")
Back to Journals » HIV/AIDS - Research and Palliative Care » Volume 7
HIV-1 subtype characteristics of infected persons living in southwestern Greece
Authors Davanos N, Panos G, Gogos C, Mouzaki A
Received 18 June 2015
Accepted for publication 9 September 2015
Published 11 December 2015 Volume 2015:7 Pages 277—283
DOI https://doi.org/10.2147/HIV.S90755
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Bassel Sawaya
Nikolaos Davanos,1 George Panos,2 Charalambos A Gogos,2 Athanasia Mouzaki1
1Division of Hematology, Department of Internal Medicine, Faculty of Medicine, 2Division of Infectious Diseases, Department of Internal Medicine, University Hospital, University of Patras, Patras, Greece
Background: The rapid replication rate of HIV-1, coupled with a high mutation rate and recombination, is the underlying force driving its genetic diversity. In the infected individual, a population of highly related but nonidentical strains exists. At the population level, multiple subtypes often cocirculate, leading to the generation of intersubtype recombinant forms. As a result, the geographic distribution of subtypes and recombinant forms is complex and uneven. Genetic subtyping of HIV-1 isolates has been shown to be helpful for understanding the genetic evolution, the worldwide spread of the virus, and the evaluation of drug resistance.
Materials and methods: We determined the genetic heterogeneity of HIV-1 group M in southwestern Greece. Protease and partial reverse-transcriptase sequences were generated from 150 HIV-1-infected individuals attending the Division of Infectious Diseases of Patras University Hospital, Greece, from 2006 to 2012, and analyzed using online subtyping tools and phylogenetic methods.
Results: The majority of the infected individuals were male (77%). HIV-1 subtype A1 was responsible for 51.3% of infections, followed by subtypes B (34%), G (4%), F1 (2%), and the circulating recombinant forms 02_AG (2.7%), 14_BG (1.3%), 35_AD (1.3%), and 01_AE (0.7%). Additionally, we identified three cases with a recombinant B/CRF02_AG strain (2%) and one with a recombinant G/GRF_AG strain. Sexual transmission was responsible for 96.3% of cases. Heterosexual transmission was responsible for 70.2% of subtype-A1 infections, whereas subtype B was transmitted by men who have sex with men in 75.5% of cases. Protease substitutions I13V, E35D, M36I, R57K, H69K, and L89M, which serve as drug-resistance support mutations in subtype B, were present in the majority of subtype-A1 sequences of the population.
Conclusion: HIV-1 infection in southwestern Greece is sexually transmitted and highly heterogeneous. Subtype A1 has surpassed subtype B, and is the most prevalent strain. In the population studied, subtype A1 exhibited certain polymorphisms in the protease region, which may serve as drug-resistance support mutations in subtype B.
Keywords: HIV-1 infection, transmission, subtype, sequence, genetic signatures, epidemiology
Introduction
Various mechanisms guiding HIV-1 evolution within an infected individual lead to viral genetic heterogeneity. The high mutation rates, resulting from reverse-transcriptase (RT) lack of proofreading activity coupled with a rapid replication rate and recombination, ensure that a number of highly related but nonidentical variants are produced.1–3 As a result, at the population level, HIV-1 comprises groups, multiple subtypes, and intersubtype recombinant forms.
From the four known circulating HIV-1 groups (M, O, N, and P), group M is responsible for more than 95% of the global pandemic. It is divided into subtypes A, B, C, D, F, G, H, J, and K.4 These are operational taxonomic units of HIV-1 that are approximately the same genetic distance from one another.5 Subtypes A and F are further divided into sub-subtypes (A1–A5 and F1 and F2). In recent years, populations of intersubtype circulating recombinant forms (CRFs; http://www.hiv.lanl.gov/content/sequence/HIV/CRFs/CRFs.html) and multiple unique recombinant forms have been identified.
The geographic distribution of HIV-1 group M subtypes and intersubtype CRFs is heterogeneous.6–10 The strains that predominate in different parts of the world differ significantly. For example, subtype B, which accounts for 10% of global HIV-1 infections, predominates in the developed world (North America, Western and Central Europe, Japan, Latin America, and Australia), and is the primary target for infection monitoring, antiretroviral drug development, and treatment. However, the predominant clades in the rest of the world, where the majority of HIV-1 infections occur, are non-B variants. For example, the variant responsible for 50% of infections globally is subtype C, which predominates in sub-Saharan countries, eastern and southern Africa, and India. Subtype A represents approximately 12% of HIV-1 infections worldwide, and is prevalent in areas of central and eastern Africa and in Eastern European countries.11
Various factors, including increasing population mobility due to globalization and immigration coupled with differences in subtype “fitness” and transmission rates, ensure that the worldwide distribution of HIV-1 variants remains complex and uneven. This has become evident in the developed Western world, where the proportion of new non-B infections is rising.12–15 Furthermore, it is also evident that HIV-1 increases its genetic diversity globally via continuous mixing of the existing variants, the generation of new recombinant forms, and recombination between already recombinant strains. Additionally, the use of highly active antiretroviral therapy with nucleoside RT inhibitors (NRTIs) and non-NRTIs (NNRTIs), as well as protease inhibitors (PIs), though reducing HIV-1 transmission, morbidity, and mortality, has led to the emergence of drug-resistant variants.8,16
Based on this knowledge, one can assume that such variations may lead to problems with the use of laboratory diagnostic and viral load-measurement assays, as well as the prescription of antiretroviral drugs, since certain HIV-1 non-B subtypes carry mutations or polymorphisms that may render them less-sensitive to specific therapies.17,18 For such reasons, as well as for epidemiological purposes, it is important to monitor circulating variants in distinct geographic areas.
Genotypic analysis of HIV-1 isolates is the method of choice for evaluating drug resistance, as well as understanding the genetic evolution and worldwide spread of the virus. Partial sequencing of the polymerase gene of the virus (protease and partial RT regions) using automated genotypic assays for antiretroviral resistance testing is usually performed, and the generated sequences are subtyped by means of HIV-sequence databases and bioinformatic tools, as well as phylogenetic analysis.19,20
In this study, we investigated the prevalence of HIV-1 subtypes in southwestern Greece from 2006 to 2012. Samples of individuals with established HIV-1 infection attending the Division of Infectious Diseases of Patras University Hospital (PUH), Greece, were studied and the subtyping results were analyzed.
Materials and methods
The data analyzed in this study are part of a service provided by the Division of Hematology, Medical School, University of Patras, to the Division of Infectious Diseases of PUH. The demographic and virologic parameters, dates of diagnosis, and the nationality of cases are sent to the Hellenic Center for Disease Control and Prevention, and are recorded in an electronic database after anonymous matching using birth dates, initials, and sex. This has been approved by the Hellenic Data Protection Authority. For the purpose of the current study, no informed consent was needed.
The study group included 150 individuals with established HIV-1 infection attending the Division of Infectious Diseases of PUH from 2006 to 2012. All patients had a viral load of at least 1×103 copies/mL and were either treatment-naïve individuals (n=112) or antiretroviral drug-treated individuals experiencing virologic failure (n=38). Peripheral blood samples were collected in ethylenediaminetetraacetic acid-coated tubes. Plasma was separated by centrifugation at 2,500 rpm for 15 minutes. Genotypic analysis, including RNA extraction, reverse transcription, polymerase chain reaction, and sequencing of the HIV-1 protease and partial RT regions of the pol gene, was performed at the Division of Hematology, Medical School, University of Patras, using the HIV-1 TruGene (TG) genotyping kit (Siemens AG, Munich, Germany) and the ViroSeq (VS) HIV-1 genotyping system for antiretroviral drug-resistance testing (Abbott Laboratories, Abbott Park, IL, USA), according to the manufacturers’ instructions. Samples collected from 2006 to 2009 (n=53) were genotyped using the TG method, whereas samples collected from 2010 to 2012 (n=97) were genotyped using the VS method.
The nucleotide sequences generated were used for subtype analysis, using the National Center for Biotechnology Information HIV-subtyping tool (http://www.ncbi.nlm.nih.gov/projects/genotyping/formpage.cgi) and the Rega HIV-1-subtyping tool version 2.0 (http://www.bioafrica.net/rega-genotype/html). Identification of amino acid substitutions was performed using the Stanford University HIV database program (http://hivdb.stanford.edu). Subsequent analysis, including multiple-sequence alignment with MUSCLE21 (multiple-sequence alignment with high accuracy and high throughput) and phylogenetic analysis with the minimum-evolution method,22 was performed using the Molecular Evolutionary Genetics Analysis 5 program (MEGA5, www.megasoftware.net).23
Results
Population characteristics and subtype distribution
The epidemiological characteristics of the population studied are shown in Table 1. The majority of the infected individuals were male (77%). The transmission route was established for 134 of 150 cases (16 unknown cases). Sexual transmission was responsible for 96.3% of all cases, and was almost equally distributed between heterosexual transmission (50%) and transmission by men who have sex with men (MSM; 46.3%). Additionally, there were five cases (3.7%) of transmission via intravenous drug use (IVDU). The country of origin for the majority of the subjects was Greece (82.7%).
 | Table 1 Epidemiological characteristics of the population studied |
The online subtype analysis of the 150 sequences revealed 77 cases with subtype A1 (51.3%), 51 cases with subtype B (34%), three cases with subtype F1 (2%), six cases with subtype G (4%), one case with CRF01_AE (0.7%), four cases with CRF02_AG (2.7%), two cases with CRF14_BG (1.3%), two cases with CRF35_AD (1.3%), three cases with a recombinant B/CRF02_AG strain (2%), and one case with a recombinant G/CRF02_AG strain (0.7%).
The epidemiological characteristics of each subtype identified in this study are shown in Table 2. The prevalent route of transmission for the major strains identified in the population studied was heterosexual for subtype A1 (70.2%), MSM for subtype B (75.5%), and heterosexual for subtype G (83.3%). The transmission route for the remaining strains was almost equally distributed between heterosexual transmission and MSM transmission, with the exception of CRF14_BG, which was transmitted by IVDU in both cases (100%).
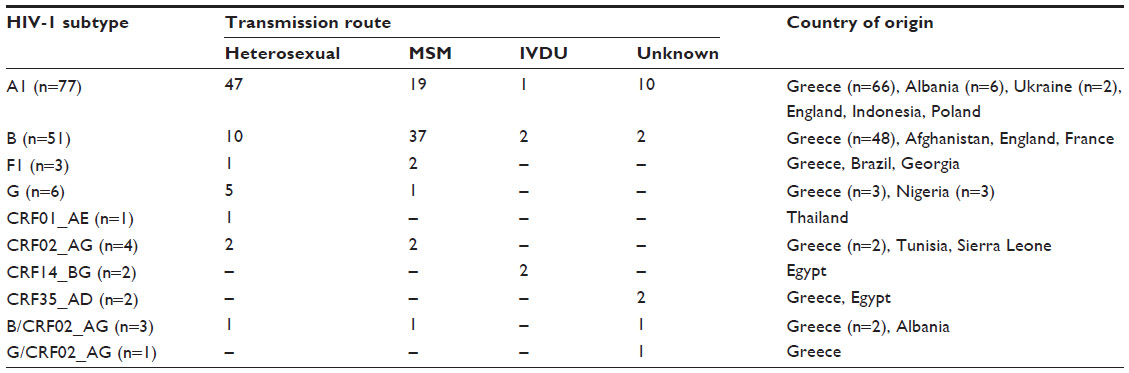 | Table 2 Epidemiological characteristics of identified subtypes |
The majority of individuals infected with subtype A1 were from Greece, Albania, and Ukraine. The majority of individuals infected with subtype B were from Greece. Two of three individuals infected with subtype F1 were from Brazil and Georgia. Three of the individuals infected with subtype G (50%) were from Africa (Nigeria). The individual infected with CRF01_AE was from Thailand. Two of the individuals infected with CRF02_AG were from Africa (Tunisia, Sierra Leone). Both CRF_BG individuals and one CRF35_AD individual were from Egypt.
Drug resistance and other mutations
The genotypic analysis of the 150 sequences revealed 26 cases with drug-resistance mutations (DRMs), 22 of which were from patients experiencing virologic failure due to exposure to antiretroviral drugs. The remaining four cases with DRMs were from treatment-naïve individuals, suggesting transmitted resistance.
The 22 cases experiencing virologic failure presented with the following DRMs: M46I, F53LY, I54LTV, G73ST, L76V, V82AT, I84V, I185V, N88D, and L90M for PIs; L100I, K103NS, V179F Y181C, G190AS, V106A, K103N, and P225H for NNRTIs; and M41L, K65R, D67GN, T69D, K70ER, L74V, Y115F, F116Y, Q151M, M184V, L210W, T215CDFIY, and K219EQR for NRTIs. The four treatment-naïve cases presented with the following DRMs: I84V for PIs, K103N for NNRTIs, and T215S and K219Q for NRTIs.
Among the 26 DRM cases, 24 had DRMs for NRTIs, 16 had DRMs for NNRTIs, and 12 had DRMs for PIs. The most common DRM for each antiretroviral agent was M184V for NRTIs (79%), K103N for NNRTIs (56%), and M46I and I54V for PIs (75%).
All subtype-A1 sequences presented a certain divergence from the standard HIV-1 subtype-B sequence (HIV-1 HXB2), used as a reference by the automated genotypic assays for antiretroviral resistance testing, which was not present in the other subtypes identified in the population studied. This involved six protease amino acid substitutions (I13V, E35D, M36I, R57K, H69K, and L89M), which are drug-resistance support mutations in subtype B.24 Specifically, the E35D and M36I substitutions were present in 100% of cases, followed by substitutions H69K (99%), I13V (96%), L89M (95%), and R57K (93%), indicating that these sequence alterations may occur as genetic signatures in subtype A1. Less frequent substitutions identified in the subtype-A1 sequences of the study group involved I15V, G16E, R41K, K45R, and K70R (<60% of cases). Finally, several polymorphic sites were identified, with L63PSA being the most prevalent (96% of cases). A similarly consistent substitution pattern was not observed for HIV-1 subtype-A1 RT sequences or subtype B and other strain sequences.
Phylogenetic analysis
For phylogenetic analysis, the sequences of this study were divided into two groups based on the method they were generated with (the TG method generates sequences of 919 nucleotides in length, whereas the VS method generates sequences of 1,302 nucleotides in length). The first group included the 53 sequences generated with the TG method and the second group included the 97 sequences generated with the VS method. Sequences containing DRM (14 for TG and 12 for VS) were excluded from the analysis. The DRM-free sequences from each group (39 for TG and 85 for VS) were aligned, and two phylogenetic trees were constructed (Figures 1 and 2). The analysis showed that all subtypes and CRFs create separate and distinct clusters with each other. Within the A1-subtype cluster, the majority of sequences from Greece and Albania created a separate cluster from subtype-A1 sequences from Ukraine (Figure 2).
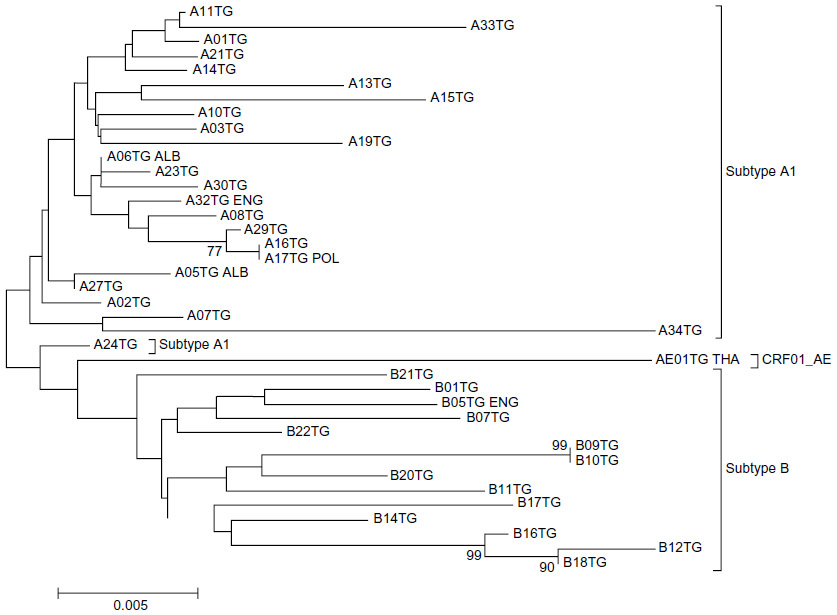 | Figure 1 Evolutionary relationships of HIV-1 sequences generated with the TruGene (TG) method. |
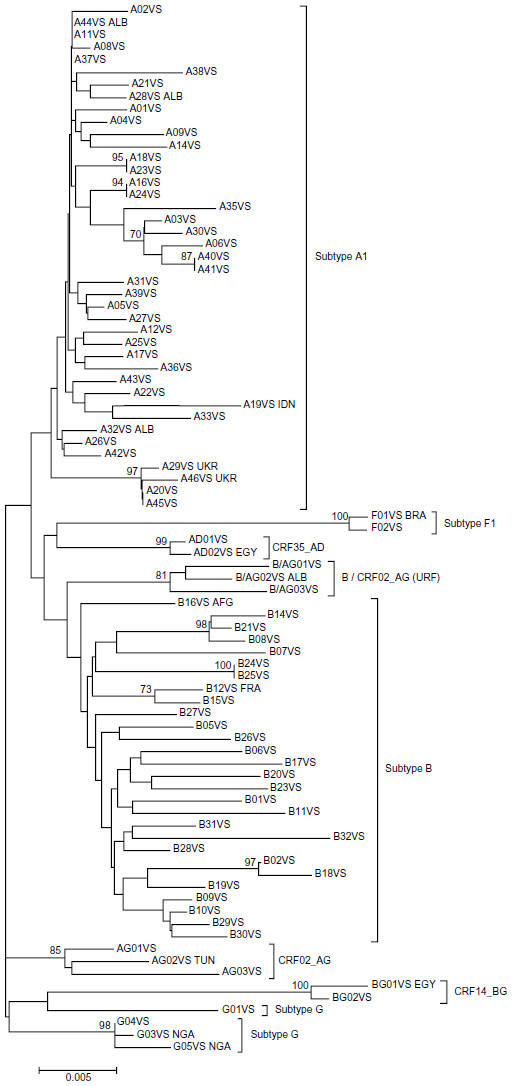 | Figure 2 Evolutionary relationships of HIV-1 sequences generated with the ViroSeq (VS) method. |
Discussion
The genetic diversity of HIV-1, comprised of multiple subtypes and intersubtype recombinant forms, poses a challenge to the evaluation of drug resistance. The rapid replication rate of HIV-1, coupled with a high mutation rate and recombination, is the underlying forces driving its diversity. In the infected individual, a population of highly related but nonidentical strains exists. At the population level, multiple subtypes often cocirculate, leading to the generation of intersubtype recombinant forms. As a result, the geographic distribution of subtypes and recombinant forms is complex and uneven. Genetic subtyping of HIV-1 isolates has been shown to be helpful for understanding the genetic evolution and worldwide spread of the virus and the evaluation of drug resistance. HIV-1 drug-resistance testing by sequencing of RT and protease regions of the polymerase gene is an important aspect of the management of infected individuals. Furthermore, the interpretation of the polymerase sequences generated by such tests can lead to an accurate subtype characterization using online bioinformatic programs and phylogenetic methods.
In the present study, we evaluated the HIV-1 subtype characteristics of infected individuals living in southwestern Greece and attending the Division of Infectious Diseases of PUH from 2006 to 2012. The majority of the infected individuals were Greek males. Sexual transmission was responsible for 96.3% of all cases, and was almost equally distributed between heterosexual transmission and transmission by MSM. In this respect, the epidemiology of this region differs from the rest of the country, where sexual transmission by MSM predominates (cf official report of the Greek Ministry of Health).31 In very few cases (3.7%), transmission was via IVDU. Our results show that subtype A1 is the predominant subtype in southwestern Greece, with a prevalence of 51.3% among infected long-term residents.
The distribution pattern of HIV-1 group M subtypes in southwestern Greece is most likely the result of an initially prevalent route of a homosexually transmitted subtype-B strain from Western Europe, combined with a heterosexually transmitted subtype-A1 strain from Africa that has been established in the population (founder effect), and in recent years has led to coprevalence of the two subtypes.24 The presence of a small number of other subtypes, CRFs, and unique recombinant forms is indicative of viral migration due to trafficking. This observation supports the case that HIV-1 group M subtypes segregate by means of diversified risk behavior rather than as a result of virologic attributes.11
Two recently published studies examining the effect of the HIV-1 subtype on virological response to combination antiretroviral therapy in white patients of Swiss25 or Greek26 origin reported higher incidence of virological failure in patients with subtype B compared to patients with subtype-A1 infections. The HIV-1 sequences examined in the present study showed various substitutions when compared to the consensus subtype-B sequence, but only subtype-A1 protease sequences exhibited a certain divergence from the HIV-1 HXB2 sequence. Protease substitutions I13V, E35D, M36I, R57K, H69K, and L89M, which are drug-resistance support mutations in subtype B, were present in the majority of subtype-A1 sequences of the population studied, suggesting that these sequence alterations may occur as natural polymorphisms, and may serve as genetic signatures. The existence of subtype-specific polymorphisms interfering with the selection of an appropriate therapeutic strategy should be evaluated toward the implementation of presubtyping for the clinical management of HIV-1-infected individuals.
Conclusion
In the present study, we evaluated the HIV-1 subtype characteristics of infected individuals living in southwestern Greece. This is the first study to show elevated levels of subtype-A1 infections in southwestern Greece, compared to subtype B, with a prevalence of 51.3% among infected long-term residents, confirming a trend reported earlier24 on increasing prevalence of HIV-1 subtype A in Greece. Finally, we report that subtype A1 naturally exhibits protease polymorphisms, which may serve as drug-resistance support mutations in subtype B.
Acknowledgments
We thank the medical and administrative personnel of the Division of Infectious Diseases of PUH, for providing epidemiological information for the study subjects. The data analyzed in this study are part of a service provided by the Division of Hematology, Medical School, University of Patras, to the Division of Infectious Diseases of PUH. The materials required for this service are financed by the Hellenic Centre for Disease Control and Prevention (KEELPNO), Greece. KEELPNO had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author contributions
ND performed the assays, codesigned the study, and cowrote the paper, GP provided epidemiological information for the study subjects and critically revised the paper, CAG codesigned the study and critically revised the paper, and AM codesigned the study and cowrote the paper. All authors contributed toward data analysis, drafting and critically revising the paper and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Gao F, Chen Y, Levy DN, Conway JA, Kepler TB, Hui H. Unselected mutations in the human immunodeficiency virus type 1 genome are mostly nonsynonymous and often deleterious. J Virol. 2004;78:2426–2433. | |
Levy DN, Aldrovandi GM, Kutsch O, Shaw GM. Dynamics of HIV-1 recombination in its natural target cells. Proc Natl Acad Sci U S A. 2004;101:4204–4209. | |
Perelson AS, Neumann AU, Markowitz M, Leonard JM, Ho DD. HIV-1 dynamics in vivo: virion clearance rate, infected cell life-span, and viral generation time. Science. 1996;271:1582–1586. | |
Robertson DL, Anderson JP, Bradac JA, et al. HIV-1 nomenclature proposal. Science. 2000;288:55–56. | |
Taylor BS, Sobieszczyk ME, McCutchan FE, Hammer SM. The challenge of HIV-1 subtype diversity. N Engl J Med. 2008;358:1590–1602. | |
Peeters M, Sharp PM. Genetic diversity of HIV-1: the moving target. AIDS. 2000;14 Suppl 3:S129–S140. | |
Peeters M, Toure-Kane C, Nkengasong JN. Genetic diversity of HIV in Africa: impact on diagnosis, treatment, vaccine development and trials. AIDS. 2003;17:2547–2560. | |
Wainberg M. HIV-1 subtype distribution and the problem of drug resistance. AIDS. 2004;18 Suppl 3:S63–S68. | |
McCutchan FE. Global epidemiology of HIV. J Med Virol. 2006; 78 Suppl 1:S7–S12. | |
Hemelaar J, Gouws E, Ghys PD, Osmanov S. Global and regional distribution of HIV-1 genetic subtypes and recombinants in 2004. AIDS. 2006;20:W13–W23. | |
Buonaguro L, Tornesello ML, Buonaguro FM. Human immunodeficiency virus type 1 subtype distribution in the worldwide epidemic: pathogenetic and therapeutic implications. J Virol. 2007;81:10209–10219. | |
Couturier E, Damond F, Roques P, et al. HIV-1 diversity in France, 1996–1998. AIDS. 2000;14:289–296. | |
Tatt ID, Barlow KL, Clewley JP, Gill ON, Parry JV. Surveillance of HIV-1 subtypes among heterosexuals in England and Wales, 1997–2000. J Acquir Immune Defic Syndr. 2004;36:1092–1099. | |
Fleury H, Recordon-Pinson P, Caumont A, et al. HIV type 1 diversity in France, 1999–2001: molecular characterization of non-B HIV type 1 subtypes and potential impact on susceptibility to antiretroviral drugs. AIDS Res Hum Retroviruses. 2003;19:41–47. | |
Wheeler WH, Ziebell RA, Zabina H, et al. Prevalence of transmitted drug resistance associated mutations and HIV-1 subtypes in new HIV-1 diagnoses, US – 2006. AIDS. 2010;24:1203–1212. | |
Clavel F, Hance AJ. HIV drug resistance. N Engl J Med. 2004;350:1023–1035. | |
Gonzalez LM, Brindeiro RM, Tarin M, et al. In vitro hypersusceptibility of human immunodeficiency virus type 1 subtype C protease to lopinavir. Antimicrob Agents Chemother. 2003;47:2817–2822. | |
Santoro MM, Alteri C, Ronga L, et al. Comparative analysis of drug resistance among B and the most prevalent non-B HIV type 1 subtypes (C, F, and CRF02 AG) in Italy. AIDS Res Hum Retroviruses. 2012;28:1285–1293. | |
Rozanov M, Plikat U, Chappey C, Kochergin A, Tatusova T. A web-based genotyping resource for viral sequences. Nucleic Acids Res. 2004;32:W654–W659. | |
de Oliveira T, Deforche K, Cassol S, et al. An automated genotyping system for analysis of HIV-1 and other microbial sequences. Bioinformatics. 2005;21:3797–3800. | |
Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. | |
Rzhetsky A, Nei M. A simple method for estimating and testing minimum evolution trees. Mol Biol Evol. 1992;9:945–967. | |
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28:2731–2739. | |
Paraskevis D, Magiorkinis E, Magiorkinis G, et al. Increasing prevalence of HIV-1 subtype A in Greece: estimating epidemic history and origin. J Infect Dis. 2007;196:1167–1176. | |
Scherrer AU, Ledergerber B, von Wyl V, et al. Improved virological outcome in white patients infected with HIV-1 non-B subtypes compared to subtype B. Clin Infect Dis. 2011;53:1143–1152. | |
Paraskevis D, Touloumi G, Bakoyannis G, et al. Effect of HIV type 1 subtype on virological and immunological response to combination antiretroviral therapy: evidence for a more rapid viral suppression for subtype A than subtype B-infected Greek individuals. AIDS Res Hum Retroviruses. 2013;29:461–469. | |
Felsenstein J. Confidence limits on phylogenies: an approach using the bootstrap. Evolution. 1985;39:783–791. | |
Tamura K, Nei M, Kumar S. Prospects for inferring very large phylogenies by using the neighbor-joining method. Proc Natl Acad Sci U S A. 2004;101:11030–11035. | |
Nei M, Kumar S. Molecular Evolution and Phylogenetics. New York: Oxford University Press; 2000. | |
Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987;4:406–425. | |
Official report of the Greek Ministry of Health: HIV/AIDS Surveillance in Greece: Available from: http://www.keelpno.gr/Portals/0/Αρχεία/HIV/2014/Epidimiologiko_2013_final.pdf. Accessed August 31, 2015. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.