif J. Human peripheral blood leucocyte non-obese diabetic-severe combined immunodeficiency interleukin-2 receptor gamma chain gene mouse model of xenogeneic graft-versus-host-like disease and the role of host major histocompatibility complex. Clin Exp Immunol. 2009;157(1):104–118. doi:10.1111/j.1365-2249.2009.03933.x
44. King M, Pearson T, Shultz LD, Leif J, Bottino R, Trucco M. A new Hu-PBL model for the study of human islet alloreactivity based on NOD-scid mice bearing a targeted mutation in the IL-2 receptor gamma chain gene. Clin Immunol. 2008;126(3):303–314. doi:10.1016/j.clim.2007.11.001
45. Thangavelu G, Blazar BR. Achievement of tolerance induction to prevent acute graft-vs.-host disease. Front Immunol. 2019;10:309. doi:10.3389/fimmu.2019.00309
46. Tobin LM, Healy ME, English K, Mahon BP. Human mesenchymal stem cells suppress donor CD4(+) T cell proliferation and reduce pathology in a humanized mouse model of acute graft-versus-host disease. Clin Exp Immunol. 2013;172(2):333–348. doi:10.1111/cei.12056
47. Amorin B, Alegretti AP, Valim V, et al. Mesenchymal stem cell therapy and acute graft-versus-host disease: a review. Hum Cell. 2014;27(4):137–150. doi:10.1007/s13577-014-0095-x
48. Tago Y, Kobayashi C, Ogura M, et al. Human amnion-derived mesenchymal stem cells attenuate xenogeneic graft-versus-host disease by preventing T cell activation and proliferation. Sci Rep. 2021;11(1):2406. doi:10.1038/s41598-021-81916-y
49. Cahill EF, Tobin LM, Carty F, Mahon BP, English K. Jagged-1 is required for the expansion of CD4+ CD25+ FoxP3+ regulatory T cells and tolerogenic dendritic cells by murine mesenchymal stromal cells. Stem Cell Res Ther. 2015;6(1):19. doi:10.1186/s13287-015-0021-5
50. Melief SM, Schrama E, Brugman MH, et al. Multipotent stromal cells induce human regulatory T cells through a novel pathway involving skewing of monocytes toward anti-inflammatory macrophages. Stem Cells. 2013;31(9):1980–1991. doi:10.1002/stem.1432
51. Koreth J, Ritz J. Tregs, HSCT, and acute GVHD: up close and personal. Blood. 2013;122(10):1690–1691. doi:10.1182/blood-2013-07-514125
52. Dong W, Wu X, Ma S, et al. The mechanism of anti-PD-L1 antibody efficacy against PD-L1-negative tumors identifies NK cells expressing PD-L1 as a cytolytic effector. Cancer Discov. 2019;9(10):1422–1437. doi:10.1158/2159-8290.CD-18-1259
53. Bruner RJ, Farag SS. Monoclonal antibodies for the prevention and treatment of graft-versus-host disease. Semin Oncol. 2003;30(4):509–519. doi:10.1016/S0093-7754(03)00250-1
54. Choi SW, Reddy P. Current and emerging strategies for the prevention of graft-versus-host disease. Nat Rev Clin Oncol. 2014;11(9):536–547. doi:10.1038/nrclinonc.2014.102
55. Ng J, Hynes K, White G, et al. Immunomodulatory properties of induced pluripotent stem cell-derived mesenchymal cells. J Cell Biochem. 2016;117(12):2844–2853. doi:10.1002/jcb.25596
56. El-Sherbiny YM, El-Jawhari JJ, Moseley TA, McGonagle D, Jones E. T cell immunomodulation by clinically used allogeneic human cancellous bone fragments: a potential novel immunotherapy tool. Sci Rep. 2018;8(1):13535. doi:10.1038/s41598-018-31979-1
57. Steinman L. A brief history of TH17, the first major revision in the TH1/TH2 hypothesis of T cell–mediated tissue damage. Nat Med. 2007;13(2):139–145. doi:10.1038/nm1551
58. Cheung TS, Bertolino GM, Giacomini C, Bornhäuser M, Dazzi F, Galleu A. Mesenchymal stromal cells for graft versus host disease: mechanism-based biomarkers. Front Immunol. 2020;11:1338. doi:10.3389/fimmu.2020.01338
59. Kim KW, Moon SJ, Park MJ, et al. Optimization of adipose tissue-derived mesenchymal stem cells by rapamycin in a murine model of acute graft-versus-host disease. Stem Cell Res Ther. 2015;6(1):202. doi:10.1186/s13287-015-0197-8
60. Gartlan KH, Markey KA, Varelias A, Bunting MD, Koyama M, Kuns RD. Tc17 cells are a proinflammatory, plastic lineage of pathogenic CD8+ T cells that induce GVHD without antileukemic effects. Blood. 2015;126(13):1609–1620. doi:10.1182/blood-2015-01-622662
61. Esplugues E, Huber S, Gagliani N, et al. Control of TH17 cells occurs in the small intestine. Nature. 2011;475(7357):514–518. doi:10.1038/nature10228
62. Capitini CM, Herby S, Milliron M, Anver MR, Mackall CL, Fry TJ. Bone marrow deficient in IFN-{gamma} signaling selectively reverses GVHD-associated immunosuppression and enhances a tumor-specific GVT effect. Blood. 2009;113(20):5002–5009. doi:10.1182/blood-2008-11-187385
63. Merli P, Caruana I, De Vito R, et al. Role of interferon-γ in immune-mediated graft failure after allogeneic hematopoietic stem cell transplantation. Haematologica. 2019;104(11):2314–2323. doi:10.3324/haematol.2019.216101
64. Alvarado LJ, Huntsman HD, Cheng H, et al. Eltrombopag maintains human hematopoietic stem and progenitor cells under inflammatory conditions mediated by IFN-γ. Blood. 2019;133(19):2043–2055. doi:10.1182/blood-2018-11-884486
65. Aggarwal S, Pittenger MF. Human mesenchymal stem cells modulate allogeneic immune cell responses. Blood. 2005;105(4):1815–1822. doi:10.1182/blood-2004-04-1559
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.