")
Back to Journals » Neuropsychiatric Disease and Treatment » Volume 15
HO-1 protects the nerves of rats with cerebral hemorrhage by regulating the PI3K/AKT signaling pathway
Authors Zhao Q, Qu R, Teng L, Yin C, Yuan Y
Received 4 December 2018
Accepted for publication 24 April 2019
Published 28 May 2019 Volume 2019:15 Pages 1459—1468
DOI https://doi.org/10.2147/NDT.S197030
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Yuping Ning
Qingping Zhao,1,* Rongbo Qu,2,* Lu Teng,1 Changyou Yin,1 Yuan Yuan1
1Department of Neurosurgery, The Affiliated Yantai Yuhuangding Hospital of Qingdao University, Yantai City, Shandong Province 264000, People’s Republic of China; 2Department of Neurosurgery, Yantai Affiliated Hospital of Binzhou Medical University, Yantai City, Shandong Province 264100, People’s Republic of China
*These authors contributed equally to this work
Objective: This study aimed to investigate the neuroprotective effect of heme oxygenase-1 (HO-1) on the PI3K/AKT signaling pathway in rats with cerebral hemorrhage.
Materials and methods: Adult male Sprague-Dawley rats were randomly divided into: a sham group, a model group and an HO-1 inhibitor group (ZnPP group). Functional defects after surgery were scored according to the Longa5 standard. Hemotoxylin and eosin staining was used to detect whether the model was constructed successfully. Superoxide dismutase (SOD) vitality and malondialdehyde (MDA) content were calculated by the xanthine oxidase method and thiobarbituric acid method, respectively. Blood-brain barrier permeability was measured by Evans Blue. Apoptosis was detected by terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick end labeling assay. The expression of Bcl-2 and BAX was evaluated by immunohistochemistry and the expression of PI3K, p-PI3K, AKT and p-AKT was tested by Western blotting.
Results: The rat intracerebral hemorrhage model was successfully constructed. Compared with the model group, the bleeding in the ZnPP group was more serious, the cell edema and deformation were aggravated, and the neurological deficit score in the rat was significantly increased. In addition, the content of Evans blue, MDA, the number of apoptotic cells, the water content of brain tissue and the expression of BAX were significantly increased, while the SOD activity and the expressions of Bcl-2, p-PI3K and p-AKT protein were decreased.
Conclusion: HO-1 could protect the nerves of rats with cerebral hemorrhage by regulating the PI3K/AKT signaling pathway.
Keywords: PI3K/AKT signaling pathway, HO-1, neuroprotection, hemorrhage
Introduction
Spontaneous intracerebral hemorrhage (ICH) refers to primary non-traumatic intracerebral hemorrhage, which accounts for approximately 15% of stroke.1 It is a common clinical neurological disease with high incidence, high disability rate and high mortality, and seriously threatens human health.2,3 Cerebral hemorrhage-induced neuronal apoptosis and secondary cerebral edema play an important role in neurological impairment.4 However, the pathophysiological mechanism of ICH has not been fully elucidated.
Previously, many researches have studied the neuroprotective effects and made major breakthroughs. For instance, Laabich et al have found that AIP acts as a neuroprotective agent against N-methyl-D-aspartate-induced retinal neuronal cell death.5 Also, the estrogen receptor selective modulator tamoxifen citrate has been confirmed to have neuroprotective effects in an acute cerebral ischemia model.6 Moreover, erythropoietin has neuroprotective effects on immature cerebral oxygen-induced cell death.7 Multiple pathways have also been found to play an important role in neuroprotection. Estradiol preconditioning could promote dopamine neuron survival by activating ER-α and increasing Akt and GSK3-β phosphorylation.8 Besides, naringenin could play a neuroprotective role in stroke rats by suppressing the NF-κB signaling pathway.9
Recently, many scholars have found that heme oxygenase-1 (HO-1) has neuroprotective effects.10–12 However, the mechanism of HO-1 and related pathways in cerebral hemorrhage have not been thoroughly studied. Therefore, we constructed the rat model ICH. The expression of HO-1 in the cerebral hemorrhage model was studied, and the related mechanisms of protein, pathway and neurological function were studied by inhibiting HO-1 expression.
Materials and methods
Regents
DMSO and ZnPP regents were purchased from Sigma Aldrich (St. Louis, MO, USA). Trizol reagent was purchased from Invitrogen (Thermo Fisher ScientificWaltham, MA, USA) and PCR kit was also purchased from Thermo Fisher Scientific. Hematoxylin and eosin (HE) staining kit, EB kit, DAB kit and BCA Protein Assay Kit were purchased from Beyotime Biotechnology (Shanghai, China).
Experimental design and groups
A total of 45 adult male Sprague Dawley rats (about 260 g) were purchased from Beijing Weitong Lihua Experimental Animal Technology Co, Ltd. Rats were placed at 23±1°C with lights on from 6:00 to 18:00.13 The rats were separated into a sham group, a model group (DMSO group) and an HO-1 inhibitor group (ZnPP group) with 15 rats in each group. All animals were treated in accordance with the Guide for the Care and Use of Laboratory Animals, and all experiments were approved and performed according to the guidelines of the Ethics Committee of the Affiliated Yantai Yuhuangding Hospital of Qingdao University.
ICH models
Rats were anesthetized with pentobarbital (40 mg/kg). All operations were carried out under aseptic conditions. Rats were placed in a stereotactic frame (Kopf Instruments, Tujunga, CA, USA). A stereotactically guided needle was placed into the right basal ganglia (3.5 mm beside to bregma, depth 5.5 mm below the surface to midline). Then 100 μL of fresh autologous non-heparinized arterial blood was infused (10 μL/min) using a microsyringe pump. After completing the injection, the needle was removed. The burr hole was sealed with bone wax and the scalp wound was sutured.14
A sustained release pump was placed in the abdominal cavity of each rat after successful ICH modeling. A slow-release pump with 2 ml DMSO solution containing ZnPP (0.01 mg/kg) was used in the HO-1 inhibitor group. A slow-release pump with 2 ml of DMSO solution was used in the model group. Similar surgical procedures were processed for sham-operated animals (sham group) except that no blood was injected.
qRT-PCR assay for the expression of HO-1
The expression level of HO-1 was examined by qRT-PCR. The brief process was introduced as follows. The brain tissue was rapidly ground in liquid nitrogen, then trizol reagent was added. Then, the sample was transferred to the EP tube, and total RNA was extracted. The RNA concentration was measured and adjusted. The cDNA was obtained by reverse transcription. The quantitative PCR program was started at 95°C for 5 mins, and the amplification procedure was 95°C for 15 seconds, 60°C for 15 seconds, and 72°C for 50 seconds for a total of 40 cycles. Glycerol phosphate dehydrogenase (GAPDH) was used as an internal reference. The primers were as follows: HO-1, (upstream) 5ʹ-CACGGCCATGACCACTTTC-3ʹ, (downstream) 5ʹ-GGAACAGAGTGGCTCCAACAA-3ʹ; GAPDH, (upstream) 5ʹ-CCTGCAAATGAGCCCCAGCCTTC-3ʹ, (downstream) 5ʹ-TGCTGGCGCTGATACGTCGT-3ʹ.15
Neurological score
After awakening, the symptoms of neurological deficits, including emotion, cognition and movement were observed in the ischemic animal models. Postoperative functional deficits were scored according to the Longa 5-point scale (Table 1). The model with more than 1 point was regarded to be successfully built.16
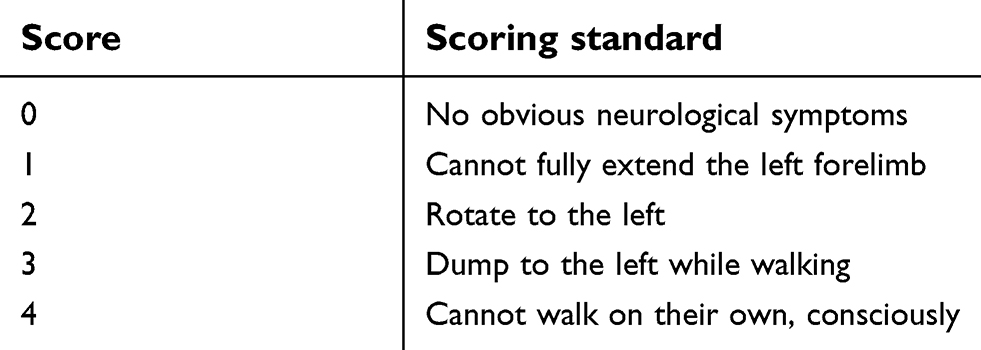 | Table 1 Criteria for functional defects after surgery |
Pathological observation of brain tissue
The rats were anesthetized and underwent intracardiac perfusion with 4% paraformaldehyde in physiological saline. The brain was taken out and fixed in 4% paraformaldehyde, dehydrated, permeabilized, and encased in paraffin. The brains were then placed in OCT embedding compound and 2–4 mm thick sections were obtained on a cryostat. Then the sections were stained with hematoxylin and eosin (HE) and observed under the microscope.17
Determination of Evans blue (EB) content in brain tissue
One hour before sacrifice, 2% EB (4 mL/kg) was injected into the femoral vein. Then normal saline was rapidly perfused from the left ventricle to the right atrium until the effluent clarifies. Finally, right nucleus accumbens and the caudate nucleus brain tissue after decapitation were collected.
After weighing, brain tissue was placed in 50% trichloroacetic acid solution for homogenization. The solution was centrifuged, and then 1 mL supernatant was taken and mixed with 3 mL absolute ethanol. Fluorescence values were measured by a fluorescence spectrophotometer with an excitation wavelength of 620 nm and emission wavelength of 680 nm. The content of EB in brain tissue was measured by EB Standard to detect the permeability of the blood-brain barrier at 72 hours after surgery.18
Evaluation of brain edema
Brain water content was evaluated at 72 hours after ICH. After weighing the wet weight, the tissue was dried for 24 hours at 90°C. The weight of brain tissue was immediately reweighed by analytical balance to yield the dry weight. The percentage of brain water content was calculated using the following formula: %H2O =(1- dry weight/wet weight) ×100%.
Determination of superoxide dismutase (SOD) and MDA content in brain tissue
All rats were killed by decapitation and the whole brain was removed quickly. The cerebral cortex was taken, and 10% cerebral cortex homogenate was prepared with physiological saline, and the supernatant was taken after centrifugation. Absorbance values of cerebral cortex supernatants were measured using UV-1700 UV-Vis spectrophotometer at 550 nm. SOD vitality was detected by the xanthine oxidase method. Then SOD activity in the tissue was calculated. The absorbance values of the cerebral cortex supernatant were measured at 532 nm. Malondialdehyde (MDA) content was determined by thiobarbituric acid method.20
Terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick end labeling (TUNEL) assay
Rats were anesthetized by intraperitoneal injection with 10% chloral hydrate (300 mg/kg). They were washed twice with saline and fixed with cardiac perfusion with 4% paraformaldehyde.21 Paraffin sections of brain tissue were made and washed twice with saline. The labeling solution were added to the sections and incubated at 37°C for 2 hours. Slices were washed twice with saline. Then biotin was added and incubated for 30 minutes. Slices were colored in DAB solution and observed under a microscope. Six high-power fields were selected for each section (400×).
The expression of Bcl-2 and BAX was detected by immunohistochemistry
After successful modeling, the rats were deeply anesthetized with pentobarbital (40 mg/kg, ip) and rapidly perfused with stroke-physiological saline solution and 4% paraformaldehyde. After being cut, the brain tissue was immersed in a neutral formaldehyde solution. Then, they were blocked in a saline solution containing 5% normal goat serum. The membranes were incubated with Bax antibody, Bcl-2 antibody (purchased in Fuzhou Maixin Biotech Co., Ltd, all diluted 1:100) at 4°C overnight. After that, the membranes were incubated with ready-to-use rapid immunohistochemistry MaxVisionTM secondary antibody (Fuzhou Maixin Biotech Co., Ltd). Positive cells were observed and counted using grid counting.22
Western blot
Brain tissue was lysed in ice-cold cell lysis solution and the homogenate was centrifuged at 4°C for 15 mins. After quantifying by BCA protein assay kit (Shanghai Biyuntian Bioengineering Co., Ltd.), the concentration of total protein was adjusted to 30 μg/μL. The protein was separated by SDS-PAGE, and then transferred onto PVDF membranes. The membranes were incubated with p-PI3K (Santa Cruz Biotechnology Co., Ltd, Dallas, TX, USA), p-AKT (Santa Cruz Biotechnology Co., Ltd.), PI3K (Bioworld) and AKT (Bioworld), respectively, at 4°C overnight. After that, the membranes were incubated with alkaline phosphatase-labeled goat anti-rabbit IgG secondary antibody (Beijing Zhongshang Jinqiao Biotechnology Co., Ltd) at room temperature for 1 hour. β-actin was used as an internal reference. The protein was visualized with an imager.23
Statistical analysis
All experiments were repeated three times. All experimental data were processed with SPSS 21.0 (IBM Corporation, Armonk, NY, USA). Data representation of mean values ± standard deviation was used in this study. The comparison of data between the two groups was carried out by Kruskal-Wallis test, while one-way ANOVA was used to analyze the difference among multiple groups. When the P-value was less than 0.05, the result was considered as statistically different.
Results
Successful establishment of the ICH mode and expression of HO-1
The pathological morphology results showed that the nerve cells were in a regular pattern with the same size, the nucleus of nerve cells was observed clearly, and cell swelling was not observed in the sham group. In the model group, the hemorrhagic area and peripheral nerve cells were obviously swollen and deformed, cells were autolysis and necrosis with vacuole, the nucleus was pyknotic and the nucleolus disappeared. There was a lot of red blood cell exudation in the interstitial, and the tissue structure was loosening. Compared with the model group, the bleeding in the ZnPP group was more serious, the edema and deformation of the cells were aggravated. The membrane and nucleus had become more blurred. The number of red blood cells bleeding was increased and the organizational structure was more uneven (Figure 1A).
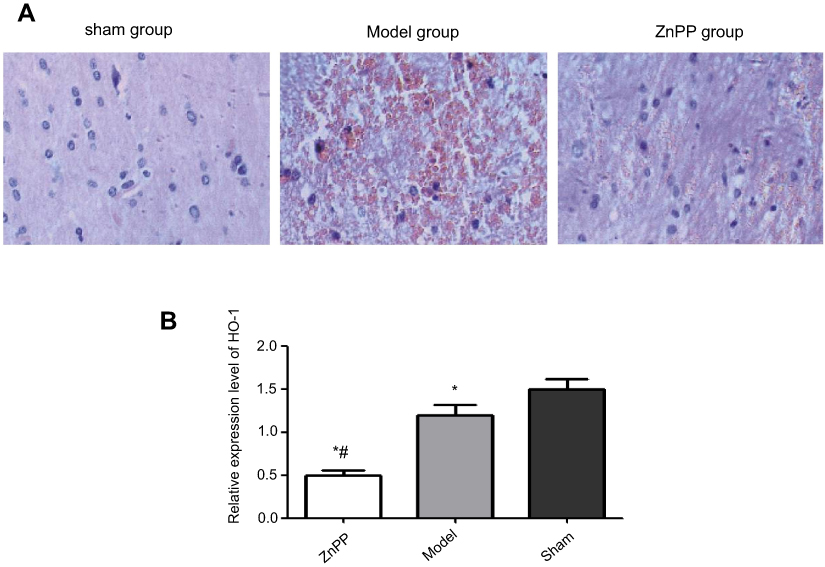 | Figure 1 Pathological morphology of rat brain tissue (×400) and expression of HO-1. (A) Pathological morphology of rat brain tissue; (B) Expression of HO-1, *p<0.05, compared with the sham group. #p<0.05, compared with the model group. |
As shown in Figure 1B, the expression of HO-1 was significantly lower after surgery. In addition, compared with the model group, the level of HO-1 was decreased in the Znpp group. We inferred that there were inhibitory effects of ZnPP against HO-1 in the injured brain tissues.
Inhibition of HO-1 aggravated neuronal damage, the content of EB and brain water
After surgery, the neural function was judged according to the Longa5 criteria. Rats in sham group had no symptoms of neurological deficit, while in model group and ZnPP group had different degrees of neurological defects. Compared with the model group, the neurological deficit scores of the ZnPP group were significantly increased (P<0.05, Figure 2A). The results indicated that neuronal damage was aggravated after cerebral hemorrhage in rats after HO-1 inhibition, and HO-1 might play a neuroprotective role in cerebral ischemia rats.
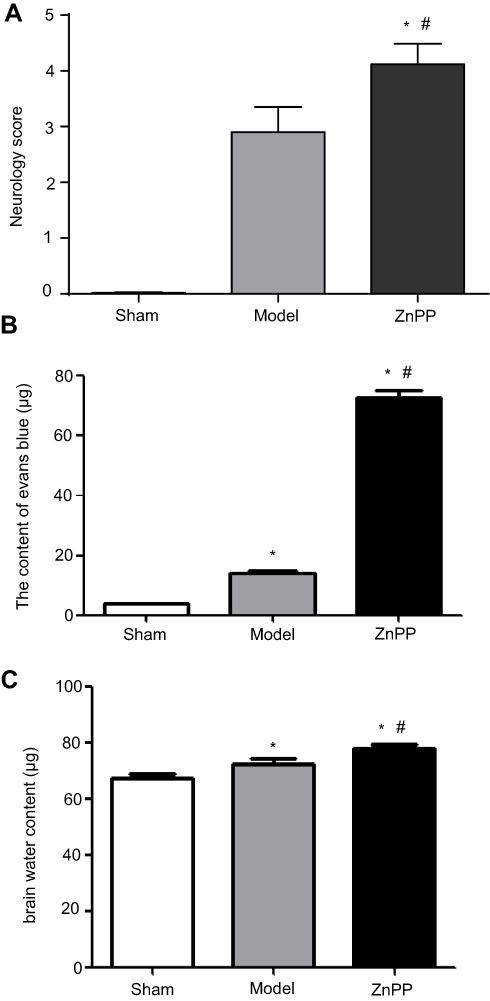 | Figure 2 Comparison of neurological deficit scores in each group and Evans blue and brain water content. (A) Neurological deficit scores; (B) Evans blue content; (C) Brain water content. *p<0.05, compared with the sham group. #p<0.05, compared with the model group. |
Compared with the sham group (4.07±0.65 μg), the content of EB in the model group (14.29±1.65 μg) and ZnPP group (26.70±1.58 μg) was significantly increased (all P<0.05, Figure 2B). In addition, the content of EB in the ZnPP group was higher than that in the model group (P<0.05). Compared with the sham operation group (67.49±2.59%), the brain water content in the model group (72.42±3.73%)and ZnPP group (77.63±2.65%) was significantly higher (P<0.05). The brain water content of the ZnPP group was significantly higher than that of the model group (P<0.05, Figure 2C). This result indicated that the inhibition of HO-1 affected the blood-brain barrier permeability and brain tissue water content of rat brain tissue.
Inhibition of HO-1 increased SOD activity and decreased MDA content
As shown in Figure 3, the SOD activity in the brain tissue of the model group was significantly decreased, while MDA content was significantly increased (P<0.05). Compared with the model group, the SOD activity was significantly decreased, while MDA content was significantly increased in ZnPP group (P<0.05).
 | Figure 3 Effect of HO-1 inhibition on SOD and MDA levels in rats with cerebral hemorrhage (A) SOD; (B) MDA. *p<0.05, compared with the sham group. #p<0.05, compared with the model group. |
Inhibition of HO-1 increased neuronal apoptosis after cerebral hemorrhage
At 48 hours after operation, the apoptosis of neuronal cells was detected by TUNEL. Sporadic, sporadic and few TUNEL positive cells were found in the brain tissue of the sham group. The number of apoptotic cells in the model group and ZnPP group were significantly increased. Compared with the model group, the number of apoptotic cells in the ZnPP group was significantly increased (Figure 4).
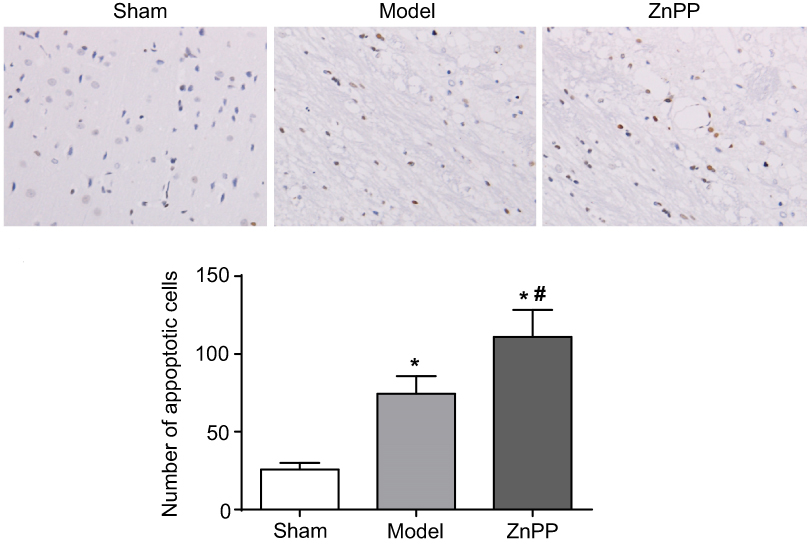 | Figure 4 Changes of neuronal apoptosis in brain tissue of rats. *p<0.05, compared with the sham group. #p<0.05, compared with the model group. The positive cells were stained brown. |
Bcl-2 and BAX were slightly expressed in the sham group, but the bcl-2/BAX ratio was always close to 1. Compared with this group, the expression levels of Bcl-2 and BAX in the model group were significantly increased (P<0.05). Compared with the model group, the positive expression of Bcl-2 in the ZnPP group was significantly decreased (P<0.05), while the expression of BAX was significantly increased (P<0.05), and the Bcl-2/BAX ratio was much less than 1 (Figure 5). The results further indicated that inhibition of HO-1 increased apoptosis in brain tissue of rats with cerebral hemorrhage.
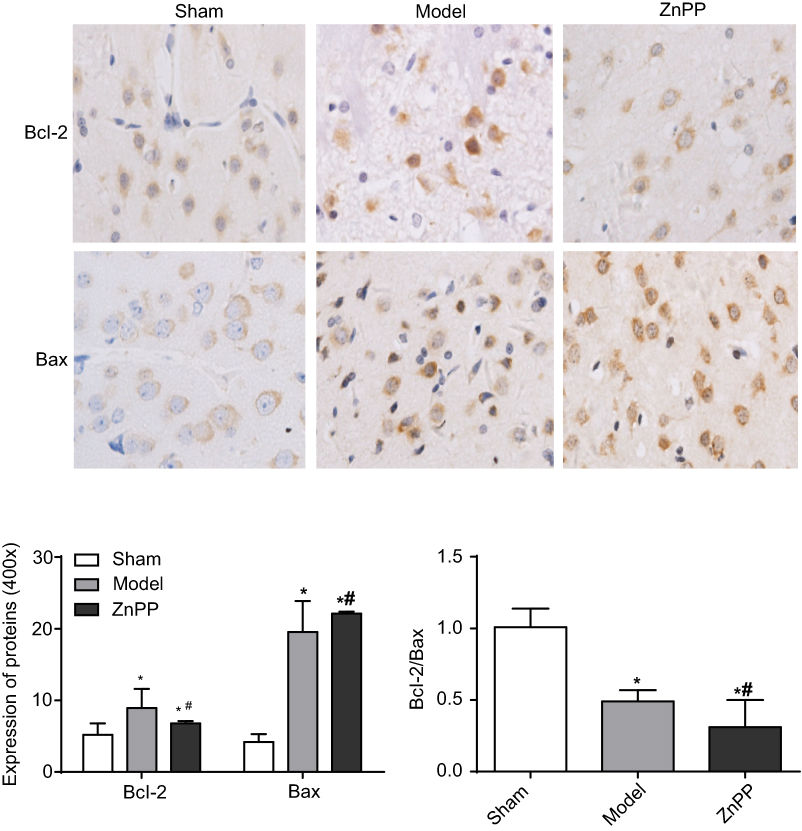 | Figure 5 The expression of Bcl-2 and BAX protein in rat brain tissue (×400). (*p<0.05 when compared with the sham group. #p<0.05 when compared with the model group.). The positive cells were stained brown. |
Inhibition of HO-1 decreased the expression of PI3K, p-PI3k
The protein expression level of PI3K, AKT, p-PI3K and p-AKT were detected at 72 hours after surgery. Compared with the sham group, there was no significant change in the protein expression level of PI3K and AKT in the model and ZnPP groups, while the protein expression level of p-PI3K and p-AKT decreased significantly (P<0.05). Compared with the model group, there was no significant change in the protein content of PI3K and AKT in the ZnPP group, while the protein expression level of p-PI3K and p-AKT decreased significantly (P<0.05, Figure 6). All of these suggested that HO-1 could alleviate neuronal damage after cerebral hemorrhage by regulating the PI3K/AKT signaling pathway.
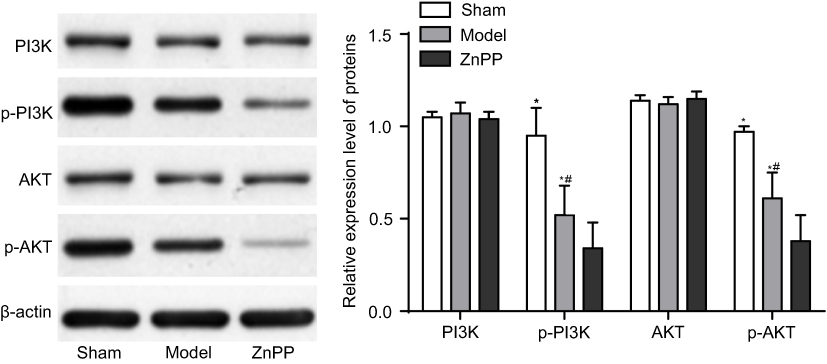 | Figure 6 The expression of PI3K, AKT, p-PI3K and p-AKT protein in rat brain tissue. *p<0.05, compared with the sham group. #p<0.05, compared with the model group. |
Discussion
HO-1 is an inducible low molecular weight stress protein that provides cellular and tissue protection in a variety of injury and disease models.19 In this study, the rat ICH model was constructed successfully. After inhibiting HO-1, the bleeding was more serious, the cell edema and deformation were aggravated, and the neurological deficit score in the rat was significantly increased. In addition, the expression of p-PI3K and p-AKT was decreased. Therefore, we speculated that HO-1 might have neuroprotective effects on rats with cerebral hemorrhage by regulating the PI3K/AKT signaling pathway.
As shown in a previous study, heme oxygenase (HO) has neuroprotective effects on oxidative stress in HT22 cells transfected with HO-1 siRNA.11 Nrf2/HO-1 could mediate neuroprotective effects of mangiferin on early brain injury after subarachnoid hemorrhage by attenuating mitochondria-associated apoptosis and neuroinflammation.10 Some scholars also have found that D163/HO-1 positive microglia and invasive macrophages show anti-inflammatory and neuroprotective phenotype after traumatic brain injury.24 Moreover, it has been confirmed that the neuroprotective effect of melatonin on ischemia is partly caused by α-7 nicotinic receptor regulation and HO-1 overexpression.25 Similarly, the neuroprotective effect of Acanthopanax senticosus is caused by HO-1 signaling in hippocampus and microglia.26 Interestingly, ischemia-induced upregulation of heme oxygenase-1 protects against apoptotic cell death and tissue necrosis.27 Moreover, ischemia-induced upregulation of heme oxygenase (HO)-1 is also critical for protecting critical perfusion flaps from apoptotic cell death and tissue necrosis.28 In addition, the cytoprotective effect of HO-1 is found after silencing HO-1 by siRNA.29 After inhibiting HO-1, the expression of BAX was significantly increased, while the expression of Bcl-2 was significantly decreased. The results further indicated that inhibition of HO-1 increased apoptosis in brain tissue of rats with cerebral hemorrhage.
Our study also found that HO-1 was involved in PI3K/AKT signaling pathway. Several articles have reported the relationship between HO-1 and PI3K/AKT signaling pathways. For instance, Feng et al30 have demonstrated that the PI3K/Akt and ERK pathways are involved in OA-induced HO-1 expression by activating Nrf2 in VSMC. Besides, the brain-protected part of sevoflurane post-treatment is confirmed to up-regulate the expression of HIF-1α and HO-1 via the PI3K/Akt pathway.31 Moreover, baicalein protects 6-OHDA-induced neurotoxicity by activating Keap1/Nrf2/HO-1 and participating in PKCα and PI3K/AKT signaling pathways.32 It is well known, the PI3K/Akt pathway has been shown to be involved in the process of degradation of extracellular matrices and chondrocyte death.33 Moreover, hypoxia confers protection against apoptosis through the PI3K/Akt pathway in endothelial progenitor cells.34 Also, Tibes et al35 have confirmed that the PI3K/Akt pathway plays an important role in acute myeloid leukemia. As one of the signal pathways for cell survival, PI3K/Akt signal transduction pathway plays an important role in cell proliferation, differentiation and inhibition of neuronal apoptosis.36 Thereby, HO-1 may protect the nerves of rats with cerebral hemorrhage by regulating PI3K/AKT signaling pathway.
By inhibiting the expression of HO-1, brain water content in cerebral ischemia rats was increased, SOD activity was down-regulated and MAD content was up-regulated in this study. The structure of the blood-brain barrier can make brain tissue less susceptible to or even harmful to harmful substances in circulating blood.37 Therefore, it could maintain the basic stability of the brain tissue environment, which has important biological significance for maintaining the normal physiological state of the central nervous system.38 SOD and MDA are important indicators for measuring oxidative damage in brain tissue.39 The main function of SOD is disproportionation of oxygen free radicals.40 The activity could reflect the body’s ability to scavenge oxygen free radicals.41 The free radicals attack the unsaturated lipids in the membrane to produce lipid peroxidation products MDA, which could reflect the degree of damage to the body caused by free radicals in the body.42
However, there were some limitations in this study. No special region of brain was researched. In the future, this will be our main research direction.
Conclusion
HO-1 can play a neuroprotective role by regulating PI3K/AKT signaling pathway and reducing apoptosis in rats with cerebral hemorrhage. It might provide a new target for protecting the nerves of patients with cerebral hemorrhage.
Ethics approval and consent to participate
This study was conducted after obtaining local ethical committee approval of The Affiliated Yantai Yuhuangding Hospital of Qingdao University and we confirm that all experiments were performed following relevant named institutional and national guidelines and regulations.
Abbreviation list
HO-1, heme oxygenase-1; HE, hemotoxylin and eosin; SOD, superoxide dismutase; MDA, malondialdehyde; EB, Evans Blue.
Acknowledgments
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Disclosure
The authors report no conflicts of interest in this work.
References
1.
2. Samarasekera N, Fonville A, Lerpiniere C, et al. Influence of intracerebral hemorrhage location on incidence, characteristics, and outcome: population-based study. Stroke. 2015;46:361–368. doi:10.1161/STROKEAHA.114.007953
3. Toyoda K, Grotta JC. Seeking best medical treatment for hyperacute intracerebral hemorrhage. Neurology. 2015;84:444–445. doi:10.1212/WNL.0000000000001221
4. Kawai T, Nakazawa H, Ida N, et al. In vivo regulation of brain-derived neurotrophic factor in dorsal root ganglia is mediated by nerve growth factor-triggered Akt activation during cystitis. PLoS One. 2013;8:e81547. doi:10.1371/journal.pone.0081547
5. Laabich A, Cooper NGF. Neuroprotective effect of AIP on N-methyl-D-aspartate-induced cell death in retinal neurons. Mol Brain Res. 2000;85:32–40.
6. Pavlov SV, Belenichev IF. Molecular and biochemical aspects of the neuroprotective effect of the selective estrogen receptor modulator tamoxifen in a model of acute cerebral ischemia. Neurochem J. 2014;8:28–32. doi:10.1134/S1819712413040077
7. Genz K, Dzietko M, Moysich A, et al. 141 neuroprotective effect of erythropoietin in oxygen-induced cell death in the immature brain. Pediatr Res. 2005;58:378. doi:10.1203/01.pdr.0000183658.17854.28
8. D’Astous M, Mendez P, Morissette M, et al. Implication of the phosphatidylinositol-3 kinase/protein kinase B signaling pathway in the neuroprotective effect of estradiol in the striatum of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mice. Mol Pharmacol. 2006;69:1492–1498. doi:10.1124/mol.105.018671
9. Raza SS, Khan MM, Ahmad A, et al. Neuroprotective effect of naringenin is mediated through suppression of NF-κB signaling pathway in experimental stroke. Neuroscience. 2013;230:157–171. doi:10.1016/j.neuroscience.2012.10.041
10. Wang Z, Guo S, Wang J, et al. Nrf2/HO-1 mediates the neuroprotective effect of mangiferin on early brain injury after subarachnoid hemorrhage by attenuating mitochondria-related apoptosis and neuroinflammation. Sci Rep. 2017;7:11883. doi:10.1038/s41598-017-12160-6
11. Kaizaki A, Tanaka S, Ishige K, Numazawa S, Yoshida T. The neuroprotective effect of heme oxygenase (HO) on oxidative stress in HO-1 siRNA-transfected HT22 cells. Brain Res. 2006;1108:39–44. doi:10.1016/j.brainres.2006.06.011
12. Arai S, Katai N, Ohta K, et al. The mechanism of neuroprotective effect of heme oxygenase on retinal ischemia-reperfusion injury in rats.
13. Zhang Y, Yang Y, Zhang GZ, et al. Stereotactic administration of edaravone ameliorates collagenase‐induced intracerebral hemorrhage in rat. CNS Neurosci Ther. 2016;22:824–835. doi:10.1111/cns.12584
14. Andaluz N, Zuccarello M, Wagner KR. Experimental animal models of intracerebral hemorrhage. Neurosurg Clin N Am. 2002;13:385–393.
15. Zhao Y, Fu B, Zhang X, et al. Paeonol pretreatment attenuates cerebral ischemic injury via upregulating expression of pAkt, Nrf2, HO-1 and ameliorating BBB permeability in mice. Brain Res Bull. 2014;109:61–67. doi:10.1016/j.brainresbull.2014.09.008
16. Wang GH, Lan R, Zhen XD, et al. An-Gong-Niu-Huang Wan protects against cerebral ischemia induced apoptosis in rats: up-regulation of Bcl-2 and down-regulation of Bax and caspase-3. J Ethnopharmacol. 2014;154:156–162. doi:10.1016/j.jep.2014.03.057
17. Alawa JN, Gideon GO, Adetiba B, Alawa CB. Comparative tissue stainability of Lawsonia inermis (Henna) and Eosin as counterstains to hematoxylin in brain tissues. Microsc Microanal. 2015;21:343–350. doi:10.1017/S1431927615000100
18. Prabhu SS, Broaddus WC, Oveissi C, Berr SS, Gillies GT. Determination of intracranial tumor volumes in a rodent brain using magnetic resonance imaging, Evans blue, and histology: a comparative study. IEEE Trans Biomed Eng. 2000;47:259–265. doi:10.1109/10.821776
19. Patel TR, Schielke GP, Hoff JT, Keep RF, Lorris Betz A. Comparison of cerebral blood flow and injury following intracerebral and subdural hematoma in the rat. Brain Res. 1999;829:125–133.
20. Hong YL, Shulan Y, Chang CY, Hu ML. Total plasma malondialdehyde levels in 16 Taiwanese college students determined by various thiobarbituric acid tests and an improved high-performance liquid chromatography-based method. Clin Biochem. 2000;33:619–625.
21. Moradian ZF, Naghdi M, Salehi P, Ajami A, Deemeh MR, Meshkibaf MH. Can SCSA and TUNEL forecast apoptosis-related motility depletion in asthenozoospermia? Andrologia. 2018;50:e13025. doi:10.1111/and.2018.50.issue-6
22. Krajewski S, Krajewska M, Shabaik A, Miyashita T, Wang HG, Reed JC. Immunohistochemical determination of in vivo distribution of Bax, a dominant inhibitor of Bcl-2. Am J Pathol. 1994;145:1323–1336.
23. Deng C, Tao R, Yu SZ, Jin H. Sulforaphane protects against 6-hydroxydopamine-induced cytotoxicity by increasing expression of heme oxygenase-1 in a PI3K/Akt-dependent manner. Mol Med Rep. 2012;5:847–851. doi:10.3892/mmr.2011.731
24. Zhang Z, Zhang ZY, Wu Y, Schluesener HJ. Lesional accumulation of CD163+ macrophages/microglia in rat traumatic brain injury. Brain Res. 2012;1461:102–110. doi:10.1016/j.brainres.2012.04.038
25. Parada E, Buendia I, León R, et al. Neuroprotective effect of melatonin against ischemia is partially mediated by alpha-7 nicotinic receptor modulation and HO-1 overexpression. J Pineal Res. 2014;56:204–212. doi:10.1111/jpi.12113
26. Jin ML, Park SY, Kim YH, Park G, Lee SJ. Acanthopanax senticosus exerts neuroprotective effects through HO-1 signaling in hippocampal and microglial cells. Environ Toxicol Pharmacol. 2013;35:335–346. doi:10.1016/j.etap.2013.01.004
27. Harder Y, Amon M, Schramm R, et al. Ischemia-induced up-regulation of heme oxygenase-1 protects from apoptotic cell death and tissue necrosis. J Surg Res. 2008;150:293–303. doi:10.1016/j.jss.2007.12.773
28. Harder Y, Amon M, Schramm R, et al. Ischemia-induced upregulation of heme oxygenase (HO)-1 protects critically perfused flaps from apoptotic cell death and tissue necrosis. J Surg Res. 2008;150:293–303.doi:10.1016/j.jss.2007.12.773
29. Tibullo D, Barbagallo I, Giallongo C, et al. Nuclear translocation of heme oxygenase-1 confers resistance to imatinib in chronic myeloid leukemia cells. Curr Pharm Des. 2013;19:2765–2770. doi:10.2174/1381612811319150012
30. Feng J, Zhang P, Chen X, He G. PI3K and ERK/Nrf2 pathways are involved in oleanolic acid-induced heme oxygenase-1 expression in rat vascular smooth muscle cells. J Cell Biochem. 2011;112:1524–1531. doi:10.1002/jcb.23065
31. Ye Z, Guo Q, Xia P, Wang N, Wang E, Yuan Y. Sevoflurane postconditioning involves an up-regulation of HIF-1α and HO-1 expression via PI3K/Akt pathway in a rat model of focal cerebral ischemia. Brain Res. 2012;1463:63–74. doi:10.1016/j.brainres.2012.04.050
32. Zhang Z, Cui W, Li G, et al. Baicalein protects against 6-OHDA-induced neurotoxicity through activation of Keap1/Nrf2/HO-1 and involving PKCα and PI3K/AKT signaling pathways. J Agric Food Chem. 2012;60:8171–8182. doi:10.1021/jf301511m
33. Chen J, Crawford R, Xiao Y. Vertical inhibition of the PI3K/Akt/mTOR pathway for the treatment of osteoarthritis. J Cell Biochem. 2013;114:245–249. doi:10.1002/jcb.24362
34. Dai T, Zheng H, Fu GS. Hypoxia confers protection against apoptosis via the PI3K/Akt pathway in endothelial progenitor cells. Acta Pharmacol Sin. 2008;29(12):1425–1431. doi:10.1111/j.1745-7254.2008.00904.x
35. Tibes R, Kornblau SM, Qiu Y, et al. PI3K/AKT pathway activation in acute myeloid leukaemias is not associated with AKT1 pleckstrin homology domain mutation. Br J Haematol. 2010;140:344–347. doi:10.1111/j.1365-2141.2007.06920.x
36. Li Q, Huai L, Zhang C, et al. Icaritin induces AML cell apoptosis via the MAPK/ERK and PI3K/AKT signal pathways. Int J Hematol. 2013;97:617–623. doi:10.1007/s12185-013-1317-9
37. Huber JD, Vangilder RL, Houser KA. Streptozotocin-induced diabetes progressively increases blood-brain barrier permeability in specific brain regions in rats. Am J Physiol Heart Circ Physiol. 2006;291:H2660. doi:10.1152/ajpheart.00489.2006
38. Spiotta AM, Stiefel MF, Gracias VH, et al. Brain tissue oxygen-directed management and outcome in patients with severe traumatic brain injury. J Neurosurg. 2010;113:571–580. doi:10.3171/2010.1.JNS09506
39. Gorman DG, Kennedy W. Impact of environmental concentrations of beta-cypermethrin on the antioxidant system in the brain and liver of zebrafish (Danio rerio). Chem Ecol. 2014;30:643–652. doi:10.1080/02757540.2014.889121
40. He Y, Wu M, Wei Z, et al. The synthesis of the catalyst imitating SOD and its functions of scavenging oxygen free radicals. J Tongji Med Univ. 1992;21:261–264.
41. Gao D. Antioxidant therapies for hypolipidemia and hyperglycemia. In: El-Missiry MA, editor. Antioxidant Enzyme. IntechOpen, 2012.
42. Al-Rawi NH. Oxidative stress, antioxidant status and lipid profile in the saliva of type 2 diabetics. Diab Vasc Dis Res. 2011;8:22–28. doi:10.1177/1479164110390243
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.