")
Back to Journals » OncoTargets and Therapy » Volume 12
HnRNPL promotes Wilms tumor progression by regulating the p53 and Bcl2 pathways
Authors Luo X , Deng C, Liu F, Liu X, Lin T, He D, Wei G
Received 26 January 2019
Accepted for publication 16 April 2019
Published 29 May 2019 Volume 2019:12 Pages 4269—4279
DOI https://doi.org/10.2147/OTT.S203046
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Sanjeev K. Srivastava
Xin Luo,1 Changkai Deng,2 Feng Liu,1,3 Xing Liu,1,3 Tao Lin,1,3 Dawei He,1,3 Guanghui Wei1,3
1Ministry of Education Key Laboratory of Child Development and Disorders, Chongqing Key Laboratory of Pediatrics, Chongqing Key Laboratory of Children Urogenital Development and Tissue Engineering, China International Science and Technology Cooperation Base of Child Development and Critical Disorders, Pediatric Research Institute, Children’s Hospital of Chongqing Medical University, Chongqing 400014, People’s Republic of China; 2Department of Pediatric Surgery, Chengdu Women and Children’s Central Hospital, Chengdu 610073, People’s Republic of China; 3Department of Pediatric Urology Surgery, Children’s Hospital of Chongqing Medical University, Chongqing 400014, People’s Republic of China
Background: Wilms tumor (WT) is the most common renal tumor in children with diffusely anaplastic or unfavorable histology, indicative of a poor prognosis. Heterogeneous nuclear ribonucleoprotein L (hnRNPL) is an RNA-binding protein (RBP) and a regulator of alternative RNA splicing that plays an important role in the occurrence and development of several cancers.
Methods: Next generation sequencing technologies was used to discovery differentially expressed genes between WT and adjacent nontumors. The gene ontology (GO) analysis was performed to uncover the biological functions of differentially expressed genes, and the kyoto encyclopedia of genes and genomes (KEGG) pathway enrichment analysis was applied to find out the related signal pathways. Expression levelsof hnRNPL with WT tissues and cells were determined by RT-qPCR.After silencing hnRNPL, the expression of hnRNPL, p53 and Bcl-2 were detected by RT-qPCR and Western blot in WT cell line. The regulatory effects of hnRNPLon proliferative and apoptotic potentials of WT cells were evaluated by MTT and flow cytometry, respectively. RNA-binding protein immuno-precipitation was used to confirm the direct interaction of hnRNPL with p53 mRNA. Mouse xenograft models ofhnRNPL knockdown were established to test the functions in the growth of WT in vivo.
Results: High levels of hnRNPL were expressed in WT tissues and cells. Functional analysis revealed that hnRNPL silencing suppressed cell proliferation and promoted cell apoptosis in WT. Molecular mechanism exploration indicated that hnRNPL directly targeted p53. Moreover, knockdown of hnRNPL inhibited the expression of p53 and Bcl2 in WT. Additionally, hnRNPL silencing inhibited the growth of xenograft tumors in vivo.
Conclusion: HnRNPL act as p53 mRNA-binding protein, which plays an important role in the proliferation and apoptosis of WT through p53 and Bcl2 pathways and these findings provide new insights into the mechanism of WT pathogenesis.
Keywords: HnRNPL, Wilms tumor, p53, Bcl2, proliferation, apoptosis
Introduction
Wilms tumor (WT) is the most common malignant tumor of the pediatric urinary system. The combination of surgery, radiation, and chemotherapy has increased the survival rate from 30% to more than 90% for many patients, but a cure remains elusive for patients with metastatic or anaplastic disease. A significant number of patients die due to postoperative recurrence, metastasis, and resistance to chemotherapeutic drugs.1
WT maintenance and disease progression are associated with the altered expression of multiple genes (WT1, WTX, CTNNB1, p53, MYCN, DROSHA, DGCR8, SIX1, and SIX2), numerous recurrent copy number aberrations, and loss of heterozygosity events.2 Wilms’ tumor 1 (WT1), located on chromosome 11p13, was first cloned in 1990 as one of the first tumor suppressor genes in WT.3 No recurrent loci for somatic WT1 mutations exist in WTs.2 Mutations in the WT1 gene occur in approximately 4–47% of WT cases, and these are mostly nonsense mutations or gene losses.4,5 The WT1 protein has become a useful marker for the diagnosis of WTs. A pediatric renal tumor which expresses WT1 protein in the nuclei of neoplastic cells is considered a WT.6,7 Subsequently, a beta-catenin proto-oncogene (CTNNB1) and the Wilms’ tumor gene on the X chromosome (WTX) have been identified in tumors.8 Combined genetic alterations of WT1, CTNNB1, and WTX have been estimated to occur in roughly one-third of WTs.9 A recent whole-exome study identified mutations in microRNA processing genes, including DROSHA and DGCR8, in 15% of WTs.10 However, the frequencies of alterations in DROSHA and DGCR8 are similarly low, leaving a significant fraction of cases without an identified genetic defect “driver”. Therefore, the identification and characterization of differential genes is of primary importance for understanding the onset and progression of tumors, ultimately leading to the recognition of potential markers and specific targets for the prevention and individualized treatment of WTs.
Currently, cancer is considered to be a disorder of the cell cycle and apoptotic mechanisms.11 Therefore, the cell cycle and apoptosis are the two hotspots of research in malignant tumors.12 Disordered cell cycle regulation, uncontrolled apoptosis, and altered cell proliferation are the most obvious characteristics of cancerous tissues.13 Many oncogenes and antioncogenes can directly regulate the cell cycle, which results in an abnormal cell cycle in which cells grow uncontrollably, with inhibited apoptosis characteristics.14
Wild-type p53 is an important tumor suppressor gene that can induce cell cycle arrest, apoptosis, differentiation, DNA repair, angiogenesis, and metastasis inhibition.15 Many malignant tumors are accompanied by missense mutations in wild-type p53 and accumulation of a large number of mutant p53 (mp53) proteins.16 Studies have shown that after the loss of the tumor suppressor function, some mp53 proteins acquire new functions, such as participating in the proliferation of cancer cells and improving the chemotherapeutic resistance of cancer cells.17,18 In WTs, expression of the p53 protein is indicative of an anaplastic pathological type.19 Additionally, 75% of anaplastic WTs have p53 gene mutations, suggesting that p53 mutation is a prognostic marker of unfavorable histology in WTs.20 Studies have shown that a favorable WT histology could develop into an unfavorable histology with dysfunctions in the p53 gene.21 Furthermore, mp53 plays an important role in the progression, recurrence, and metastasis of tumors and may be an important marker of poor prognosis in patients with WTs.
Heterogeneous nuclear ribonucleoproteins (hnRNPs) directly regulate the alternative splicing of a set of RNAs and serve as multifunctional RNA-binding proteins (RBPs) for mRNA transport, translation, and stabilization.22 This ribonucleoprotein family contains at least 20 members, named hnRNPA1 to hnRNPU. HnRNPL, an HnRNP family member, plays an important role in the occurrence and development of liver cancer, lung cancer, breast cancer, esophageal squamous cell carcinoma, colorectal cancer, bladder cancer, pancreatic cancer, and other tumors, thus supporting its clinical relevance.23,24 Importantly, both hnRNPL and its RNA targets are aberrantly expressed, which was closely related to the promotion of growth, proliferation, adhesion, invasion, and metastasis in tumor cells.25,26 Recent research has shown that downregulation of hnRNPL expression can significantly inhibit hepatoma cell proliferation and migration.27 In lung cancer, hnRNPL regulates the tumorigenic capacity of non-small cell lung cancer cells by interacting with the purine-rich regions of exon 3 of caspase-9 pre-mRNA, resulting in its degradation and affecting the expression of a caspase-9 mRNA subtype.28 Similarly, in pancreatic cancer, lncRNA uc.345 has been shown to increase the expression of hnRNPL, whereas hnRNPL knockdown significantly reduces the proliferative capacity promoted by lncRNA uc.345.29 However, there is no research on the expression and biological role of hnRNPL in the development of human WTs.
Based on the above research background, we compared the expression profiles of mRNAs in paired WT specimens and adjacent normal tissues, identifying that hnRNPL is overexpressed in WT samples and correlated with the p53 signaling pathway. Mechanistic investigations demonstrated that hnRNPL could promote WT growth and suppress apoptosis by upregulating p53 and Bcl2 expression, which might lead to new therapeutic approaches and improve the survival of children with WT.
Materials and methods
Patient tissue specimens
Tumor and paracarcinoma tissues of 50 WT patients were collected in the Department of Urology Surgery at the Children’s Hospital of Chongqing Medical University. Each specimen was immediately snap-frozen in liquid nitrogen and stored at −80°C. The histological confrmation for these samples was performed by 2 pathologists. This study was approved by the Ethics Committee of Chongqing Medical University, and written informed consent from all patients and their parents was obtained before surgery.
RNA sequencing (RNA-Seq) and bioinformatics analysis of tumor tissues
WT and adjacent nontumorous tissues from 3 patients were immediately subjected to whole-transcriptome sequencing using an Illumina Gene Expression Sample Prep Kit and a Solexa sequencing chip (Gene, Shanghai, China). Then, bioinformatics approaches, including Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses, were used to analyze genes with distinct variations to determine target RNAs and related signaling pathways.
Cell culture and transfection
A human immortalized normal kidney cell line (HK2) and WT cell line (G401) were purchased from Beijing Union Cell Resource Center. The cells were cultured under adherent conditions in F12/DMEM (Gibco, USA) and McCoy’s 5A medium (modified, Gibco, USA) supplemented with 10% FBS (Gibco, USA), 100 IU/mL penicillin and 100 µg/mL streptomycin in a 5% CO2 incubator at 37°C. The cells were observed and passaged every other day. HnRNPL-targeting siRNA and negative control (NC) siRNA sequences (Gene, Shanghai, China) are shown in Table 1. Twenty-four hours before transfection, G401 cells in the logarithmic growth phase were digested with a Trypsin-EDTA solution (Gibco, USA) and counted. Then, 1.5×105 cells were collected and plated in a 6-well plate. An siRNA transfection reagent was prepared and used according to the manufacturer’s instructions. The cells were divided into three groups as follows: an experimental group with hnRNPL siRNA transfection, a NC group with NC siRNA transfection, and a blank control group with no transfection.
 | Table 1 HnRNPL siRNA sequences |
Real-time quantitative RT-PCR
To quantitate the expression of hnRNPL, TP53, and Bcl2, we extracted total RNA from 50 patients’ samples and the G401, HK2, and G401 cell lines at 48 hrs after transfection with RP1202 (Bioteke, China). The isolated total RNA was reverse transcribed using PrimeScript RT Master Mix (Takara, Japan) for mRNA analysis according to the manufacturer’s instructions. The quantity of the mRNA was measured using SYBR Premix Ex Taq II (Takara, Japan), and the reactions were performed on a CFX96 Real-Time PCR Detection System (Bio-Rad, USA). The PCR conditions were 30 s at 95°C followed by 40 cycles at 95°C for 5 s and 60°C for 45 s. The sequence-specific primers used for HnRNPL, P53, and Bcl2 and GAPDH are shown in Table 2. The expression levels of hnRNPL, TP53, and Bcl2 mRNA were quantified relative to those of GAPDH using the 2-ΔΔCT method.
 | Table 2 Sequence-specific primers |
Western blot analysis to detect HnRNPL, p53, and Bcl2 protein expression
Cells were harvested 48 hrs after siRNA transfection, and protein was isolated with RIPA buffer (Beyotime, China). Cellular protein was extracted from each group, and the total protein content was determined according to the instructions provided with the cell protein extraction kit. The total protein samples were separated on SDS-PAGE gels and transferred onto PVDF membranes (Millipore, USA). The membranes were immunoblotted overnight at 4°C with primary antibodies against the following targets: HnRNPL (1:800; Abcam, UK), p53 (1:800; Abcam, UK), Bcl2 (1:1000; Abcam, UK), and human β-actin (1:500; Abcam, UK). The PVDF membranes were washed with TBST and then incubated with secondary HRP-conjugated goat IgG (1:5000; ZSGB-BIO,China) for 1 hr at 37°C. Signals were detected with a Bio-Rad Image Lab system (Bio-Rad, USA). The images were quantified by Image J software, and the relative protein expression levels were normalized to β-actin levels in each sample. All experiments were performed in triplicate.
MTT assay
Cells were plated in 96-well plates after transfection (104/well/100 μL; 6 wells per group) and incubated in a humidified atmosphere with 5% CO2 at 37°C. After incubation for 0, 24, 48, and 72 hes, MTT (5 mg/mL) was added to each well, and the plates were incubated for an additional 4 hes. Then, the culture medium was discarded, and 150 μL of DMSO was added. Finally, the plate was shaken at 200 rpm for 10 mins, and the optical density was determined at 490 nm on a multiwell plate reader. The background absorbance of the medium without cells was subtracted. All samples were assayed in triplicate, and the mean±SD for each experiment was calculated. Cell growth curves were plotted with time on the transverse axis and A490 on the longitudinal axis.
Apoptosis rate assay
After incubation at 37°C with 5% CO2 for 48 hrs, siRNA-transfected cells were harvested and washed twice with cold PBS. The cells were centrifuged, resuspended, and stained with Annexin V-FITC and propidium iodide from an Annexin V-FITC Apoptosis Detection Kit for 10 mins at room temperature in the dark. Then, the cells were analyzed on a flow cytometer according to the manufacturer’s protocol. All tests were repeated in triplicate.
RNA immunoprecipitation (RIP) assay
Transfected cells were lysed in complete RIP lysis buffer (Millipore,USA), and the cell lysates were stored at −80°C until use. The cell extracts were then incubated with magnetic beads conjugated to specific antibodies or control IgG (Millipore) overnight at 4°C. The beads were washed 6 times and incubated with proteinase K to remove proteins. All procedures were performed according to the manufacturer’s protocols. Finally, the purified RNA was reverse transcribed into cDNA and subjected to qRT-PCR analysis.
Xenograft tumor model establishment and analysis
All the animal experiments were approved by the Animal Ethics Committee of the Children’s Hospital of Chongqing Medical University and were in accordance with the experimental protocol of the Children’s Hospital of Chongqing Medical University. Cell lines were stably transfected with hnRNPL siRNA and NC siRNA and diluted with the appropriate cell culture medium. For tumor xenograft formation, 6×106 total cells were injected into the dorsal skin (6 points per mouse) of 4- to 5-week-old male BALB/c nude mice (n=3 per group, from the Experimental Animal Center of Chongqing Medical University, China). The mice were observed daily. After 4, 16, and 30 days, anesthetized mice were subcutaneously injected with 75 mg/kg D-luciferin (Xenogen, USA) in PBS. Bioluminescence images were acquired with an IVIS Imaging System (Xenogen) 2–5 mins after injection. Analyses were performed using Living Image software (Xenogen), which uses the photon flux of a region of interest (ROI) drawn around the bioluminescence signal to be measured. For determination of the “fold increase” above background, average background measurements were obtained using the same ROI for a corresponding region in NC mice. The data were divided by the average background measurement and normalized to the signal obtained immediately after xenograft formation (day 4). The tumors were obtained from nude mice after 1 month and weighed.
Statistical analyses
Statistical analysis was conducted using SPSS 19.0 software (IBM, USA). Gene expression is indicated as the mean value and its range of variability (lower limit, upper limit). Protein expression is indicated as the mean ± SD ( ± s). Comparison of multiple sample means was performed using . A P-value <0.05 was considered statistically significant for all tests.
± s). Comparison of multiple sample means was performed using . A P-value <0.05 was considered statistically significant for all tests.
Results
HnRNPL upregulation in human WT tissues and cells
The tumor types of all specimens were confirmed by two independent pathologists. Based on the RNA-Seq data from the clinical samples of 3 WT patients, 328 upregulated genes and 420 downregulated genes were identified in the WT tissues. A hierarchical clustering analysis showed that the gene expression pattern was changed (Figure 1A). We identified hnRNPL overexpression in the WT samples (P<0.01; Figure 1B). Furthermore, hnRNPL mRNA expression was significantly higher in WT tissues than in adjacent nontumorous tissues by qRT-PCR with 50 patients’ samples (P<0.01; Figure 1C). Then, hnRNPL was overexpression in WT cells compared with normal kidney cells (P<0.001; Figure 1D) as determined by qRT-PCR. These results were consistent with the chip sequencing results and implied that hnRNPL plays an important role in WT formation.
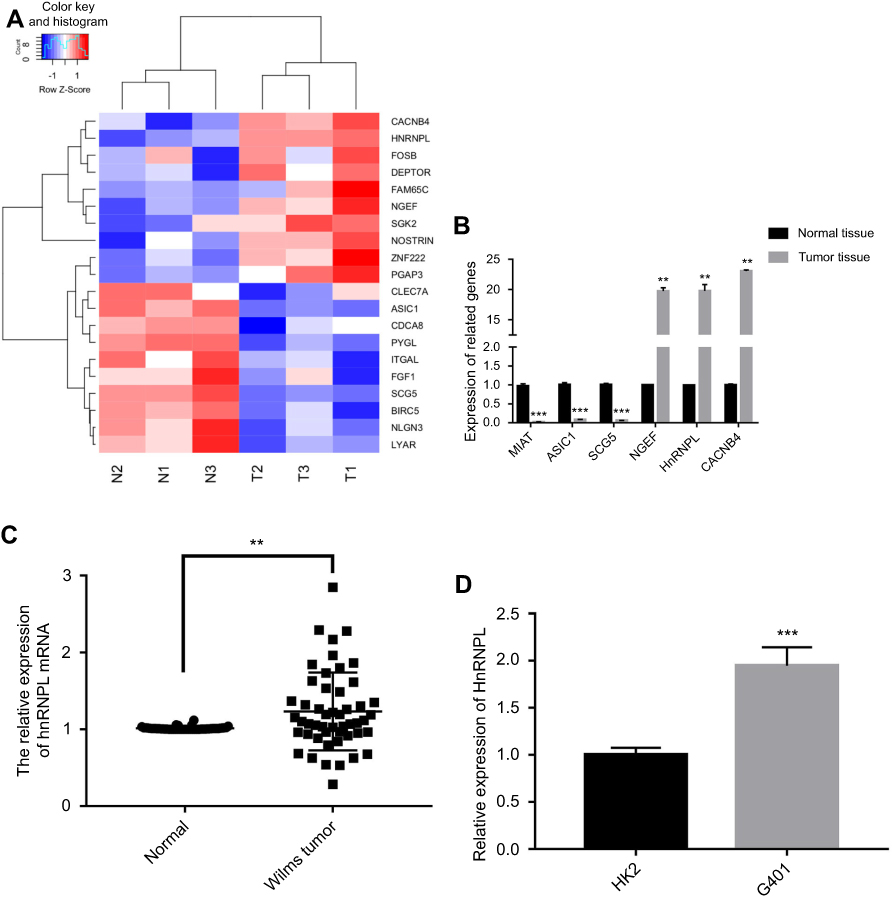 | Figure 1 HnRNPL was upregulated in human WT tissues and cells. (A) Hierarchical clustering: gene expression is similar in the tumor tissues, but hnRNPL mRNA expression is increased. (B) RNA-Seq results showing that hnRNPL mRNA is overexpression in WT tissues. (C) q-PCR results showing that the expression of hnRNPL mRNA is upregulated in 50 WT patients’ tissues. (D) q-PCR results showing that the expression of hnRNPL mRNA is upregulated in WT cells. The experiments were all repeated at least three times. **P<0.01, ***P<0.001. Abbreviations: hnRNPL, heterogeneous nuclear ribonucleoprotein L; WT, Wilms tumor; q-PCR, quantitative polymerase chain reaction. |
Differentially expressed genes participate in biological processes and the p53 signaling pathway in WTs
To systematically identify the functions of the genes differentially expressed in WTs, we performed GO and KEGG pathway enrichment analyses. GO analysis showed that the differentially expressed genes are involved in many processes, such as cell proliferation, single-organism processes, biological quality regulation, and RNA polymerase II transcriptional regulation (Figure 2A). KEGG pathway enrichment analysis showed that these differentially expressed genes are involved in some signaling pathways that are significantly correlated with tumor progression, such as leukocyte transendothelial migration, the p53 signaling pathway, and transcriptional misregulation in WT (Figure 2B).
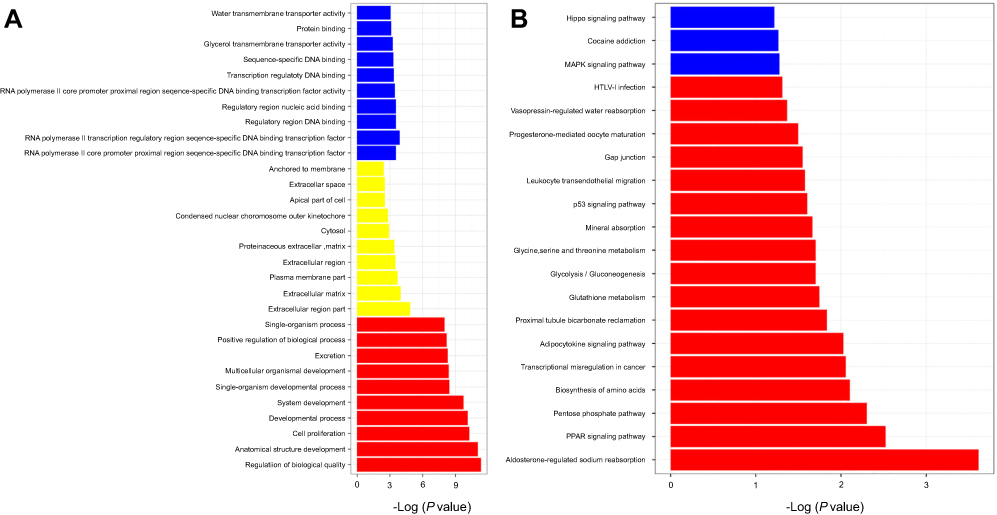 | Figure 2 Differentially expressed genes participate in the biological process of tumors. Bioinformatics analysis of differentially expressed genes ([A] GO analysis; [B] KEGG analysis). Abbreviations: GO analysis, Gene Ontology analysis; KEGG analysis, Kyoto Encyclopedia of Genes and Genomes pathway enrichment analysis. |
Relative hnRNPL, p53, and Bcl2 expression levels were significantly decreased in hnRNPL siRNA-transfected WT cells
To further investigate the effect of hnRNPL in WTs, we downregulated hnRNPL expression by transfecting tumor cells with siRNAs. Based on the bioinformatics results and previous literature, we were interested in determining whether hnRNPL could regulate the p53 signaling pathway by RNA binding or alternative splicing. qRT-PCR indicated that the expression levels of hnRNPL, p53, and Bcl2 in G401 cells were significantly lower in the hnRNPL siRNA transfection group than in the blank control and NC groups (P<0.05; Figure 3). The Western blot results were similar that siRNA transfection group is lower expression than the blank and NC groups (P<0.01; Figure 4). These results verified that hnRNPL silencing decreased p53 and Bcl2 activity, suggesting that hnRNPL could regulate p53 pathway enhancers in WTs.
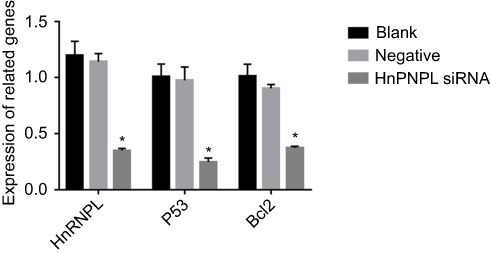 | Figure 3 Relative hnRNPL, p53, and Bcl2 expression levels were significantly decreased in hnRNPL siRNA-transfected cells. q-PCR results showing the relative mRNA expression levels of hnRNPL, p53, and Bcl2. The experiments were all repeated at least three times. *P<0.05. Abbreviations: hnRNPL, heterogeneous nuclear ribonucleoprotein L; Bcl2, B cell lymphoma 2. |
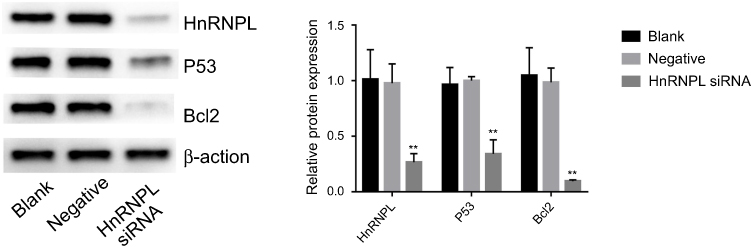 | Figure 4 P53 and Bcl2 are positively regulated by hnRNPL. Western blots of hnRNPL/p53/Bcl2 protein (typical blot for each group). The experiments were all repeated at least three times. **P<0.01. Abbreviations: hnRNPL, heterogeneous nuclear ribonucleoprotein L; Bcl2, B cell lymphoma 2. |
WT cells proliferation was significantly inhibited and apoptosis was promoted after transfection with HnRNPL siRNA
To study the functional consequences of silencing hnRNPL in WT cells, we measured cell proliferation and apoptosis. The growth of tumor cells in the hnRNPL siRNA transfection group was obviously inhibited as determined by MTT assays (P<0.05; Figure 5). Flow cytometry showed that the early apoptosis rate was significantly higher in the hnRNPL siRNA transfection group after 48 hrs than in the blank control and NC groups (P<0.001; Figure 6). Based on these data, hnRNPL silencing inhibited proliferation of WT cells and promoted cell apoptosis by reducing Bcl2 levels.
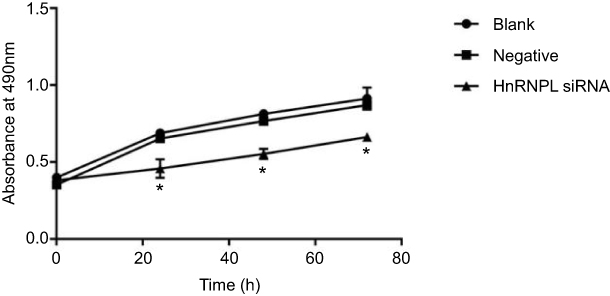 | Figure 5 Knockdown of hnRNPL inhibits G401 cells’ proliferation. MTT assay results showing that tumor cell viability was inhibited after transfection with hnRNPL siRNA for 0, 24, 48, or 72 hrs. The experiments were all repeated at least three times. *P<0.05. Abbreviation: hnRNPL, heterogeneous nuclear ribonucleoprotein L. |
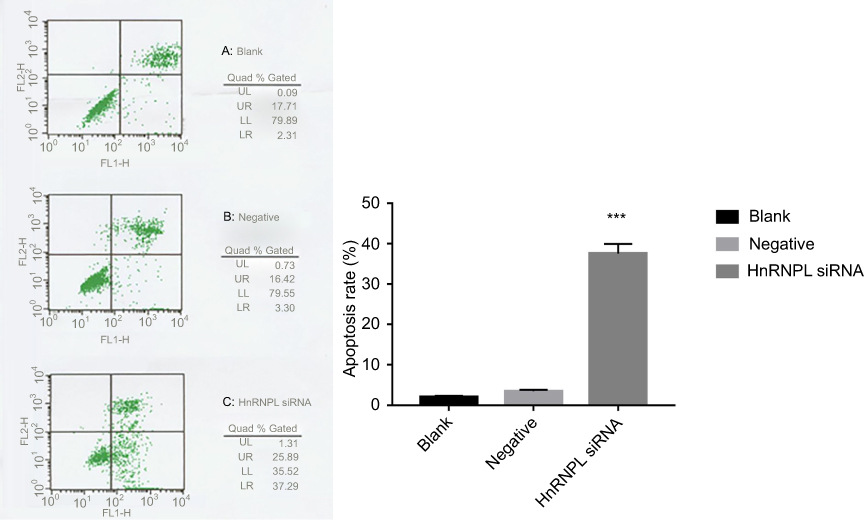 | Figure 6 Knockdown of hnRNPL promotes G401 cells’ apoptosis. Flow cytometry analysis showing the early apoptosis rate of tumor cells after hnRNPL siRNA transfection. The experiments were all repeated at least three times. ***P<0.001.Abbreviations: hnRNPL, heterogeneous nuclear ribonucleoprotein L; siRNA, small interfering RNA. |
P53 mRNA is a novel target for hnRNPL in WT
As a classic RBP, hnRNPL can regulate target genes through protein–RNA binding. To characterize the association between hnRNPL and p53, we used RIP assays to map hnRNPL–p53 interactions in WT cells. The results showed that the hnRNPL protein could directly interact with p53 mRNA in WT (Figure 7).
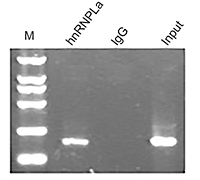 | Figure 7 P53 is directly regulated by hnRNPL. RNA immunoprecipitation showing the relationship between HnRNPL and p53.Abbreviation: hnRNPL, heterogeneous nuclear ribonucleoprotein L. |
HnRNPL promotes WT progression in vivo
Finally, to assess the impact of hnRNPL on WT progression in vivo, we subcutaneously injected G401 cells transfected with hnRNPL siRNA or NC siRNA into BALB/c nude mice. The intratumoral accumulation of hnRNPL siRNA resulted in significant suppression of tumor growth at 30 days after transplantation, as assessed by the bioluminescence imaging of xenografts (P<0.05; Figure 8A-E) and weighing of tumor tissues (P<0.05; Figure 8F). Thus, hnRNPL acts as an oncogenic factor in WT growth in vivo.
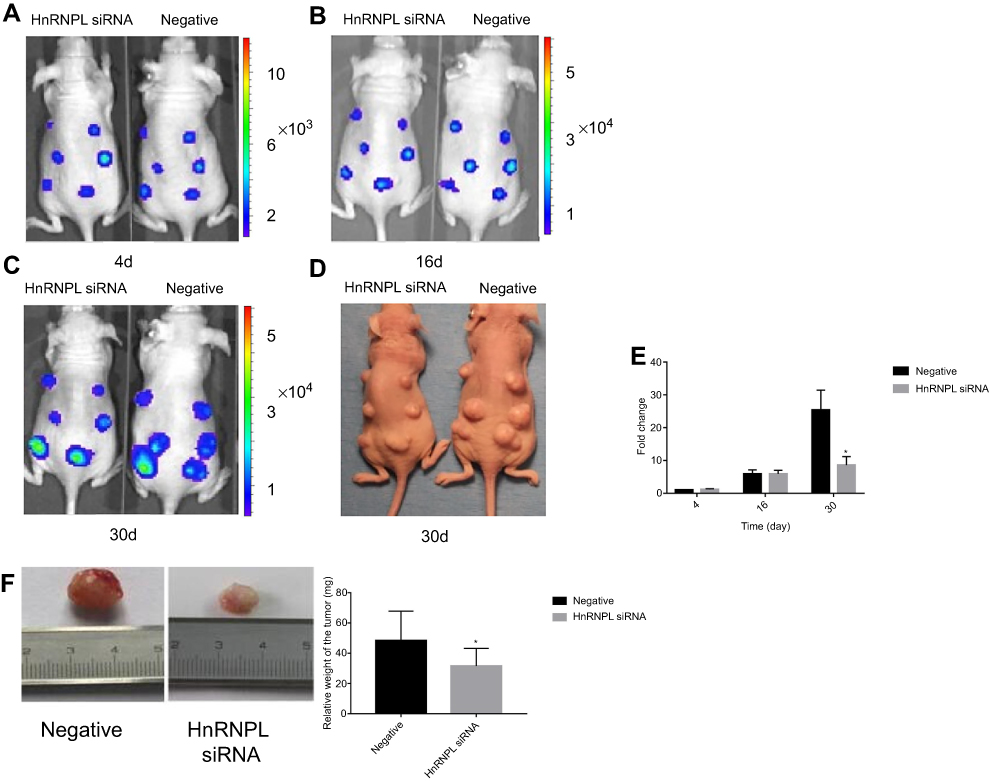 | Figure 8 HnRNPL promotes WT progression in vivo. On the indicated day after xenografting, bioluminescence images were acquired. (A, B) Representative mice injected with hnRNPL siRNA or siRNA-NC are shown in the supine position. The intensity of the signal from day 4 and 16 are displayed. (C, D) The intensity of the signal and an image from day 30 are displayed. (E) Quantitative analysis of the intensity of the signal. (F) BALB/c nude mice resected tumors from each group are shown and quantitative analysis of the weight of xenografts. *P<0.05. Abbreviations: hnRNPL, heterogeneous nuclear ribonucleoprotein L; WT, Wilms tumor; NC, negative control. |
Taken together, these data indicate that hnRNPL knockdown could inhibit proliferation and accelerate apoptosis by directly regulating the p53/Bcl2 signaling pathway in WTs. Thus, hnRNPL likely participates in the mechanism of nephroblastoma formation and may be a useful therapeutic target for WTs.
Discussion
The initiation and progression of WTs is a complex process involving multiple genes and stages, including mutation of normal genes, inactivation of tumor suppressor genes, and activation of oncogenes.30 Considering the important role of RNA editing as a posttranscriptional regulatory mechanism, it is not surprising that aberrant activation of RNA editing has emerged as a driver of cancer progression.31 The discovery of widespread deregulated RNA processing events in many human cancers indicates that transcriptome remodeling and translational deregulation are hallmarks of malignant transformation and therapeutic resistance.32 Importantly, rapid advancements in the sensitivity of sequencing technologies have facilitated the detection of rare but functionally relevant transcripts in primary human cells.32 We generated RNA expression profiles of WTs using high-throughput sequencing technology and identified 748 abnormally expressed genes. We screened an essential gene, hnRNPL, that is overexpressed in WTs and verified the results in tumor tissues and cells by qRT-PCR. Previous studies of hnRNPL in diseases have focused on its function as a spliceosome and an RBP.33,34 In addition, more than 90% of genes are shown to undergo alternative splicing, and many disease-causing mutations give rise to alternative mRNA transcripts.35,36 RBPs are highly pervasive posttranscriptional regulators involved in pathways regulating gene expression, including maturation, nuclear transport, stabilization, degradation, and translational control of RNAs.32 However, the function of hnRNPL in WTs is unknown. Here, using a combination of in vitro, in vivo, and human tumor data, we demonstrate that hnRNPL is an important modulator of WT pathogenesis and can facilitate the translation of p53 mRNA by directly binding with the p53 gene. HnRNPL knockdown decreases p53 and Bcl2 activity, induces cell apoptosis, and inhibits cell proliferation and tumor growth in WT.
P53 is a well-known tumor suppressor gene that is frequently mutated in many human cancers, and it has been extensively studied as a potential therapeutic target in WTs.20 P53 mRNA translation is regulated by some microRNAs and RBPs.37 Here, we provided evidence that hnRNPL functions as a positive regulator of p53 translation and confirmed a direct regulatory relationship between endogenous hnRNPL and p53 mRNA using RIP assays in WT cells. HnRNPL might act as an alternative spliceosome and an RBP binding to the 5ʹuntranslated region, 3ʹ end or pre-mRNA of p53 mRNA, contributing to human WT initiation and progression. Interestingly, although p53 can promote apoptosis by transcriptionally activating apoptotic genes or interacting with Bcl2 regulators and inducing cell cycle arrest,38 we found that hnRNPL knockdown results in increased apoptosis and inhibited proliferation in WT G401 cells via the blockade of Bcl2 protein which inhibits cell apoptosis by decreasing p53 levels.25 Thus, these findings demonstrate that hnRNPL promotes growth and reduces apoptosis in WTs, which supports the hypothesis that hnRNPL is closely related to WT oncogenesis.
Surprisingly, knockdown of hnRNPL could reduce the expression of p53 but inversely inhibit the growth of WTs, attributed to the fact that the p53 gene may be mutated in cancers.16,39 In fact, 75% of anaplastic nephroblastoma cases have p53 mutations, and p53 mutations could change a favorable WT histology to an unfavorable histology.40,41 Additionally, mutated p53 may be exploited by cancer cells to facilitate tumorigenesis, for example, by promoting cell proliferation, enhancing antiapoptotic activity and enhancing chemotherapy resistance.42,43 Therefore, hnRNPL might facilitate p53 mutations by splicing and editing RNA in WTs. Finally, we established a xenograft tumor model using immunodeficient mice and demonstrated that hnRNPL promoted tumorigenesis in vivo.
Conclusion
Our results have shown that hnRNPL is an oncogene that can act as a hub in the p53/Bcl2 signaling pathway, which plays an important role in the proliferation and apoptosis of WT cells. This study, for the first time, links hnRNPL to the p53 and Bcl2 signaling pathways and provides new insights into disease pathogenesis along with a useful therapeutic target for WTs.
Ethics approval and informed consent
All protocols were approved by the Ethics Committee of Chongqing Medical University, and informed consent was obtained from all patients and their parents before surgery.
Acknowledgments
This work was supported by the Children’s Hospital of Chongqing Medical University Clinical Research Projects (project no.: lcyj2014-16).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Tian F, Yourek G, Shi X, Yang Y. The development of Wilms tumor: from WT1 and microRNA to animal models. BBA - Revi Cancer. 2014;1846(1):180–187. doi:10.1016/j.bbcan.2014.07.003
2. Treger TD, Chowdhury T, Pritchard-Jones K. The genetic changes of Wilms tumour. Nature reviews. Nephrology. 2019;15(4):240–251. doi:10.1038/s41581-019-0112-0.
3. Call KM, Glaser T, Ito CY, et al.Isolation and characterization of a zinc finger polypeptide gene at the human chromosome 11 Wilms‘ tumor locus. Cell. 1990;60(3):509–520. doi:10.1016/0092-8674(90)90601-A
4. Ohlsson R, Nyström A, Pfeifer-Ohlsson S, et al. IGF2 is parentally imprinted during human embryogenesis and in the Beckwith-Wiedemann syndrome. Nat Genet. 1993;4(1):94–97. doi:10.1038/ng0593-94
5. Dewi A, Morris MR, Cooper WN, et al. Germline mutations in DIS3L2 cause the perlman syndrome of overgrowth and Wilms tumor susceptibility. Nat Genet. 2012;44(3):277. doi:10.1038/ng.1071
6. Salvatorelli L, Parenti R, Leone G, Musumeci G, Vasquez E, Magro G. Wilms tumor 1 (WT1) protein: diagnostic utility in pediatric tumors. Acta Histochem. 2015;117(4–5):367–378. doi:10.1016/j.acthis.2015.03.010
7. Parenti R, Salvatorelli L, Musumeci G, et al. Wilms’ tumor 1 (WT1) protein expression in human developing tissues. Acta Histochem. 2015;117(4–5):386–396. doi:10.1016/j.acthis.2015.03.009
8. Scott RH, Anne M, Linda B, et al. Stratification of Wilms tumor by genetic and epigenetic analysis. Oncotarget. 2012;3(3):327–335. doi:10.18632/oncotarget.468
9. Ruteshouser EC, Robinson SM, Huff V. Wilms tumor genetics: mutations in WT1, WTX, and CTNNB1 account for only about one-third of tumors. Genes Chromosomes Cancer. 2010;47(6):461–470. doi:10.1002/gcc.20553
10. Chen KS, Stroup EK, Budhipramono A, Rakheja D. Mutations in microRNA processing genes in Wilms tumors derepress the IGF2 regulator PLAG1. Genes Dev. 2018;32(15–16):996–1007. doi:10.1101/gad.313783.118
11. Devita VT, Eggermont AMM, Samuel H, Kerr DJ. Clinical cancer research: the past, present and the future. Nat Rev Clin Oncol. 2014;11(11):663–669. doi:10.1038/nrclinonc.2014.153
12. Patel JD, Lada K, Sylvia A, et al. Clinical cancer advances 2013: annual report on progress against cancer from the American society of clinical oncology. J Clin Oncol Off J Am Soc Clin Oncol. 2015;33(7):786–809. doi:10.1200/JCO.2014.59.9746
13. Senderowicz AM. Targeting cell cycle and apoptosis for the treatment of human malignancies. Curr Opin Cell Biol. 2004;16(6):670–678. doi:10.1016/j.ceb.2004.09.014
14. Livingston DM, Shivdasani R. Toward mechanism-based cancer care. Jama. 2001;285(5):588–593. dio:10.1001/jama.285.5.588
15. Williams AB, Schumacher B. p53 in the DNA-damage-repair process. Cold Spring Harb Perspect Med. 2016;6:5. doi:10.1101/cshperspect.a026070
16. Liu J, Zhang C, Hu W, Feng Z. Tumor suppressor p53 and its mutants in cancer metabolism. Cancer Lett. 2015;356(2):197–203. doi:10.1016/j.canlet.2013.12.025
17. Schulzheddergott R, Stark N, Edmunds SJ, et al. Therapeutic ablation of gain-of-function mutant p53 in colorectal cancer inhibits stat3-mediated tumor growth and invasion. Cancer Cell. 2018;34(2):298–314. doi:10.1016/j.ccell.2018.07.004
18. Schulzheddergott R, Moll U. Gain-of-function (GOF) mutant p53 as actionable therapeutic target. Cancers. 2018;10(6):188. doi:10.3390/cancers10110400
19. Bardeesy N, Falkoff D, Petruzzi MJ, et al. Anaplastic Wilms‘ tumour, a subtype displaying poor prognosis, harbours p53 gene mutations. Nat Genet. 1994;7(1):91–97. doi:10.1038/ng0594-91
20. Deng C, Dai R, Li X, Liu F. Genetic variation frequencies in Wilms‘ tumor: a meta-analysis and systematic review. Cancer Sci. 2016;107(5):690–699. doi:10.1111/cas.12910
21. Redni ST, De CB, Lopes LF, Teixeira R, Simpson A. Immunohistochemical detection of p53 protein expression as a prognostic indicator in Wilms tumor. Pediatr Blood Cancer. 2011;5(3):455. doi:10.1002/mpo.1229
22. Fei T, Chen Y, Xiao T, et al. Genome-wide CRISPR screen identifies HNRNPL as a prostate cancer dependency regulating RNA splicing. Proc Natl Acad Sci U S A. 2017;114(26):E5207. doi:10.1073/pnas.1617467114
23. Jia R, Zhang S, Liu M, et al. HnRNP L is important for the expression of oncogene SRSF3 and oncogenic potential of oral squamous cell carcinoma cells. Sci Rep. 2016;6:35976. doi:10.1038/srep35976
24. Lv D, Wu H, Xing R, et al. HnRNP-L mediates bladder cancer progression by inhibiting apoptotic signaling and enhancing MAPK signaling pathways. Oncotarget. 2017;8(8):13586–13599. doi:10.18632/oncotarget.14600
25. Kędzierska H, Piekiełko-Witkowska A. Splicing factors of SR and hnRNP families as regulators of apoptosis in cancer. Cancer Lett. 2017;396(Complete):53–65. doi:10.1016/j.canlet.2017.03.013
26. Geuens T, Bouhy D, Timmerman V. The hnRNP family: insights into their role in health and disease. Hum Genet. 2016;135(8):851–867. doi:10.1007/s00439-016-1683-5
27. Yau WY, Shih HC, Tsai MH, Sheu JC, Chen CH, Chow LP. Autoantibody recognition of an N-terminal epitope of hnRNP L marks the risk for developing HBV-related hepatocellular carcinoma. J Proteomics. 2013;94(20):346–358. doi:10.1016/j.jprot.2013.10.003
28. Goehe RW, Shultz JC, Charuta M, et al. hnRNP L regulates the tumorigenic capacity of lung cancer xenografts in mice via caspase-9 pre-mRNA processing. J Clin Invest. 2010;120(11):3923. doi:10.1172/JCI43552
29. Liu C, Wang J, Yuan X, et al. Long noncoding RNA uc.345 promotes tumorigenesis of pancreatic cancer by upregulation of hnRNPL expression. Oncotarget. 2016;7(44):71556–71566. doi:10.18632/oncotarget.12253
30. Weksberg R, Brzezinski J. Identifying new Wilms' tumour predisposition genes. The Lancet. Child & adolescent health. 2019;3(5):285-287. doi:10.1016/S2352-4642(19)30064-1
31. Gagnidze K, Rayon-Estrada V, Harroch S, Bulloch K, Papavasiliou FN. A new chapter in genetic medicine: RNA editing and its role in disease pathogenesis. Trends Mol Med. 2018;24(3):294–303. doi:10.1016/j.molmed.2018.01.002
32. Jiang Q, Crews LA, Holm F, Jamieson CHM. RNA editing-dependent epitranscriptome diversity in cancer stem cells. Nat Rev Cancer. 2017;17(6):381–392. doi:10.1038/nrc.2017.23
33. Teng F, Chen Y, Brown M, Liu XS. Abstract IA11: dependency of prostate cancer on HNRNPL and its associated RNAs. Cancer Res. 2016;76(6Supplement):IA11–IA11. doi:10.1158/1538-7445.NONRNA15-IA11
34. Chiou NT, Shankarling G, Lynch K. HnRNP L and HnRNP A1 induce extended U1 snRNA interactions with an exon to repress spliceosome assembly. Mol Cell. 2013;49(5):972–982. doi:10.1016/j.molcel.2012.12.025
35. Wang ET, Rickard S, Shujun L, et al. Alternative isoform regulation in human tissue transcriptomes. Nature. 2008;456(7221):470–476. doi:10.1038/nature07509
36. Guey-Shin W, Cooper TA. Splicing in disease: disruption of the splicing code and the decoding machinery. Nat Rev Genet. 2007;8(10):749–761. doi:10.1038/nrg2164
37. Freeman JA, Espinosa JM. The impact of post-transcriptional regulation in the p53 network. Brief Funct Genomics. 2013;12(1):46–57. doi:10.1093/bfgp/els058
38. Kim EM, Jung CH, Kim J, Hwang SG, Park J, Um HD. The p53/p21 complex regulates cancer cell invasion and apoptosis by targeting Bcl-2 family proteins. Cancer Res. 2017;77(11):3092–3100. doi:10.1158/0008-5472.CAN-16-2098
39. Marie C, Aurélien DR, Rickman DS, et al.WNT/beta-catenin pathway activation in Wilms tumors: a unifying mechanism with multiple entries?Genes Chromosomes Cancer. 2010;48(9):816–827. doi:10.1002/gcc.20686
40. Maschietto M, Williams RD, Chagtai T, et al. TP53 mutational status is a potential marker for risk stratification in Wilms tumour with diffuse anaplasia. PLoS One. 2016;9(10):e109924. doi:10.1371/journal.pone.0109924
41. Natrajan R, Williams RD, Hing SN, et al. Array CGH profiling of favourable histology Wilms tumours reveals novel gains and losses associated with relapse. J Pathol. 2010;210(1):49–58. doi:10.1002/path.2021
42. Zhang Y, Coillie SV, Fang JY, Xu J. Gain of function of mutant p53: R282W on the peak? Oncogenesis. 2016;5(2):e196. doi:10.1038/oncsis.2016.8
43. He C, Li L, Guan X, Xiong L, Miao X. Mutant p53 gain of function and chemoresistance: the role of mutant p53 in response to clinical chemotherapy. Chemotherapy. 2016;62(1):43–53. doi:10.1159/000446361
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.