")
Back to Journals » Nature and Science of Sleep » Volume 13
High-Fat Diet-Induced Mitochondrial Dysfunction Promotes Genioglossus Injury – A Potential Mechanism for Obstructive Sleep Apnea with Obesity
Authors Chen Q, Han X, Chen M, Zhao B , Sun B, Sun L , Zhang W, Yu L, Liu Y
Received 10 October 2021
Accepted for publication 14 December 2021
Published 23 December 2021 Volume 2021:13 Pages 2203—2219
DOI https://doi.org/10.2147/NSS.S343721
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ahmed BaHammam
Qingqing Chen,1,2 Xinxin Han,2 Meihua Chen,2,3 Bingjiao Zhao,1,2 Bingjing Sun,1,2 Liangyan Sun,1,2 Weihua Zhang,1,2 Liming Yu,1,2 Yuehua Liu1,2
1Department of Orthodontics, Shanghai Stomatological Hospital, Fudan University, Shanghai, People’s Republic of China; 2Shanghai Key Laboratory of Craniomaxillofacial Development and Diseases, Fudan University, Shanghai, People’s Republic of China; 3Department of Periodontology, Shanghai Stomatological Hospital, Fudan University, Shanghai, People’s Republic of China
Correspondence: Yuehua Liu; Liming Yu
Shanghai Stomatological Hospital, Fudan University, No. 356 East Beijing Road, Shanghai, 200001, People’s Republic of China
Tel +86 2163298475
Fax +86 2163614515
Email [email protected]; [email protected]
Purpose: Obesity is a worldwide metabolic disease and a critical risk factor for several chronic conditions. Obstructive sleep apnea (OSA) is an important complication of obesity. With the soaring morbidity of obesity, the prevalence of OSA has markedly increased. However, the underlying mechanism of the high relevance between obesity and OSA has not been elucidated. This study investigated the effects of obesity on the structure and function of the genioglossus to explore the possible mechanisms involved in OSA combined with obesity.
Methods: Six-week-old male C57BL/6J mice were fed high-fat diet (HFD, 60% energy) or normal diet (Control, 10% energy) for 16 weeks. The muscle fibre structure and electromyography (EMG) activity of genioglossus were measured. The ultrastructure and function of mitochondrial, oxidative damage and apoptosis in genioglossus were detected by transmission electron microscopy (TEM), qPCR, Western blotting, immunohistochemistry and TUNEL staining. We further studied the influence of palmitic acid (PA) on the proliferation and myogenic differentiation of C2C12 myoblasts, as well as mitochondrial function, oxidative stress, and apoptosis in C2C12 myotubes.
Results: Compared with the control, the number of muscle fibres was decreased, the fibre type was remarkably changed, and the EMG activity had declined in genioglossus. In addition, a HFD also reduced mitochondria quantity and function, induced excessive oxidative stress and increased apoptosis in genioglossus. In vitro, PA treatment significantly inhibited the proliferation and myogenic differentiation of C2C12 myoblasts. Moreover, PA decreased the mitochondrial membrane potential, upregulated mitochondrial reactive oxygen species (ROS) levels, and activated the mitochondrial-related apoptotic pathway in myotubes.
Conclusion: Our findings suggest that a HFD caused genioglossus injury in obese mice. The mitochondrial dysfunction and the accompanying oxidative stress were involved in the genioglossus injury, which may provide potential therapeutic targets for OSA with obesity.
Keywords: obesity, obstructive sleep apnea, genioglossus, muscle, mitochondria
Introduction
Obesity is a worldwide metabolic disease that has shown increasing morbidity.1,2 Recent studies have found that obesity characterized by an excessive accumulation of lipids in the blood and subcutaneous and visceral compartments, leads to disorders in glucose and lipid metabolism and increases the risk of several chronic conditions, notably cardiovascular and respiratory diseases.3–5 Obstructive sleep apnea (OSA) is one of the most common respiratory diseases and is characterized by intermittent and recurrent collapse of the upper airway during sleep, inducing hypoxia or apnea and causing a significant threat to the health and quality of life of the affected individuals.6,7 Obesity is highly correlated with the incidence of OSA.8,9 According to a recent epidemiological study, the prevalence of OSA was four times higher in the obese population than in the general population.10 The anatomic stenosis of the upper airway and the dysfunction of upper airway dilator muscles were reported to have important implications in the pathogenesis of OSA.11,12 High-fat diet (HFD)-induced obesity increases the accumulation of fat around the upper airway, aggravates stenosis of the upper airway, and more easily results in airflow reduction or obstruction during sleep.13–16 However, how the dysfunction of the upper airway dilator muscles in the obese population is involved in the mechanism of OSA has not been elucidated.
The genioglossus is the largest upper airway dilator muscle and plays a very important role in maintaining airway patency.17 Compared to control subjects, OSA patients show an abnormal structure and function of the genioglossus, with a higher proportion of type II fibres (fast-twitch, glycolytic), which decreases fatigue resistance in the genioglossus.18 HFD-induced obesity is a metabolic disease. The accumulation of excess free fatty acids (FFAs) in skeletal muscle could impair myogenesis, decrease skeletal muscle mass and reduce muscle strength.19,20 However, there have been few studies addressing the effects of obesity on genioglossus structure and function.
Mitochondria are the primary energy source in muscle cells. Mitochondria produce adenosine triphosphate (ATP) via the oxidative metabolism of glucose and fatty acids, which is essential for the maintenance of skeletal muscle function.21 In the context of HFD-induced obesity, excess lipids in skeletal muscle suppress glucose oxidative metabolism and lead to disturbances in mitochondrial function. A large body of studies have shown that mitochondrial damage is involved in obesity-induced skeletal muscle dysfunction, impairing mitochondrial biogenesis, lowering mitochondrial content and reducing the activity of mitochondrial enzymes.22–24 Moreover, mitochondrial damage was also found in the genioglossus of chronic intermittent hypoxia (CIH) rats.25 Therefore, it is essential for us to investigate the potential influence of obesity on the mitochondria in the genioglossus, which will be beneficial to better understand the underlying mechanism of OSA that occurs with obesity.
Mitochondrial dysfunction contributes to the excessive accumulation of reactive oxygen species (ROS) and oxidative stress, which causes oxidative damage to major cellular structures (in particular, the plasma membrane, proteins and DNA) and leads to apoptosis.26 In HFD-induced obesity, oxidative damage accompanied by mitochondrial impairment has been found, which is a critical factor for skeletal muscle dysfunction.27,28 Our previous work that focused on OSA demonstrated that ROS overproduction caused by hypoxia contributed to pyroptosis in myoblasts and might be a critical pathogenesis of OSA.29
Therefore, we hypothesized that mitochondrial dysfunction and oxidative stress may be involved in the effects of HFD-induced obesity in the genioglossus, which is an important pathogenesis of OSA combined with obesity. To address this hypothesis, we examined the changes in genioglossus activity and structure, as well as the mitochondrial ultrastructure and function, in HFD-induced obese mice. In addition, the underlying preliminary mechanisms have been investigated, including oxidative stress and apoptosis in the genioglossus. We also used palmitic acid (PA), one of the most abundant saturated free fatty acids (FFAs) in humans, to mimic HFD-induced obesity in an in vitro study.30 We studied the influence of PA on the proliferation and myogenic differentiation of C2C12 myoblasts, as well as mitochondrial function, oxidative stress, and apoptosis in C2C12 myotubes.
Materials and Methods
Animals
Six-week-old male C57BL/6J mice were provided by Shanghai Model Organisms Center and housed under controlled environmental conditions (22 ± 2 °C, 50% ± 5% humidity, 12:12 h light/dark cycle). The mice were randomly divided into two groups: the control group (normal diet, 10% energy; Research diet D12450J) and the HFD group (60% energy; Research diet D12492). All mice had free access to food and water under specific pathogen-free (SPF) conditions, and the mice body weights were recorded weekly. The animal study was approved by the Animal Welfare and Ethics Group, Department of Laboratory Animal Science at Fudan University. All procedures were performed according to Laboratory animal – Guideline for ethical review of animal welfare (GB/T 35892–2018, standardization administration of the People’s Republic of China). After 16 weeks, the mice were euthanized, and samples were collected. Before sample collection, the mice were fasted overnight and anaesthetized with chloral hydrate. Blood was collected from the retro-orbital plexus and centrifuged at 3000 rpm for 10 minutes. Plasma was collected and used for clinical biochemical analyses. Additionally, each mouse genioglossus was harvested for subsequent analysis.
Blood Biochemical Assay
Chemical analysis of the serum samples (n = 6) was carried out with an automatic biochemistry analyser (Chemray 800, Rayto Life and Analytical Sciences Co., Shenzhen, China). The following parameters were evaluated: triglycerides (TGs) and cholesterol (CHOL). The levels of FFAs were measured with commercial kits (Nanjing Jiancheng Bioengineering Institute, Nanjing, China).
Intraperitoneal Glucose Tolerance Test (IPGTT) and Insulin Tolerance Test (IPITT)
At the end of the 16-week experiment, the IPGTT and IPITT were performed. For the IPGTT, mice were intraperitoneally injected with glucose (Sigma–Aldrich, St. Louis, MO, USA; 1.5 g/kg body weight) after 12 h of fasting. For the IPITT, mice were intraperitoneally injected with insulin (Sigma–Aldrich, St. Louis, MO, USA; 0.5 U/kg body weight) after 4 h of fasting. Blood samples (n = 6) were taken from the tail vein, and the blood glucose levels were measured at 0, 15, 30, 60, and 120 minutes using a glucometer (Roche, USA).
Electromyography (EMG)
EMG of the genioglossus muscle was acquired and analysed as described in our previous study.31 Briefly, six mice from each group were first anaesthetized with 1% pentobarbital, and then the genioglossus was exposed. EMG electrodes (AD Instrument, Australia) were inserted into the genioglossus, and the EMG signal was amplified and recorded (LabChart 8, AD Instrument, filter frequency was 1–1000 Hz, time constant: 1 s).
Histological Examination and Immunohistochemical Assay
The genioglossus samples were harvested and immediately fixed in ice-cold 4% paraformaldehyde solution. The histopathological tests were performed using standard laboratory procedures. Briefly, the genioglossus samples were embedded in paraffin and cut into 5 μm sections, which were mounted onto glass slides for haematoxylin and eosin (H&E), Masson’s trichrome, myosin heavy chain (MyHC)-slow, MyHC-fast, 4-hydroxynonenal (4-HNE) and 8-hydroxy-2ʹ-deoxyguanosine (8-OHdG) staining. For histological examinations, genioglossus sections were stained with micrographs of H&E and Masson’s trichrome staining (Solarbio, Beijing, China). For tissue immunohistochemistry, after being deparaffinized and rehydrated, the genioglossus sections were boiled in 10 mM citrate buffer for 10 minutes, processed with 0.25% Triton X-100 for 30 minutes and blocked with a 5% bovine serum albumin (BSA) solution at room temperature for 30 minutes. Next, the sections were incubated overnight at 4 °C with primary antibodies against MyHC-slow (1:1000, Abcam, UK), MyHC-fast (1:1000, Abcam, UK), 4-HNE (1:200, Abcam, UK) and 8-OHdG (1:200, Abcam, UK). Then, staining was carried out with an immunodetection kit and DAB peroxidase substrate kit according to the manufacturer’s instructions (ZSGB-BIO, Beijing, China). Micrographs were acquired with a light microscope (DM2500, Leica, Wetzlar, Germany). Three to ten fields of view were randomly selected for quantification analysis.
Transmission Electron Microscopy (TEM)
The genioglossus samples were sliced into several tissue pieces (each 1 × 1×1 mm) and fixed in 2.5% glutaraldehyde at 4 °C. Then, the samples were incubated in 0.1 M phosphate buffer with 1% osmium tetroxide for 2 h, dehydrated, embedded in epoxy resin, and polymerized for 48 h at 60 °C. Subsequently, the sections were cut into ultrathin sections (50–70 nm), stained with uranyl acetate-lead citrate, and examined with a transmission electron microscope (JEOL-1200, JEOL electron, Japan).
Mitochondrial DNA (mtDNA) Copy Number Determination
The genomic DNA of genioglossus samples (n = 6) was isolated with the DNA Extraction Kit (Tiangen, DP304, Beijing, China) according to the manufacturer’s instructions. QPCR was used to detect the copy numbers of nuclear DNA and mtDNA with SuperReal PreMix Plus (Tiangen, FP205, Beijing, China) on a LightCycler 480 System (Roche, Basel, Switzerland). The relative copy number of mtDNA was determined by the comparative cycle threshold (Ct) value of nuclear DNA. The primers of nuclear DNA: (F) CGTGGGCTCCAGCATTCTA and (R) TCACCAGTCATTTCTGCCTTTG; and the primers of mtDNA: (F) CGAAAGGACAAGAGAAATAGAG and (R) GAACAAGGTTTTAAGTCTTACGCA.
Cell Culture and Palmitic Acid Treatment
C2C12 mouse myoblasts from the Cell Bank of Chinese Academy of Sciences Shanghai Institute of Cell Biology (Shanghai, China) were cultured in growth medium (GM) (high-glucose DMEM with 10% FBS (Gibco, MA, USA) and 1% penicillin/streptomycin (Gibco, MA, USA)) at 37 °C in 5% CO2/95% air. For myogenic differentiation, when C2C12 myoblasts were 100% confluent, GM was switched to high-glucose DMEM containing 2% horse serum (Gibco, Thermo Fisher Scientific, Waltham, MA, USA) and 1% penicillin/streptomycin for 5 days, with medium change every 2 days.
In vitro, PA was used to mimic the high-fat environment. PA (Sigma–Aldrich, St. Louis, MO, USA) was dissolved in absolute ethanol to generate a 100 mM stock solution after filtration through a 0.22 μm PES membrane. Then, the PA stock solution was added to serum-free DMEM containing 5% BSA (Sigma–Aldrich, St. Louis, MO, USA) at final concentrations of 100 μM, 300 μM and 500 μM at 37 °C with continuous shaking for 2 h. Serum-free DMEM containing 5% BSA with absolute ethanol was used as the control. To examine the effect of PA on the proliferation of C2C12, the C2C12 myoblasts were treated with different concentrations of PA for 6 h to 48 h. To assess the effect of PA on the myogenic differentiation of C2C12, the C2C12 myoblasts were treated with different concentrations of PA for 2 days during cellular differentiation. To investigate the effect of PA on the mitochondrial function of C2C12 myotubes, after 5 days of myogenic differentiation, the myotubes were treated with different concentrations of PA for 2 days. In some experiments, in order to detect the role of PA-induced mitochondrial ROS in the damage of C2C12 myotubes, Szeto-Schiller (SS) peptide SS–31 (Selleckchem, Houston, TX, USA) was used to treat C2C12 myotubes at 10μM for 2days. SS–31 is reported to target mitochondria and reduce mitochondrial ROS.32,33
Cell Viability Assay, Lactic Dehydrogenase (LDH) Measurements and Cell Morphological Imaging
Cell viability was determined using a Cell Counting Kit-8 assay (CCK-8, Dojindo, Japan). In brief, C2C12 myoblasts were seeded in 96-well plates at a density of 5×103 cells per well and allowed to attach for 24 h. Then, the C2C12 myoblasts were treated with PA at different concentrations (100 μΜ, 300 μM and 500 μM). After treatment for 0 h, 6 h, 12 h, 24 h, and 48 h, the cells were incubated with CCK-8 for 1 h, and the absorbance values at 450 nm were measured with an Epoch 2 microplate spectrophotometer (BioTek, VT, USA). After treatment with PA for 24 h, the morphological changes in the C2C12 myoblasts were detected with a Leica DMi8 microscope (Leica, Wetzlar, Germany).
Cytoplasmic LDH was released into the medium following cell death. The amount of LDH in the extracellular medium was determined using a commercial LDH kit (Beyotime, Nanjing, China). Briefly, C2C12 myoblasts were seeded in 96-well plates and treated with PA for 24 h. Then, the culture supernatants were collected and centrifuged at 400 × g for 5 minutes. LDH levels were detected according to the manufacturer’s protocol, and the absorbance at 495 nm was measured with an Epoch 2 microplate spectrophotometer.
Flow Cytometry Detection of Apoptosis
Cell apoptosis was measured using an Annexin V-FITC/propidium iodide (PI) apoptosis detection kit (Beyotime, Nanjing, China). After treatment with PA for 24 h, C2C12 myoblasts were harvested and incubated with annexin V-FITC and PI for 15 minutes according to the manufacturer’s instructions. The rate of apoptosis was measured using flow cytometry (ACEA NovoCyte, Hangzhou, China) and analysed with Novo Express software 1.4.1 (ACEA Biosciences).
Immunofluorescence Cell Staining
C2C12 myoblasts were incubated with different concentrations of PA during cellular differentiation for 2 days. Then, the myotubes were washed with PBS, fixed in 4% paraformaldehyde for 15 minutes, permeabilized with 0.25% Triton X-100 for 10 minutes and blocked with 5% BSA for 1 h at room temperature. Subsequently, the cells were incubated overnight at 4 °C with primary antibodies against MyHC (1:10, MF20 DSHB, USA). The following day, the cells were incubated with the secondary antibody for 1 h, and the nuclei were counterstained with DAPI for 5 minutes at room temperature. Finally, fluorescent images were captured using a fluorescence microscope (DMi8, Leica, Wetzlar, Germany).
MitoTracker Green and Mitochondrial Membrane Potential (MMP) Fluorescent Staining
After myogenic differentiation of C2C12 for 5 days, the myotubes were treated with different concentrations of PA for 2 days. Then, C2C12 myotubes were incubated with MitoTracker green dye (Beyotime, Nanjing, China) at 37 °C for 30 minutes to observe changes in mitochondrial morphology. The nuclei were counterstained with DAPI for 5 minutes at 37 °C. Images were immediately acquired with a fluorescence microscope (DMi8, Leica, Wetzlar, Germany). Moreover, the MMP was evaluated using a tetraethylbenzimidazolylcarbocyanine iodide kit (JC-1, Beyotime, Nanjing, China) according to the manufacturer’s instructions. C2C12 myotubes were incubated with different concentrations of PA for 2 days. Then, the myotubes were stained with 10 μM JC-1 for 20 minutes at 37 °C. The nuclei were counterstained with DAPI for 5 minutes at 37 °C, and images were immediately acquired with a fluorescence microscope.
Mitochondrial ROS Determination
Mitochondrial ROS was evaluated using the mitochondrial superoxide indicator MitoSOX Red (Invitrogen, Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer’s instructions. Briefly, after myogenic differentiation of C2C12 for 5 days, the myotubes were treated with PA or SS-31 for 2 days. Then, C2C12 myotubes were washed twice with PBS and incubated in 5 μM MitoSOX Red for 30 minutes at 37 °C. The nuclei were counterstained with DAPI for 5 minutes at 37 °C, and images were immediately acquired with a fluorescence microscope.
Apoptosis Assay
A TUNEL assay was performed to detect apoptotic cells in genioglossus tissues or C2C12 myotubes using a One-Step TUNEL Assay Kit (Beyotime, Shanghai, China) according to the manufacturer’s instructions. Briefly, the fixed genioglossus sections or C2C12 myotubes were incubated with TUNEL reaction mixture for 30 minutes at 37 °C and then counterstained with 4ʹ,6-diamidino-2-phenylindole (1:10000, DAPI, Abcam, UK) for 5 minutes. Images were acquired with a fluorescence microscope (DM2500, Leica, Wetzlar, Germany).
Western Blotting
Total protein was extracted from genioglossus tissues or C2C12 myotubes using RIPA buffer (Beyotime, Nanjing, China) with protease and phosphatase inhibitors. To detect the protein levels of Cyt-c in the mitochondria and cytoplasm of genioglossus tissues, mitochondrial and cytoplasm proteins were isolated using a mitochondria isolation kit (Beyotime, Nanjing, China). Protein concentration was determined with a bicinchoninic acid (BCA) Protein Assay kit (Thermo Fisher Scientific, Waltham, MA, USA). Total protein was separated by 10% SDS polyacrylamide gel electrophoresis (SDS–PAGE) and transferred onto polyvinylidene fluoride (PVDF) membranes using a Bio–Rad system (Hercules, CA). The membranes were incubated overnight at 4 °C with primary antibodies. Antibodies against DRP1 (1:1000), cleaved caspase-3 (1:1000) and Bax (1:1000) were purchased from Cell Signaling Technology (BSN, USA). Antibodies against MFN2 (1:300), Bcl-2 (1:1000), Cyt-c (1:1000) and Cox IV (1:1000) were purchased from Proteintech Group (PA, USA). Antibodies against MyoG (1:500), MyoD1 (1:500) and GAPDH (1:500) were purchased from Santa Cruz Biotechnology (TX, USA). The MyHC antibody (1:60) was purchased from Developmental Studies Hybridoma Bank (IA, USA). The OXPHOS antibody (1:1000) was purchased from Abcam (Cambridge, UK). Horseradish peroxidase‑conjugated secondary antibodies (1:10000, Cell Signaling Technology, BSN, USA) were applied the following day and incubated with the membranes at room temperature for 1.5 h. Then, the membranes were developed with a chemiluminescent substrate kit (Thermo Fisher Scientific, Waltham, MA, USA), and the bands were detected with Amersham Imager 600 (GE Health care, IL, USA). GAPDH was used as an internal control in the total protein and cytoplasm protein, Cox IV was used as an internal control in the mitochondria protein.
Statistical Analysis
Data were analysed using GraphPad Prism 7.0 (GraphPad Software, San Diego, CA, USA) and expressed as the mean ± SD. Differences between groups were determined using Student’s t-test or one-way ANOVA. A p value< 0.05 was considered statistically significant.
Results
Effects of HFD on Glucose and Lipid Metabolism in Mice
The body weights of the HFD mice significantly increased after 16 weeks, and these mice were markedly heavier than the control group (p < 0.001) (Figure 1A). Moreover, the serum TG, FFA and CHOL levels were all elevated in the HFD group (Figure 1B–D). In addition, the IPGTT and the IPITT were performed, and the data showed that HFD induced systemic insulin resistance, with a significant reduction in glucose tolerance and insulin sensitivity (Figure 1E and F).
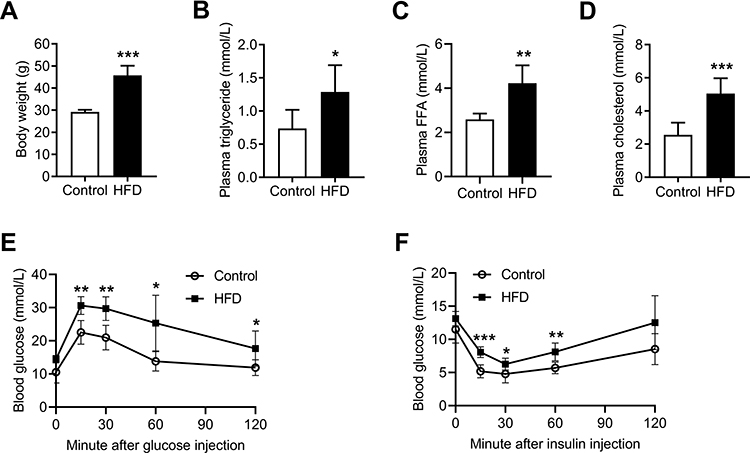 |
Figure 1 HFD induced obesity and caused glucose and lipid metabolism disorders in C57BL/6J mice. (A) Body weights of control and HFD-fed mice at the end of 16 weeks. (B) Plasma triglyceride, (C) plasma FFA, and (D) plasma cholesterol of the control and HFD-fed mice at the end of 16 weeks. (E and F) IPGTT and IPITT of the control and HFD-fed mice at the end of 16 weeks. Data are represented as the mean ± SD. *p < 0.05, **p < 0.01, ***p < 0.001 compared to the control. |
Effects of HFD on Genioglossus Structure and Activity
The genioglossus is the major upper airway muscle and plays an important role in the maintenance of upper airway patency. The structure of the genioglossus was detected using H&E and Masson’s trichrome staining and MyHC-slow or MyHC-fast immunohistochemistry assays (Figure 2A). H&E-stained sections of the genioglossus showed that the diameters of the muscle fibres in the HFD group were increased, while the number of muscle fibres was significantly less than that in the control group (p < 0.05) (Figure 2B and C). Masson’s trichrome staining showed that a HFD enhanced collagen content in the genioglossus compared with the control (p < 0.01) (Figure 2D). Then, the genioglossus was subjected to immunohistochemical staining for the determination of muscle fibre types, which showed that the numbers of slow-twitch fibres decreased and the fast-twitch fibres increased in the HFD group (p < 0.001) (Figure 2E).
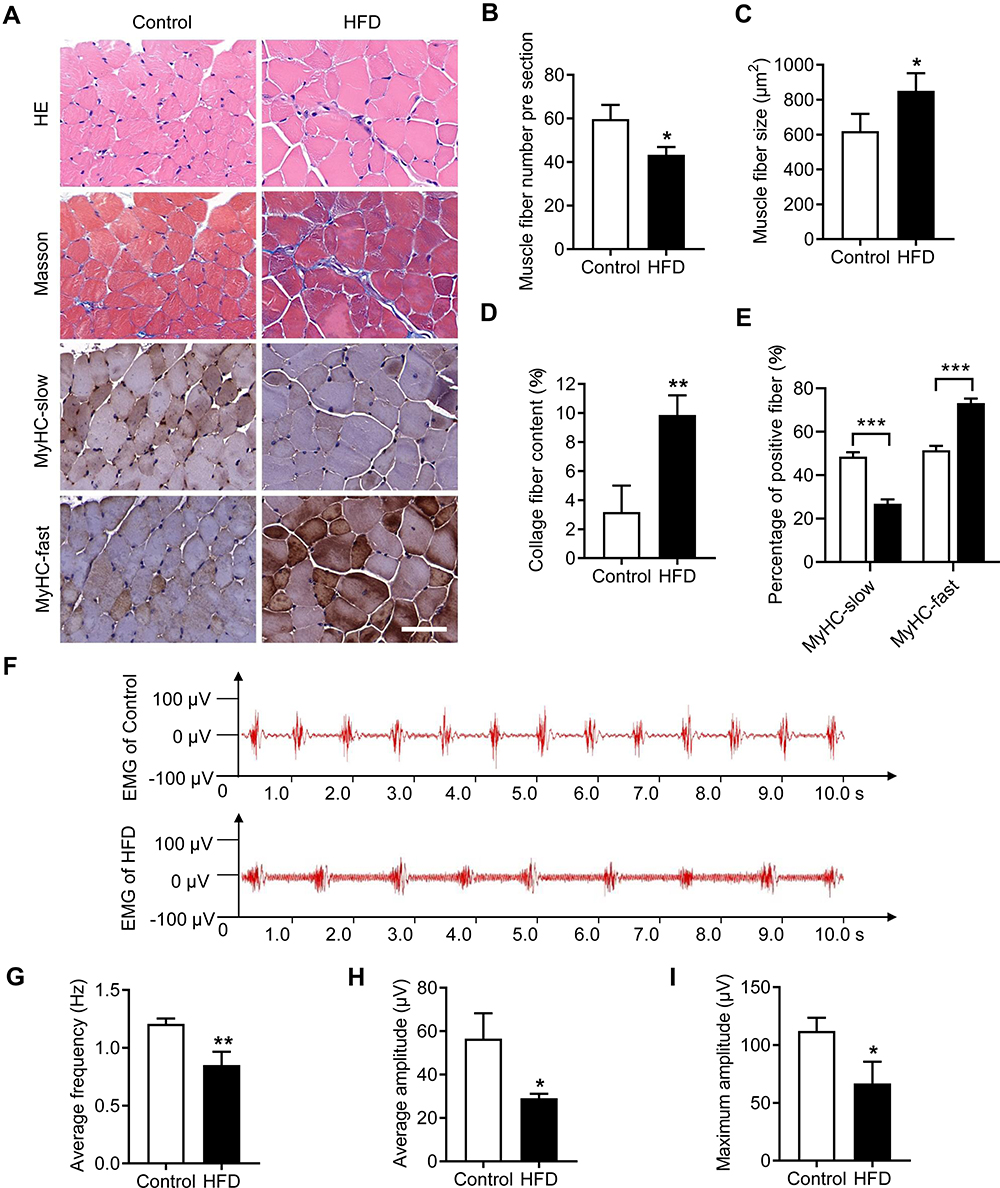 |
Figure 2 HFD impaired muscle structure and function in the genioglossus. (A) Representative images of genioglossus sections from control and HFD-fed mice with H&E and Masson’s trichrome staining and MyHC-slow or MyHC-fast immunohistochemical staining. (B) Quantification of muscle fibre number, (C) muscle fibre size, (D) collagen fibre content and (E) percentage of MyHC-slow and MyHC-fast per visual field. (F) Typical EMG in the genioglossi of control and HFD mice. (G) The average frequency, (H) average amplitude and (I) maximum amplitude of the genioglossus by EMG. Data are represented as the mean ± SD. *p < 0.05, **p < 0.01, ***p < 0.001 compared to the control. Scale bar is 50 μm. |
To study the influence of a HFD on the function of the genioglossus, we measured EMG activity, as depicted in Figure 2F. The average frequency of the genioglossus by EMG in the HFD group was approximately 0.85 Hz, which is significantly lower than that of the control (1.2 Hz, p < 0.01) (Figure 2G). Moreover, both the average amplitude and maximum amplitude declined in the HFD group (p < 0.05) (Figure 2H and I).
Effects of HFD on Mitochondrial Function in the Genioglossus
Mitochondria are the main energy source in skeletal muscle. We investigated the effects of HFD-induced obesity on the mitochondria of the genioglossus by TEM. For the control group, the genioglossus showed regular myofibril structure and normal mitochondrial morphology, with a large number of cristae. The HFD group showed serious damage to the genioglossus ultrastructure, with vague myofibril structure and a decrease in the number of mitochondria (Figure 3A and B). The mitochondria in the HFD group were swollen, and the cristae were partially disintegrated (Figure 3A and C). We also evaluated the copy number of mtDNA in the genioglossus and the result showed that the relative mtDNA level was significantly decreased in the HFD group (Figure 3D). In addition, the expression levels of mitochondrial fission protein DRP1 and fusion protein MFN2 were measured by Western blot. As shown in Figure 3E and F, the basal expression level of DRP1 was elevated, and the expression of MFN2 was inhibited, indicating an imbalance in mitochondrial fusion and fission in the genioglossus of the HFD group. To further investigate the effect of HFD on the function of mitochondria in the genioglossus, the mitochondrial respiratory chain complexes were assessed by Western blotting. In the HFD mice, the expression of mitochondrial respiratory chain complexes III and V were downregulated (Figure 3G and H).
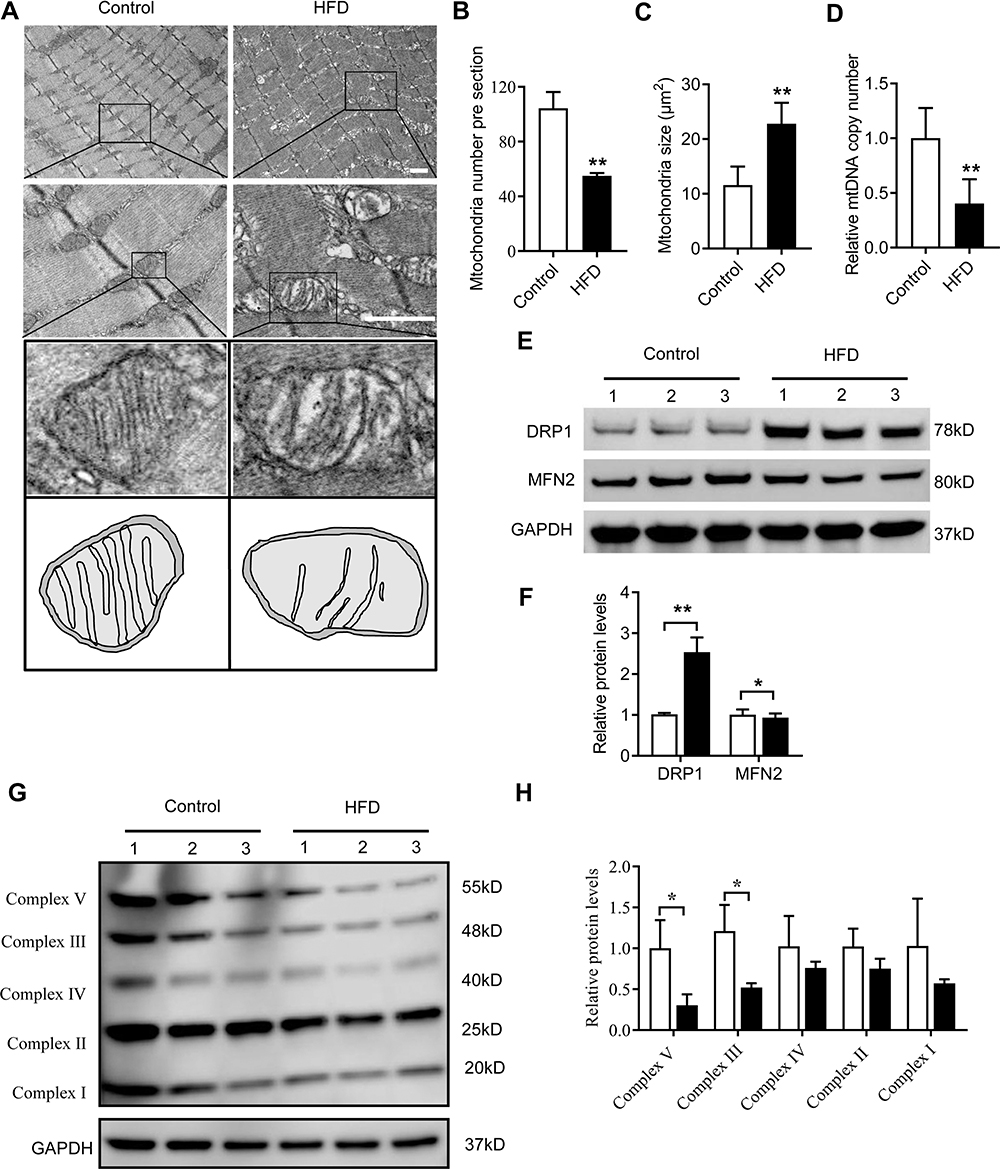 |
Figure 3 HFD induced mitochondrial damage and dysfunction in the genioglossus. (A) Mitochondrial morphology and size in the genioglossus of control and HFD-fed mice determined by TEM. (B) Quantification of mitochondrial number and (C) mitochondrial size per visual field. (D) Relative mtDNA copy number in the genioglossi of control and HFD mice. (E) Protein expression of DRP1 and MFN2 measured by Western blot analysis in the genioglossus of control and HFD mice (F). (G and H) Protein expression of mitochondrial respiratory chain complexes I–V measured by Western blot analysis in the genioglossus of control and HFD mice. Data are represented as the mean ± SD. *p < 0.05, **p < 0.01 compared to the control. Scale bars are 1 μm. |
Effects of HFD on Oxidative Stress and Apoptosis in the Genioglossus
To study the effects of a HFD on oxidative stress in the genioglossus, the protein levels of 4-HNE and 8-OHdG, markers of oxidative stress-induced lipid peroxidation and DNA damage, were detected using immunohistochemical staining. The results showed that the numbers of 4-HNE-positive fibres (Figure 4A and B) and 8-OHdG-positive fibres (Figure 4C and D) were substantially elevated in the genioglossus of the HFD group, which indicated that the level of oxidative stress was elevated.
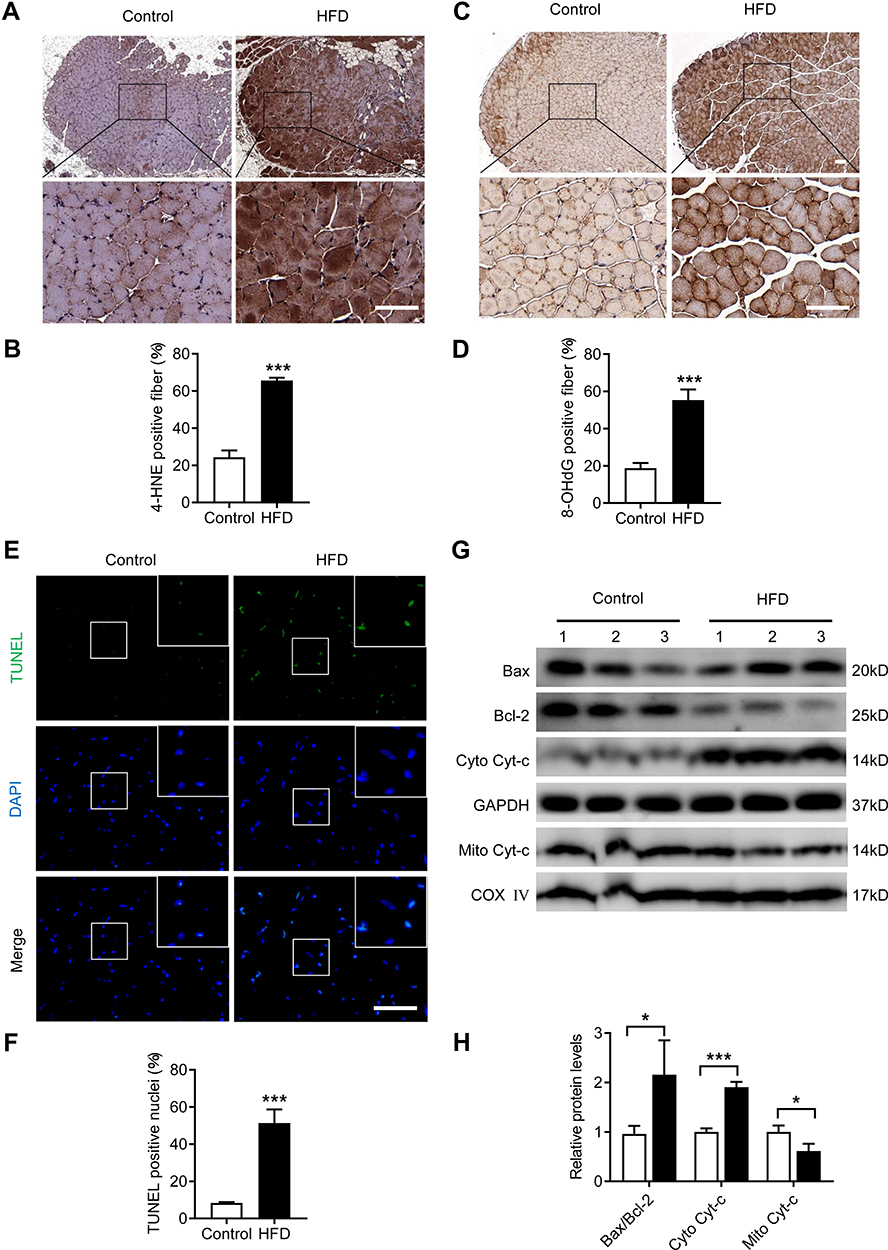 |
Figure 4 HFD led to oxidative stress and apoptosis in the genioglossus. (A) Representative images of genioglossus sections from control and HFD mice after 4-HNE immunohistochemical staining. (B) Quantification of the percentage of 4-HNE-positive fibres per visual field. (C) Representative images of genioglossus sections from control and HFD mice after 8-OHdG immunohistochemical staining. (D) Quantification of the percentage of 8-OHdG -positive fibres per visual field. (E) Representative images of TUNEL staining in the genioglossus. (F) Percentage of TUNEL-positive nuclei per visual field. (G and H) Protein expression of Bax, Bcl-2, Cyto Cyt-c and Mito Cyt-c measured by Western blot analysis in the genioglossus of control and HFD mice. Data are represented as the mean ± SD. *p < 0.05, ***p < 0.001 compared to the control. Scale bars are 50 μm. |
To investigate the effects of a HFD on apoptosis in the genioglossus, TUNEL staining was performed, which labelled the DNA fragmentation of apoptotic cells. The results showed that the number of TUNEL-positive cells in the HFD group significantly increased compared with those in the control group (p < 0.001) (Figure 4E and F). Moreover, we also analysed the expression of the mitochondria-related apoptotic proteins Cyt-c, Bax (a proapoptotic protein) and Bcl-2 (an antiapoptotic protein) in the genioglossus by Western blotting. Compared with control mice, the protein level of cytoplasm Cyt-c (cyto Cyt-c) was increased and mitochondria Cyt-c (mito Cyt-c) was decreased in genioglossus of the HFD group, indicating that Cyt-c was released from the mitochondria into cytoplasm. A HFD also induced an elevated level of protein Bax/Bcl-2 (Figure 4G and H).
Effects of PA on the Viability and Apoptosis in C2C12 Myoblasts
Myoblast proliferation is essential for skeletal muscle renewal and regeneration. To investigate the effects of PA on myoblast proliferation, C2C12 myoblasts were treated with different doses of PA for 6, 12, 24 and 48 hours, and CCK-8 assays were performed. As shown in Figure 5A, PA at 100 μM did not affect the viability of C2C12 myoblasts. However, at 300 μM, the cell morphologies changed and the percent of surviving cells was reduced to 62.31% and 54.48% that of untreated cells at 24 h and 48 h, respectively (p < 0.001). More importantly, PA at 500 μM exhibited a dramatic cytotoxic effect on C2C12 myoblasts, with a large portion of round and floating cells in the medium (Figure 5C), and the percent of surviving cells was only 23.57% and 11.93% that of untreated cells at 24 h and 48 h, respectively (p < 0.001). The cytotoxic effects of PA on C2C12 myoblasts were also measured by an LDH assay. After treatment with PA at 300 μM and 500 μM for 24 h, the LDH leakage in the medium was elevated, exhibiting concentration-dependent cytotoxicity (p < 0.001) (Figure 5B).
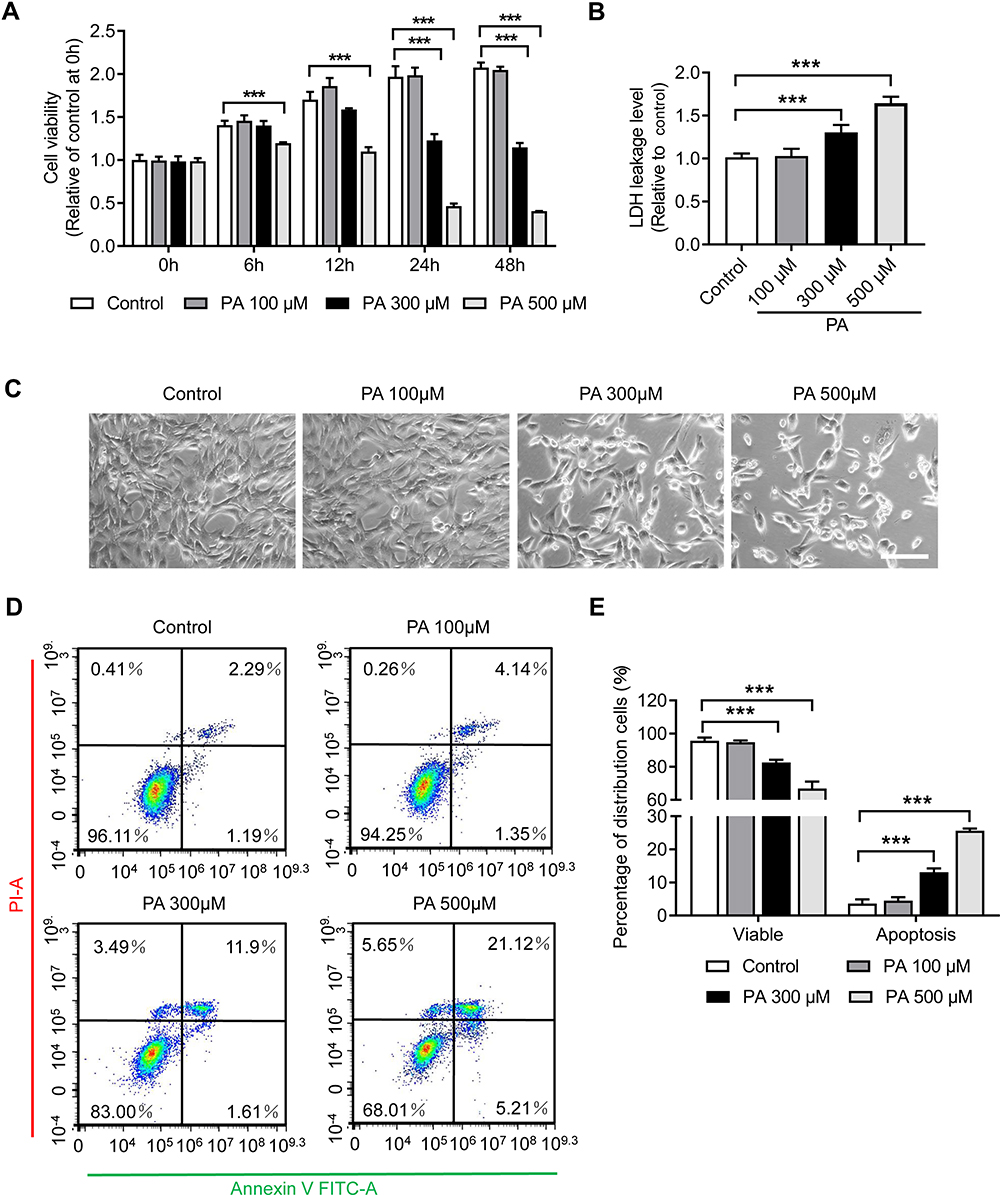 |
Figure 5 PA inhibited the proliferation and viability of C2C12 myoblasts. (A) The viability of C2C12 myoblasts was measured via CCK-8 assay after PA treatment. (B) LDH release in the medium of C2C12 myoblasts after PA treatment for 24 hours. (C) Optical micrographs of C2C12 myoblast morphology and quantity changes after PA treatment for 24 hours. (D) Apoptosis of C2C12 myoblasts was analysed using Annexin V-FITC/PI staining by flow cytometry after PA treatment for 24 hours. (E) Quantification of the percentage of viable (Annexin V-FITC negative and PI negative) cells and apoptotic (Annexin V-FITC positive) cells. Data are represented as the mean ± SD. ***p < 0.001 compared to the control. Scale bars are 100 μm. |
To further investigate the effects of a HFD on myoblast damage, apoptosis in C2C12 myoblasts was detected using Annexin V-FITC/PI staining and flow cytometry. Consistent with the findings of the CCK-8 and LDH assays, the results indicated that the number of apoptotic cells was significantly increased after treatment with PA at 300 μM and 500 μM for 24 h (p < 0.001), and no changes were found with PA treatment at 100 μM (Figure 5D and E).
Effects of PA on the Myogenic Differentiation of C2C12 Myoblasts
Myogenic differentiation is another important characteristic of myoblasts and plays a key role in the regeneration of skeletal muscles. To investigate whether PA affects myogenic differentiation, C2C12 myoblasts were incubated with different concentrations of PA during cellular differentiation for 2 days. The results of myotube immunofluorescence staining demonstrated that treatment with PA at 300 μM and 500 μM significantly prevented C2C12 differentiation, with the number of myotubes decreasing and the fusion of the myotubes becoming disrupted (p < 0.01) (Figure 6A and B). Moreover, the relative protein levels of the key myogenic differentiation markers MyoD1, MyoG and MyHC were all downregulated significantly following treatment with 300 μM PA (Figure 6C and D).
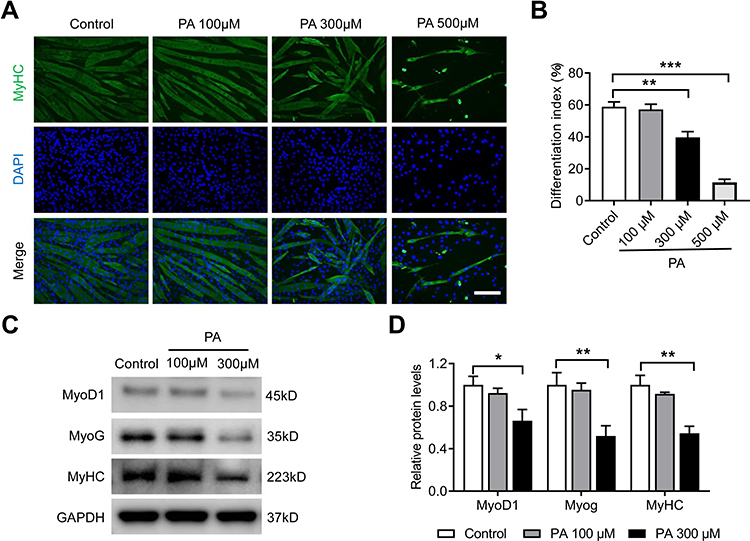 |
Figure 6 PA inhibited myogenic differentiation of C2C12 myoblasts. C2C12 myoblasts were treated with different concentrations of PA for 2 days during myogenic differentiation. (A) The differentiation of C2C12 myoblasts was assessed using immunofluorescence staining with MyHC (green) and DAPI (blue). (B) The differentiation index is presented as the percentage of MyHC-positive nuclei to the total number of nuclei. (C and D) Protein expression of MyoD1, MyoG and MyHC measured by Western blot analysis. Data are represented as the mean ± SD. *p < 0.05, **p < 0.01, ***p < 0.001 compared to the control. Scale bars are 50 μm. |
Effects of PA on Mitochondrial Function in C2C12 Myotubes
We evaluated the effects of PA on mitochondrial morphology in C2C12 myotubes using MitoTracker green. As shown in Figure 7A and C, the green fluorescence intensity of MitoTracker was markedly decreased after treatment with PA at 300 μM, indicating decreased mitochondrial mass. Then, the expression of DRP1 and MFN2 was measured by Western blot. Consistent with the in vivo findings, the expression level of DRP1 was significantly elevated after 300 μM PA treatment (p < 0.01) (Figure 7E and F). Moreover, the MMP was measured with JC-1 kits, which reflect the function of mitochondrial energy metabolism. For the untreated myotubes, the red fluorescence of JC-1 aggregates represented potential-dependence in the mitochondria to produce adenosine triphosphate (ATP). Conversely, after treatment with 300 μM PA, the green fluorescence of the JC-1 monomer was detected, and the ratio of red/green fluorescence intensity significantly decreased, which indicated mitochondrial membrane depolarization (Figure 7B and D). In addition, we observed a decrease in the expression of mitochondrial respiratory chain complexes III and V at 300 μM PA treatment (p < 0.01) (Figure 7G and H). Together, these findings suggest that PA can lead to mitochondrial damage and impair mitochondrial respiratory function.
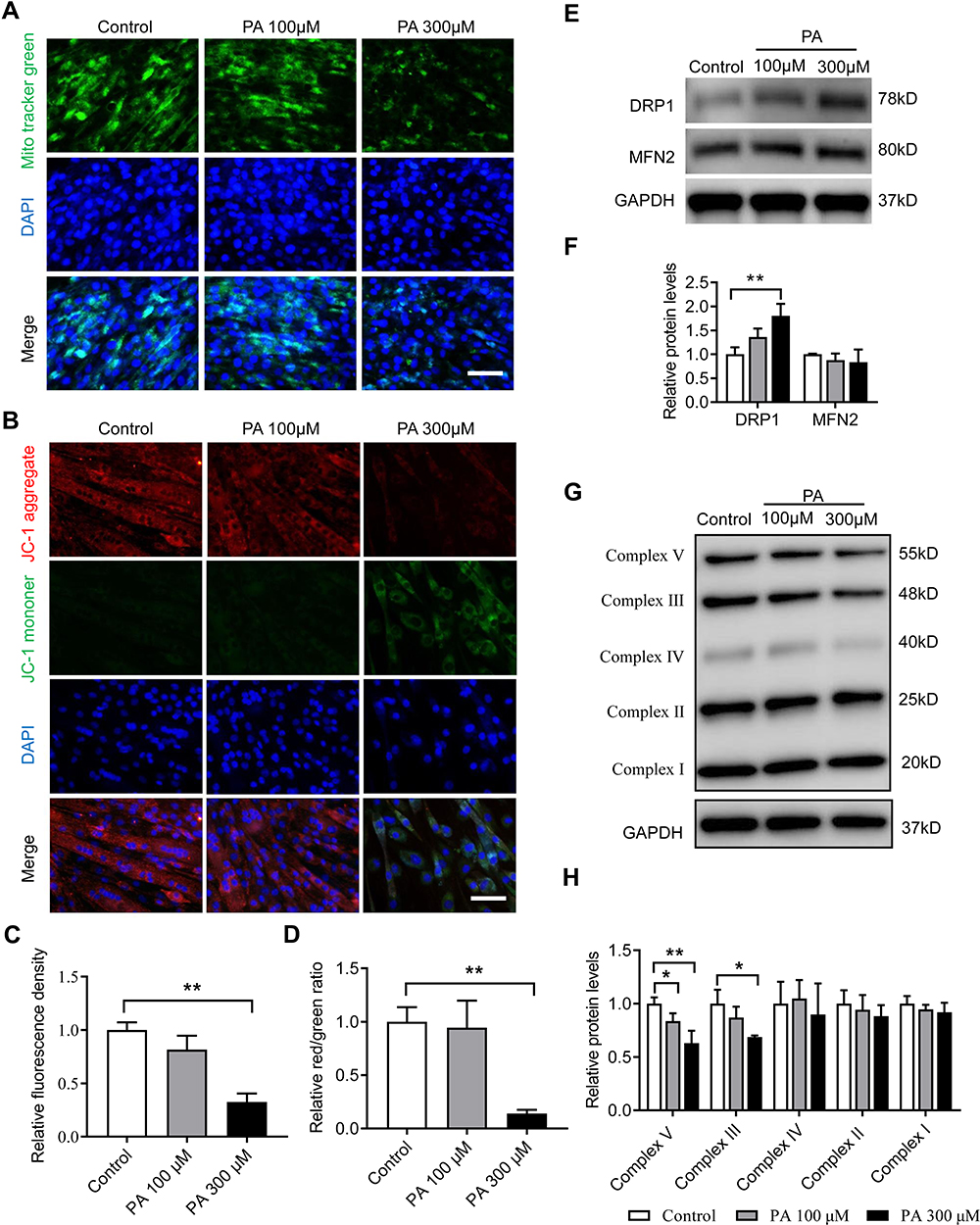 |
Figure 7 PA induced mitochondrial dysfunction in C2C12 myotubes. C2C12 myotubes were treated with different concentrations of PA for 2 days. (A) Representative images of C2C12 myotubes with MitoTracker green staining. (B) The mitochondrial membrane potential of C2C12 myotubes was assessed using fluorescence staining with a JC-1 assay. (C) Relative fluorescence intensity of MitoTracker green. (D) Relative red/green ratio of (B). (E and F) Protein expression of DRP1 and MFN2 in C2C12 myotubes measured by Western blot analysis. (G and H) Protein expression of mitochondrial respiratory chain complexes I–V measured by Western blot analysis. Data are represented as the mean ± SD. *p < 0.05, **p < 0.01 compared to the control. Scale bars are 50 μm. |
Effects of PA on Mitochondrial ROS and Apoptosis in C2C12 Myotubes
Mitochondrial dysfunction can lead to ROS overproduction, causing damage to cells and tissues.26 To evaluate the effects of PA on mitochondrial ROS levels, MitoSOX Red (a mitochondrial superoxide indicator) was used for immunofluorescence staining. PA at 300 μM significantly increased mitochondrial ROS accumulation in C2C12 myotubes (p < 0.001) (Figure 8A and B). ROS are known to activate apoptotic proteins and promote cell apoptosis. After treatment with 300 μM PA, the number of TUNEL-positive cells was significantly increased, and the expression of the mitochondria-related apoptotic proteins Cyt-c, Bax/Bcl-2 and cleaved caspase-3 was notably elevated in C2C12 myotubes (Figure 8C–F). SS–31 is reported to be an antioxidant targeting mitochondria.32,33 SS–31 treatment significantly reduced the mitochondrial ROS accumulation in C2C12 myotubes induced by PA (Figure 8A and B). In addition, compared with the 300 μM PA-treated group, SS–31 treatment reduced the number of TUNEL-positive cells (Figure 8C and D) and downregulated the protein level of cleaved caspase-3 (Figure 8E and F), indicating SS–31 could partly alleviate the apoptotic in C2C12 myotubes caused by PA.
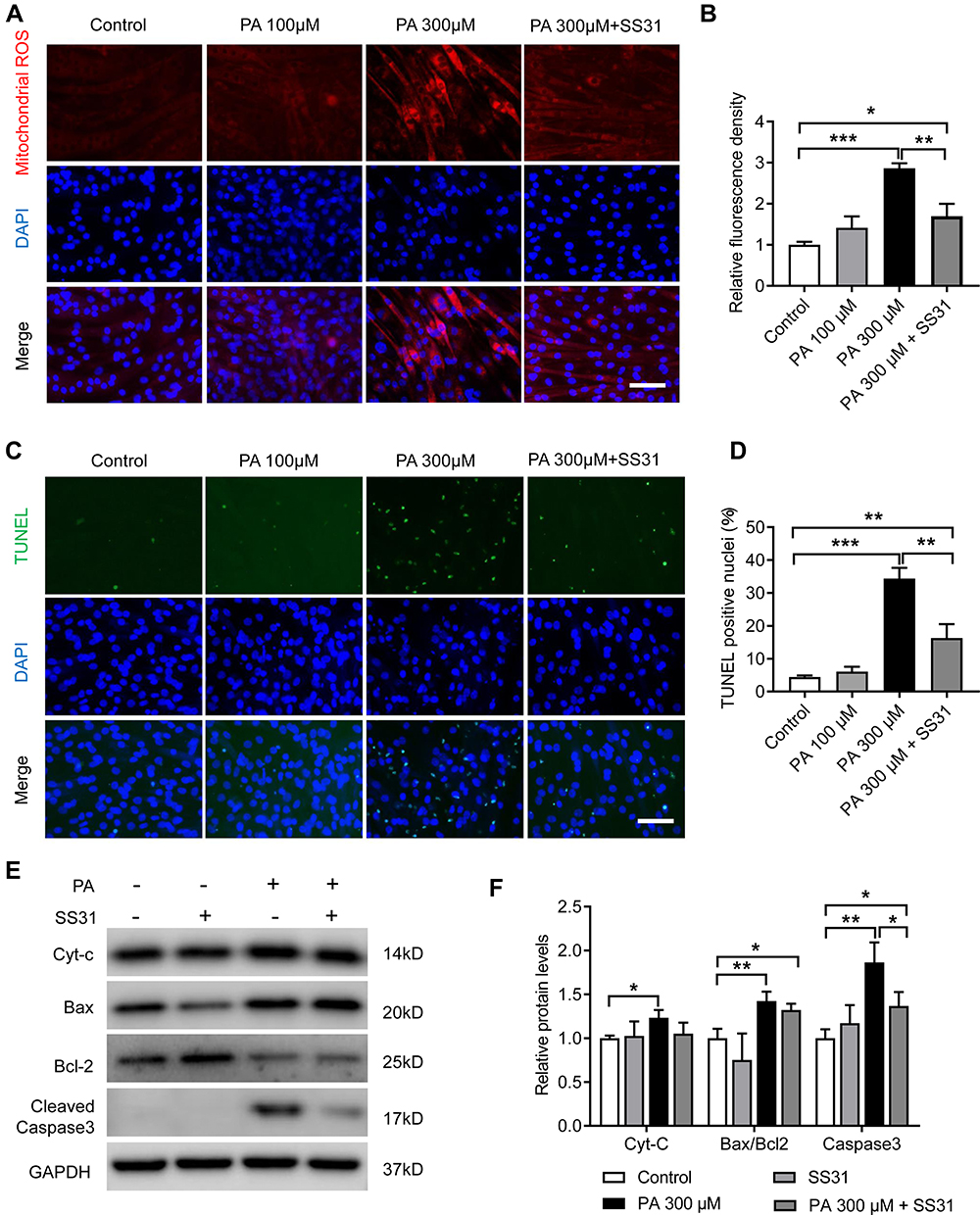 |
Figure 8 PA increased mitochondrial ROS and apoptosis in C2C12 myotubes. C2C12 myotubes were treated with PA or SS-31 for 2 days. (A and B) Fluorescence images and analyses of mitochondrial ROS with MitoSOX red in C2C12 myotubes. (C) Representative images of TUNEL staining in C2C12 myotubes. (D) Quantification of the percentage of TUNEL-positive nuclei per visual field. (E and F) Protein expression of Cyt-c, Bax, Bcl-2 and cleaved caspase-3 measured by Western blot analysis in C2C12 myotubes. Data are represented as the mean ± SD. *p < 0.05, **p < 0.01, ***p < 0.001. Scale bars are 50 μm. |
Discussion
The dysfunction of upper airway dilator muscles, especially the genioglossus, is thought to be the main pathogenesis of OSA. The HFD-induced obese mice in our study showed that genioglossus injury was accompanied by mitochondrial dysfunction, oxidative stress and apoptosis, which might be an important mechanism of OSA co-occurring with obesity.
In the present study, mice exhibited a significant increase in body weight and plasma levels of FFAs after 16 weeks of HFD administration. Skeletal muscles play an important role in lipid metabolism, while the excessive accumulation of lipids can impair oxidative capacities, promote lipotoxicity and lead to changes in the structure and function of skeletal muscles.34 As predicted, we discovered changes in genioglossus muscle fibres of HFD mice, with a decrease in the number of muscle fibres and an increase in the collagen content.
In addition, the muscle fibre type, with a dynamic and plastic structure, is of importance to muscle development, metabolism and functional adaptations. Slow-twitch fibres have more mitochondria and exhibit high oxidative capacity and fatigue resistance. In contrast, fast-twitch fibres have lower mitochondria levels and oxidative capacity. Studies have demonstrated that obese individuals have a decreased proportion of slow-twitch fibres and an increased proportion of fast-twitch fibres in skeletal muscles.35,36 The genioglossus in patients with OSA have also been reported to have a higher proportion of fast-twitch fibres and be more vulnerable to fatigue.37 In this study, we confirmed that a HFD caused significant changes in fibre types in the genioglossus, with the percentage of slow-twitch fibres declining and the percentage of fast-twitch fibres increasing. Changes in muscle fibre structure could lead to impairment of muscle function. Consistently, we found that the EMG activity of the genioglossus was significantly diminished in HFD-induced obese mice.
Mitochondria are the main energy source in skeletal muscle and play a critical role in maintaining contractility.38 The muscle fibre structure and fibre type have been reported to be significantly associated with mitochondrial dysfunction.39 Overaccumulation of FFAs could induce excessive ceramide levels,40 which were reported to ultimately cause mitochondrial dysfunction and lipotoxicity.41,42 In this study, we found that HFD-induced obesity led to mitochondrial damage; as the number of mitochondria decreased, they became swollen, and the cristae ruptured.
Mitochondria in skeletal muscle are highly dynamic, and mitochondrial biogenesis, fusion, fission and mitophagy are all involved in the regulation of mitochondrial morphology and function. Recent studies have shown that disorder in the balance between mitochondrial fission and fusion is implicated in mitochondrial damage and dysfunction in skeletal muscle in obesity.43,44 We detected a decrease in the mitochondrial fusion protein MFN2 and an increase in the fission protein DRP1 in the genioglossi of HFD-induced obese mice, indicating that obesity impaired mitochondrial fusion and induced excessive mitochondrial fission in the genioglossus. Our in vitro data support this conclusion. PA-treated C2C12 myotubes exhibited a decrease in mitochondrial mass and an increase in protein DRP1 expression, coupled with decreases in the MMP and mitochondrial oxidative phosphorylation (OXPHOS) complexes III and V, which are consistent with the observation in vivo. Mitochondrial dysfunction has been suggested to be one of the key processes during obesity that leads to skeletal muscle injury.45 Considering the importance of the mitochondria on oxidative metabolism and energy generation, we speculated that mitochondrial damage and dysfunction could ultimately lead to decreased oxidative capacities and fatigue resistance in the genioglossus.
Mitochondrial dysfunction can trigger apoptosis by regulating apoptosis-related genes, such as those in the Bcl-2 family.46 In the genioglossus, more TUNEL-positive apoptotic cells were observed in HFD-induced obese mice. Moreover, the expression levels of pro-apoptosis proteins (Cyt-c and cleaved caspase-3) were upregulated, while the expression of an apoptosis-inhibiting protein (Bcl-2) was downregulated in the genioglossus of obese mice. In support of these findings, our in vitro study demonstrated that high levels of PA significantly inhibited cellular proliferation and myogenic differentiation in C2C12 myoblasts. Moreover, PA exposure stimulated cell apoptosis in both C2C12 myoblasts and myotubes.
Mitochondrial impairment contributes to excessive ROS generation and oxidative stress, which is an important mechanism of tissue injury and apoptosis.47 Oxidative stress is a critical factor for skeletal muscle injury in obesity.48 ROS overproduction was also found in the genioglossus of rats with chronic intermittent hypoxia.49 In this study, we indeed detected the oxidative damage in the genioglossus of HFD-induced obese mice, manifested by the dramatic upregulation of 4-HNE-positive staining muscle fibres and 8-OHdG-positive fibres. Consistent with the in vivo findings, we also found increased production of mitochondrial ROS in C2C12 myotubes after PA treatment. Furthermore, SS-31, an antioxidant targeting mitochondria, reduced the accumulation of mitochondrial ROS and ultimately alleviated the apoptotic in C2C12 myotubes caused by PA.
In conclusion, our data indicate that a HFD could decrease EMG activity and muscle fibre mass, cause marked changes in muscle fibre type, lead to ultrastructural damage and increase apoptosis in the genioglossus. Most impressively, this study identifies that genioglossus injury might be mainly mediated by mitochondrial disturbance and the accompanying oxidative stress in HFD-induced obese mice, which may provide potential therapeutic targets for OSA co-occurrence with obesity.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This project was supported by NSFC grants 81771109 and 82071153 to Yuehua Liu, 81901031 to Xinxin Han and 81701011 to Bingjiao Zhao; the National Science Foundation of Shanghai (grant no. 19ZR1445400 to Xinxin Han); the Youth Program of Shanghai Municipal Health and Family Planning Commission grant (grant no. 20184Y0228 to Qingqing Chen).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Reilly JJ, El-Hamdouchi A, Diouf A, et al. Determining the worldwide prevalence of obesity. Lancet. 2018;391(10132):1773–1774. doi:10.1016/S0140-6736(18)30794-3
2. Salah RO, Ghandour R, Husseini A. Prevalence of overweight, obesity, and associated factors among adolescents in the occupied Palestinian territory: a cross-sectional study. Lancet. 2021;398(Suppl 1):S46. doi:10.1016/S0140-6736(21)01532-4
3. Rodgers GP, Gibbons GH. Obesity and hypertension in the Time of COVID-19. JAMA. 2020;324(12):1163–1165. doi:10.1001/jama.2020.16753
4. Zheng Y, Phillips CL, Sivam S, et al. Cardiovascular disease in obesity hypoventilation syndrome - A review of potential mechanisms and effects of therapy. Sleep Med Rev. 2021;60:101530. doi:10.1016/j.smrv.2021.101530
5. Friedman AN, Kaplan LM, le Roux CW, et al. Management of obesity in adults with CKD. J Am Soc Nephrol. 2021;32(4):777–790. doi:10.1681/ASN.2020101472
6. Gottlieb DJ, Punjabi NM. Diagnosis and management of obstructive sleep apnea: a review. JAMA. 2020;323(14):1389–1400. doi:10.1001/jama.2020.3514
7. Li Y, Wang Y. Obstructive sleep apnea-hypopnea syndrome as a novel potential risk for aging. Aging Dis. 2021;12(2):586–596. doi:10.14336/AD.2020.0723
8. Peppard PE, Young T, Barnet JH, et al. Increased prevalence of sleep-disordered breathing in adults. Am J Epidemiol. 2013;177(9):1006–1014. doi:10.1093/aje/kws342
9. Ischander MM, Lloyd RJ. Severe paediatric obesity and sleep: a mutual interactive relationship! J Sleep Res. 2021;30(3):e13162. doi:10.1111/jsr.13162
10. Erridge S, Moussa O, McIntyre C, et al. Obstructive sleep apnea in obese patients: a uk population analysis. Obes Surg. 2021;31(5):1986–1993. doi:10.1007/s11695-020-05196-7
11. Horner RL. Pathophysiology of obstructive sleep apnea. J Cardiopulm Rehabil Prev. 2008;28(5):289–298. doi:10.1097/01.HCR.0000336138.71569.a2
12. Perger E, Taranto-Montemurro L. Upper airway muscles: influence on obstructive sleep apnoea pathophysiology and pharmacological and technical treatment options. Curr Opin Pulm Med. 2021;27(6):505–513. doi:10.1097/MCP.0000000000000818
13. Sung CM, Tan SN, Shin MH, et al. The site of airway collapse in sleep apnea, its associations with disease severity and obesity, and implications for mechanical interventions. Am J Respir Crit Care Med. 2021;204(1):103–106. doi:10.1164/rccm.202011-4266LE
14. Lee RW, Vasudavan S, Hui DS, et al. Differences in craniofacial structures and obesity in Caucasian and Chinese patients with obstructive sleep apnea. Sleep. 2010;33(8):1075–1080. doi:10.1093/sleep/33.8.1075
15. Katz SL, Blinder H, Naik T, et al. Does neck circumference predict obstructive sleep apnea in children with obesity? Sleep Med. 2021;78:88–93. doi:10.1016/j.sleep.2020.12.018
16. Brennick MJ, Pack AI, Ko K, et al. Altered upper airway and soft tissue structures in the New Zealand Obese mouse. Am J Respir Crit Care Med. 2009;179(2):158–169. doi:10.1164/rccm.200809-1435OC
17. Luu BL, Saboisky JP, McBain RA, et al. Genioglossus motor unit activity in supine and upright postures in obstructive sleep apnea. Sleep. 2020;43(6):zsz316. doi:10.1093/sleep/zsz316.
18. McSharry D, O’Connor C, McNicholas T, et al. Genioglossus fatigue in obstructive sleep apnea. Respir Physiol Neurobiol. 2012;183(2):59–66. doi:10.1016/j.resp.2012.05.024
19. Geiger AE, Daughtry MR, Yen CN, et al. Dual effects of obesity on satellite cells and muscle regeneration. Physiol Rep. 2020;8(15):e14511. doi:10.14814/phy2.14511
20. Pattanakuhar S, Pongchaidecha A, Chattipakorn N, et al. The effect of exercise on skeletal muscle fibre type distribution in obesity: from cellular levels to clinical application. Obes Res Clin Pract. 2017;11(5 Suppl 1):112–132. doi:10.1016/j.orcp.2016.09.012
21. Rahman FA, Quadrilatero J. Mitochondrial network remodeling: an important feature of myogenesis and skeletal muscle regeneration. Cell Mol Life Sci. 2021;78(10):4653–4675. doi:10.1007/s00018-021-03807-9
22. Fu X, Zhu M, Zhang S, et al. Obesity impairs skeletal muscle regeneration through inhibition of AMPK. Diabetes. 2016;65(1):188–200. doi:10.2337/db15-0647
23. Pileggi CA, Parmar G, Harper ME. The lifecycle of skeletal muscle mitochondria in obesity. Obes Rev. 2021;22(5):e13164. doi:10.1111/obr.13164
24. Song K, Zhang Y, Ga Q, et al. Increased insulin sensitivity by high-altitude hypoxia in mice with high-fat diet-induced obesity is associated with activated AMPK signaling and subsequently enhanced mitochondrial biogenesis in skeletal muscles. Obes Facts. 2020;13(5):455–472. doi:10.1159/000508112
25. Meng Y, Li W, Zou Y, et al. How Does chronic intermittent hypoxia influence upper airway stability in rats? Nat Sci Sleep. 2020;12:749–758. doi:10.2147/NSS.S249948
26. Pang B, Chan WS, Chan CB. Mitochondria homeostasis and oxidant/antioxidant balance in skeletal muscle-do myokines play a role? Antioxidants. 2021;10(2):179.
27. Bonnard C, Durand A, Peyrol S, et al. Mitochondrial dysfunction results from oxidative stress in the skeletal muscle of diet-induced insulin-resistant mice. J Clin Invest. 2008;118(2):789–800. doi:10.1172/JCI32601
28. Liu S, Yang D, Yu L, et al. Effects of lycopene on skeletal muscle-fiber type and high-fat diet-induced oxidative stress. J Nutr Biochem. 2021;87:108523. doi:10.1016/j.jnutbio.2020.108523
29. Yu LM, Zhang WH, Han XX, et al. Hypoxia-induced ROS contribute to myoblast pyroptosis during obstructive sleep apnea via the NF-kappaB/HIF-1alpha signaling pathway. Oxid Med Cell Longev. 2019;2019:4596368. doi:10.1155/2019/4596368
30. Fatima S, Hu X, Huang C, et al. High-fat diet feeding and palmitic acid increase CRC growth in beta2AR-dependent manner. Cell Death Dis. 2019;10(10):711. doi:10.1038/s41419-019-1958-6
31. Zhu LY, Yu LM, Zhang WH, et al. Aging induced p53/p21 in genioglossus muscle stem cells and enhanced upper airway injury. Stem Cells Int. 2020;2020:8412598. doi:10.1155/2020/8412598
32. Zhao K, Zhao GM, Wu D, et al. Cell-permeable peptide antioxidants targeted to inner mitochondrial membrane inhibit mitochondrial swelling, oxidative cell death, and reperfusion injury. J Biol Chem. 2004;279(33):34682–34690. doi:10.1074/jbc.M402999200
33. Yang DQ, Zuo QN, Wang T, et al. Mitochondrial-targeting antioxidant SS-31 suppresses airway inflammation and oxidative stress induced by cigarette smoke. Oxid Med Cell Longev. 2021;2021:6644238. doi:10.1155/2021/6644238
34. Mengeste AM, Rustan AC, Lund J. Skeletal muscle energy metabolism in obesity. Obesity. 2021;29(10):1582–1595. doi:10.1002/oby.23227
35. Umek N, Horvat S, Cvetko E. Skeletal muscle and fiber type-specific intramyocellular lipid accumulation in obese mice. Bosn J Basic Med Sci. 2021:21(6):730. doi:10.17305/bjbms.2021.5876
36. Duan Y, Li F, Tan B, et al. Metabolic control of myofibers: promising therapeutic target for obesity and type 2 diabetes. Obes Rev. 2017;18(6):647–659. doi:10.1111/obr.12530
37. Skelly JR, O’Connell RA, Jones JF, et al. Structural and functional properties of an upper airway dilator muscle in aged obese male rats. Respiration. 2011;82(6):539–549. doi:10.1159/000332348
38. Conley KE, Lindstedt SL, Hoppeler HH. Mitochondria to motion: optimizing oxidative phosphorylation to improve exercise performance. J Exp Biol. 2016;219(Pt 2):243–249. doi:10.1242/jeb.126623
39. Schiaffino S, Reggiani C. Fiber types in mammalian skeletal muscles. Physiol Rev. 2011;91(4):1447–1531. doi:10.1152/physrev.00031.2010
40. Zhang Y, Rao E, Zeng J, et al. Adipose fatty acid binding protein promotes saturated fatty acid-induced macrophage cell death through enhancing ceramide production. J Immunol. 2017;198(2):798–807. doi:10.4049/jimmunol.1601403
41. Reginato A, Veras A, Baqueiro M, et al. The role of fatty acids in ceramide pathways and their influence on hypothalamic regulation of energy balance: a systematic review. Int J Mol Sci. 2021;22(10):5357. doi:10.3390/ijms22105357
42. Bekhite M, Gonzalez-Delgado A, Hubner S, et al. The role of ceramide accumulation in human induced pluripotent stem cell-derived cardiomyocytes on mitochondrial oxidative stress and mitophagy. Free Radic Biol Med. 2021;167:66–80. doi:10.1016/j.freeradbiomed.2021.02.016
43. Houzelle A, Jorgensen JA, Schaart G, et al. Human skeletal muscle mitochondrial dynamics in relation to oxidative capacity and insulin sensitivity. Diabetologia. 2021;64(2):424–436. doi:10.1007/s00125-020-05335-w
44. Fealy CE, Grevendonk L, Hoeks J, et al. Skeletal muscle mitochondrial network dynamics in metabolic disorders and aging. Trends Mol Med. 2021;27(11):1033–1044. doi:10.1016/j.molmed.2021.07.013
45. Tran L, Langlais PR, Hoffman N, et al. Mitochondrial ATP synthase beta-subunit production rate and ATP synthase specific activity are reduced in skeletal muscle of humans with obesity. Exp Physiol. 2019;104(1):126–135. doi:10.1113/EP087278
46. Bock FJ, Tait S. Mitochondria as multifaceted regulators of cell death. Nat Rev Mol Cell Biol. 2020;21(2):85–100. doi:10.1038/s41580-019-0173-8
47. Sun Y, Ge X, Li X, et al. High-fat diet promotes renal injury by inducing oxidative stress and mitochondrial dysfunction. Cell Death Dis. 2020;11(10):914. doi:10.1038/s41419-020-03122-4
48. Pinho RA, Sepa-Kishi DM, Bikopoulos G, et al. High-fat diet induces skeletal muscle oxidative stress in a fiber type-dependent manner in rats. Free Radic Biol Med. 2017;110:381–389. doi:10.1016/j.freeradbiomed.2017.07.005
49. Wang W, Ding W, Huang H, et al. The role of mitophagy in the mechanism of genioglossal dysfunction caused by chronic intermittent hypoxia and the protective effect of adiponectin. Sleep Breath. 2021;25(2):931–940. doi:10.1007/s11325-020-02211-0
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.