")
Back to Journals » Vascular Health and Risk Management » Volume 16
High Lipoprotein(a) Levels as a Predictor of Major Adverse Cardiovascular Events in Hospitalized-Acute Myocardial Infarction Patients
Authors Sumarjaya IDGD, Nadha IKB, Lestari AAW
Received 5 October 2019
Accepted for publication 3 March 2020
Published 8 April 2020 Volume 2020:16 Pages 125—132
DOI https://doi.org/10.2147/VHRM.S233503
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Daniel Duprez
Video abstract presented by Anak Agung Wiradewi Lestari.
Views: 379
I Dewa Gde Dwi Sumarjaya,1 I Ketut Badjra Nadha,2 Anak Agung Wiradewi Lestari3
1Specialist Program of Cardiology and Vascular Medicine, Faculty of Medicine, Universitas Udayana, Sanglah General Hospital, Bali, Indonesia; 2Department of Cardiology and Vascular Medicine, Faculty of Medicine, Universitas Udayana, Sanglah General Hospital, Bali, Indonesia; 3Department of Clinical Pathology, Faculty of Medicine, Universitas Udayana, Sanglah General Hospital, Bali, Indonesia
Correspondence: Anak Agung Wiradewi Lestari
Department of Clinical Pathology, Faculty of Medicine, Universitas Udayana, Sanglah General Hospital, Bali, Indonesia
Email [email protected]
Background: Risk stratification models with incorporation of biochemical markers have received attention recently. In acute myocardial infarction (AMI) one such marker is lipoprotein(a) (Lp(a)). Lp(a) has prothrombotic and proinflammatory properties. High levels of Lp(a) probably contribute to the additional adverse effects in AMI, as it enhances the damaging effect of acute thrombosis. This study aimed to evaluate serum Lp(a) as a predictor of major adverse cardiovascular events (MACE) in hospitalized-acute myocardial infarction patients.
Methods: A prospective cohort study was conducted at Sanglah Hospital, Denpasar, during June–August 2018, among 66 people by consecutive sampling. Samples that met the inclusion and exclusion criteria were examined for serum Lp(a) at the time of admission and the occurrence of MACE during hospitalization was observed. Data regarding serum Lp(a), demography, smoking history, dyslipidemia, hypertension, diabetes mellitus, and MACE were collected. Log rank test and Cox proportional hazards regression were conducted with SPSS version 20 for Windows.
Results: During observation, MACE occurred in 25 (38%) patients, including cardiogenic shock in 7 (10.6%) patients, heart failure in 20 (30.3%) patients, cardiovascular death in 5 (7, 6%) patients, malignant arrhythmias in 5 (7.6%) patients, and postinfarction angina in 5 (7.6%) patients. After the Log rank test, a significant difference in survival was observed (p = 0.001) between groups of high Lp(a) (survival rate of 60.6 hours; 95% CI 43.3– 77.9) and low Lp(a) (average survival of 104.3 hours, 95% CI 91.4– 117.2). The hazard ratio of high Lp(a) against MACE was 4.63 (p=0.002), and it increased to 4.69 in multivariate analysis with Cox proportional hazards regression test (p=0.003).
Conclusion: The high level of Lp(a) in AMI patients was a risk factor for the occurrence of MACE during hospitalization. Patients with high Lp(a) also had worse survival compared to patients with low Lp(a).
Keywords: acute myocardial infarction, lipoprotein(a), MACE
Introduction
Risk factors for cardiovascular disease and stroke have been thoroughly researched over the past years. The rise of aging population and the build-up of behavioral risk factors including unhealthy diet, physical inactivity, excess body weight, and non-cardiovascular comorbidities all had inevitably contributed to atherosclerotic cardiovascular disease, including coronary artery disease and stroke.1 Despite advancement of knowledge in risk factors and prevention through pharmacological intervention such as low density lipoprotein (LDL)-lowering therapy, there is still a challenge regarding the so called “residual risk”. Genome wide association study revealed that the “residual risk” was associated with several biomarkers, one in particular involved in atherosclerotic disease was lipoprotein(a) (Lp(a)).2
Study in the effort to find a potential biomarker has been growing in the past decade. The current reference of ideal biomarker was troponin’s role in diagnosis of acute myocardial infarction (AMI).3 Research in the field of biomarkers related to necrosis, inflammation, ischemia and myocardial stretch in the AMI has attracted much attention. To date, there have been many biomarkers studies to predict the long term major adverse cardiovascular events (MACE) in patients with AMI. It is hoped that biomarkers could provide earlier information about the overall risk of subsequent occurrence of MACE, therefore reducing morbidity and mortality from MACE in patients with AMI.4,5
From epidemiological studies, it has been shown that an increase in Lp(a) levels was associated with an increased incidence of coronary heart disease (CHD), especially if the levels exceed 50 mg/dL. Lp(a) is a family of low-density lipoproteins and contains apolipoprotein B100 (apoB) and apolipoprotein A (apoA) molecules. Both of these components contribute to the prothrombotic and proinflammatory effect of Lp(a). Although pathophysiologically rational, however, up until now the existing studies have still been unclear whether Lp(a) posed additional risk in populations with existing cardiovascular disease. Several studies have suggested that there was a relationship between Lp(a) levels and secondary risk in individuals who have CHD (or other atherosclerotic conditions), while some studies show no relationship. Up until the present day, very little is known about Lp(a) as a risk predictor for MACE after AMI events nor its incorporation to the existing scoring systems.6–8
Over the past few decades, various types of clinical risk stratification have been introduced to assess and classify patient’s risk so that high-risk AMI patients could be advised to have an early revascularization. Some well-known tools came from the glycoprotein IIb/IIIa in Platelet Unstable angina: Receptor Suppression Using Integrilin Therapy (PURSUIT), Thrombolysis in Myocardial Infarction (TIMI) and the Global Registry of Acute Cardiac Events (GRACE).9 However, none of these scoring systems include biomarkers (except troponin) to increase its value in predicting high-risk patients. According to the aforementioned, this study aimed to evaluate the relationship between blood levels of Lp(a) and the occurrence of MACE in AMI patients during hospitalization at Sanglah General Hospital, Bali, Indonesia.
Materials and Methods
Subject Selection
An observational study with a prospective cohort design was carried out from May 1st to August 30th, 2018 at Sanglah General Hospital, Bali, Indonesia. Patients with AMI (NSTEMI and STEMI) hospitalized during the study period were recruited. The sample was recruited by consecutive sampling and had to meet the inclusion and exclusion criteria. Inclusion criteria were all AMI patients aged ≥18 years admitted to Emergency Department of Sanglah Hospital, Denpasar. The exclusion criteria included patients with sepsis, patients with malignancy, patients with end-stage renal failure, and patients with chronic heart failure (CHF). Patients included in the study were properly informed regarding the study and signed an informed consent. The researcher filled out the research questionnaire based on history, physical examination and supporting examination of each sample. Data regarding age, gender, smoking habit, history of previous diseases such as hypertension, diabetes mellitus, chronic kidney disease (exclude ESRD), height and weight measurement, and lipid profile were collected. STEMI was defined as the presence of persistent chest pain, ST-elevation on electrocardiogram (ECG) in particular lead to indicate infarction process, and positive cardiac marker (Troponin I and Troponin T) during admission. NSTEMI was determined by the presence of persistent chest paint, without ST-elevation on ECG, and if there was elevation of cardiac marker. All patients were managed with standard therapy according to the clinical pathway at Sanglah General Hospital, Denpasar. Patients were followed-up during hospitalization for the occurrence of MACE, including death. MACE was the primary end point, and survival during hospitalization was the secondary end point. The protocol of the study had been reviewed thoroughly according to the Declaration of Helsinki and given approval by ethic committee of Universitas Udayana/Sanglah General Hospital (2412/UN14.2.2.VII.14/LP/2018).
Lp(a) Evaluation
Sampling Lp(a) was carried out in the Emergency Department (ED) of Sanglah Hospital at the time of the patient’s arrival. Patients were divided into two groups, the group of AMI patients with high initial Lp(a) serum and low initial Lp(a) serum according to the cut-off value calculated by Receiver Operating Characteristic (ROC) curve analysis. The Lp(a) quantification was assessed using immunoturbidimetry method and presented in mg/dL.
Data Analysis
Data were analyzed using SPSS version 20 for Windows and depicted in percentage, mean, standard deviation, confidence interval (CI), and significant value (p) based on data analysis. In order to obtain the cut-off value of Lp(a), ROC curve analysis was conducted to provide area under the curve (AUC), sensitivity, and specificity. The mortality risk was evaluated from the Hazard Ratio (HR) by using Kaplan-Meier curve from these predictive factors. A p-value less than 0.05 was considered statistically significant.
Result
There were 66 people with AMI who were involved in this study consisting of 53 males (80.3%) and 13 females (19.7%). STEMI were found among 42 (63.6%) patients and NSTEMI 24 (36.4%) patients. The average age in this study was 59.23 ± 10.3 years (Table 1). All patients were examined for serum Lp(a) and carefully observed for occurrence of MACE during hospitalization. The study population was grouped into two groups based on the level of Lp(a) as a predictor of MACE in accordance with the results of the ROC curve analysis (Figure 1). Based on the ROC curve analysis, the best cut-off point was >10.25 mg/dL with an area under the curve (AUC) of 0.787 (80% sensitivity and 63.4% specificity; p≤0.0001) (Figure 1).
 |
Figure 1 The ROC curve in determining the cut-off point of Lp(a) level as a MACE predictor. |
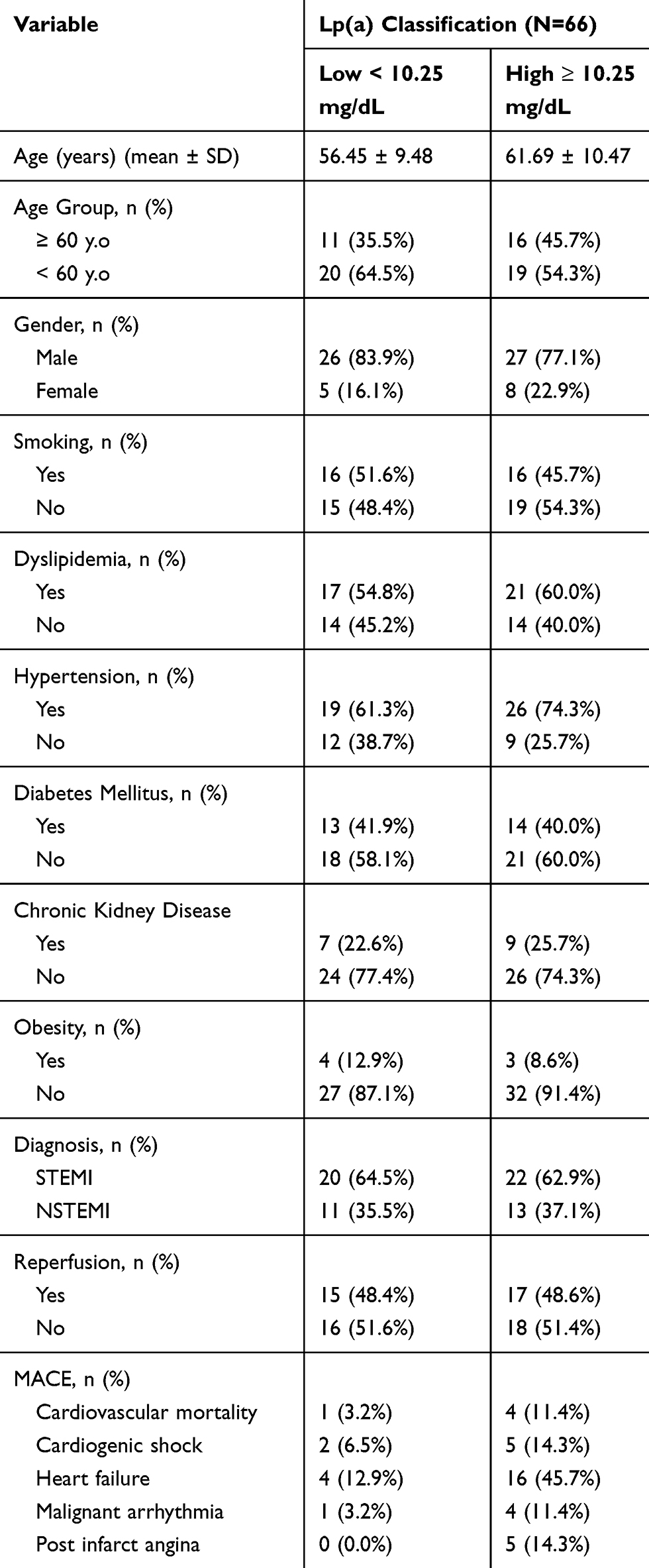 |
Table 1 The Baseline Characteristics of the Study Group |
In order to determine the effect of high serum levels of Lp(a) (≥10.25 mg/dL) on MACE, bivariate analysis was conducted. Lp(a) was used as an independent factor and MACE as a dependent factor with the Kaplan-Meier survival estimation method. Figure 2 shows a graph of the estimated MACE in AMI patients based on Lp(a) levels. After a Log rank test, there were significant differences in survival (p = 0.001) between groups of patients with high serum Lp(a) (mean MACE 60,571 hours; CI95% 43,278–77,865) compared to patients with low serum Lp(a) (mean MACE 104,339 hours; CI95% 91,432–117,246) (Figure 2).
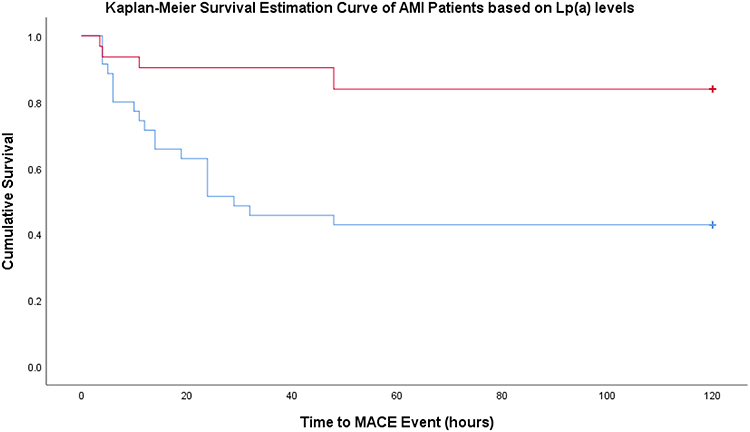 |
Figure 2 Kaplan-Meier Survival Estimation Curve in AMI based on serum levels of Lp(a). Blue lines indicate group with high Lp(a) and red lines indicate group with low Lp(a). |
Multivariate analysis with Cox proportional hazards regression was performed to control variables considered as confounders in this study including age, sex, hypertension, diabetes mellitus, dyslipidemia, smoking, obesity, a history of chronic kidney disease, and reperfusion therapy (Table 2). The results of multivariate analysis are shown in Table 2 which shows that high Lp(a) levels were shown to be independent predictors of MACE in AMI patients during hospitalization, with adjusted HR values of 4.685 (95% CI 1.668–13.158; p = 0.003). This result showed that after controlling for other confounding factors, the risk of MACE in AMI patients with high Lp(a) levels was almost 5-fold compared to patients with low Lp(a) levels (Table 2).
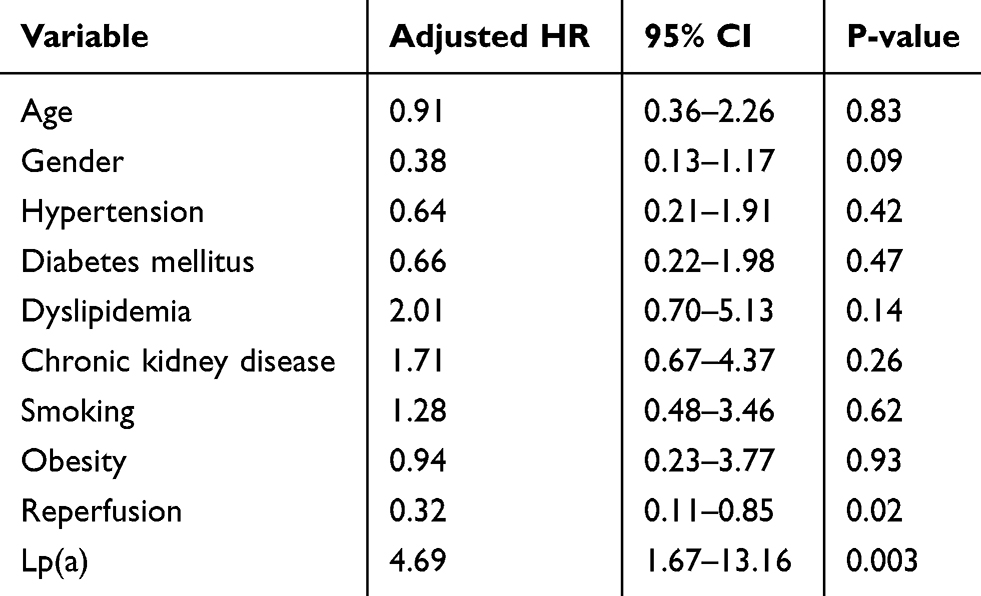 |
Table 2 Cox Regression Analysis of Lp(a) Levels as Independent Predictors of MACE in AMI Patients During Hospitalization |
Discussion
The unique structure of Lp(a) that resembles plasminogen is thought to have a large role in the pathophysiology of AMI. The structure can bind to fibrin and receptors on endothelial cells which will interfere clot lysis thus increase the tendency of thrombosis.10–12 The oxidative form of Lp(a) can weaken the fibrinolytic system by decreasing the activation of plasminogen, increasing the production of vasodilator inhibitory agents in endothelial cells thus affecting vasodilation.12–14 Lp(a) has also been shown to interact with other molecules in a plethora of pathways related to thrombosis including inhibition of activation of transforming growth factor-β (TGF-β) and platelet activation.
It is thought that the mechanism of Lp(a) inhibits the generation of TGF-β as it competes with the binding of plasminogen to cells, and reduces its conversion to plasmin (plasmin required in activation of Latent TGF-β).15 High blood levels of Lp(a) decrease the levels of the migration inhibitor TGF-beta and it enhances smooth muscle cell proliferation and migration into the intima, thus further enhancing the formation of atheroma plaque.16,17 Lp(a)'s influence on platelet activation is still under debate. Platelets are key players in formation of arterial thrombus. Studies have demonstrated that Lp(a) had an inhibitory effect on platelet activation as it decreases platelet activation induced by collagen, ADP, or platelet-activating factor.18–21 Studies that support Lp(a) activate platelet cames from the results that apo(a) and Lp(a) enhanced platelet aggregation and granule release.22 Another study found that apo(a) and Lp(a) induced platelet aggregation in response to lower than aggregate dose of arachidonic acid.23 Perhaps, the controversy of Lp(a)'s effect on platelet responsiveness depends on the balance of activating factors.24 After all, existing epidemiological research to date shows that high Lp(a) levels are independent predictors of CVD risk and MACE.10–14
This study involved 66 samples taken by consecutive sampling with a total of 53 males (80.3%) and 13 females (19.7%). STEMI (63.6%) was found more than NSTEMI (36.4%). The proportion of males and the diagnosis of STEMI were dominant in this study which is similar to the existing large registry. According to the Global Registry of Acute Coronary Events (GRACE) and the French registry of ACS (ONACI) the highest percentage of AMI is STEMI and male predominant, which concludes male sex as one of the risk factors.25,26
Several risk factors were recorded in this study such as age, smoking, hypertension, diabetes mellitus (DM), history of chronic kidney disease (CKD), dyslipidemia, and obesity. There were 27 (40.9%) people aged ≥60 years, 32 (48.5%) smokers, 45 (68.2%) with hypertension, 27 (40.9%) with DM, 16 (24.2%) with CKD, 38 (57.6%) people with dyslipidemia and 7 (10.6%) with obesity. Our study followed the patients during hospitalization for acute myocardial infarction. Short period of hospitalization probably masked the effect of these traditional risk factors for survival during the acute episode of acute myocardial infarct. Most study as opposed to other study that conduct follow-up for at least 30 days or more. Additionally, our study also had a relatively small sample and perhaps it needed more power to detect a lower degree of association. However, we could only confirm this presumption with a larger and better designed study.
Reperfusion is an attempt to save the myocardium from ischemia due to occlusion of coronary vessels in the heart. The procedure was conducted in STEMI patients with an onset <12 hours, or >12 hours if still within high-risk criteria. Reperfusion measures were also carried out in NSTEMI patients with high-risk criteria based on clinical or GRACE scores. The number of subjects who underwent reperfusion therapy in this study were 32 (48.5%) people, 11 (16.7%) underwent fibrinolytic of which 8 (12.1%) people were successful and as many as 3 (4.5%) people failed. There were 20 (30.3%) people who underwent PCI.
Major cardiovascular events observed in this study included cardiovascular death, heart failure, cardiogenic shock, malignant arrhythmias, and post-infarct angina. There were 25 (37.9%) people with MACE, the most frequent was heart failure in 20 (30.3%) patients, followed by cardiogenic shock in 7 (10.6%) patients, malignant arrhythmias in 5 (7.6%) and angina post-infarction in 5 (7.6%). There were 9 (13.6%) patients with more than one MACE. In this study, 5 (7.6%) patients experienced mortality.
The cut-off value for Lp(a) in this study was slightly lower than in previous studies. The research conducted by Peng et al used a particle-enhanced turbidimetric immunoassay to determine the concentration of blood Lp(a) (Sekisui Medical Co., Tokyo, Japan). The cut-off point of Lp(a) for predicting MACE in 1 year of post-STEMI obtained in their study was 17.6 mg/dL (AUC 0.764, sensitivity 65.71% and specificity 77.14%).27 Another study by Mitsuda et al in predicting MACE within 3 years, used Lp(a) with cut-off point of 19 mg/dL (AUC 0.674, sensitivity 69.2% and specificity 62%). Mitsuda et al used a different method in measuring serum Lp(a), the fixed-rate immunonephelometric method using JEOL JCA-BM8000 clinical chemistry analyzer (JEOL Inc., Tokyo, Japan).28 Note that the cut-off points obtained by both researchers were based on long-term prediction of MACE. Other studies also used higher cut-offs (>30 mg/dL), but these values were based on the prediction of coronary heart disease rather than MACE in acute AMI conditions.29–31 Currently, there is no definite cut-off point for Lp(a) as a predictor of in-hospital MACE for AMI patients.
This study showed that AMI patients with high serum Lp(a) (> 10.25 mg/dL) had worse survival during the treatment period than AMI patients with low serum Lp(a). Besides, AMI patients with high serum Lp(a) were 4.63 times more likely to develop MACE during hospitalization. After controlling for confounders, the risk of MACE in AMI patients with high Lp(a) increased to 4.69 times. One study in China by Peng et al conducted an investigation that involved 175 STEMI patients who underwent PCI and determined the risk of MACE for 1 year based on the serum levels of Lp(a). The sample was divided with the cut-off value determined with ROC curve results which was 17.6 mg/dL. The research concluded that patients with high levels of Lp(a) were at a risk approximately 6.4 times higher to experience MACE events than patients with low Lp(a) levels (p <0.001). Kaplan-Meier survival analysis showed lower survival for patients with high serum Lp(a) compared to low Lp(a) (log rank p <0.001).27
A similar study by Mitsuda et al with a longer time-span (3 years) in 176 STEMI patients after PCI assessed occurrence of MACE and cerebrovascular events. ROC curve analysis obtained a cut-off value of 19 mg/dL. The results also concluded that patients with high Lp(a) levels were at high risk of MACE and cerebrovascular events (HR 1.030, 95% CI 1.011–1.048, p = 0.002) and had a worse survival (log rank p = 0.034).28
The study by Ikenaga et al assessed Lp(a) serum a week after AMI (total 410 patients) and divided it into two groups, high (> 40 mg/dL) and low (≤40 mg/dL). The MACE incidence for 5 years was significantly higher in the group with high Lp(a) levels compared to group with low Lp(a) levels. The incidence of new lesions requiring revascularization was also reported to be higher in group with high Lp(a) levels. In this study, the Odds Ratio for MACE event was 1.64 (95% CI 1.31–2.06 and p <0.001).31
Numerous meta-analyses agreed that elevated Lp(a) levels were proven as a marker of increased cardiovascular risk.32–38 Some metanalyses also showed that Lp(a) was a risk factor for ischemic stroke.39,40 Although there are differences among various methods of Lp(a) determination, it remains clear that elevated Lp(a) levels are mildly associated with cardiovascular event for a broad spectrum of subgroups. The significant increase of Lp(a) was also an associated risk for MACE.36,38 However, regarding the issue whether Lp(a) could improve cardiovascular risk prediction has been largely unaddressed. Recently, Willeit et al conducted an assessment of whether Lp(a) could improve CVD risk prediction by addition of Lp(a) to Framingham risk score and Reynolds risk score variables in Bruneck study.41 Albeit, due to its cost and with only marginal improvement in prediction, it is still lacking a reason to determine Lp(a) routinely in the setting of primary prevention. However, results of this study and previous research suggest that in the setting of secondary prevention and AMI patients in a hospital setting, it has shown promise in predicting MACE occurrence.
Lp(a) has prothrombotic and proinflammatory properties. High levels of Lp(a) probably contribute to the additional adverse effect in AMI as it enhances the damaging effect of acute thrombosis and will augment the occurrence of MACE.17,42-44 Looking at the pathogenicity structure of Lp(a), it can be concluded that these compounds can be used as acute biomarkers in the pathogenesis of AMI and provide good prognostic information in high-risk populations. The assessment of serum Lp(a) can be used as predictor of MACE or could be incorporated into the existing risk stratification so that it can increase the accuracy in detection of high-risk patients where reperfusion could improve outcome.
The limitation of this study is that the selection of research subjects was carried out by consecutive sampling (non-probability sampling), research was only carried out in one research location, and included a relatively small sample size. There was no universal cut-off point for serum Lp(a) in the acute phase. Therefore, cut-off determination by researchers could lead to bias, and comparison between research is difficult. Measurement of serum Lp(a) was done only once, thus, pattern of changes could not be assessed. In this study, there was no comparison between serum Lp(a) and cardiac troponin levels in predicting MACE post-AMI, due to limitations in the examination of cardiac troponin. Besides, there was no evaluation on echocardiography in the assessment of heart function, especially left ventricle, which could influence the risk of MACE post-AMI. The overall treatment and medication used by the patients during the study, although it would be similar as it followed hospital protocol, also plays a role in the overall outcome, therefore it might have confounded the results. Finally, future study with a larger sample, and preferably multicenter involvement, needs to be conducted to confirm this finding.
Conclusion
AMI patients with high serum Lp(a) had poorer survival during hospitalization compared to patients with low Lp(a). The high level of Lp(a) in AMI patients was also a risk factor for the occurrence of MACE during hospitalization.
Ethics and Consent Statements
Ethical approval has been obtained from the Ethics Committee of Clinical Research, Faculty of Medicine, Universitas Udayana/Sanglah General Hospital, Bali, Indonesia, based on COPE prior to the study.
Acknowledgments
We would like to show our gratitude to Dr. I Putu Yuda Prabawa, S.Ked for criticizing the earliest draft of the manuscript and who provided more in-depth insight regarding statistical analysis. We also thank all the other parties and colleagues for their comments on the manuscript. An abstract of this paper was presented at the 28th ASMIHA Conference as a poster presentation with interim findings. The poster was published in “Original Research Abstracts” section in European Heart Journal Supplements; 2019;21(Supplement F):F33-F114 with DOI: 10.1093/eurheartj/suz182.
Author Contributions
All authors contributed to data analysis, drafting and revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Funding
This study was self-funded by authors.
Disclosure
All authors declare that they have no conflicts of interest regarding this work.
References
1. Riska Pertiwi GA, Ngurah Aryawangsa AA, Prabawa IPY, et al. Factors associated with visit-to-visit variability of blood pressure in hypertensive patients at a primary health care service, Tabanan, Bali, Indonesia. Fam Med Community Heal. 2018;6(4):191–199. doi:10.15212/FMCH.2018.0124
2. Tada H, Takamura M, Kawashiri M-A. Lipoprotein(a) as an old and new causal risk factor of atherosclerotic cardiovascular disease. J Atheroscler Thromb. 2019;26(7):583–591. doi:10.5551/jat.RV17034
3. Wiryadana KA, Supadmanaba IGP, Samatra IDPG. Progress and potential roles blood biomarkers of ischemic stroke in clinical setting. Indones J Biomed Sci. 2017;11(2):19–29. doi:10.15562/ijbs.v11i2.138
4. Abu-Assi E, López-López A, González-Salvado V, et al. The risk of cardiovascular events after an acute coronary event remains high, especially during the first year, despite revascularization. Rev Española Cardiol. 2016;69(1):11–18. doi:10.1016/j.recesp.2015.06.016
5. Loria V, Leo M, Biasillo G, Dato I, Biasucci LM. Biomarkers in acute coronary syndrome. Biomark Insights. 2008;3:453–468. doi:10.4137/BMI.S588
6. Gómez M, Valle V, Arós F, et al. Oxidized LDL, lipoprotein (a), and other emergent risk factors in acute myocardial infarction (FORTIAM study). Rev Española Cardiol. 2009;62(4):373–382. doi:10.1016/S0300-8932(09)70894-2
7. Morita Y, Himeno H, Yakuwa H, Usui T. Serum lipoprotein(a) level and clinical coronary stenosis progression in patients with myocardial infarction. Circ J. 2006;70(2):156–162. doi:10.1253/circj.70.156
8. Schwartz GG, Ballantyne CM, Barter PJ, et al. Association of lipoprotein(a) with risk of recurrent ischemic events following acute coronary syndrome: analysis of the dal-outcomes randomized clinical trial. JAMA Cardiol. 2018;3(2):164–168. doi:10.1001/jamacardio.2017.3833
9. Yan AT, Yan RT, Tan M, et al. Risk scores for risk stratification in acute coronary syndromes: useful but simpler is not necessarily better. Eur Heart J. 2007;28(9):1072–1078. doi:10.1093/eurheartj/ehm004
10. O’Donoghue ML, Morrow DA, Cannon CP, et al. Multimarker risk stratification in patients with acute myocardial infarction. J Am Heart Assoc. 2016;5(5):e002586.
11. Oemrawsingh RM, Akkerhuis KM, Umans VA, et al. Cohort profile of BIOMArCS: the BIOMarker study to identify the acute risk of a coronary syndrome-a prospective multicentre biomarker study conducted in the Netherlands. BMJ Open. 2016;6(12):e012929–e012929. doi:10.1136/bmjopen-2016-012929
12. McCormick SPA. Lipoprotein(a): biology and clinical importance. Clin Biochem Rev. 2004;25(1):69–80.
13. Kostner KM, Marz W, Kostner GM. When should we measure lipoprotein (a)? Eur Heart J. 2013;34(42):3268–3276. doi:10.1093/eurheartj/eht053
14. Tsimikas S. Lipoprotein(a): novel target and emergence of novel therapies to lower cardiovascular disease risk. Curr Opin Endocrinol Diabetes Obes. 2016;23(2):157–164. doi:10.1097/MED.0000000000000237
15. Kojima S, Harpel PC, Rifkin DB. Lipoprotein (a) inhibits the generation of transforming growth factor beta: an endogenous inhibitor of smooth muscle cell migration. J Cell Biol. 1991;113(6):1439–1445. doi:10.1083/jcb.113.6.1439
16. Manocha A, Srivastava LM. Lipoprotein (a): a unique independent risk factor for coronary artery disease. Indian J Clin Biochem. 2016;31(1):13–20. doi:10.1007/s12291-015-0483-3
17. Malaguarnera M, Vacante M, Russo C, et al. Lipoprotein(a) in cardiovascular diseases. Biomed Res Int. 2012;2013:650989.
18. Barre DE. Lipoprotein (a) reduces platelet aggregation apo(a)-mediated decreases in production via thromboxane A 2. Platelets. 1998;9(2):93–96. doi:10.1080/09537109876852
19. Barre D. Apolipoprotein (a) mediates the lipoprotein (a)-induced biphasic shift in human platelet cyclic AMP. Thromb Res. 2003;112(5–6):321–324. doi:10.1016/j.thromres.2004.01.002
20. Barre DE. Arginyl-glycyl-aspartyl (RGD) epitope of human apolipoprotein (a) inhibits platelet aggregation by antagonizing the IIb subunit of the fibrinogen (GPIIb/IIIa) receptor. Thromb Res. 2007;119(5):601–607. doi:10.1016/j.thromres.2006.04.013
21. Tsironis L, Mitsios J, Milionis H, Elisaf M, Tselepis A. Effect of lipoprotein (a) on platelet activation induced by platelet-activating factor: role of apolipoprotein (a) and endogenous PAF-acetylhydrolase. Cardiovasc Res. 2004;63(1):130–138. doi:10.1016/j.cardiores.2004.03.005
22. Rand ML, Sangrar W, Hancock MA, et al. Apolipoprotein(a) enhances platelet responses to the thrombin receptor-activating peptide SFLLRN. Arterioscler Thromb Vasc Biol. 1998;18(9):1393–1399. doi:10.1161/01.ATV.18.9.1393
23. Martínez C, Rivera J, Loyau S, et al. Binding of recombinant apolipoprotein(a) to human platelets and effect on platelet aggregation. Thromb Haemost. 2001;85(4):686–693. doi:10.1055/s-0037-1615654
24. Boffa MB, Koschinsky ML. Lipoprotein (a): truly a direct prothrombotic factor in cardiovascular disease? J Lipid Res. 2016;57(5):745–757. doi:10.1194/jlr.R060582
25. Donataccio MP, Puymirat E, Vassanelli C, et al. Presentation and revascularization patterns of patients admitted for acute coronary syndromes in france between 2004 and 2008 (from the national observational study of diagnostic and interventional cardiac catheterization [ONACI]). Am J Cardiol. 2014;113(2):243–248. doi:10.1016/j.amjcard.2013.09.014
26. Fox KAA, Eagle KA, Gore JM, Steg PG, Anderson FA. The global registry of acute coronary events, 1999 to 2009-GRACE. Heart. 2010;96(14):1095–1101. doi:10.1136/hrt.2009.190827
27. Peng Y, Shao Z, Jiang J, et al. Association between serum lipoprotein (a) levels and major adverse cardiovascular events in Chinese patients with ST-segment elevation myocardial infarction. Int J Clin Exp Med. 2017;10(1):897–904.
28. Mitsuda T, Uemura Y, Ishii H, et al. Lipoprotein(a) levels predict adverse vascular events after acute myocardial infarction. Heart Vessels. 2016;31(12):1923–1929. doi:10.1007/s00380-016-0823-0
29. Cho JY, Jeong MH, Ahn Y, et al. High lipoprotein(a) levels are associated with long-term adverse outcomes in acute myocardial infarction patients in high killip classes. Korean Circ J. 2010;40(10):491–498. doi:10.4070/kcj.2010.40.10.491.
30. Maranhão RC, Carvalho PO, Strunz CC, Pileggi F. Lipoprotein (a): structure, pathophysiology and clinical implications. Arq Bras Cardiol. 2014;103(1):76–84. doi:10.5935/abc.20140101
31. Ikenaga H, Ishihara M, Inoue I, et al. Usefulness of Lipoprotein (a) for predicting progression of non-culprit coronary lesions after acute myocardial infarction. Circ J. 2011;75(12):2847–2852. doi:10.1253/circj.CJ-11-0365
32. Waldeyer C, Makarova N, Zeller T, et al. Lipoprotein(a) and the risk of cardiovascular disease in the European population: results from the biomarCaRE consortium. Eur Heart J. 2017;38(32):2490–2498. doi:10.1093/eurheartj/ehx166
33. Danesh J, Collins R, Peto R. Lipoprotein(a) and coronary heart disease. Circ. 2000;102(10):1082–1085. doi:10.1161/01.CIR.102.10.1082
34. Craig WY, Neveux LM, Palomaki GE, Cleveland MM, Haddow JE. Lipoprotein(a) as a risk factor for ischemic heart disease: metaanalysis of prospective studies. Clin Chem. 1998;44(11):2301–2306. doi:10.1093/clinchem/44.11.2301
35. Genser B, Dias KC, Siekmeier R, Stojakovic T, Grammer T, Maerz W. Lipoprotein (a) and risk of cardiovascular disease–a systematic review and meta analysis of prospective studies. Clin Lab. 2011;57(3–4):143–156.
36. Lan Y, Zhao X, Wang X, et al. Lipoprotein(a) as a risk factor for predicting coronary artery disease events: a meta-analysis. Biomark J. 2018;4(3):17.
37. Wang Z, Zhai X, Xue M, Cheng W, Hu H. Prognostic value of lipoprotein (a) level in patients with coronary artery disease: a meta-analysis. Lipids Health Dis. 2019;18(1):150. doi:10.1186/s12944-019-1092-6
38. Erqou S, Kaptoge S, Perry PL, Di Angelantonio E, Thompson A, Danesh J. Emerging Risk Factors Collaboration. Lipoprotein(a) concentration and the risk of coronary heart disease, stroke, and nonvascular mortality. JAMA. 2009;302(4):412.
39. Nave AH, Lange KS, Leonards CO, et al. Lipoprotein (a) as a risk factor for ischemic stroke: a meta-analysis. Atherosclerosis. 2015;242(2):496–503. doi:10.1016/j.atherosclerosis.2015.08.021
40. Smolders B, Lemmens R, Thijs V. Lipoprotein (a) and stroke. Stroke. 2007;38(6):1959–1966. doi:10.1161/STROKEAHA.106.480657
41. Willeit P, Kiechl S, Kronenberg F, et al. Discrimination and net reclassification of cardiovascular risk with lipoprotein(a). J Am Coll Cardiol. 2014;64(9):851–860. doi:10.1016/j.jacc.2014.03.061
42. Kamiński KA, Tycińska AM, Stepek T, et al. Natural history and risk factors of long-term mortality in acute coronary syndrome patients with cardiogenic shock. Adv Med Sci. 2014;59(2):156–160. doi:10.1016/j.advms.2013.12.003
43. Kaulgud RS, Vijayalxmi PB, Arun BS, Supriya RR, Vigneshwar M. Novel biomarkers for risk stratification of acute coronary syndromes. Int J Biomed Res. 2015;6(8):539. doi:10.7439/ijbr.v6i8.2346
44. Nordestgaard BG, Chapman MJ, Ray K, et al. Lipoprotein(a) as a cardiovascular risk factor: current status. Eur Heart J. 2010;31(23):2844–2853. doi:10.1093/eurheartj/ehq386.
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.