")
Back to Journals » OncoTargets and Therapy » Volume 7
hnRNP C1/C2 and Pur-beta proteins mediate induction of senescence by oligonucleotides homologous to the telomere overhang
Authors Mulnix R, Pitman R, Retzer A, Bertram C, Arasi K, Crees Z, Girard J, Uppada S, Stone A, Puri N
Received 16 September 2013
Accepted for publication 2 November 2013
Published 18 December 2013 Volume 2014:7 Pages 23—32
DOI https://doi.org/10.2147/OTT.S54575
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Richard E Mulnix,1,* Ryan T Pitman,1 Allison Retzer,2 Ceyda Bertram,1 Kavin Arasi,2 Zachary Crees,2 Jennifer Girard,2 Srijayaprakash B Uppada,1 Amanda L Stone,1 Neelu Puri1,*
1Department of Biomedical Sciences, University of Illinois at Chicago, Rockford, IL, USA; 2College of Medicine, University of Illinois at Chicago, Rockford, IL, USA
*These authors contributed equally to this work
Background: Experimental disruption of the telomere overhang induces a potent DNA damage response and is the target of newly emerging cancer therapeutics. Introduction of T-oligo, an eleven-base oligonucleotide homologous to the 3'-telomeric overhang, mimics telomere disruption and induces DNA damage responses through activation of p53, p73, p95/Nbs1, E2F1, pRb, and other DNA damage response proteins. ATM (ataxia telangiectasia mutated) was once thought to be the primary driver of T-oligo-induced DNA damage responses; however, recent experiments have highlighted other key proteins that may also play a significant role.
Methods: To identify proteins associated with T-oligo, MM-AN cells were treated with biotinylated T-oligo or complementary oligonucleotide, cell lysates were run on SDS-PAGE (sodium dodecyl sulfate polyacrylamide gel electrophoresis), and the protein bands observed after treatment of cells with T-oligo or complementary oligonucleotide were analyzed using mass spectrometry. To study the effect of T-oligo on expression of hnRNP C1/C2 (heterogeneous nuclear ribonucleoprotein C1 and C2) and purine-rich element binding proteins (Pur proteins), cells were treated with T-oligo, and immunoblotting experiments were performed. To determine their role in senescence, cells were treated with shRNA (short hairpin ribonucleic acid) against these proteins, and senescence was studied using the senescence associated beta-galactosidase assay.
Results: Using mass spectrometry, RNA-binding hnRNP C1/C2 and DNA-binding Pur proteins were found to associate with T-oligo. hnRNP C1/C2 exhibited increased expression (3.6–12.0-fold) in non-small-cell lung cancer (NSCLC) and in melanoma cells (4.5–5.2-fold), and Pur proteins exhibited increased expression of 2.2-fold in NSCLC and 2.0-fold in melanoma cells after T-oligo treatment. Experimental knockdown of hnRNP C1/C2 and Pur-beta completely abrogated T-oligo induced senescence in both MU melanoma and H358 NSCLC cells. Additionally, knockdown of Pur-beta prevented T-oligo-induced phosphorylation of p53, hypophosphorylation of pRb, and upregulation of E2F1, p21, and p53.
Conclusion: These novel findings highlight proteins essential to T-oligo's anticancer effects that may be of interest in telomere biology and cancer therapeutics.
Keywords: T-oligo, melanoma, NSCLC, p53, p21, pRb
Introduction
Telomeres are highly specialized multimeric structures located at the ends of chromosomes and are structurally and functionally distinct from the rest of the chromosome. Their major function is to shield the terminal ends of chromosomal DNA from damage by forming T-loops, thus preventing their false recognition as DNA double-strand breaks. The 3′ end of each telomere consists of a single stranded overhang of TTAGGG tandem repeats,1 which are incorporated into the T-loop structure and protected by the shelterin protein complex. When this protein complex is absent, the telomere overhang is exposed, resulting in DNA damage responses (DDR) such as apoptosis and senescence.1,2
Exogenous addition of T-oligo, an eleven-base oligonucleotide homologous to the telomere overhang, has been shown to induce DDR through p95/Nbs1, E2F1, p16INK4A, p53, and pRb,2–7 similar to those seen after disruption of the telomere.3,7 Previous studies implicated WRN (Werner syndrome protein) and ATM (ataxia telangiectasia mutated) as essential mediators of T-oligo-induced DDR.8,9 The WRN exonuclease facilitates non-homologous end-joining, DNA replication, and base-excision repair.10 In the absence of WRN, senescence rapidly develops due to critically shortened telomeres.11 Activated ATM phosphorylates DDR signaling proteins to induce cell cycle arrest, DNA repair and/or apoptosis12 through p53, CHK2, and γ-H2AX.12–14 However, T-oligo has also been shown to function independently of DNA damage signaling pathways via cdk2.15 Nevertheless, despite multiple studies demonstrating T-oligo induced DDR, T-oligo’s specific mechanism of action is still unclear.
In addition to the use of T-oligo as a tool to study novel mechanisms of telomere-induced DDR, T-oligo has been proposed as a possible therapeutic modality in multiple cancerous cell types. T-oligo displays preferentially robust apoptotic and senescent effects on malignant cells, relative to their normal cell counterparts.5,6,16,17 Additionally, we propose the use of T-oligo as a tool to identify new targets for cancer therapeutics.
In the present study, we identified proteins which co-precipitated with biotinylated T-oligo and streptavidin-coated beads by mass spectrometry. Several proteins associated with T-oligo were identified, including heterogeneous nuclear ribonucleoprotein C1 and C2 (hnRNP C1/C2) and the purine-rich element binding proteins alpha and beta (Pur-alpha and Pur-beta). hnRNP C1/C2 are integral members of the telomerase holoenzyme18 that may mediate the interaction between telomerase and the telomere.19 hnRNP C1/C2 binds to chromatin in a DNA-damage-dependent manner, implicating it in DNA repair and/or damage responses and has also been linked to cancer.20,21 Further, hnRNP C1/C2 may play an important role in regulating p53 transcription in response to cytostatic drugs.22 Pur-alpha and Pur-beta are single-stranded DNA binding proteins, which form a heterodimeric complex23 and regulate DNA replication and transcription.24 Both Pur-alpha and Pur-beta have been linked with acute myelogenous leukemia and brain tumors.25,26 Moreover, Pur-alpha and Pur-beta may function in a pathway involving pRb, which plays an important role in senescence.25,27,28 Therefore, in this study, we investigated the potential roles of hnRNP C1/C2, as well as Pur-alpha and Pur-beta, in T-oligo-induced DDR and suggest their role in T-oligo-mediated senescence.
Materials and methods
Cell culture, reagents and antibodies
MU and MM-AN melanoma cell lines which are p53 proficient29 and deficient,5 respectively, were obtained as described earlier,5,30,31 and maintained at 37°C and 7% CO2 in MEM (minimum essential medium) (Thermo Fisher Scientific, Pittsburg, PA, USA; Cat No 10-010-CV), supplemented with 10% (volume/volume [v/v]) fetal bovine serum (FBS) (Atlanta Biologicals, Lawrenceville, GA, USA) and 1% (v/v) antibiotic-antimycotic. H358 and SW1573 NSCLC cells were obtained from American Type Culture Collection (ATCC, Manassas, VA, USA) and cultured according to ATCC directions. Briefly, H358 cells were maintained at 37°C and 7% CO2 and cultured in RPMI (Roswell Park Memorial Institute)-1640 medium (Thermo Fisher Scientific, Pittsburg, PA, USA; Cat No SH30027) supplemented with 10% FBS, 100 units/mL penicillin, 100 μg/mL streptomycin, 0.01 M HEPES (hydroxyethyl piperazineethanesulfonic acid), and 1 mM sodium pyruvate, while SW1573 cells were cultured in MEM supplemented with 10% (v/v) FBS, 100 units/mL penicillin, 100 μg/mL streptomycin, and 1 mM nonessential amino acids. All cell culture reagents were obtained from Invitrogen (Grand Island, NY, USA), unless otherwise noted. Cells were prepared for experimentation by detachment with trypsin, collection in media and centrifugation at 350 × g for 3 minutes. Cells were then resuspended, counted and plated in 35 mm × 10 mm tissue culture dishes for experimentation.
DNA oligonucleotides homologous to the 3′ overhang sequence, T-oligo (pGTTAGGGTTAG), and complementary to this sequence (pCTAACCCTAAC) (complementary oligo nucleotide) were obtained from Midland Certified Reagent Company (Midland, TX, USA). All short hairpin ribonucleic acid (shRNA) including anti-hnRNP C shRNA, anti-Pur-beta shRNA, and control shRNA were obtained from Sigma-Aldrich (St Louis, MO, USA).
Antibodies for immunoblotting against p21, E2F1, hnRNP C1/C2 (Santa Cruz Biotechnology, Santa Cruz, CA, USA), p53, phospho-p53, pRb, and p21 (Cell Signaling, Danvers, MA, USA) were used in 2% bovine serum albumin in TBST (Tris-buffered saline with Tween) according to the manufacturers’ recommendations. Pur-beta was a generous gift from Dr Robert Kelm, from the University of Vermont.32 Beta-actin antibody, anti-mouse, and anti-rabbit secondary antibodies, 5-Bromo-4-chloro-3-indolyl beta-D-galactopyranoside, propidium iodide, and all other chemicals were obtained from Sigma-Aldrich.
Mass spectrometry and immunoprecipitation
To identify proteins associated with T-oligo, MM-AN melanoma cells were treated with 40 μM biotinylated T-oligo or complementary oligonucleotide for 6 hours. Cells were collected and lysed, and the lysates were incubated with streptavidin agarose-coated beads to isolate the biotinylated T-oligo or complementary oligonucleotide. Non-biotinylated T-oligo or complementary oligonucleotide were added in 10.0-fold excess. Supernatant, containing T-oligo-associated proteins, was then eluted and run on an SDS (sodium dodecyl sulfate) tris-glycine 6%–12% gradient gel and stained with Coomassie Brilliant Blue. Protein bands present (~50–37 kDa) in T-oligo-treated lysates (Figure S1) were cut from the gel and sent to the University of Illinois at Chicago Proteomics Core Laboratory for identification, and were analyzed using the Applied Biosystems 4700 MALDI-TOF/TOF MS (matrix-assisted laser desorption/ionization-time of flight mass spectrometry) unit. For protein identification, two peptides per protein, with 90% peptide confidence and 90% protein confidence thresholds, were used. A relevant literature search and Ingenuity Pathways Analysis (Ingenuity Systems, Redwood, CA, USA) were utilized to identify targets for validation and further research.
Immunoblotting
H358 and SW1573 NSCLC cell lines, and MM-AN and MU melanoma cells were plated at 1.5 × 105 cells per 35 mm × 10 mm tissue culture dish in their respective media. After 24 hours, the media was replaced with a diluent (sterile water), 40 μM of a complementary oligonucleotide, or 40 μM of T-oligo. The cells were then incubated for 6, 18, 24, or 48 hours after treatment. After which, cells were washed twice with phosphate buffered saline (PBS) and cell lysates were collected using 200 μL lysis buffer (20 mM Tris-HCL pH 8.0) containing 150 mM NaCl, 100 mM NaF, 1% NP-40, 10% glycerol, 10 μM sodium orthovanadate, and 1X Complete Protease Inhibitors (Roche Diagnostics, Indianapolis, IN, USA). A sonicator was then used to homogenize cell lysates, and protein concentration was determined using the Bio-Rad protein assay utilizing a Coomassie Brilliant Blue acidic dye with quantification by spectrophotometric measurement at 595 nm (Hercules, CA, USA). From each sample, 50 μg of protein was run on an SDS-PAGE (sodium dodecyl sulfate polyacrylamide gel electrophoresis) gel, and blots were probed with antibodies for hnRNP C1/C2, and Pur-alpha or Pur-beta. Immunoblots were then visualized using Western Lightning ECL (Perkin Elmer, Waltham, MA, USA). Densitometry was performed with Image J software, and figures display representative immunoblots from experiments done in triplicate.
Transfection of shRNA vectors
To demonstrate the role of Pur-beta and hnRNP C1/C2 proteins in T-oligo responses in H358 and MU cells, the knockdown of these proteins was performed using the shRNA vectors for hnRNP C1/C2 (SHCLNV-NM_031314 clone TRCN0000006644 Sigma, St Louis, MO, USA), shRNA control (SHC002V Sigma), or Pur-beta (SHCLNV-NM_033224 clone TRCN0000155904), according to the manufacturer’s instructions. Briefly, the cells were plated at 50,000 cells per well in a 12-well plate and grown for 24 hours. The cells were then transfected using virus particles and incubated for 24 hours at 37°C. The multiplicity of infection of the virus particles used was determined to be 5, which was obtained by titration experiments. The transfected cells used for the study were selected based on puromycin resistance. Knockdown was validated by immunoblotting after 24 hours.
Identification of senescent cells
MU and H358 cells were plated at 25,000 cells per 60 mm × 15 mm dish in triplicate and transfected after 24 hours with hnRNP C1/C2 or Pur-beta shRNA vectors. Following which, the cells were treated with T-oligo for 1 week as described above. Cells were then washed twice in PBS, fixed at room temperature using 3% formaldehyde for 5 minutes, and washed twice with PBS. The beta-galactosidase solution was prepared as described previously,33 and cells were incubated for 24 hours at 37°C. Cells were then visualized at 40× magnification on a Nikon Diaphot (Nikon, Mellville, NY, USA) and counted by a blinded observer using 15 different microscopic fields for each sample. Cells that stained blue were considered positive for senescence-associated beta-galactosidase. Statistical analyses between various treatment groups were carried out with Statistical Product and Service Solutions (SPSS) Statistics (SPSS Inc., Chicago, IL, USA) 17.0 software using a one-way ANOVA (analysis of variance) with post hoc analysis. Significance was established at α=0.05. Photographic images of the beta-galactosidase-positive cells were taken using a 10× objective lens on an Olympus CK2 microscope.
Results
Screening and identification of proteins associated with T-oligo
Previous studies have implicated WRN and ATM as important mediators of T-oligo-induced DDR; however, recent results indicate that other proteins may also play a significant role in T-oligo-mediated responses.10,12,15 To further investigate the mechanism behind the DDR related to T-oligo exposure, we treated MM-AN melanoma cells with biotinylated T-oligo or complementary oligonucleotide, and proteins associated with T-oligo were analyzed by mass spectrometry, as described in the methods section. This study identified proteins, such as hnRNP A, hnRNP C1/C2, hnRNP D, VDAC 1 and 2, MSI-1, Pur-alpha, and Pur-beta, which are known to be involved in DDR22,25,34–40 (Table S1). In the present study, hnRNP C1/C2, Pur-alpha and Pur-beta, identified by Ingenuity Pathways Analysis, were further investigated due to their key roles in nucleic acid binding, regulation of transcription/translation, and association with telomeres and DDR.18,21,24,26,32
T-oligo increases hnRNP C1/C2, Pur-alpha, and Pur-beta expression
To investigate the roles of hnRNP C1/C2, Pur-alpha, and Pur-beta as elements in T-oligo-induced DDR, melanoma (MU and MM-AN) and NSCLC (H358 and SW1573) cell lines were exposed to T-oligo or a complementary oligonucleotide and subsequently immunoblotted. Treatment of melanoma cells with T-oligo revealed a marked increase of hnRNP C1/C2 expression in MU cells (5.2-fold) at 6 hours of exposure (Figure 1A), with corresponding increases in hnRNP C1/C2 at 18 hours in MM-AN cells (4.5-fold) (Figure 1B). Furthermore, in NSCLC cells at 18 hours, there was also an increase in hnRNP C1/C2 in H358 (3.6-fold) (Figure 1C) and SW1573 (12.0-fold) (Figure 1D), and a prolonged increase in expression of hnRNP C1/C2 in SW1573 (3.0-fold) extending up to 24 hours (Figure 1D). MM-AN and H358 cells were then immunoblotted to evaluate expression of Pur-alpha and Pur-beta after exposure to T-oligo. Pur-alpha and Pur-beta were upregulated at 24 hours (2.0-fold) in MM-AN cells (Figure 2A), with a similar increase in both Pur-alpha and Pur-beta at 18 hours in H358 cells (2.2-fold) (Figure 2B).
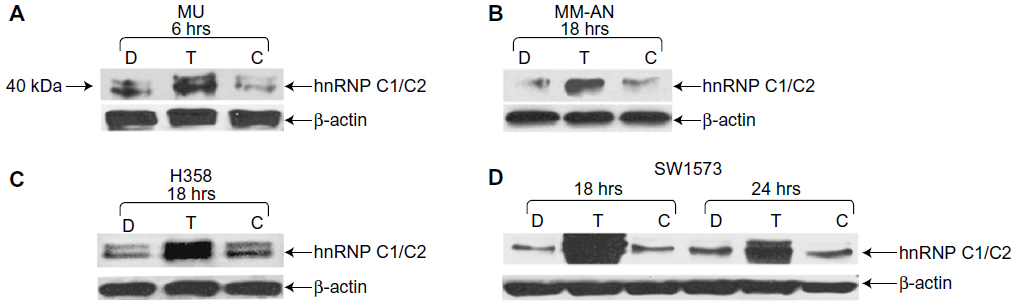 | Figure 1 Exposure to T-oligo increased the expression of hnRNP C1/C2. H358, SW1573, MU, and MM-AN cancer cell lines were treated with either diluent, T-oligo, or complementary oligonucleotide for the indicated periods of time and then immunoblotted for hnRNP C1/C2. |
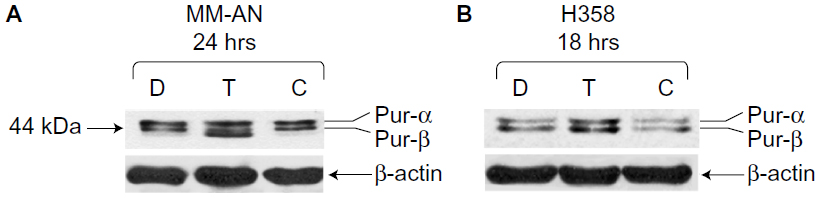 | Figure 2 Upregulation of Pur-alpha and Pur-beta induced by T-oligo in MM-AN and H358 cells, as shown by immunoblotting. |
hnRNP C1/C2 and Pur-beta knockdown decreases T-oligo-induced senescence
To validate the role of hnRNP C1/C2 and Pur-beta in T-oligo-induced DDR, we utilized anti-hnRNP C1/C2 and anti-Pur-beta shRNA vectors to create hnRNP C1/C2 and Pur-beta-deficient H358 and MU cells. As seen in Figure 3A, a significant decrease in hnRNP C1/C2 expression (3.5-fold) in H358 cells and Pur-beta expression (3.0-fold) in MU cells was documented after 24 hours of transfection. Additionally, we observed no increase in Pur-beta expression upon exposure to T-oligo in Pur-beta-deficient MU cells (Figure 3A). hnRNP C1/C2 or Pur-beta deficient H358 or MU cells were then exposed to 40 μM T-oligo for 1 week, and subsequently stained for senescence-associated beta-galactosidase as described previously.7 Knockdown of Pur-beta reduced the ability of T-oligo to induce senescence by 19.3-fold in MU cells (Figure 3B and C), in comparison to cells exposed to T-oligo after transfection with control shRNA. Similarly, abrogation of hnRNP C1/C2 expression by shRNA reduced the ability of T-oligo to induce senescence in H358 cells by 14.8-fold (Figure 3D) and in MU cells by 12.5-fold, in comparison with cells exposed to T-oligo after transfection with control shRNA, and representative images have been presented (Figure 3B and D).
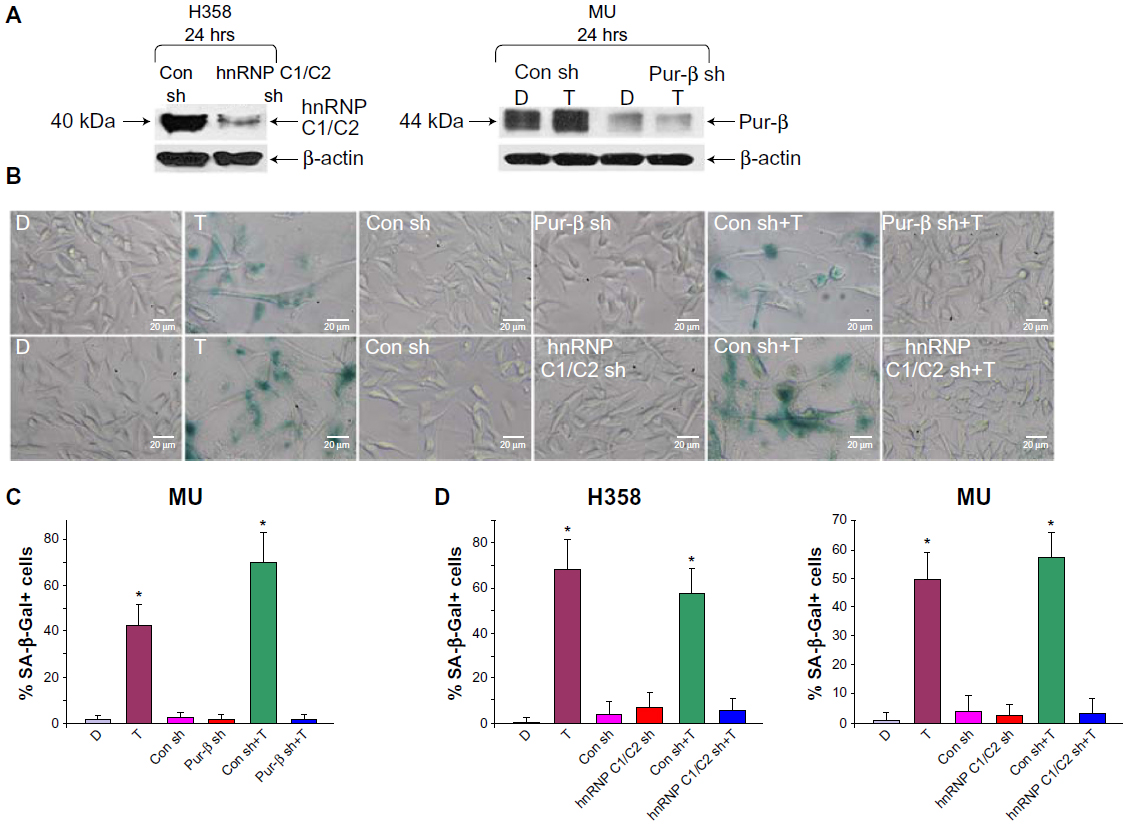 | Figure 3 Knockdown of hnRNP C1/C2 and Pur-beta decreased T-oligo-induced senescence in H358 and MU cells. |
Pur-beta knockdown decreases T-oligo-induced hypophosphorylation of pRb and increases p53 and phospo-p53 expression
Exposure to T-oligo is well known to induce upregulation and phosphorylation of p53 and hypophosphorylation of pRb.4,6,7,16 At present, the mechanism of T-oligo-driven phosphorylation of p53 is unclear. To determine the role of Pur-beta in T-oligo-induced p53 phosphorylation, we immunoblotted for p53, phospho-p53(Ser-15) and E2F1 in Pur-beta proficient/deficient MU cells. T-oligo, in Pur-beta proficient cells, induced upregulation of E2F1 (6.5-fold), phospho-p-53(Ser-15) (6.0-fold), and p53 (2.0-fold) (Figure 4A). T-oligo also induced upregulation of p21 (1.7-fold), as well as hypophosphorylation of pRb (2.0-fold) in Pur-beta proficient cells (Figure 4B). Treatment with T-oligo, in Pur-beta deficient MU cells, failed to induce p53 phosphorylation, increase p53 expression, or increase E2F1 expression (Figure 4A). Furthermore, T-oligo failed to upregulate p21 or induce hypophosphorylation of pRb, which are important drivers of senescence in cancerous cells7 (Figure 4B). This suggests that Pur-beta may play a role in T-oligo induced senescence and apoptosis.
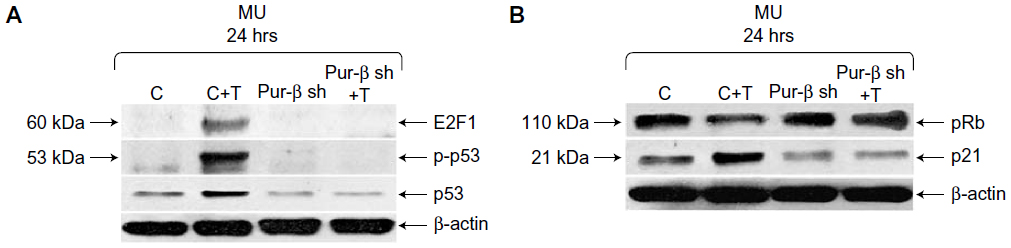 | Figure 4 Knockdown of Pur-beta prevents T-oligo-induced upregulation of senescence-associated proteins in MU melanoma cells. |
Discussion
This study identifies hnRNP C1/C2 and Pur-beta as proteins associated with T-oligo, which may also play a role in mediating T-oligo-induced anticancer effects, and sheds light on the possible mechanism of action of T-oligo. These proteins may be novel targets for further investigation into telomere biology and cancer therapeutics.
Our results confirm previous studies by demonstrating an increase in E2F1, p53, phospho-p53, and p21 expression, as well as hypophosphorylation of pRb in cells treated with T-oligo.3,4,8 T-oligo can induce DDR in a p53-dependent or independent manner.41 The knockdown of both p53 and pRb halts senescence induced by T-oligo,4 suggesting that these two proteins are essential for T-oligo-induced senescence. However, we previously demonstrated the ability of the p53 homologue, p73, to mediate the effects of T-oligo in cancer cell lines with a nonfunctional p53.3,5 Furthermore, E2F1 may be important in mediating the effects of T-oligo in a p53-independent manner.3 While the downstream effects of T-oligo are well known, target proteins involved in mediating these effects are unclear. WRN and ATM have previously been implicated in initiating DDR;8,10 however, recent evidence indicates that these effects may also occur independently of ATM.15 Moreover, the exonuclease activity of WRN is length dependent and requires greater than 35 nucleotides of single-stranded DNA for its activity.42 Therefore, it is important to investigate other proteins that may be involved in T-oligo function.
hnRNPs are RNA binding proteins that associate with heterogeneous nuclear RNA and appear to influence messenger RNA (mRNA) trafficking, pre-mRNA processing, and other aspects of mRNA metabolism.18,43 All hnRNPs are found within the nucleus, where they mediate translocation of a wide range of RNA transcripts and proteins between the nucleus and the cytoplasm.44 In cancer cells, hnRNP C1/C2 modulates p53 expression through a regulatory element within p53 mRNA.22 Further, hnRNP C1/C2, in conjunction with other hnRNPs, assists in the formation of the telomerase holoenzyme, and may mediate binding to the telomere.18,19 In the present study, we found that hnRNP C1/C2 associates directly, or through other telomere binding proteins, with T-oligo. Previous studies have demonstrated that T-oligo induces DDR in cancer cells,5,45 and it has also been suggested that hnRNP C1/C2 migrates to sites of DNA damage.46 These and the present study suggest that exposure to T-oligo, which is homologous to the telomere 3′ overhang, could signal DNA damage and result in recruitment of hnRNP C1/C2. Additionally, studies indicate that T-oligo increases TRF-2 (telomeric repeat-binding factor 2), which may bind and stabilize hnRNP C1/C2 as well as cause its upregulation.18,19,29 hnRNP C1/C2 may also mediate retention of T-oligo in the nucleus,3 bind other vital proteins to T-oligo, or may be recruited away from the telomerase holoenzyme leading to telomere overhang exposure and induction of DDR. We propose that hnRNP C1/C2 may be essential to T-oligo-induced senescence, through its regulation of p53 mRNA.22 However, the cause of hnRNP C1/C2 upregulation and recruitment upon exposure to T-oligo, which may be due to a transcriptional or translational response, needs further investigation.
The Pur family proteins bind purine-rich elements in single-stranded DNA and play key roles in transcriptional and translational regulation, as well as mRNA trafficking in a variety of cell types.26 Further, Pur proteins may play a role in pathways that inhibit oncogenic transformation, as seen in acute myelogenous leukemia with associated deletions in Pur-alpha and Pur-beta.25 In the present study, both Pur-alpha and Pur-beta were upregulated in MM-AN and H358 cells after exposure to T-oligo. Furthermore, knockdown of Pur-beta prevented T-oligo-induced senescence, pRb hypophosphorylation, and p53 phosphorylation, as well as upregulation of E2F1, p21, and p53 expression. Earlier studies have shown that T-oligo-induced senescent cells exhibit p53 activation in addition to induction and upregulation of its downstream target p21 and subsequent reduction of pRb phosphorylation.4,7 However, the cause of Pur-alpha and Pur-beta upregulation in response to T-oligo is unknown. T-oligo is known to activate ATM,3 which may in turn cause upregulation of Pur-beta, which consequently heterodimerizes with Pur-alpha, which binds to hypophosphorylated pRb,25,27 possibly stabilizing it and driving cellular senescence.7 Additionally, our results suggest that Pur-beta is essential for T-oligo-induced DDR, and suggests for the first time, to the best of our knowledge, a connection between Pur-beta, p53, and downstream DDR. Furthermore, Pur-beta could be a new target for anticancer treatment and a valuable tool in the investigation of telomere-related DDR. Further investigations into the importance of other T-oligo-associated proteins are ongoing and include assessment of the requirement for hnRNP C1/C2, Pur-alpha, and Pur-beta in T-oligo-induced inhibition of cellular proliferation.
Conclusion
The present study identifies T-oligo-associated proteins that may have a role in T-oligo’s anticancer effects. This study may also help in understanding the anticancer mechanism of action of T-oligo, and generate further research into a new line of telomere-based anticancer agents that specifically target cancer cells and leave normal cells minimally affected. T-oligo represents a novel method for treating cancer and promises to generate new ideas for developing a wide range of telomere-based anticancer agents.
Acknowledgments
This study was funded by the University of Illinois at Chicago Cancer Center 2010 Pilot Grant Program. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Disclosure
The authors have no conflicts of interest in this work.
References
Tian X, Chen B, Liu X. Telomere and telomerase as targets for cancer therapy. Appl Biochem Biotechnol. 2010;160:1460–1472. | |
Rankin AM, Faller DV, Spanjaard RA. Telomerase inhibitors and ‘T-oligo’ as cancer therapeutics: contrasting molecular mechanisms of cytotoxicity. Anticancer Drugs. 2008;19:329–338. | |
Eller MS, Puri N, Hadshiew IM, Venna SS, Gilchrest BA. Induction of apoptosis by telomere 3′ overhang-specific DNA. Exp Cell Res. 2002;276:185–193. | |
Li GZ, Eller MS, Hanna K, Gilchrest BA. Signaling pathway requirements for induction of senescence by telomere homolog oligonucleotides. Exp Cell Res. 2004;301:189–200. | |
Puri N, Eller MS, Byers HR, Dykstra S, Kubera J, Gilchrest BA. Telomere-based DNA damage responses: a new approach to melanoma. FASEB J. 2004;18:1373–1381. | |
Longe HO, Romesser PB, Rankin AM, et al. Telomere homolog oligonucleotides induce apoptosis in malignant but not in normal lymphoid cells: mechanism and therapeutic potential. Int J Cancer. 2009;124:473–482. | |
Li GZ, Eller MS, Firoozabadi R, Gilchrest BA. Evidence that exposure of the telomere 3′ overhang sequence induces senescence. Proc Natl Acad Sci U S A. 2003;100:527–531. | |
Eller MS, Li GZ, Firoozabadi R, Puri N, Gilchrest BA. Induction of a p95/Nbs1-mediated S phase checkpoint by telomere 3′ overhang specific DNA. FASEB J. 2003;17:152–162. | |
Sarkar S, Faller DV. T-oligos inhibit growth and induce apoptosis in human ovarian cancer cells. Oligonucleotides. 2011;21:47–53. | |
Eller MS, Liao X, Liu S, et al. A role for WRN in telomere-based DNA damage responses. Proc Natl Acad Sci U S A. 2006;103:15073–15078. | |
Crabbe L, Jauch A, Naeger CM, Holtgreve-Grez H, Karlseder J. Telomere dysfunction as a cause of genomic instability in Werner syndrome. Proc Natl Acad Sci U S A. 2007;104:2205–2210. | |
Lee JH, Paull TT. Activation and regulation of ATM kinase activity in response to DNA double-strand breaks. Oncogene. 2007;26:7741–7748. | |
Burma S, Chen BP, Murphy M, Kurimasa A, Chen DJ. ATM phosphorylates histone H2AX in response to DNA double-strand breaks. J Biol Chem. 2001;276:42462–42467. | |
Canman CE, Lim DS, Cimprich KA, Taya Y, Tamai K, Sakaguchi K, et al. Activation of the ATM kinase by ionizing radiation and phosphorylation of p53. Science. 1998;281:1677–1679. | |
Rankin AM, Sarkar S, Faller DV. Mechanism of T-oligo-induced cell cycle arrest in Mia-PaCa pancreatic cancer cells. J Cell Physiol. 2012;227:2586–2594. | |
Yaar M, Eller MS, Panova I, et al. Telomeric DNA induces apoptosis and senescence of human breast carcinoma cells. Breast Cancer Res. 2007;9:R13. | |
Ruden M, Puri N. Novel anticancer therapeutics targeting telomerase. Cancer Treat Rev. 2013;39:444–456. | |
Ford LP, Wright WE, Shay JW. A model for heterogeneous nuclear ribonucleoproteins in telomere and telomerase regulation. Oncogene. 2002;21:580–583. | |
Ford LP, Suh JM, Wright WE, Shay JW. Heterogeneous nuclear ribonucleoproteins C1 and C2 associate with the RNA component of human telomerase. Mol Cell Biol. 2000;20:9084–9091. | |
Park YM, Hwang SJ, Masuda K, et al. Heterogeneous nuclear ribonucleoprotein C1/C2 controls the metastatic potential of glioblastoma by regulating PDCD4. Mol Cell Biol. 2012;32:4237–4244. | |
Lee SY, Park JH, Kim S, Park EJ, Yun Y, Kwon J. A proteomics approach for the identification of nucleophosmin and heterogeneous nuclear ribonucleoprotein C1/C2 as chromatin-binding proteins in response to DNA double-strand breaks. Biochem J. 2005;388:7–15. | |
Christian KJ, Lang MA, Raffalli-Mathieu F. Interaction of heterogeneous nuclear ribonucleoprotein C1/C2 with a novel cis-regulatory element within p53 mRNA as a response to cytostatic drug treatment. Mol Pharmacol. 2008;73:1558–1567. | |
Kelm RJ Jr, Elder PK, Strauch AR, Getz MJ. Sequence of cDNAs encoding components of vascular actin single-stranded DNA-binding factor 2 establish identity to Puralpha and Purbeta. J Biol Chem. 1997;272:26727–26733. | |
Ramsey JE, Kelm RJ Jr. Mechanism of strand-specific smooth muscle alpha-actin enhancer interaction by purine-rich element binding protein B (Purbeta). Biochemistry. 2009;48:6348–6360. | |
Lezon-Geyda K, Najfeld V, Johnson EM. Deletions of PURA, at 5q31, and PURB, at 7p13, in myelodysplastic syndrome and progression to acute myelogenous leukemia. Leukemia. 2001;15:954–962. | |
Johnson EM, Daniel DC, Gordon J. The Pur protein family: genetic and structural features in development and disease. J Cell Physiol. 2013;228:930–937. | |
Johnson EM, Chen PL, Krachmarov CP, et al. Association of human Pur alpha with the retinoblastoma protein, Rb, regulates binding to the single-stranded DNA Pur alpha recognition element. J Biol Chem. 1995;270:24352–24360. | |
Campisi J, d’Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol. 2007;8:729–740. | |
Pitman R, Wojdyla L, Puri N. Mechanism of DNA damage responses induced by exposure to an oligonucleotide homologous to the telomere overhang in melanoma. Oncotarget. 2013;4:761–767. | |
Ramirez-Montagut T, Andrews DM, Ihara A, et al. Melanoma antigen recognition by tumour-infiltrating T lymphocytes (TIL): effect of differential expression of melan-A/MART-1. Clini Exp Immunol. 2000;119:11–18. | |
Pandolfi F, Trentin L, Boyle LA, et al. Expression of cell adhesion molecules in human melanoma cell lines and their role in cytotoxicity mediated by tumor-infiltrating lymphocytes. Cancer. 1992;69:1165–1173. | |
Kelm RJ Jr, Cogan JG, Elder PK, Strauch AR, Getz MJ. Molecular interactions between single-stranded DNA-binding proteins associated with an essential MCAT element in the mouse smooth muscle alpha-actin promoter. J Biol Chem. 1999;274:14238–14245. | |
Dimri GP, Lee X, Basile G, et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci U S A. 1995;92:9363–9367. | |
Patry C, Bouchard L, Labrecque P, et al. Small interfering RNA-mediated reduction in heterogeneous nuclear ribonucleoparticule A1/A2 proteins induces apoptosis in human cancer cells but not in normal mortal cell lines. Cancer Res. 2003;63:7679–7688. | |
Wang W, Martindale JL, Yang X, Chrest FJ, Gorospe M. Increased stability of the p16 mRNA with replicative senescence. EMBO Rep. 2005;6:158–164. | |
Liu S, Ishikawa H, Tsuyama N, et al. Increased susceptibility to apoptosis in CD45(+) myeloma cells accompanied by the increased expression of VDAC1. Oncogene. 2006;25:419–429. | |
Cheng EH, Sheiko TV, Fisher JK, Craigen WJ, Korsmeyer SJ. VDAC2 inhibits BAK activation and mitochondrial apoptosis. Science. 2003;301:513–517. | |
Sureban SM, May R, George RJ, et al. Knockdown of RNA binding protein musashi-1 leads to tumor regression in vivo. Gastroenterology. 2008;134:1448–1458. | |
He Y, Brown MA, Rothnagel JA, Saunders NA, Smith R. Roles of heterogeneous nuclear ribonucleoproteins A and B in cell proliferation. J Cell Sci. 2005;118:3173–3183. | |
DeGeorge BR Jr, Gao E, Boucher M, et al. Targeted inhibition of cardiomyocyte Gi signaling enhances susceptibility to apoptotic cell death in response to ischemic stress. Circulation. 2008;117:1378–1387. | |
Ohashi N, Yaar M, Eller MS, Truzzi F, Gilchrest BA. Features that determine telomere homolog oligonucleotide-induced therapeutic DNA damage-like responses in cancer cells. J Cell Physiol. 2007;210:582–595. | |
Machwe A, Xiao L, Orren DK. Length-dependent degradation of single-stranded 3′ ends by the Werner syndrome protein (WRN): implications for spatial orientation and coordinated 3′ to 5′ movement of its ATPase/helicase and exonuclease domains. BMC Mol Biol. 2006;7:6. | |
Chaudhury A, Chander P, Howe PH. Heterogeneous nuclear ribonucleoproteins (hnRNPs) in cellular processes: Focus on hnRNP E1’s multifunctional regulatory roles. RNA. 2010;16:1449–1462. | |
Kohler A, Hurt E. Exporting RNA from the nucleus to the cytoplasm. Nat Rev Mol Cell Biol. 2007;8:761–773. | |
Ohashi N, Yaar M, Eller MS, Truzzi F, Gilchrest BA. Features that determine telomere homolog oligonucleotide-induced therapeutic DNA damage-like responses in cancer cells. J Cell Physiol. 2007;210:582–595. | |
Hossain MN, Fuji M, Miki K, Endoh M, Ayusawa D. Downregulation of hnRNP C1/C2 by siRNA sensitizes HeLa cells to various stresses. Mol Cell Biochem. 2007;296:151–157. | |
Seko Y, Azmi H, Fariss R, Ragheb JA. Selective cytoplasmic translocation of HuR and site-specific binding to the interleukin-2 mRNA are not sufficient for CD28-mediated stabilization of the mRNA. J Biol Chem. 2004;279:33359–33367. | |
Tenenbaum G, Yuval R, Elbaz G, Bar-Eli M, Weinberg R. The relationship between cognitive characteristics and decision making. Can J Appl Physiol. 1993;18:48–62. | |
Luo LZ, Jin HW, Huang HQ. Transferrin-cisplatin specifically deliver cisplatin to HepG2 cells in vitro and enhance cisplatin cytotoxicity. J Proteomics. 2012;77:237–250. |
Supplementary materials
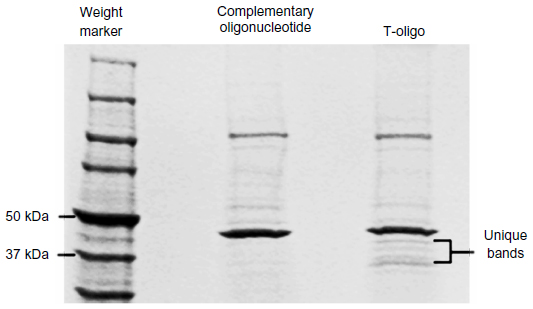 | Figure S1 SDS-PAGE analysis of T-oligo-associated proteins. |
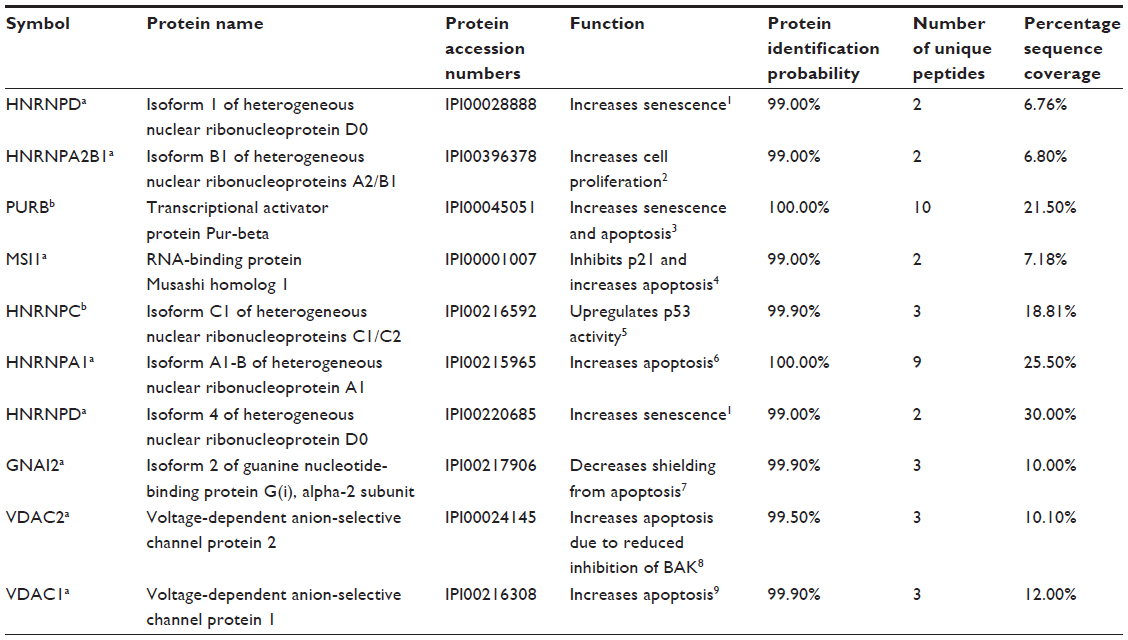 | Table S1 List of proteins identified by mass spectrometry |
References
Wang W, Martindale JL, Yang X, Chrest FJ, Gorospe M. Increased stability of the p16 mRNA with replicative senescence. EMBO Rep. 2005;6:158–164. | |
He Y, Brown MA, Rothnagel JA, Saunders NA, Smith R. Roles of heterogeneous nuclear ribonucleoproteins A and B in cell proliferation. J Cell Sci. 2005;118:3173–3183. | |
Lezon-Geyda K, Najfeld V, Johnson EM. Deletions of PURA, at 5q31, and PURB, at 7p13, in myelodysplastic syndrome and progression to acute myelogenous leukemia. Leukemia. 2001;15:954–962. | |
Sureban SM, May R, George RJ, et al. Knockdown of RNA binding protein musashi-1 leads to tumor regression in vivo. Gastroenterology. 2008;134:1448–1458. | |
Christian KJ, Lang MA, Raffalli-Mathieu F. Interaction of heterogeneous nuclear ribonucleoprotein C1/C2 with a novel cis-regulatory element within p53 mRNA as a response to cytostatic drug treatment. Mol Pharmacol. 2008;73:1558–1567. | |
Patry C, Bouchard L, Labrecque P, et al. Small interfering RNA-mediated reduction in heterogeneous nuclear ribonucleoparticule A1/A2 proteins induces apoptosis in human cancer cells but not in normal mortal cell lines. Cancer Res. 2003;63:7679–7688. | |
DeGeorge BR Jr, Gao E, Boucher M, et al. Targeted inhibition of cardiomyocyte Gi signaling enhances susceptibility to apoptotic cell death in response to ischemic stress. Circulation. 2008;117:1378–1387. | |
Cheng EH, Sheiko TV, Fisher JK, Craigen WJ, Korsmeyer SJ. VDAC2 inhibits BAK activation and mitochondrial apoptosis. Science. 2003;301:513–517. | |
Liu S, Ishikawa H, Tsuyama N, et al. Increased susceptibility to apoptosis in CD45(+) myeloma cells accompanied by the increased expression of VDAC1. Oncogene. 2006;25:419–429. |
 © 2013 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2013 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.