")
Back to Journals » Journal of Inflammation Research » Volume 15
HSP70 Ameliorates Septic Acute Kidney Injury via Binding with TRAF6 to Inhibit of Inflammation-Mediated Apoptosis
Authors Zhang Y, Song C, Ni W, Pei Q, Wang C, Ying Y, Yao M
Received 16 December 2021
Accepted for publication 25 March 2022
Published 5 April 2022 Volume 2022:15 Pages 2213—2228
DOI https://doi.org/10.2147/JIR.S352717
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Ning Quan
Yiqiu Zhang,1,* Chenlu Song,1,* Wei Ni,1,2 Qing Pei,1 Caixia Wang,1 Youguo Ying,3 Min Yao1
1Department of Plastic and Reconstructive Surgery, Shanghai Ninth People’s Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, People’s Republic of China; 2Wuhan National Laboratory for Optoelectronics, Huazhong University of Science and Technology, Wuhan, People’s Republic of China; 3Department of Intensive Care Unit, Shanghai Ninth People’s Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Min Yao; Youguo Ying, Shanghai Ninth People’s Hospital, Shanghai Jiao Tong University School of Medicine, 639 Zhizaoju Road, Shanghai, 200011, People’s Republic of China, Email [email protected]; [email protected]
Purpose: Acute kidney injury (AKI) is one of the most severe complications of sepsis, the pathological features of which are excessive inflammation and programmed cell death of resident renal cells. Heat shock protein 70 (HSP70) is a critical stress protein for repressing inflammation, however, its role in AKI is not fully understood. The current study aimed to determine the protective effect of HSP70 on septic AKI and its underlying mechanisms.
Methods: Hsp70.1 knockout and wildtype mice were used for creating sepsis model by cecal ligation and puncture (CLP). Renal function, histological changes, pro-inflammatory cytokines, and apoptosis were analyzed with H&E, PAS, ELISA, western-blot, and immunofluorescence. Moreover, the effects of HSP70 on renal proximal tubular epithelial (HK-2) cells with LPS were assessed by measuring the levels of nuclear factor kappa B (NF-κB) signaling and downstream cytokines, viability, and apoptosis using western-blot, qRT-PCR, flow-cytometry, and immunofluorescence. Immunoprecipitate and immunoblotting were used for determining the interaction of HSP70 with tumor necrosis factor receptor-associated factor 6 (TRAF6). Exogenous HSP70 was applied to further identify its biological significance at the cellular and animal level.
Results: Hsp70.1 deficiency significantly aggravated renal dysfunction with increasing serum levels of BUN, SCr, kidney injury molecule-1 (KIM-1), and neutrophil gelatinase-associated lipocalin (NGAL), and shortened survival in CLP mice. Furthermore, hsp70.1 knockout caused renal-tissue structural damage, especially proximal tubular, and inflammatory cascade and increased apoptotic cells, along with elevated Bax, caspase 3 and cleaved caspase 3, as well as decreased Bcl2 in vivo and vitro. Significantly, HSP70 directly interacted with TRAF6 in HK-2 cells, leading to suppression of inflammatory response and apoptosis. Moreover, exogenous HSP70 alleviated renal damage, decreased apoptosis and elevated survival rate in septic AKI in vivo and vitro.
Conclusion: Our findings demonstrated that HSP70 played a critical role in sepsis-induced AKI via interaction with TRAF6 and inhibiting inflammation and apoptosis.
Keywords: heat shock protein 70, sepsis, acute kidney injury, tumor necrosis factor receptor-associated factor 6, nuclear factor kappa B, apoptosis
Introduction
Sepsis is a dysregulated systemic response syndrome to infection, leading to extensive tissue damage and multiple organs dysfunction. In sepsis, acute kidney injury (AKI) is one of the most severe and common diseases.1,2 It was reported that in intensive care units, approximately 22–67% septic patients would progress into AKI, which shortened their survival time and increased economic burden.3 The current therapies of AKI included renal replacement therapy (RRT) and supportive treatments, such as antimicrobial therapy and restoration of microcirculatory perfusion. However, the survival outcome is far from satisfaction with the mortality rate over 50%.4 Therefore, in order to find more effective treatments, it is of great significances to explore the underlying pathological mechanism of septic AKI, as well as potential biological targets.
A lot of researches indicated that abnormal inflammatory response was involved in the development of AKI.5,6 As a known, NF-κB signaling acted as an important factor to AKI. In classical pathway, pathogen-associated molecular patterns (PAMPs) in circulation, with endotoxin lipopolysaccharide (LPS) included, interact with toll-like receptors (TLR) on tubular cells and myeloid differentiation protein 88 (MyD88), followed by the recruitment of IL-1 receptor associated kinase (IRAK) and tumor necrosis factor receptor-associated factor 6 (TRAF6). IκB kinase α/β (IKKα/β) is stimulated to phosphorylate and subsequently degrade IκBα. Then NF-κB dimers (p65, p50) translocate to the nucleus and induce the inflammatory gene transcription (tnf-α, il-6).7–9 These circulatory cytokines in turn could bind receptors on cell membranes to amplify the inflammatory response and promote renal cell apoptosis, leading to the occurrence of septic AKI.10,11
Heat shock protein 70 (HSP70), one of major chaperone proteins, plays a pivotal role in refolding, disassembly, and transport of proteins in normal cell growth and pathophysiological conditions. HSP70 is encoded by two genes (hsp70.1, hsp70.3) that are 99% homology and separated by 7 kb12 in mouse. The hsp70.1 gene was the main function part of Hsp7013,14 Current studies have demonstrated that the induction of HSP70 could prevent the activation of NF-κB cascade by increasing IκBα synthesis in human bronchial epithelial cells and squamous cell carcinoma.15,16 It was reported that HSP70 protected liver from I/R injury via suppressing NF-κB signaling pathway, and ultimately prolonged subjects’ survival time.17,18 In vitro experiments showed that upregulation of HSP70 in kidney had a positive effect on cell proliferation by decreasing oxidative stress via MEK/ERK signaling pathway.19 In the model of ischemic tubular injury, HSP70 overexpression was able to suppress tubular cell apoptosis through downregulating mitochondrial Bax and caspase 3.20 However, the role of HSP70 still remains unclear in septic AKI. Therefore, to solve the problem, in vivo and in vitro experiments were conducted in this study to explore the effect of HSP70 on septic AKI and its underlying molecular mechanism.
Materials and Methods
Reagents
Recombinant human HSP70 (HSP70) protein was purchased from R&D Systems (AP-100-100, Minneapolis, MN). The human renal proximal tubular epithelial (HK-2) cells and human embryonic kidney (HEK) 293T cells were obtained from Cell bank of Chinese Academy Science (China). Lipopolysaccharide (LPS) was achieved from Sigma-Aldrich (USA).
Animals
The present study was approved by the Animal Ethics Committee of the Shanghai Ninth People’s Hospital. All mice were kept on the C57BL/6 background and maintained in a standard, specific pathogen-free facility environment (22–24 °C, 12 h light/dark cycle) with free access to food and water ad libitum. HSP70 (hsp70.1) knockout mice were generated by Cyagen Biosciences Inc. through CRISPR/Cas9-mediated genome editing (Serial Number: KOCMP-15511-Hsp70.1). C57BL/6 mice were purchased from Shanghai Laboratory Animal Center, Chinese Academy of Sciences, China. Animal model of sepsis was induced by cecal ligation and puncture (CLP) as described in previous studies.21 The experimental mice (6–8 weeks old, 20–25 g) were anesthetized by intraperitoneal injection of 1% sodium pentobarbital (100 mg/kg). Midline laparotomy was performed (approximately 1 cm). The cecum was ligated below the ileocecal valve by 4–0 silk ligatures and punctured using a 21-gauge needle. Then the cecum was extruded a few of feces and was returned to the abdomen. The incision was closed. The sham mice underwent the same surgical procedures without ligation and puncture. After the operation, all mice received fluid resuscitation with 0.9% saline (mg/kg) by subcutaneous injection. Mice were injected through inner canthus veniplex with recombinant human HSP70 protein of 50 μg/kg alone or at 15 min before CLP. Blood and renal tissue were collected from all mice at 24 h after the operation. Meanwhile, survival analyses were performed regularly for consecutive 5 days.
A total of 72 HSP70 gene mice were used with CLP, including hsp70.1+/+, hsp70.1+/−, and hsp70.1−/− (each group N = 8). In another experiment, a total of 150 C57BL/6 mice were randomly assigned into five groups (n = 10) as control (no surgery), sham, HSP70, CLP, HSP70+CLP. All experiments were repeated for three times.
Histology and TUNEL Assay
Mouse kidney tissues were fixed in 4% paraformaldehyde for 24 h, paraffin-embedded, then sectioned at 5 μm and stained with hematoxylin-eosin (HE) staining and periodic acid-Schiff (PAS) staining. An optical microscope (Nikon TE2000-U; Japan) was used for histological evaluations. The apoptosis of renal tissues was evaluated using Terminal deoxynucleotidyl transferase dUTP-mediated nick-end labeling (TUNEL) assay kit (Sigma, USA). Briefly, after above tissue treatment, slices were labeled with terminal deoxynucleotidyl transferase and nucleotides including tetramethylrhodamine-labeled dUTP in a TUNEL reaction solution and observed by fluorescence microscopy (Olympus, Japan).
Cell Culture and Transfection
The human renal proximal tubular epithelial (HK-2) cells and human embryonic kidney (HEK) 293T cells were cultured in DMEM (Dulbecco’s modified Eagle’s medium) (Gibco, California, USA) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin (Thermo Fisher Scientific, USA) in a humidified atmosphere at 37°C with 5% CO2. To establish sepsis model in vitro, HK-2 cells were treated with LPS (0, 0.5, 1, 5, 10 μg/mL) for 6–12-24 h. To evaluate the protective effect of HSP70, HK-2 cells were pretreated with HSP70 (0, 0.5, 1, 2.5, 5 μg/mL) for 15 min before LPS stimulation (5 μg/mL).
For HSP70 gene knockdown, HK-2 was transfected with the short hairpin RNA (shRNA) targeting HSP70 (shHSP70-1, shHSP70-2, shHSP70-3) and negative control (sh-NC) according to the manufacturer’s instructions (GenePharma, Shanghai, China). The targeting sequence was 5’-GGUCCUAAGAAUCGUUCAATT-3’ (shHSP70-1), 5’-CUCCCUUUGAGCAUUGAAUTT-3’ (shHSP70-2), 5’-GGAGCUGACAAGUACUUGUTT-3’ (shHSP70-3) and 5’-UUCUCCGAACGUGUCACGUTT-3’ (sh-NC) respectively. For overexpression, HEK293T cells were transfected with lentiviral plasmid pLenti-CMV-HSP70-GFP-Puro and two lentiviral packaging plasmids (psPAX2 and pMD2.G) with empty vector as a control (Public Protein/Plasmid Library, Nanjing, China) using Lipofectamine 3000 (Invitrogen, Carlsbad, CA). After 48 h, the HEK293T cells supernatant was collected, filtered and concentrated to infect HK-2 cells for establishing stable HSP70-overexpression cell lines.
Cell Proliferation Assay
Cell viability was measured by Cell Counting Kit-8 (CCK8, Beyotime, Shanghai, China). In brief, cells were seeded in 96-well plate. At incubation of various time (0, 24, 48, 72, 96 h), add 10 μL CCK8 solution to each well and incubate for 2 h at 37 °C. Then absorbance at 450nm were measured by an Infinite 200 PRO microplate reader (Tecan, Austria). Crystal violet staining is a quick and reliable method for screening cell survival and death in molecular studies.22 HK-2 cells were seeded in a 6-well plate and incubated for 2–3 days at 37 °C until reaching 40–50% confluence. Then the medium was removed and the cells were washed twice using PBS. After that, cells were dyed with 1 mL 0.5% crystal violet staining solution for 20 minutes at room temperature (RT). Lastly, the cells were incubated in 1 mL methanol for 20 min, and then washed using PBS before taking the images. Quantifications of density were calculated using ImageJ software (National Institutes of Health, MD, USA).
Reverse Transcription‐quantitative PCR (RT‐qPCR)
The TRIzol Reagent (Invitrogen, USA) was used to isolate the total RNA. PrimeScript™ RT Master Mix kit (Takara, Kusatsu, Shiga, Japan) was applied for reverse transcription. The qPCR was performed using SYBR Green PCR Kit (Qiagen, Valencia, CA). The sequences of primers were purchased from Sangon Biotech (Shanghai, China) in Table 1. The relative expression of TNF-α and IL-6 was calculated using the 2−ΔΔCt method with endogenous GAPDH as the control.
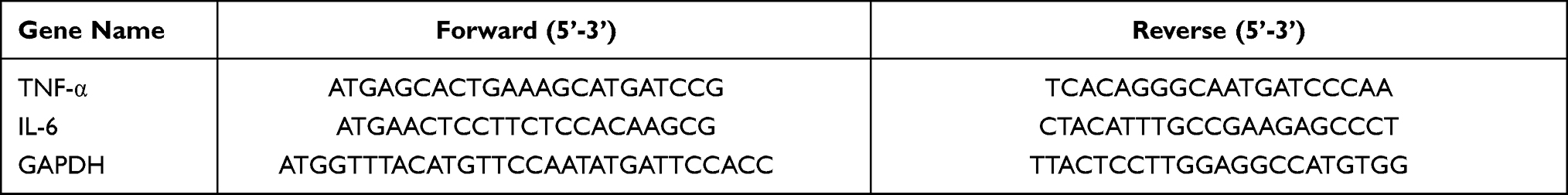 |
Table 1 Lists of Primers Sequences in RT-qPCR |
Western Blotting
Both kidney tissue and HK-2 cells were lysed in RIPA lysis buffer containing protease inhibitor cocktail. Protein concentrations were measured using a BCA protein assay kit (Invitrogen, USA). Protein samples in 10% sodium dodecyl sulfate-polyacrylamide difluoride (SDS-PAGE) membranes were separated and then transferred to polyvinylidene fluoride membranes (PVDF) (Millipore, MA). After blocking by 5% non-fat milk at RT for 1 h, membranes were incubated with primary antibodies against Hsp70.1 antibody (1:1000; PA5-97846, ThermoFisher), HSP70 (1:1000; ab79852, Abcam), TRAF6 (1:1000; ab137452, Abcam), GAPDH (1:1000; 9485, Abcam), IκBα (1:1000; ab32518, Abcam), p-IκBα (1:1000; ab133462, Abcam), p65 (1:1000; ab32536, Abcam), p-p65 (1:1000; ab222494, Abcam), TNF-α (1:1000; ab183218, Abcam), IL-6 (1:1000; ab259341, Abcam), Bax (1:1000; ab32503, Abcam), Bcl2 (1:1000; ab194583, Abcam), caspase 3 (1:1000; ab184787, Abcam) overnight at 4 °C. After washing with Tris-Buffered Saline Tween-20 (TBST) buffer on the next day, the membranes were incubated in secondary antibodies (Goat Anti-Rabbit IgG, 1:5000, ab205718, Abcam) at RT for 2 h. At last, protein bands were treated with chemiluminescence detection kit (Thermo Fisher Scientific, USA) and detected by Fusion-capture system (Fusion, France).
Renal Function Assessment and Enzyme-Linked Immunosorbent Assay (ELISA)
Blood samples were collected at 24 h after CLP and then centrifuged at 3000 rpm for 15 min at room temperature. Supernatant liquid was obtained and kept in - 80 °C refrigerator. Blood urea nitrogen (BUN) and serum creatinine (SCr) were determined by chromatometry with assay kits (BioAssay Systems, Hayward, CA). Serum kidney injury molecule‐1 (KIM-1; Cat#MKM100), neutrophil gelatinase‐associated lipocalin (NGAL; Cat#MLCN20), TNF-α (Cat#MTA00B) and IL-6 (Cat#M6000B) were examined by commercial ELISA kits (R&D Systems, Minneapolis, MN). HK-2 cells were pretreated with HSP70 and LPS stimulation, and cell Supernatants were collected at 24 h and centrifuged following above methods. TNF-α (Cat#DTA00D) and IL-6 (Cat#QK206) were examined by ELISA kits (R&D Systems, Minneapolis, MN).
Flow Cytometry
The Annexin V, 633 Apoptosis Assay Kit (Dojindo, Japan) was used to detect the apoptotic fraction of HK-2 cells. Briefly, after 24 h treatment of LPS (5 μg/mL), HK-2 cells were collected, centrifugated and then added with 5 μL Annexin V solution for 20 min. Then propidium iodide (PI) solution was added and cells were incubated for 10 min. The fluorescence values were assessed by FACS Calibur system (BD Biosciences, NJ, USA). The flow cytometry data was analyzed using Flowjo software Version 10 (Treestar, USA).
Immunoprecipitation and Co-Immunoprecipitation Assays
HEK293T cells were transfected with the plasmids of FLAG-HSP70 and HA-TRAF6 (Public Protein/Plasmid Library, Nanjing, China). After 24 h, cells were harvested with RIPA buffer and incubated with 1 μg anti-HA antibody (ab1424, Abcam) and 20 μL Protein A/G beads (Santa Cruz Biotechnology, USA) overnight at 4 °C. To detect the endogenous interaction of HSP70 and TRAF6, HK-2 cells were lysed and incubated with 3 μg anti-IgG (ab172730, Abcam) or anti-HSP70 (ab5442, Abcam) antibody and 20 μL Protein A/G beads overnight at 4 °C. Proteins were analyzed by Western blotting followed by an anti-HA (1:1000) and anti-FLAG (1:1000; F7425, Sigma) antibody. Endogenous proteins were detected using anti-TRAF6 antibody (1:1000; ab137452, Abcam).
Immunofluorescence (IF)
Immunofluorescence of proximal tubules (PTs)’ marker aquaporin-1 (AQP-1) was observed to identify histopathological changes of both luminal and basal borders.23 The fixative tissues were sectioned out, permeabilized with 0.1% Tween-20 and washed with PBS for 5 min. Then sections were blocked with 2% BSA for 20 min and incubated with anti-AQP-1 antibody (1:100; ab168387, Abcam) for 60 min. After washing, tissue sections were incubated with anti-Alexa Fluor 555 antibody (1:500; ab150078, Abcam) for 60 min and washed with PBS. Sections were incubated with phalloidin-iFluor 488 reagent (1:20; ab176753, Abcam) for 60 min to stain actin filament. The cell nuclei were stained with DAPI (1:100; #62248, Thermo Fisher Scientific, USA) for 5 min and washed by PBS. Slices were viewed using fluorescence microscopy (Olympus, Japan).
HK-2 cells were fixed on slices and blocked. The cells were incubated with anti-Mouse HSP70 (1:200; ab2787) and anti-Rabbit TRAF6 (1:200; ab137452, Abcam) antibodies or anti-Rabbit p65 antibody (1:200; ab32536, Abcam) overnight at 4 °C. Then anti-Mouse Alexa Fluor 555 (1:1000; ab150118, Abcam), anti-Rabbit Alexa Fluor 488 (1:1000; ab150077, Abcam) and anti-Rabbit Alexa Fluor 555 (1:1000; ab150078, Abcam) antibodies were added as second antibodies at RT for 1 h. Cell nuclei were stained with DAPI. Images were taken by confocal microscopy (LSM710, Carl Zeiss, Germany) or fluorescence microscopy (Olympus, Japan).
Statistical Analysis
Continuous variables were presented as the Mean ± SD. Differences between groups were analyzed using the Mann–Whitney test. Survival curves was carried out by Kaplan-Meier method and the survival differences were measured using the Log rank test. P values < 0.05 were considered to be statistically significant. All experiment data were analyzed using the GraphPad Prism version 8.0 software (GraphPad, San Diego, CA, USA).
Results
HSP70 Deficiency Aggravates Renal Damage in Sepsis-Induced AKI in vivo
To evaluate the protective effects of HSP70 on AKI, animal model with sepsis was established in the hsp70.1−/− mice using CLP. The hsp70.1−/− mice started to die at 4 h as earliest and all of mice died by 40 h post-CLP. The survival rate in hsp70.1−/− was significantly lower, compared with that in the hsp70.1+/+ group, (Figure 1A, P < 0.001). To assess the function of kidney, BUN, SCr, KIM-1, and NGAL were measured by chromatometry and ELISA. These protein levels were dramatically increased in hsp70.1−/− mice with CLP, compared to that in the controls (Figure 1B–E, P < 0.001). To estimate whether HSP70 deficiency was able to amplify the inflammatory cascades induced by CLP, the systemic levels of inflammatory mediators were determined. As expected, the levels of TNF-α and IL-6 in hsp70.1−/− group were significantly elevated compared to that in the hsp70.1+/+ group (Figure 1F, G, P < 0.001). Meanwhile, kidney tissues in HSP70 deficiency mice showed remarkably increased damages characteristics histologically with tubular brush border loss, proximal tubular dilatation and vacuolization by HE and PAS staining, but hsp70.1+/+mice showed a moderate dilated tubular only (Figure 1H and I).
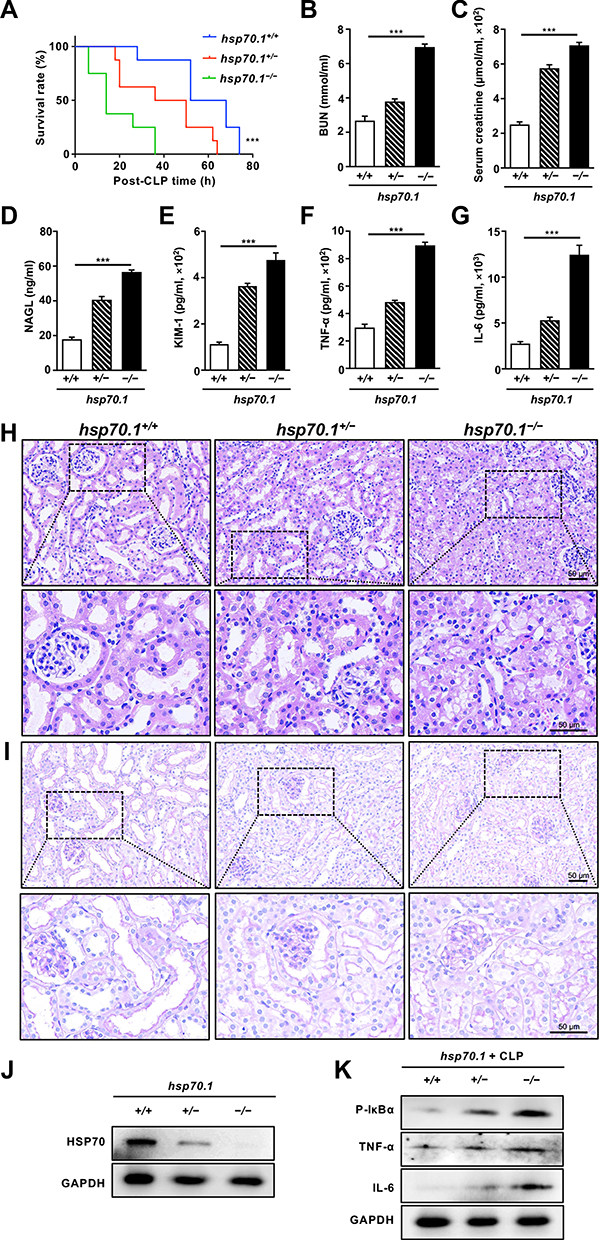 |
Figure 1 Effects of HSP70 deficiency on renal damage in vivo. Abbreviations: h, hour; CLP, cecal ligation and puncture; BUN, blood urea nitrogen; SCr, serum-creatinine; NGAL, neutrophil gelatinase‐associated lipocalin; KIM-1, kidney injury molecule‐1; TNF-α, tumor necrosis factor-α; IL-6, interleukin 6; HE, hematoxylin-eosin; PAS, periodic acid-Schiff; WB, Western blotting. Notes: HSP70 gene knockout mice were treated with CLP and estimated for survival rate (A). At 24 h post-CLP, serum BUN (B) and SCr (C) were determined by chromatometry. Serum NAGL (D), KIM-1 (E), TNF-α (F) and IL-6 (G) were measured by ELISA. Renal histological changes were observed by HE and PAS staining (H and I). The protein level of HSP70 and CLP-induced inflammatory factors (p-IκBα, TNF-α and IL-6) in hsp70.1−/−mice was confirmed by WB (J and K). Data are presented as mean ± SD of data. All experiments were repeated for three times. N = 8. ***P< 0.001 hsp70.1−/− vs hsp70.1+/+ group. Bar = 50 μm. |
AQP-1, as a water transporter, highly expressed in the proximal tubular epithelial cells.24,25 In the Figure 2A, the red staining of AQP-1 was observed in PTs using IF. Hsp70.1−/− mice tissue revealed proximal tubular destruction and elevated expressions of AQP-1. Furthermore, in hsp70.1−/− mice, the protein level of HSP70 showed dramatically suppressed using Western blotting (Figure 1J). Meanwhile, inflammatory mediators p-IκBα, TNF-α and IL-6 were increased under CLP (Figure 1K).
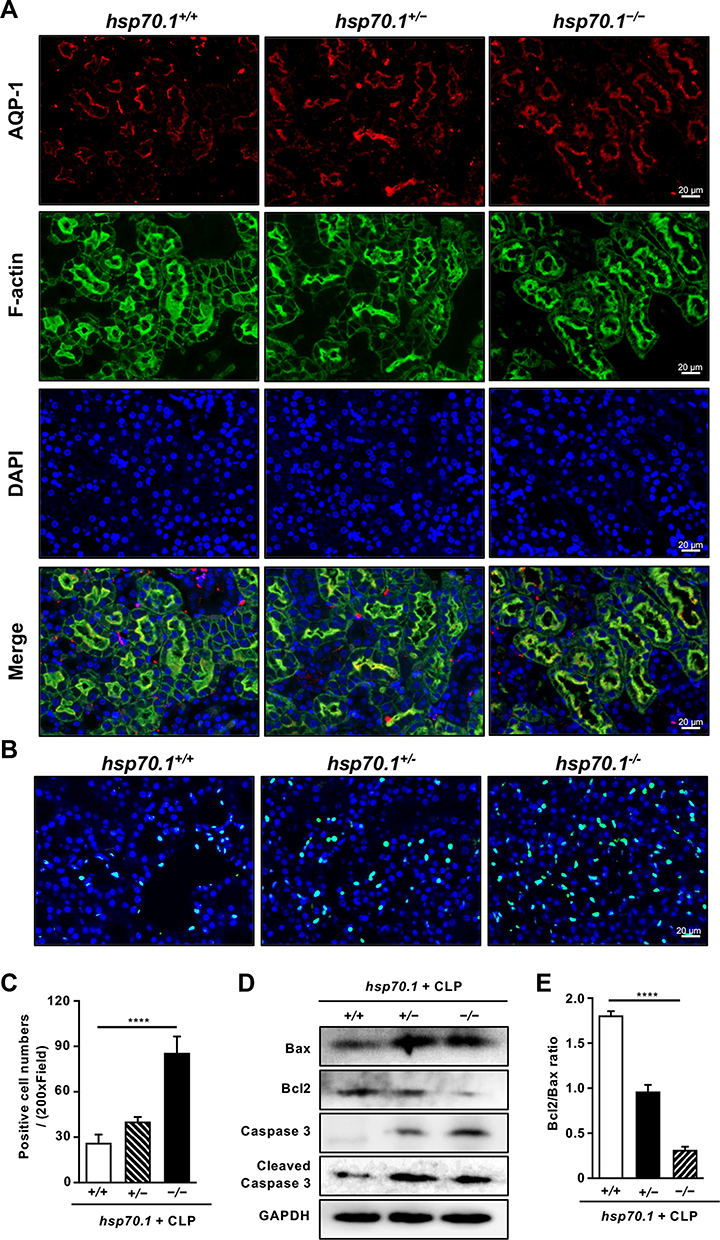 |
Figure 2 HSP70 deficiency aggravates the proximal tubulars’ injury and apoptosis in septic AKI in vivo. Abbreviations: CLP, cecal ligation and puncture; AQP-1, aquaporin-1; TUNEL, Terminal deoxynucleotidyl transferase dUTP-mediated nick-end labeling; WB, Western blotting. Notes: At 24 h post-CLP of gene knockout mice, the level of specific marker AQP-1 was detected in proximal tubulars by IF (A). AQP-1, red; F-actin, green; DAPI, blue. The level of dead cells was assessed by TUNEL staining (B and C). TUNEL positive cell, green; DAPI, blue. The apoptotic protein levels of Bax, Bcl2, caspase 3 and cleaved caspase 3 were measured by WB (D). Bcl2/Bax ratio was calculated (E). Data are presented as mean ± SD. All experiments were repeated for three times. N = 8. ****P< 0.0001 hsp70.1−/− vs hsp70.1+/+ group. Bar = 20 μm. |
In addition, cell apoptosis is the other important mechanism during pathological process of AKI occurrence. Using TUNEL staining, the numbers of positive cells were increased to 4 times of that in hsp70.1+/+ mice (Figure 2B and C, P < 0.0001). The protein levels of Bax, caspase 3, cleaved caspase 3 were up-regulated and Bcl2 was down-regulated in gene deletion group (Figure 2D). Bcl2/Bax ratio was presented in Figure 2E. All results illustrated that HSP70 deficiency aggravated renal dysfunction, pathological damage, inflammatory reaction, and tissue apoptosis in septic AKI.
HSP70 Suppresses NF‐κB Signaling-Induced Inflammation and Apoptosis of LPS-Induced Sepsis in vitro
To construct the septic model in vitro, HK-2 cells were stimulated with different concentrations of LPS (Figure 3A). The relative levels of TNF-α were much higher at 24 h than that at 6 and 12 h, and there was no significant difference between the protein levels at 5 μg/mL and that at 10 μg/mL. Thus, 5 μg/mL was chosen as the optimal concentration for inducing TNF-α at 24 h. The treatment of exogenous HSP70 significantly reduced the levels of NF‐κB signaling protein (IκBα, p-IκBα, p65, p-p65) and the downstream of inflammatory factors including TNF-α and IL-6, in a dose-dependent manner after LPS stimulation (Figure 3B). TNF-α and IL-6 were also downregulated in cell supernatant (Figure 3C and D). The results indicated that NF‐κB signaling pathway-mediated inflammation involved in the effect of HSP70 on renal sepsis. To determine whether exogenous HSP70 regulates cell apoptosis, the cell survival was analyzed by CCK8 and flow cytometry. HK-2 cells’ viability was increased by the HSP70 treatment after LPS stimulation (Figure 3E, P < 0.01). Under induction of LPS, HSP70 intervention obviously declined apoptotic cells using the Annexin-V and PI staining (Figure 3F and G, P < 0.05).
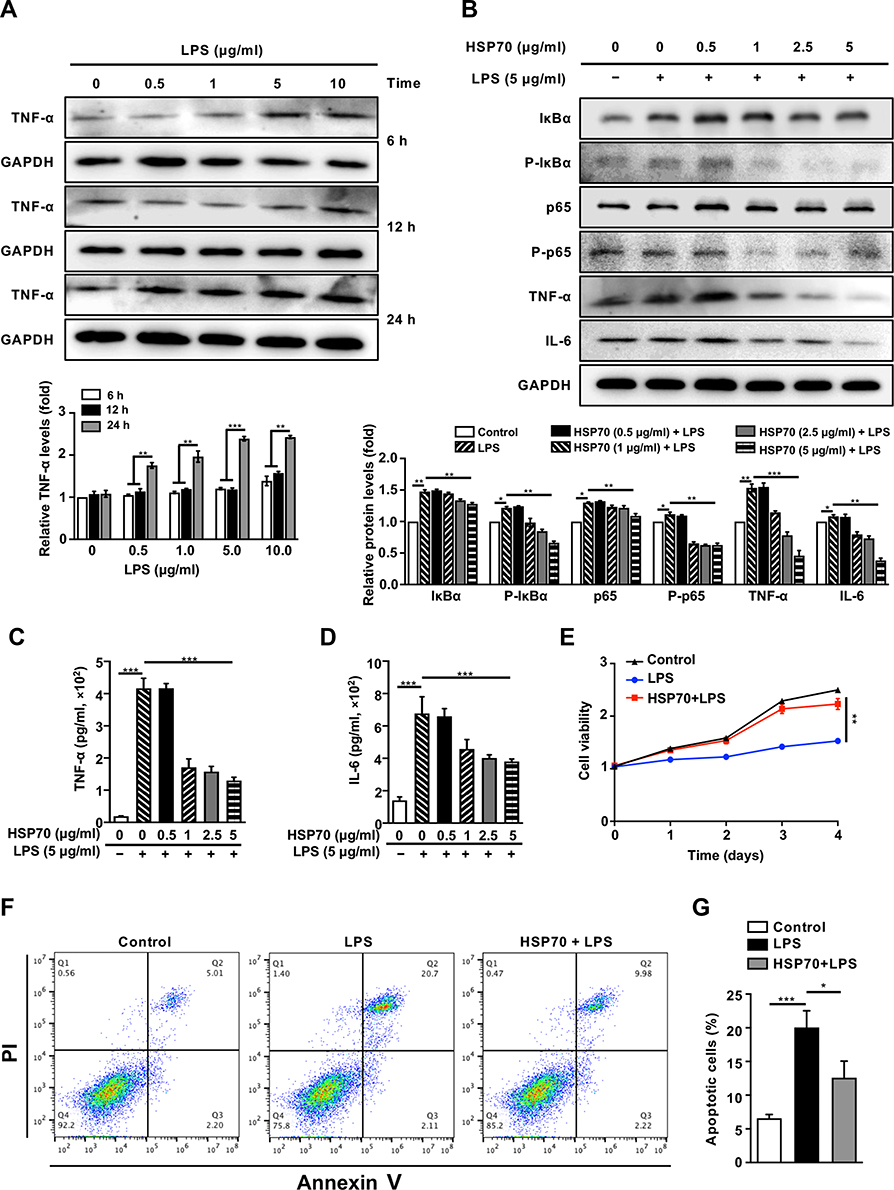 |
Figure 3 Inhibition of exogenous HSP70 on NF‐κB signaling pathway and apoptosis of LPS-induced sepsis in vitro. Abbreviations: h, hour; HK-2, human renal proximal tubular epithelial; LPS, lipopolysaccharide; TNF-α, tumor necrosis factor-α; IL-6, interleukin 6; WB, Western blotting; CCK8, cell counting kit-8; PI, propidium iodide. Notes: HK-2 cells were treated with different concentrations of LPS to construct the septic model. The level of TNF-α was measured by WB (A). In HK-2 cells pre-treated with various concentrations of exogenous HSP70 after LPS stimulation (5 μg/mL) for 24 h, the levels of TNF-α, and other proteins of NF‐κB signaling (such as IκBα, p-IκBα, p65, p-p65 and IL-6) were measured by WB (B). Cytokines (TNF-α, IL-6) of cell supernatant were measured by ELISA (C and D). In HSP70 (5 μg/mL) intervention before LPS stimulation (5 μg/mL), cell viability was evaluated by CCK8 (E), and dead cells were analyzed by flow cytometry with Annexin-V and PI staining (F and G). Data are presented as mean ± SD. Triplicate experiments were repeated for three times. *P< 0.05, **P< 0.01, ***P< 0.001. |
To further explore the underlying mechanism in HK-2 cells, hsp70 gene was knockdown and overexpressed for determining cell function and screening signaling pathway. As shown in Figure 4A, the level of HSP70 was dramatically declined by shRNA or increased by overexpression. Furthermore, HSP70 knockdown significantly increased the protein levels of NF‐κB signaling pathways (TRAF6 and p-IκBα) and related inflammatory mediators (TNF-α, IL-6). In contrast, HSP70 overexpression led to the reduction of aforementioned proteins (Figure 4B). Additionally, the gene expressions of TNF-α and IL-6 were consistent with the pattern of proteins (Figure 4C and D, P < 0.01). To confirm whether endogenous HSP70 could modulate the cell apoptosis, we assessed the cell survival and apoptotic proteins. In shRNA transfection cells under LPS stimulation, cell viabilities were reduced using CCK8 assay and crystal violet staining (Figure 4E and F, P < 0.01), which were reversed in HSP70 OE cells (Figure 4G and H, P < 0.01). Under LPS stimulation, apoptotic cells were distinctly diminished in HSP70 overexpressed cells and increased in HSP70 knockdown cells using flow cytometry (Figure 4I and J, P < 0.01). The levels of apoptosis-related proteins, including Bax, caspase 3, and cleaved caspase 3, were reduced, then Bcl2 was elevated in HSP70 overexpressed cells using WB (Figure 4K). Bcl2/Bax ratio was presented in Figure 4L. The data demonstrated that HSP70 noteworthily suppressed NF‐κB signaling pathway and cell apoptosis in septic HK-2 cells.
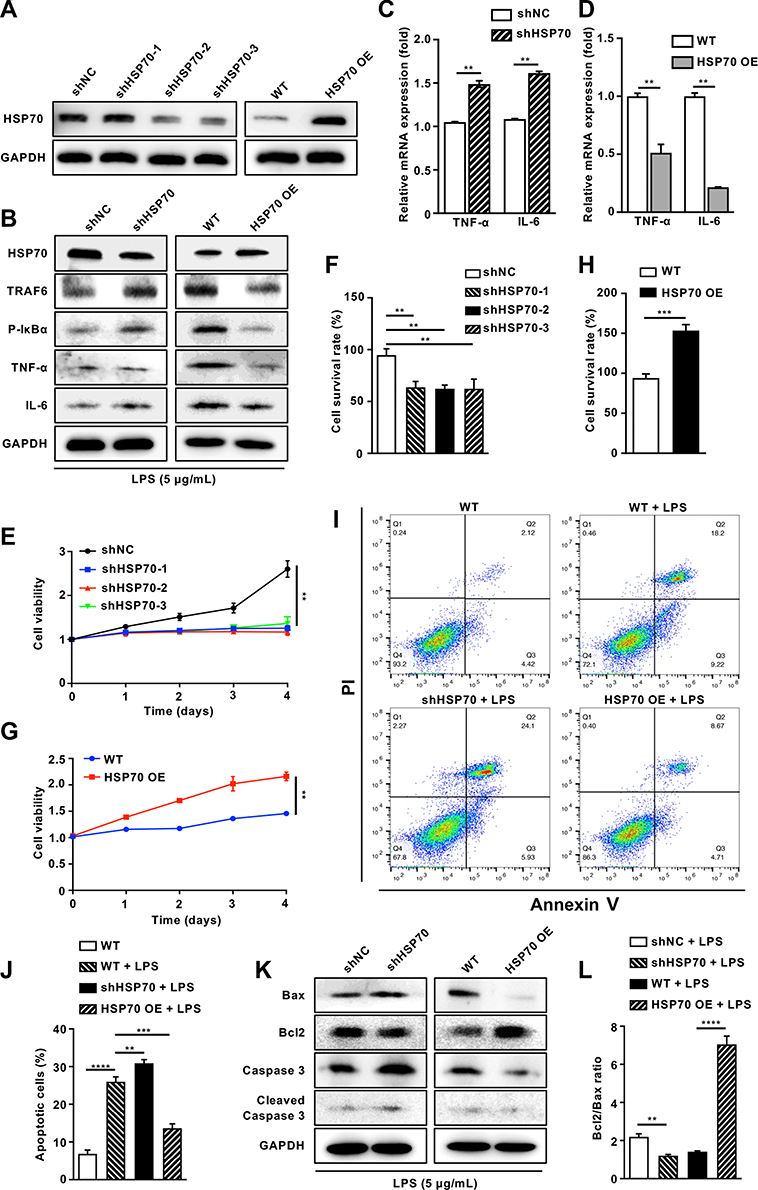 |
Figure 4 Knockdown or overexpression of HSP70 mediates NF‐κB signaling pathway and apoptosis of LPS-induced sepsis in vitro. Abbreviations: h, hour; HK-2, human renal proximal tubular epithelial; LPS, lipopolysaccharide; TNF-α, tumor necrosis factor-α; IL-6, interleukin 6; WB, Western blotting; CCK8, cell counting kit-8; PI, propidium iodide. Notes: HK-2 cells with gene knockdown and overexpress of HSP70 were established and the protein level of HSP70 was confirmed with WB (A). After LPS stimulation (5 μg/mL) for 24 h, the proteins of HSP70, NF‐κB signaling pathways (TRAF6 and p-IκBα) and related inflammatory mediators (TNF-α, IL-6) were measured by WB (B), the mRNA expressions of TNF-α and IL-6 were measured by qRT-PCR (C, D), the viability was measured by a CCK8 assay (E and G) and crystal violet staining (F and H) in HSP70 knockdown and overexpress HK-2 cells. Apoptotic cells were analyzed by flow cytometry with Annexin-V and PI staining under LPS stimulation (5 μg/mL) for 24 h (I and J). The levels of apoptosis-related protein Bax, Bcl2, caspase 3 and cleaved caspase 3 were measured by WB after LPS stimulation (5 μg/mL) for 24 h (K). Bcl2/Bax ratio was calculated (L). Data are presented as mean ± SD. Triplicate experiments were repeated for three times. **P< 0.01, ***P< 0.001, ****P< 0.0001. |
HSP70 Inhibits NF‐κB-Mediated Inflammation Through Binding to TRAF6 in vitro
HSP70, as a chaperone protein, plays the biological role via targeting other proteins, thus we explored whether HSP70 bound toTRAF6 that was upstream of NF‐κB. HA-TRAF6 and FLAG-HSP70 were co-expressed in HEK293T cells, then IP was performed with anti-HA antibody. The band of FLAG-HSP70 was seen with antibody against FLAG as shown in Figure 5A. Moreover, IP was performed by anti-HSP70 antibody in HK-2 cells. The TRAF6-HSP70 binding was also observed by anti-TRAF6 antibody (Figure 5B). In addition, the merged image of yellow cytoplasm indicated there was an interaction between HSP70 and TRAF6 by IF in HK-2 cells (Figure 5C). The results demonstrated that HSP70 can bind to TRAF6. Following, we measured p65 level in the downstream of NF-κB using IF. Under LPS stimulation, the red staining of p65 was increased in nucleus and reduced in cytoplasm in HK-2 cells. HSP70 overexpression remarkably reduced p65 aggregation of cell nucleus, indicating the inhibition of NF-κB signaling pathway (Figure 5D). The results confirmed that HSP70 could diminish NF-κB signaling through binding to TRAF6.
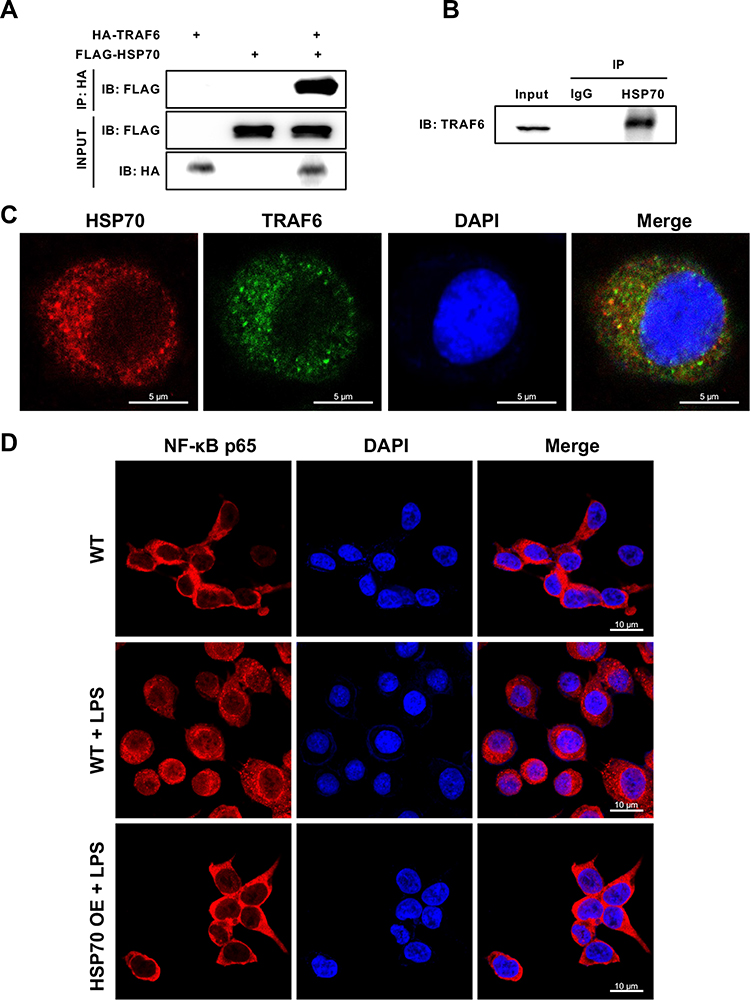 |
Figure 5 HSP70 inhibits NF‐κB-mediated inflammation through binding to TRAF6 in vitro. Abbreviations: IP, Immunoprecipitation; WB, Western blotting; IF, immunofluorescence. Notes: HA-TRAF6 and FLAG-HSP70 were co-expressed in HEK293T cells by plasmid transfection. Then IP was performed with an anti-HA antibody. After 24 h, the protein of FLAG-HSP70 was detected using anti-FLAG antibody by WB (A). In HK-2 cells, IP was performed by anti-HSP70 antibody. The protein of TRAF6 was measured using anti-TRAF6 antibody by WB (B). Images of HSP70 and TRAF6 in HK-2 cells were observed by IF (C). HSP70, red; TRAF6, green; DAPI, blue. The p65 level in the downstream of NF-κB pathway was detected by IF in HK-2 cells under LPS stimulation (5 μg/mL) for 24 h (D). p65, red; DAPI, blue. Triplicate experiments were repeated for three times. h: hour. Bar = 5 μm C, 10μm D. |
Exogenous HSP70 Ameliorates Renal Damage via Inhibition of Tissue Apoptosis in Sepsis‐induced AKI
To verify above results, recombinant human HSP70 protein was used in control and CLP group. As shown in Figure 6A, CLP mice started to die at 24 h and all died by 72 h post-CLP. Intervention of HSP70 significantly extended the survival time that caused by CLP (P < 0.05). However, the effects were not obvious in HSP70 group. Meanwhile, HSP70 treatment attenuated levels of BUN, SCr, KIM-1 and NGAL after CLP detected using chromatometry and ELISA, as well as TNF-α and IL-6 (Figure 6B–G, P < 0.01). Using HE and PAS staining, CLP mice exhibited exudation of fluid and erythrocyte, loss of brush border and dilatation of proximal tubular. These histological destructions were obviously improved in HSP70+CLP group and non-significant in HSP70 group (Figure 6H and I). Furthermore, the levels of p-IκBα, TNF-α, and IL-6 were elevated post-CLP, which was reversed by treatment with HSP70 (Figure 6L). Lastly, the TUNEL-positive cells were diminished in HSP70+CLP group, along with protein levels of Bax, caspase 3, and cleaved caspase 3 (Figure 6J,K and M, P < 0.01). The change of Bcl2 conflicted with aforementioned. Bcl2/Bax ratio was presented in Figure 6N. The results demonstrated that exogenous HSP70 played a critical role in protecting renal function from CLP-induced septic AKI through inhibition of apoptosis and inflammation.
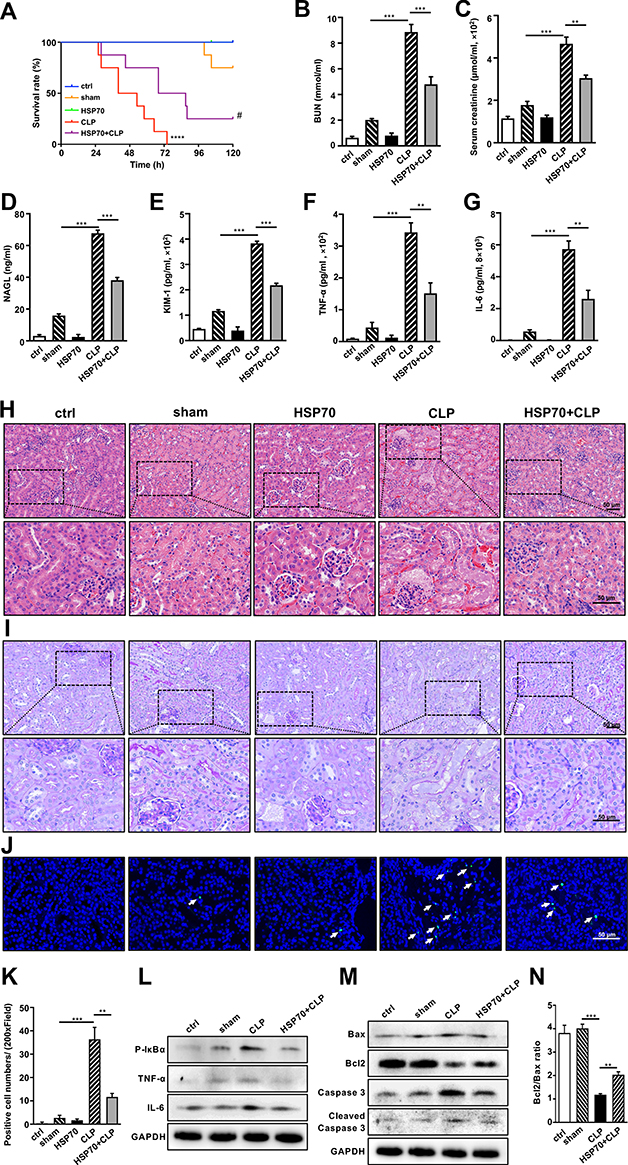 |
Figure 6 Exogenous HSP70 ameliorates renal damage via inhibition of inflammation-mediated apoptosis in sepsis‐induced AKI in vivo. Abbreviations: h, hour; CLP, cecal ligation and puncture; BUN, blood urea nitrogen; SCr, serum-creatinine; NGAL, neutrophil gelatinase‐associated lipocalin; KIM-1, kidney injury molecule‐1; TNF-α, tumor necrosis factor-α; IL-6, interleukin 6; HE, hematoxylin-eosin; PAS, periodic acid-Schiff; TUNEL, Terminal deoxynucleotidyl transferase dUTP-mediated nick-end labeling; WB, Western blotting. Notes: C57BL/6 mice, randomly divided into five groups (control, sham, HSP70, CLP, HSP70+CLP), were assessed for survival rate (A). At 24 h post-surgery, serum BUN (B) and SCr (C) were determined by chromatometry with kits. Serum NAGL (D), KIM-1 (E), TNF-α (F) and IL-6 (G) were measured by ELISA. Renal histological changes were observed by HE and PAS staining (H and I). The number of dead cells was assessed by TUNEL staining (J and K). The arrows indicated positive staining in kidney. TUNEL positive cell, green; DAPI, blue. The protein levels of p-IκBα, TNF-α and IL-6 were confirmed by WB (L). The apoptosis-related proteins of Bax, Bcl2, caspase 3 and cleaved caspase 3 were measured by WB (M). Bcl2/Bax ratio was calculated (N). Data were presented as mean ± SD. All experiments were repeated for three times. N = 10. ** P< 0.01, *** P< 0.001, **** P< 0.0001 CLP vs sham group, HSP70+CLP vs CLP group. # P< 0.05 HSP70+CLP vs CLP group. Bar = 50 μm. |
Discussion
Sepsis refers to life-threatening organ dysfunction that is the most common cause of acute kidney injury. Despite the underlying mechanism of septic AKI is not fully elucidated, it is reported that the systemic inflammatory response plays a pivotal role in pathological progress of disease.26,27 Our data demonstrated that HSP70 significantly alleviated renal dysfunction, tissue structural damage, inflammatory cascade, and prolonged survival through inhibiting NF-κB signaling activities and inflammation-related apoptosis in septic AKI. To our knowledge, this is the first study to report that HSP70 exerted protective function, as an inhibitor of inflammation and apoptosis, through interaction with TRAF6 in septic AKI in vivo and in vitro (Figure 7).
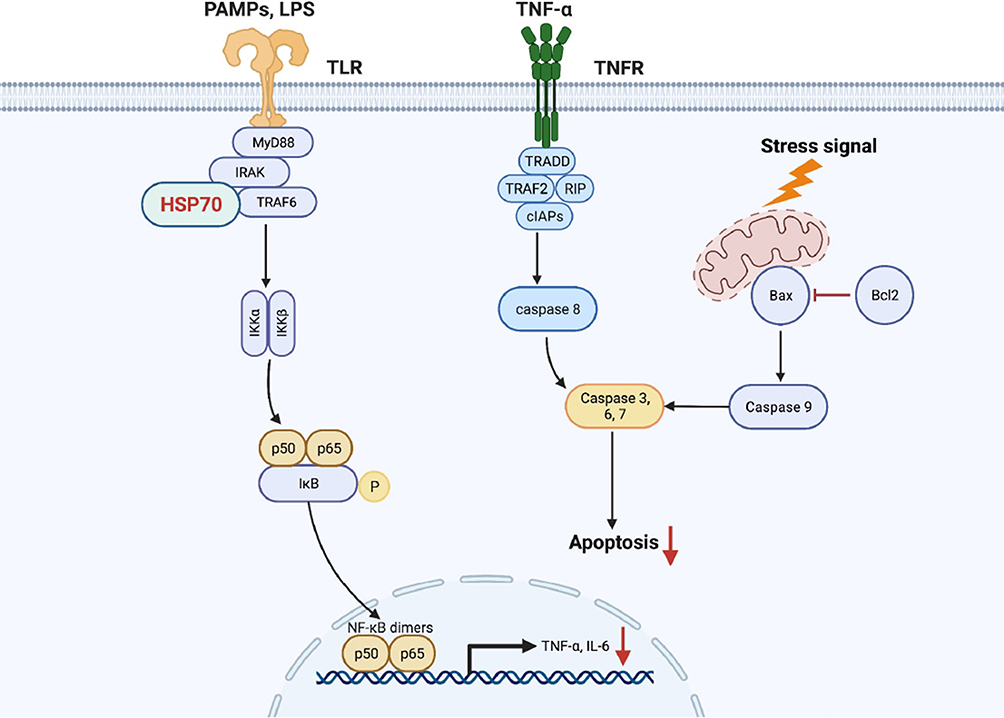 |
Figure 7 The proposed mechanism by that HSP70 ameliorates inflammation and apoptosis in renal tubular epithelial cells in septic acute kidney injury. Notes: HSP70 interacted with TRAF6 and repressed NF-κB signaling activities (IκBα, p-IκBα, p65, p-p65), thereby alleviating p65 translocation to the nucleus and downstream inflammatory proteins (TNF-α, IL-6). Subsequently, pro-apoptotic Bax was suppressed as well as caspase 3 and cleaved caspase 3, and anti-apoptotic Bcl2 was increased, inhibiting the process of cell apoptosis. |
Heat shock proteins (HSPs) are highly conserved proteins that act as molecular chaperones for protein folding, membrane translocation and intracellular assembly. Among them, HSP70 is known as an important protein for its beneficial effect combating the oxygen free radicals, inflammation, toxics and ischemia.28–30 A number of literatures have been addressed to the role of HSP70 in AKI. HSP70 was abundantly expressed in the Brown Norway rats, and for that reason, the rats showed strong resistance to renal dysfunction and morphological changes from ischemic injury.31 In addition, HSP70 overexpression was able to ameliorate renal tubular damage in ischemia/reperfusion (I/R) injury.20 However, no studies were conducted to explore the concrete effect of HSP70 in AKI that induced by sepsis. Here, with the model of CLP-sepsis, HSP70-deficient mice showed increased mortality, abnormal kidney functions (BUN, SCr, KIM-1, NGAL), and destructive pathological changes especially in proximal tubulars. The overexpression or intervention of HSP70 greatly improved cell survival in LPS-induced HK-2 cells. These data indicated that HSP70 presented the protective effect in septic AKI in vivo and in vitro. In addition, HSP70 prevented renal inflammatory response via inhibiting NF-κB signaling pathway. In detail, in vivo and in vitro experiments showed that HSP70 reduced NF-κB inflammatory proteins (TRAF6, IκBα, p-IκBα, p65, p-p65) and its related inflammatory cytokines (TNF-α and IL-6). These results were consistent with other experiments that heat shock protein 70 could suppress the activation of NF-κB signaling. In LPS-induced septic shock, extracellular HSP70 protected against myocardial and hepatic dysfunction, with associated inhibition of inflammatory mediators including TNF-α and iNOS, through the MAPK/NF-κB pathway.32 The induction of HSP70 has been demonstrated to be protective in ischemic acute renal failure via inhibition of NF-κB inflammatory activation and cell death pathway.33 Moreover, as shown in our experiments, overexpression of HSP70 strongly alleviated the translocation of p65 from the cytoplasm to the nucleus under LPS stimulation. Similar results were also verified in other experiments. Previous studies showed that HSP70 inhibited the nuclear translocation of p65 and prevented the degradation of IκBα to terminate inflammatory responses in LPS-induced macrophage and dendritic cells.34,35 Therefore, we verified the essential role of HSP70 in diminishing renal damages and inflammatory cascade in septic acute kidney injury.
The major activation of NF-κB signaling pathway depends on recruitment of TRAF6, which subsequently phosphorylates IκBα and translocates p65/p50 to the nucleus, leading to the gene transcription of tnf-α and il-6. Indeed, our data showed that HSP70 could interact with TRAF6 to downregulate NF-κB signaling pathway in renal proximal tubular epithelial (HK-2) cells. Some studies have verified the relationship between HSP70 and TRAF6.Huaqun Chen took the first to report that HSP70 interacted with TRAF6-C domain and inhibited its ubiquitination to prevent LPS-induced NF-κB activation in macrophages.36 The finding was also supported by Li-Chao Wang’s investigation that HSP70-TRAF6 binding blocked K63-linked ubiquitination of TRAF6 and inactivated NF-κB signaling pathway in neuroinflammation pathogenesis.37 Based on above convincing discoveries, we could reasonably presume that HSP70 may inhibit ubiquitination of TRAF6 to prevent downstream inflammatory response in septic AKI, which will be carried out in the subsequent study.
As well known, inflammatory infiltration in turn was able to induce the initiation and progress of apoptosis in AKI. The extrinsic apoptosis pathway is initiated by death receptors on cell membrane with TNF-α, FasL, TRAKIL.38 TNF-α, as a popular apoptosis stimulus in AKI, binds TNF receptor (TNFR), leading to the recruitment of TNFR1-associated death domain (TRADD), TNFR-associated factor 2 (TRAF2), cellular inhibitor of apoptosis proteins (cIAPs), receptor-interacting protein (RIP), and the activation of the initiator caspase 8.39 And that, the intrinsic pathway of apoptosis is mediated by mitochondrial dysfunction in response to intracellular stimuli such as hypoxia, oxide free radical and calcium overload.40 Subsequently, the activation of pro-apoptotic Bax neutralizes anti-apoptotic Bcl2, leading to destruction of mitochondrial function and membrane integrity, and auto-activation of initiator caspase 9. The upstream initiators result in the stimulation of downstream executor caspase-3, −6 and −7 for substrate cleavage, inducing cell death.41,42 Cell apoptosis has been defined as a key factor in the pathogenesis of septic acute kidney injury. The suppression of apoptosis could improve the subjects’ survival time in sepsis.43,44 Apoptotic tubular cells were obviously observed in kidney biopsy of septic patients using TUNEL and caspase-3 staining, in consistent with that in mice model of CLP-induced AKI.45,46 In this study, in vivo and in vitro experiments showed that amounts of apoptotic cells were viewed in renal tissue sections and cells of HSP70 deficiency. Overexpression or intervention of HSP70 presented the protective role against acute kidney injury due to its inhibition on cell apoptosis, and this could be confirmed by the levels of apoptosis-related proteins. The findings demonstrated that abundant HSP70 greatly decreased levels of Bax, caspase 3 as well as cleaved caspase 3, increased levels of Bcl2 and ratio of Bcl2/Bax. The protective mechanism of HSP70 is potentially due to the suppression of inflammatory cytokine TNF-α and intracellular stimulus, thereby inhibiting apoptosis and cell death in septic AKI. Based on current experiments, the strong evidence demonstrated that HSP70 ameliorates renal damage via inhibition of cell apoptosis in septic AKI.
Conclusion
In summary, our findings revealed that HSP70 alleviated renal damage via interaction with TRAF6 to inhibit of inflammation-mediated cell apoptosis in sepsis-induced acute kidney injury. Current research demonstrated that HSP70 could be a potential therapeutic target for septic AKI.
Data Sharing Statement
The original data are included in the article, and further information can be in contact with the corresponding author.
Ethics Statement
The animal experiments in this study were reviewed and approved by the Animal Ethics Committee of the Shanghai Ninth People’s Hospital. The protocols and animal maintenance were carried out according to the guidelines of Laboratory Animal care and Use Committee of Shanghai Ninth People’s Hospital.
Acknowledgments
We would like to thank Pro. Jinke Cheng for their technical support.
Author Contributions
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work.
Funding
This study was supported by the National Natural Science Foundation of China (No. 81772118).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Peerapornratana S, Manrique-Caballero CL, Gomez H, Kellum JA. Acute kidney injury from sepsis: current concepts, epidemiology, pathophysiology, prevention and treatment. Kidney Int. 2019;96(5):1083–1099. doi:10.1016/j.kint.2019.05.026
2. Gando S, Shiraishi A, Yamakawa K, et al. Role of disseminated intravascular coagulation in severe sepsis. Thromb Res. 2019;178:182–188. doi:10.1016/j.thromres.2019.04.025
3. Skube SJ, Katz SA, Chipman JG, Tignanelli CJ. Acute Kidney Injury and Sepsis. Surg Infect (Larchmt). 2018;19(2):216–224. doi:10.1089/sur.2017.261
4. Dellepiane S, Marengo M, Cantaluppi V. Detrimental cross-talk between sepsis and acute kidney injury: new pathogenic mechanisms, early biomarkers and targeted therapies. Crit Care. 2016;20:20. doi:10.1186/s13054-016-1198-4
5. Zhou Y, Xu W, Zhu H. CXCL8(3-72) K11R/G31P protects against sepsis-induced acute kidney injury via NF-kappaB and JAK2/STAT3 pathway. Biol Res. 2019;52(1):29. doi:10.1186/s40659-019-0236-5
6. Kaushal GP. Autophagy protects proximal tubular cells from injury and apoptosis. Kidney Int. 2012;82(12):1250–1253. doi:10.1038/ki.2012.337
7. Qian Y, Commane M, Ninomiya-Tsuji J, Matsumoto K, Li X. IRAK-mediated translocation of TRAF6 and TAB2 in the interleukin-1-induced activation of NFkappa B. J Biol Chem. 2001;276(45):41661–41667. doi:10.1074/jbc.M102262200
8. Kopp E, Medzhitov R. Recognition of microbial infection by toll-like receptors. Curr Opin Immunol. 2003;15(4):396–401. doi:10.1016/S0952-7915(03)00080-3
9. Plociennikowska A, Hromada-Judycka A, Borzecka K, Kwiatkowska K. Co-operation of TLR4 and raft proteins in LPS-induced pro-inflammatory signaling. Cell Mol Life Sci. 2015;72(3):557–581. doi:10.1007/s00018-014-1762-5
10. Bellomo R, Kellum JA, Ronco C, et al. Acute kidney injury in sepsis. Intensive Care Med. 2017;43(6):816–828. doi:10.1007/s00134-017-4755-7
11. Gomez H, Kellum JA. Sepsis-induced acute kidney injury. Curr Opin Crit Care. 2016;22(6):546–553. doi:10.1097/MCC.0000000000000356
12. Hunt C, Calderwood S. Characterization and sequence of a mouse hsp70 gene and its expression in mouse cell lines. Gene. 1990;87(2):199–204. doi:10.1016/0378-1119(90)90302-8
13. Wilhide ME, Tranter M, Ren X, et al. Identification of a NF-kappaB cardioprotective gene program: NF-kappaB regulation of Hsp70.1 contributes to cardioprotection after permanent coronary occlusion. J Mol Cell Cardiol. 2011;51(1):82–89. doi:10.1016/j.yjmcc.2011.03.011
14. Van Molle W, Wielockx B, Mahieu T, et al. HSP70 protects against TNF-induced lethal inflammatory shock. Immunity. 2002;16(5):685–695. doi:10.1016/S1074-7613(02)00310-2
15. Lee KH, Lee CT, Kim YW, Han SK, Shim YS, Yoo CG. Heat shock protein 70 negatively regulates the heat-shock-induced suppression of the IkappaB/NF-kappaB cascade by facilitating IkappaB kinase renaturation and blocking its further denaturation. Exp Cell Res. 2005;307(1):276–284. doi:10.1016/j.yexcr.2005.03.014
16. King TA, Ghazaleh RA, Juhn SK, Adams GL, Ondrey FG. Induction of heat shock protein 70 inhibits NF-kappa-B in squamous cell carcinoma. Otolaryngol Head Neck Surg. 2005;133(1):70–79. doi:10.1016/j.otohns.2004.04.038
17. Uchinami H, Yamamoto Y, Kume M, et al. Effect of heat shock preconditioning on NF-kappaB/I-kappaB pathway during I/R injury of the rat liver. Am J Physiol Gastrointest Liver Physiol. 2002;282(6):G962–971. doi:10.1152/ajpgi.00466.2001
18. Dokladny K, Lobb R, Wharton W, Ma TY, Moseley PL. LPS-induced cytokine levels are repressed by elevated expression of HSP70 in rats: possible role of NF-kappaB. Cell Stress Chaperones. 2010;15(2):153–163. doi:10.1007/s12192-009-0129-6
19. Wang Z, Jin H, Li C, Hou Y, Mei Q, Fan D. Heat shock protein 72 protects kidney proximal tubule cells from injury induced by triptolide by means of activation of the MEK/ERK pathway. Int J Toxicol. 2009;28(3):177–189. doi:10.1177/1091581809337418
20. Wang Z, Gall JM, Bonegio RG, et al. Induction of heat shock protein 70 inhibits ischemic renal injury. Kidney Int. 2011;79(8):861–870. doi:10.1038/ki.2010.527
21. Wei S, Gao Y, Dai X, et al. SIRT1-mediated HMGB1 deacetylation suppresses sepsis-associated acute kidney injury. Am J Physiol Renal Physiol. 2019;316(1):F20–F31. doi:10.1152/ajprenal.00119.2018
22. Feoktistova M, Geserick P, Leverkus M. Crystal violet assay for determining viability of cultured cells. Cold Spring Harb Protoc. 2016;2016(4):pdbprot087379. doi:10.1101/pdb.prot087379
23. Kumaran GK, Hanukoglu I. Identification and classification of epithelial cells in nephron segments by actin cytoskeleton patterns. FEBS J. 2020;287(6):1176–1194. doi:10.1111/febs.15088
24. Ma T, Yang B, Gillespie A, Carlson EJ, Epstein CJ, Verkman AS. Severely impaired urinary concentrating ability in transgenic mice lacking aquaporin-1 water channels. J Biol Chem. 1998;273(8):4296–4299. doi:10.1074/jbc.273.8.4296
25. Vallon V, Verkman AS, Schnermann J. Luminal hypotonicity in proximal tubules of aquaporin-1-knockout mice. Am J Physiol Renal Physiol. 2000;278(6):F1030–1033. doi:10.1152/ajprenal.2000.278.6.F1030
26. Glodowski SD, Wagener G. New insights into the mechanisms of acute kidney injury in the intensive care unit. J Clin Anesth. 2015;27(2):175–180. doi:10.1016/j.jclinane.2014.09.011
27. Murugan R, Karajala-Subramanyam V, Lee M, et al. Acute kidney injury in non-severe pneumonia is associated with an increased immune response and lower survival. Kidney Int. 2010;77(6):527–535. doi:10.1038/ki.2009.502
28. Feder ME, Hofmann GE. Heat-shock proteins, molecular chaperones, and the stress response: evolutionary and ecological physiology. Annu Rev Physiol. 1999;61:243–282. doi:10.1146/annurev.physiol.61.1.243
29. Srivastava P. Roles of heat-shock proteins in innate and adaptive immunity. Nat Rev Immunol. 2002;2(3):185–194. doi:10.1038/nri749
30. Polla BS, Bachelet M, Elia G, Santoro MG. Stress proteins in inflammation. Ann N Y Acad Sci. 1998;851:75–85. doi:10.1111/j.1749-6632.1998.tb08979.x
31. Basile DP, Donohoe D, Cao X, Van Why SK. Resistance to ischemic acute renal failure in the Brown Norway rat: a new model to study cytoprotection. Kidney Int. 2004;65(6):2201–2211. doi:10.1111/j.1523-1755.2004.00637.x
32. Hsu JH, Yang RC, Lin SJ, et al. Exogenous heat shock cognate protein 70 pretreatment attenuates cardiac and hepatic dysfunction with associated anti-inflammatory responses in experimental septic shock. Shock. 2014;42(6):540–547. doi:10.1097/SHK.0000000000000254
33. Jo SK, Ko GJ, Boo CS, Cho WY, Kim HK. Heat preconditioning attenuates renal injury in ischemic ARF in rats: role of heat-shock protein 70 on NF-kappaB-mediated inflammation and on tubular cell injury. J Am Soc Nephrol. 2006;17(11):3082–3092. doi:10.1681/ASN.2005101077
34. Shi Y, Tu Z, Tang D, et al. The inhibition of LPS-induced production of inflammatory cytokines by HSP70 involves inactivation of the NF-kappaB pathway but not the MAPK pathways. Shock. 2006;26(3):277–284. doi:10.1097/01.shk.0000223134.17877.ad
35. Tanaka T, Shibazaki A, Ono R, Kaisho T. HSP70 mediates degradation of the p65 subunit of nuclear factor kappaB to inhibit inflammatory signaling. Sci Signal. 2014;7(356):ra119. doi:10.1126/scisignal.2005533
36. Chen H, Wu Y, Zhang Y, et al. Hsp70 inhibits lipopolysaccharide-induced NF-kappaB activation by interacting with TRAF6 and inhibiting its ubiquitination. FEBS Lett. 2006;580(13):3145–3152. doi:10.1016/j.febslet.2006.04.066
37. Wang LC, Liao LX, Lv HN, et al. Highly selective activation of heat shock protein 70 by allosteric regulation provides an insight into efficient neuroinflammation inhibition. EBioMedicine. 2017;23:160–172. doi:10.1016/j.ebiom.2017.08.011
38. Guicciardi ME, Gores GJ. Life and death by death receptors. FASEB J. 2009;23(6):1625–1637. doi:10.1096/fj.08-111005
39. Boatright KM, Salvesen GS. Mechanisms of caspase activation. Curr Opin Cell Biol. 2003;15(6):725–731. doi:10.1016/j.ceb.2003.10.009
40. Kroemer G, Galluzzi L, Brenner C. Mitochondrial membrane permeabilization in cell death. Physiol Rev. 2007;87(1):99–163. doi:10.1152/physrev.00013.2006
41. Pistritto G, Trisciuoglio D, Ceci C, Garufi A, D’Orazi G. Apoptosis as anticancer mechanism: function and dysfunction of its modulators and targeted therapeutic strategies. Aging-Us. 2016;8(4):603–619. doi:10.18632/aging.100934
42. Degterev A, Boyce M, Yuan J. A decade of caspases. Oncogene. 2003;22(53):8543–8567. doi:10.1038/sj.onc.1207107
43. Lee SY, Lee YS, Choi HM, et al. Distinct pathophysiologic mechanisms of septic acute kidney injury: role of immune suppression and renal tubular cell apoptosis in murine model of septic acute kidney injury. Crit Care Med. 2012;40(11):2997–3006. doi:10.1097/CCM.0b013e31825b912d
44. Zhang B, Zeng M, Li M, et al. Protopine protects mice against LPS-induced acute kidney injury by inhibiting apoptosis and inflammation via the TLR4 signaling pathway. Molecules. 2019;25(1):15. doi:10.3390/molecules25010015
45. Lerolle N, Nochy D, Guerot E, et al. Histopathology of septic shock induced acute kidney injury: apoptosis and leukocytic infiltration. Intensive Care Med. 2010;36(3):471–478. doi:10.1007/s00134-009-1723-x
46. Zhu Y, Fu Y, Lin H. Baicalin inhibits renal cell apoptosis and protects against acute kidney injury in pediatric sepsis. Med Sci Monit. 2016;22:5109–5115. doi:10.12659/MSM.899061
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.