")
Back to Journals » OncoTargets and Therapy » Volume 12
HGF/MET Regulated Epithelial-Mesenchymal Transitions And Metastasis By FOSL2 In Non-Small Cell Lung Cancer
Authors Yin J, Hu W, Fu W, Dai L, Jiang Z, Zhong S, Deng B, Zhao J
Received 29 May 2019
Accepted for publication 17 September 2019
Published 5 November 2019 Volume 2019:12 Pages 9227—9237
DOI https://doi.org/10.2147/OTT.S217595
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Federico Perche
Jun Yin,1,* Weimin Hu,2,* Wenfan Fu,1 Lu Dai,1 Zeyong Jiang,1 Shengpeng Zhong,1 Boyun Deng,1 Jian Zhao1
1Department of Chest Surgery, Affiliated Cancer Hospital and Institute of Guangzhou Medical University, Guangzhou, Guangdong 510095, People’s Republic of China; 2Department of Abdominal Surgery, Affiliated Cancer Hospital and Institute of Guangzhou Medical University, Guangzhou, Guangdong 510095, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Jian Zhao
Department of Chest Surgery, Affiliated Cancer Hospital and Institute of Guangzhou Medical University, Guangzhou, Guangdong 510095, People’s Republic of China
Email [email protected]
Background: HGF/MET has been found to be associated with non-small cell lung cancer (NSCLC). However, the underlying molecular mechanisms of HGF/MET involved in regulating the metastasis of NSCLC remain unclear.
Methods: The effect of HGF/MET and FOSL2 on cell migration and invasion were assessed by transwell and scratch assays. HGF/MET-induced phosphorylation and upregulation of FOSL2 was analyzed by RT-PCR and Western blotting. Regulatory effects of FOSL2 on SNAI2 transcription were detected by chromatin immunoprecipitation (ChIP) and dual-Luciferase reporter assays. The correlations of FOSL2 expression with clinical outcomes were assessed in 56 NSCLC patients.
Results: HGF/MET induced the phosphorylation and upregulation of FOSL2 by ERK1/2 kinase, FOSL2 promoted the transcription of SNAI2 by binding with the SNAI2 promoter, and SNAI2 subsequently promoted the epithelial-mesenchymal transition (EMT), invasion, and migration of NSCLC cells. According to the clinical correlation analysis in NSCLC, high expression of FOSL2 correlated with advanced tumor stage and metastasis.
Conclusion: Our studies propose that the regulatory mechanisms of the HGF/MET-induced cascade pathway is mediated by FOSL2 in NSCLC metastasis and suggested that FOSL2 could potentially be employed as a prognostic biomarker and potential therapeutic target of NSCLC metastasis.
Keywords: non-small cell lung cancer, metastasis, HGF/MET, FOSL2, SNAI2
Introduction
Lung cancer is the leading cause of cancer-related death, and the primary histological type of lung cancer is classified as non-small cell lung cancer (NSCLC) (75–85%), of which adenocarcinoma and squamous carcinoma are the two most common subtypes.1–3 For NSCLC, high metastatic potential is the most important biological feature and the main cause of death, and the 5-year survival rate of NSCLC is less than 20%.4 Accordingly, a better understanding of the pathophysiology of NSCLC metastasis has led to the development of targeted agents that promise to improve these outcomes.5 However, the molecular mechanism underlying NSCLC metastasis remains unclear.
NSCLC is a malignant neoplasm originating from epithelial cells, and the regulation of epithelial-mesenchymal transitions (EMT) has been implicated in the detachment of epithelial-derived cancer cells from the primary focus and the formation of distant metastasis, including NSCLC.6,7 EMT refers to the biological process in which epithelial cells lose their epithelial phenotypes through specific procedures and transform them into mesenchymal cells. The main characteristics of EMT include the decrease of cell adhesion molecules E-cadherin, the transformation of the cytokeratin cytoskeleton into the Vimentin-based cytoskeleton and the morphological characteristics of mesenchymal cells.8 Through the role of EMT, cells acquire invasion and migration abilities, which play important roles in embryonic development, wound healing and tumor metastasis.9,10 In recent years, a series of EMT-associated transcription factors (EMT-TFs) (including TWIST, ZEB1, ZEB2, SNAI1 and SNAI2) have been found to be directly involved in the regulation of EMT and tumor metastasis.11–13 Although it has been found that EMT is closely related to the occurrence of metastasis, the regulatory mechanisms of EMT in NSCLC remain unclear.
At present, abnormal expressions and mutations of hepatocyte growth factor (HGF) and its receptor tyrosine kinase (MET), was found in NSCLC. XL184 (Cabozantinib) is a small-molecule kinase inhibitor with potent activity toward MET, as well as a number of other receptor tyrosine kinases, including VEGFR, RET, KIT, AXL, and FLT. It has also been found that HGF/MET are closely related to the metastasis of NSCLC.14–16 In this study, we found that HGF/MET induced the phosphorylation and upregulation of FOSL2. We also found that FOSL2 regulated the EMT, invasion, and migration by transcriptional regulation of SNAI2. In addition, clinical analysis found that the expression levels of FOSL2 in NSCLC tissues were associated with metastases and short overall survival. These results not only shed light on the understanding of the mechanisms of metastasis in NSCLC, but also led to the discovery of a biomarker that could be used for the identification of NSCLC patients with poor prognosis.
Methods
Cell Culture And Transfections
Human adenocarcinoma cells A549 and squamous carcinoma cells SK-MES-1 were purchased commercially (ATCC cell lines) and cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum at 37°C in a humidified atmosphere with 5% CO2. The expression plasmid of FOSL2 was constructed by inserting cDNA into the pCDNA3.1 plasmid, and the shRNA of FOSL2 was designed for lentivirus production. Plasmids and siRNAs were transfected into cells by Lipofectamine 2000 (Invitrogen), and shRNA lentiviral vectors were transfected into cells by lentivirus. Cells were harvested 48 or 72 hrs after transfection for analysis.
Tissue Sample Collection
Primary NSCLC tissues were collected from the Affiliated Cancer Hospital and Institute of Guangzhou Medical University (Guangzhou, China) with informed consent and Institutional Review Board (IRB) permission. Tissue sample collection was conducted in accordance with the Declaration of Helsinki. 56 NSCLC patients were recruited into this study. All of the following criteria were met: patients who suffered from primary NSCLC; a histological diagnosis of NSCLC with at least one measurable lesion; a TNM clinical stage of I to IV. Fresh NSCLC tissues were obtained at surgery or by aspiration biopsy and immediately snap-frozen in liquid nitrogen and stored at −80°C until use. All clinical and biological data on these samples were available. All patients provided written informed consent, and the collection of NSCLC tissues for research purposes was approved by the relevant human research ethics committees of the Affiliated Cancer Hospital and Institute of Guangzhou Medical University (Approval no. (2014) 100).
Real-Time Quantitative PCR (RT-PCR)
Total RNA of cells were extracted by Trizol (Invitrogen). 1 µg total RNA was used for cDNA synthesis using a Reverse Transcription Kit (Takara), then cDNA was used for RT-PCR using SYBR Green Realtime PCR Master Mix (TOYOBO). Real-time quantitative polymerase chain reaction (RT-PCR) was performed by using ABI ViiATM7Dx Real-Time PCR System (Life Technologies). Expression levels of mRNA were normalized by GAPDH.
The mRNA RT-PCR primers were listed here:
E-cadherin, forward 5′-GGGGTCTGTCATGGAAGGTG-3′ and reverse 5′-CAAAATCCAAGCCCGTGGTG-3′;
Vimentin, forward 5′-AAGTCCGCACATTCGAGCAA-3′ and reverse 5′-GGTGGACGTAGTCACGTAGC-3′;
TWIST, forward 5′-CGGCCAGGTACATCGACTTC-3′ and reverse 5′-CAGAGGTGTGAGGATGGTGC-3′;
ZEB1, forward 5′-GATGCGAAACGCGAGGTTTT-3′ and reverse 5′-CTTTCACTGCTCCTCCCTGG-3′;
ZEB2, forward 5′-TCCCAGAGAGAAACTTGGCG-3′ and reverse 5′-CCTGGGATTGGCTTGTTTGC-3′;
SNAI1, forward 5′-TGCCAATGCTCATCTGGGAC-3′ and reverse 5′-GACATTCGGGAGAAGGTCCG-3′;
SNAI2, forward 5′-TCATCTTTGGGGCGAGTGAG-3′ and reverse 5′-TGTCCTTGAAGCAACCAGGG-3′;
JUN, forward 5′-TCCTGCCCAGTGTTGTTTGT-3′ and reverse 5′-GACTTCTCAGTGGGCTGTCC-3′;
JUNB, forward 5′-CCTACCGGAGTCTCAAAGCG-3′ and reverse 5′-TTGGTGTAAACGGGAGGTGG-3′;
JUND, forward 5′-GAAAGTCCTCAGCCACGTCA-3′ and reverse 5′-GAGCGAGATCGAGGAAAGGG-3′;
FOS, forward 5′-CAGACTACGAGGCGTCATCC-3′ and reverse 5′-AGTTGGTCTGTCTCCGCTTG-3′;
FOSB, forward 5′-AGGAAGAGGAGAAGCGAAGGG-3′ and reverse 5′-AGAGAGAAGCCGTCAGGTTG-3′;
FOSL1, forward 5′-GTGGTTCAGCCCGAGAACTT-3′ and reverse 5′-CGGGCTGATCTGTTCACAAG-3′;
FOSL2, forward 5′-AGTGACTCATCTCGGGCAGA-3′ and reverse 5′-GCGTCCCTTACAGCACTTCT-3′;
GAPDH, forward 5′-TGACTTCAACAGCGACACCCA-3′ and reverse 5′-CACCCTGTTGCTGTAGCCAAA-3′.
Western Blotting
The cells were harvested and lysed by RIPA buffer for 30 min at 4°C. 50 µg heat-denatured proteins were loaded into 15% SDS–PAGE for SDS-polyacrylamide gel electrophoresis, and then transferred to polyvinylidene difluoride membrane for Western blotting analysis. After blocking nonspecific binding sites with 5% (wt/vol) nonfat milk, 0.1% (vol/vol) Tween-20 diluted in Tris (pH 7.8)-buffered saline, rabbit polyclonal anti-FOSL2 (Cell Signaling, 1:1000 dilutions), anti-ERK1/2 (p44/42 MAPK, Cell Signaling, 1:1000 dilutions), anti-phospho-ERK1/2 (phospho-p44/42 MAPK, T202/T204, Cell Signaling, 1:1000 dilutions), anti-SNAI2 (Cell Signaling, 1:1000 dilutions) and anti-GAPDH (Cell Signaling, 1:1000 dilutions) primary antibody was used to incubate overnight at 4°C. Then, HRP (horseradish peroxidase) conjugate goat-anti-rabbit secondary antibody (Cell Signaling, 1:1000 dilutions) was used to incubate for 4 hrs. The bound antibodies were detected using ECL Plus Western Blotting Detection system (GE Healthcare). GAPDH was used as an internal control.
Transwell Assay
The invasion capability of cells was detected using transwell-chamber culture systems (Becton Dickinson). After 48 h incubation at 37°C, the cells were transferred into the upper chamber of the transwell with matrigel (1×105 cells per well in an 8 μm 24-well transwell). Following 24 h incubation at 37°C, cells on the upper surface of the upper chamber (non-invasion cells) were removed by cotton swabs, and cells on the lower surface of the filters were fixed and stained with Giemsa stain. The number of invaded cells was counted under a light microscope (Leica).
Cell Migration Assay (Wound Healing Assay)
The migratory ability of cells was assessed by the wound healing assay. Cells were grown to confluence in medium containing 10% FBS. A uniform scratch defect was created across the monolayer using a pipette tip. Wells were then washed with PBS, followed by the addition of serum-free medium. Plates were imaged at 0 h, 24 h and 48 h, and the degree the cells at the scratch margin had migrated close to the initial defect was assessed, and images of the microspheres were captured using a microscope (Leica).
Chromatin Immunoprecipitation (Chip)
For ChIP analysis, cells grown on a 6-well plate were processed as described in the ChIP Assay kit protocol (Millipore). Firstly, the chromatin DNA was extracted and broken into fragments of 200–400 bp in length by sonication. Then, the chromatin fragments were immunoprecipitated with the following antibodies: IgG (Cell Signaling) and anti-FOSL2 (Cell Signaling). Finally, the precipitated DNA fragments were measured by RT-PCR. To normalize PCR efficiency, the intensity of the PCR products from the chromatin immunoprecipitates were normalized against the intensity of the PCR products of the genomic DNA input amplified by the same primer pairs.
Primers specific for SNAI2 promoter region were listed here:
SNAI2 promoter, forward 5′-GCCTGCCTTTAGAGGGCTAC-3′ and reverse 5′-TGGCATCTGGAGAGGTTTGC-3′.
Dual-Luciferase Reporter Assays
Luciferase reporter plasmids of the SNAI2 upstream regulation region were constructed as pGL2-SNAI2 plasmids. The pGL2-control plasmids were used as negative controls. Cells were transfected with 500 ng/well of pGL2 plasmid and 100 ng/well of pRL-TK using Lipofectamine (Invitrogen). After 24 hrs, cells were transfected with the FOSL2 plasmids. After another 48 hrs, cells were harvested and analyzed for luciferase activity using the Dual Luciferase Reporter Assay (Promega) in a Clarity Luminescence Microplate Reader (BioTek). The relative luciferase activity (firefly luciferase) was normalized to pRL-TK activity (Renilla luciferase). Results were expressed as a fold induction over that of empty pGL2 activity.
Statistical Analysis
All values were expressed as mean ± standard deviation (SD) from at least three separate experiments. Student’s unpaired t-test, chi-square test, and Kaplan-Meier survival analysis were performed using SPSS 21.0 statistical software (IBM). A two-tailed P value test was used in all analyses, and difference was considered as statistically significant if the P value was less than 0.05 (P<0.05).
Results
HGF/MET Promoted EMT, Invasion And Migration Of NSCLC Cells By Activating SNAI2 Expression
As adenocarcinoma and squamous carcinoma are the two most common subtypes of NSCLC, we used the lung adenocarcinoma cell line, A549, and the lung squamous carcinoma cell line, SK-MES-1, to study the effect of HGF/MET on EMT. First, we found that exogenous HGF decreased the expression of epithelial marker E-cadherin and increase the expression of the mesenchymal marker, Vimentin, in A549 and SK-MES-1 cells. Moreover, the MET inhibitor (XL184) inhibited the regulatory effect of exogenous HGF on E-cadherin and Vimentin expressions, suggesting that HGF/MET was involved in the regulation of EMT in NSCLC cells (Figure 1A).
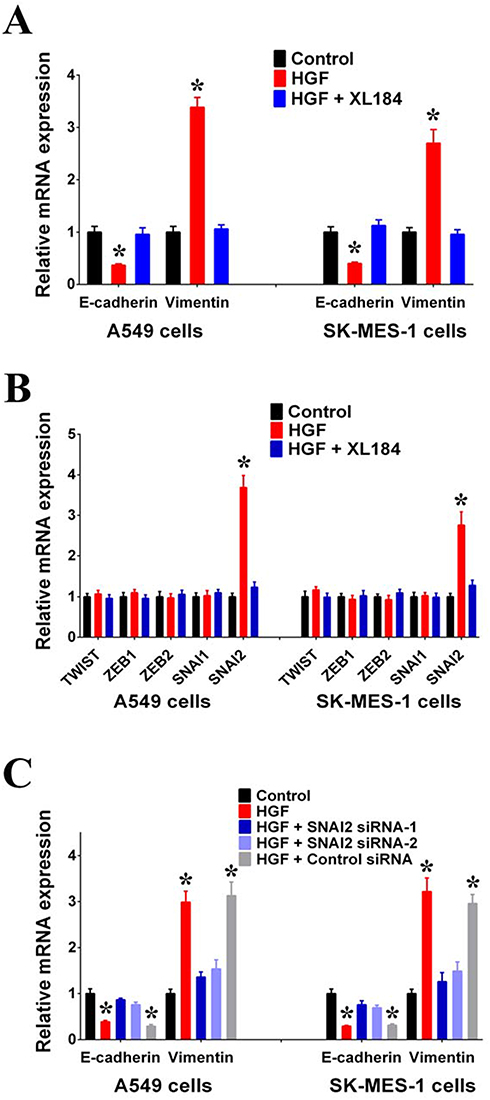 |
Figure 1 HGF/MET can induce EMT of NSCLC cells by activating SNAI2 expressions. (A) Expressions of E-cadherin and Vimentin in A549 and SK-MES-1 cells with or without exogenous HGF treatment (50 μg/L, 72 hrs) were measured by RT-PCR. MET inhibitor (XL184, 10 μmol/L) can inhibit the regulating effect of exogenous HGF. (B) Expressions of TWIST, ZEB1, ZEB2, SNAI1 and SNAI2 in A549 and SK-MES-1 cells with or without exogenous HGF treatment (50 μg/L, 72 hrs) were measured by RT-PCR. MET inhibitor (XL184, 10 μmol/L) can inhibit the regulating effect of exogenous HGF. (C) HGF/MET regulated the expression of E-cadherin and Vimentin by SNAI2 in A549 and SK-MES-1 cells. (n=3, *P < 0.05). |
It has been reported that the EMT is regulated by EMT-TFs, such as TWIST, ZEB1, ZEB2, SNAI1 and SNAI2.11 In order to detect which EMT-TF was involved in the regulation of HGF/MET-induced EMT, we detected the effect of exogenous HGF on the expressions of EMT-TFs. We found that the addition of HGF to A549 and SK-MES-1 cells increased the expression of SNAI2 compared with untreated A549 and SK-MES-1 cells. Additionally, the MET inhibitor, XL184, inhibited the effect of exogenous HGF on SNAI2 expression (Figure 1B). We also observed that knockdown of SNAI2 by siRNAs inhibited the effect of exogenous HGF on decreasing E-cadherin and increasing Vimentin in A549 and SK-MES-1 cells (Figure 1C).
Next, we used Transwell and wound healing assays to detect the effects of HGF/MET on the invasion and migration of NSCLC cells. The addition of exogenous HGF increased the invasion and migration of A549 and SK-MES-1 cells, while the addition of the MET inhibitor, XL184, blocked the effect (Figure 2A and B). Furthermore, we also found that knockdown of SNAI2 by siRNAs inhibited the effect of exogenous HGF on the invasion and migration of A549 and SK-MES-1 cells (Figure 2A and B). Therefore, these results indicated that HGF/MET regulated the EMT, invasion and migration of NSCLC cells via SNAI2 expression.
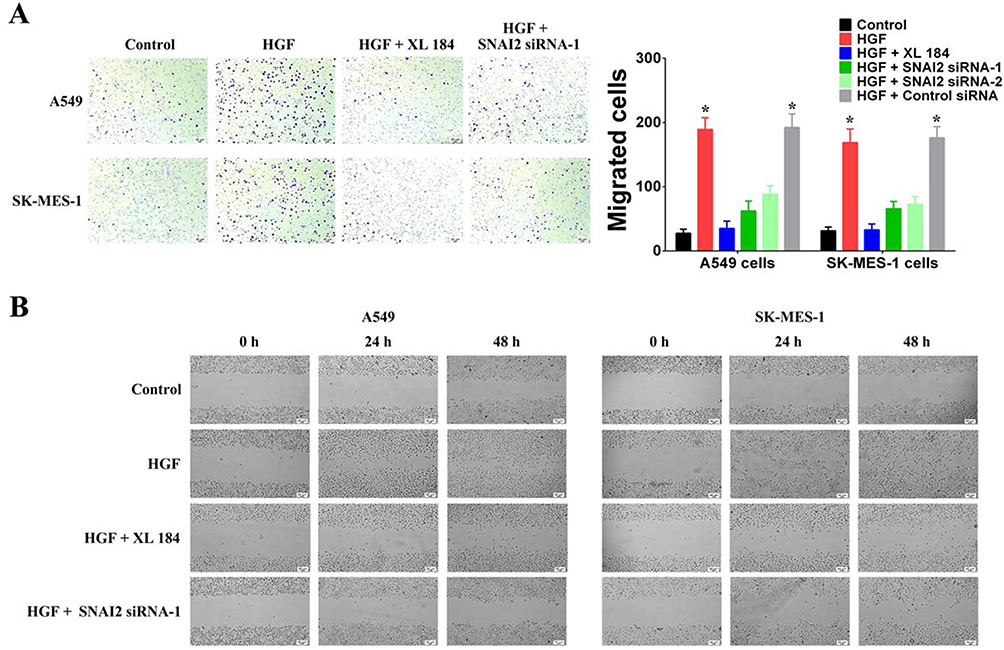 |
Figure 2 HGF/MET can induce invasion and migration of NSCLC cells by SNAI2. (A) Cell invasion of A549 and SK-MES-1 cells with or without HGF treatment (50 μg/L, 72 hrs) was detected by transwell-chamber culture systems. MET inhibitor (XL184, 10 μmol/L) can inhibit the regulating effect of exogenous HGF. HGF/MET regulated the invasion by SNAI2. Bar graphs show the number of invaded cells. (B) Cell migration of A549 and SK-MES-1 cells with or without HGF treatment (50 μg/L, 72 hrs) was detected by the scratch assay. MET inhibitor (XL184, 10 μmol/L) can inhibit the regulating effect of exogenous HGF. HGF/MET regulated the migration by SNAI2. (n=3, *P < 0.05). |
HGF/MET Regulated The Transcription Of SNAI2 By FOSL2
In order to investigate the mechanism of HGF/MET regulation on SNAI2 expression, we searched for putative transcription factor binding sites at the upstream regulation region of SNAI2 and identified AP1 binding sites. Various heterodimers of the JUN and FOS family proteins, including JUN, JUNB, JUND, FOS, FOSB, FOSL1, and FOSL2 bind AP1 sites, and we found that the addition of exogenous HGF increased the expressions of FOSL2. However, the MET inhibitor (XL184), blocked the regulatory effect of exogenous HGF on FOSL2 expression (Figure 3A). According to the results of chromatin immunoprecipitation assays, exogenous HGF enriched FOSL2 on the promoter of SNAI2 and the addition of the MET inhibitor inhibited the process; thus, suggesting that FOSL2 binds the SNAI2 promoter (Figure 3B). By using the dual-luciferase reporter assay, it was demonstrated that exogenous HGF enhanced the transcriptional activity of the SNAI2 promoter and the knockdown of FOSL2 by siRNAs inhibited that effect. Thus, FOSL2 can activate the transcriptional activity of the SNAI2 promoter (Figure 3C). Moreover, we also found that knockdown of FOSL2 by siRNAs inhibited the effect of exogenous HGF on the expressions of SNAI2 in A549 and SK-MES-1 cells (Figure 3D). Together, these data demonstrated that FOSL2 was involved in the regulation of HGF/MET-induced SNAI2 expression in NSCLC cells.
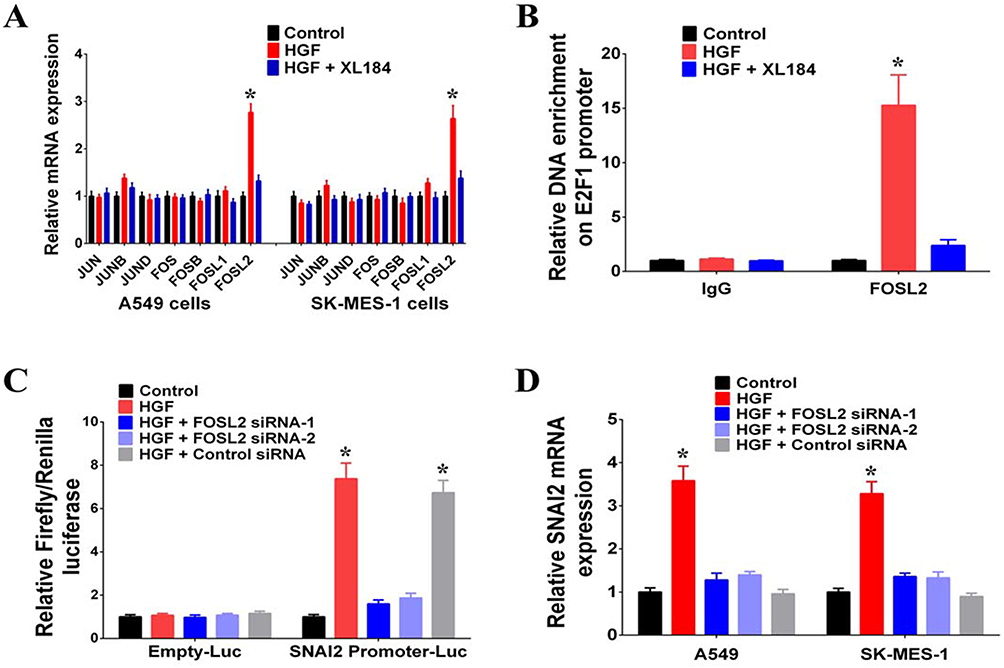 |
Figure 3 HGF/MET regulated SNAI2 expression by FOSL2. (A) Expressions of JUN, JUNB, JUND, FOS, FOSB, FOSL1 and FOSL2 in A549 and SK-MES-1 cells with or without exogenous HGF treatment (50 μg/L, 72 hrs) were measured by RT-PCR. MET inhibitor (XL184, 10 μmol/L) can inhibit the regulating effect of exogenous HGF. (B) FOSL2 signal in the SNAI2 promoter region was detected by chromatin immunoprecipitation (ChIP) assay. (C) The transcriptional activation of SNAI2 by FOSL2 was detected by luciferase assays. (D) HGF/MET regulated the expression of SNAI2 by FOSL2 in A549 and SK-MES-1 cells. (n=3, *P < 0.05). |
To determine whether FOSL2 contributed in a critical way to mediating HGF/MET-induced EMT, invasion and migration, we first regulated FOSL2 expression using plasmids or shRNAs that achieved FOSL2 overexpression or knockdown, respectively, in A549 and SK-MES-1 cells (Figure 4A). It was found that overexpression of FOSL2 decreased E-cadherin and increased Vimentin (Figure 4B). Moreover, it was observed that knockdown of FOSL2 inhibited the effect of exogenous HGF on decreasing E-cadherin and increasing Vimentin (Figure 4B). According to the results of transwell and wound healing assays, it was found that overexpression of FOSL2 increased the invasion and migration of A549 and SK-MES-1 cells (Figure 4C and D). In addition, knockdown of FOSL2 inhibited the effect of exogenous HGF on invasion and migration (Figure 4C and D). These results revealed that the roles of HGF/MET in inducing EMT, invasion and migration were achieved through the upregulation of FOSL2 which promoted the transcription of SNAI2.
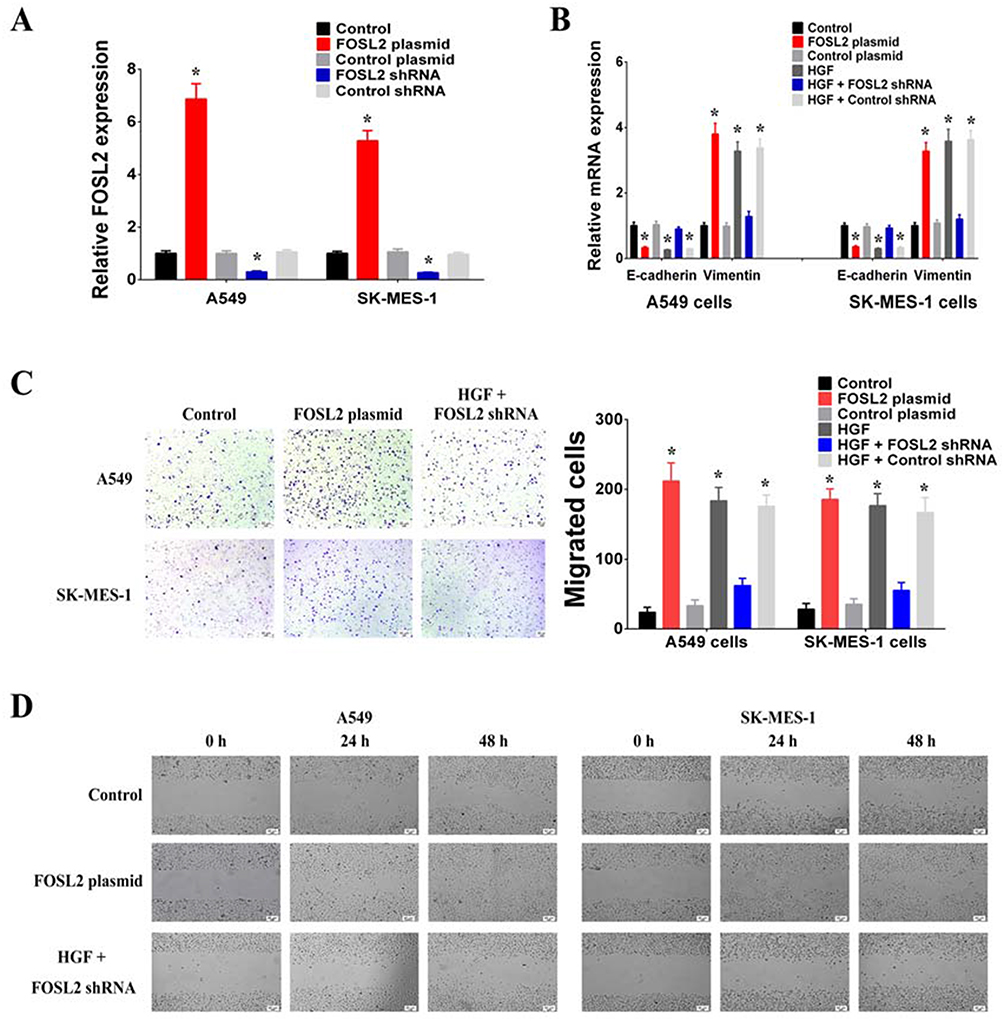 |
Figure 4 HGF/MET regulated EMT, invasion and migration of NSCLC cells by FOSL2. (A) The expressions of FOSL2 were increased after transfection with FOSL2 plasmid and decreased after transfection with FOSL2 shRNA in A549 and SK-MES-1 cells. (B) HGF/MET regulated the expression of E-cadherin and Vimentin by FOSL2 in A549 and SK-MES-1 cells. (C) HGF/MET regulated the invasion by FOSL2 in A549 and SK-MES-1 cells. Bar graphs show the number of invaded cells. (D) HGF/MET regulated the migration by FOSL2 in A549 and SK-MES-1 cells. (n=3, *P < 0.05). |
HGF/MET Induced Phosphorylation And Upregulation Of FOSL2 By Activating ERK1/2
To determine the mechanism underlying the upregulation of FOSL2 by HGF/MET, we further characterized the signaling pathways activated by HGF/MET. We used selective inhibitors, including a PI3K inhibitor (LY294002), an ERK1/2 inhibitor (U0126) and a p38 MAPK inhibitor (SB203580) to clarify the HGF/MET-triggered signaling pathways mediating the upregulation of FOSL2. The results demonstrated that treatment of A549 and SK-MES-1 cells with the ERK1/2 inhibitor (U0126) completely abolished exogenous HGF, induced the upregulation of FOSL2, while not affecting other signaling pathways, including p38 MAPK and PI3K (Figure 5A).
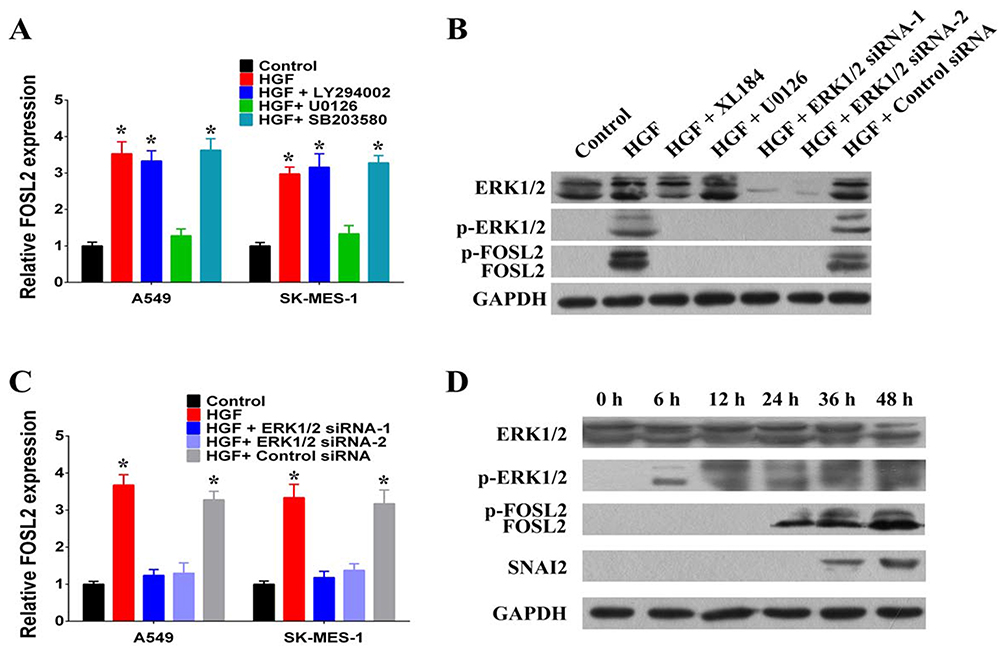 |
Figure 5 HGF/MET induced upregulation and phosphorylation of FOSL2 by activating ERK1/2. (A) The effect of PI3K inhibitor (LY294002, 20 μmol/L), ERK1/2 inhibitor (U0126, 10 μmol/L) and p38 MAPK inhibitor (SB203580, 20 μmol/L) on HGF induced upregulation of FOSL2 in A549 and SK-MES-1 cells. (B) HGF/MET regulated upregulation and phosphorylation of FOSL2 by activating ERK1/2. (C) HGF/MET regulated the upregulation of FOSL2 by ERK1/2 in A549 and SK-MES-1 cells. (D) Time series analysis of HGF/MET regulating ERK1/2, FOSL2 and SNAI2. (n=3, *P < 0.05). |
Exogenous HGF treatment induced the phosphorylation of ERK1/2, and MET inhibition inhibited the HGF-induced phosphorylation of ERK1/2 (Figure 5B). Remarkably, exogenous HGF also increased the phosphorylation and upregulation of FOSL2, while the MET (XL184) and ERK1/2 inhibitors (U0126) inhibited HGF-induced phosphorylation and upregulation of FOSL2 (Figure 5B). Moreover, knockdown of ERK1/2 by siRNA also blocked HGF-induced phosphorylation and upregulation of FOSL2; thus, demonstrating that activation of ERK1/2 was required for this effect (Figure 5B and C). In addition, through time series analyses, ERK1/2 and FOSL2 were phosphorylated at 6 hrs after exogenous HGF treatment, FOSL2 was upregulated at 12 hrs after exogenous HGF treatment, and SNAI2 was upregulated at 24 hrs after exogenous HGF treatment (Figure 5D). Therefore, these results pointed to a critical role for ERK1/2-induced phosphorylation and upregulation of FOSL2 upon HGF/MET-induced SNAI2 expression.
Clinical Analysis Of FOSL2 Expressions In NSCLC Samples
As described above, we found that FOSL2 plays an important role in promoting EMT, invasion and migration, which likely contribute to enhanced tumor metastasis. Therefore, we were interested in relating the FOSL2 expression levels to the properties of clinical cases of NSCLC and measured the mRNA levels of FOSL2 in 56 pairs of adjacent and primary NSCLC samples. The results showed that the expression levels of FOSL2 were significantly higher in primary tissues (C tissues) compared with adjacent tissues (NC tissues) (Figure 6A). Then, for the statistical analysis, these NSCLC samples were divided into two groups depending on the mRNA levels of FOSL2, high FOSL2 and low FOSL2. The association between the FOSL2 expression levels and the clinicopathologic features of NSCLC patients are listed in Table 1. Patients with high FOSL2 were associated with higher TNM stages (P<0.05) and metastasis (P<0.05) than patients with low FOSL2. Kaplan-Meier curves demonstrated that patients with high FOSL2 had shorter mean overall survivals (OS) than patients with low FOSL2, and the difference was statistically significant (P<0.05) (Figure 6B). These findings suggest that FOSL2 expressions were significantly associated with tumor metastasis and shortened survival time in NSCLC patients.
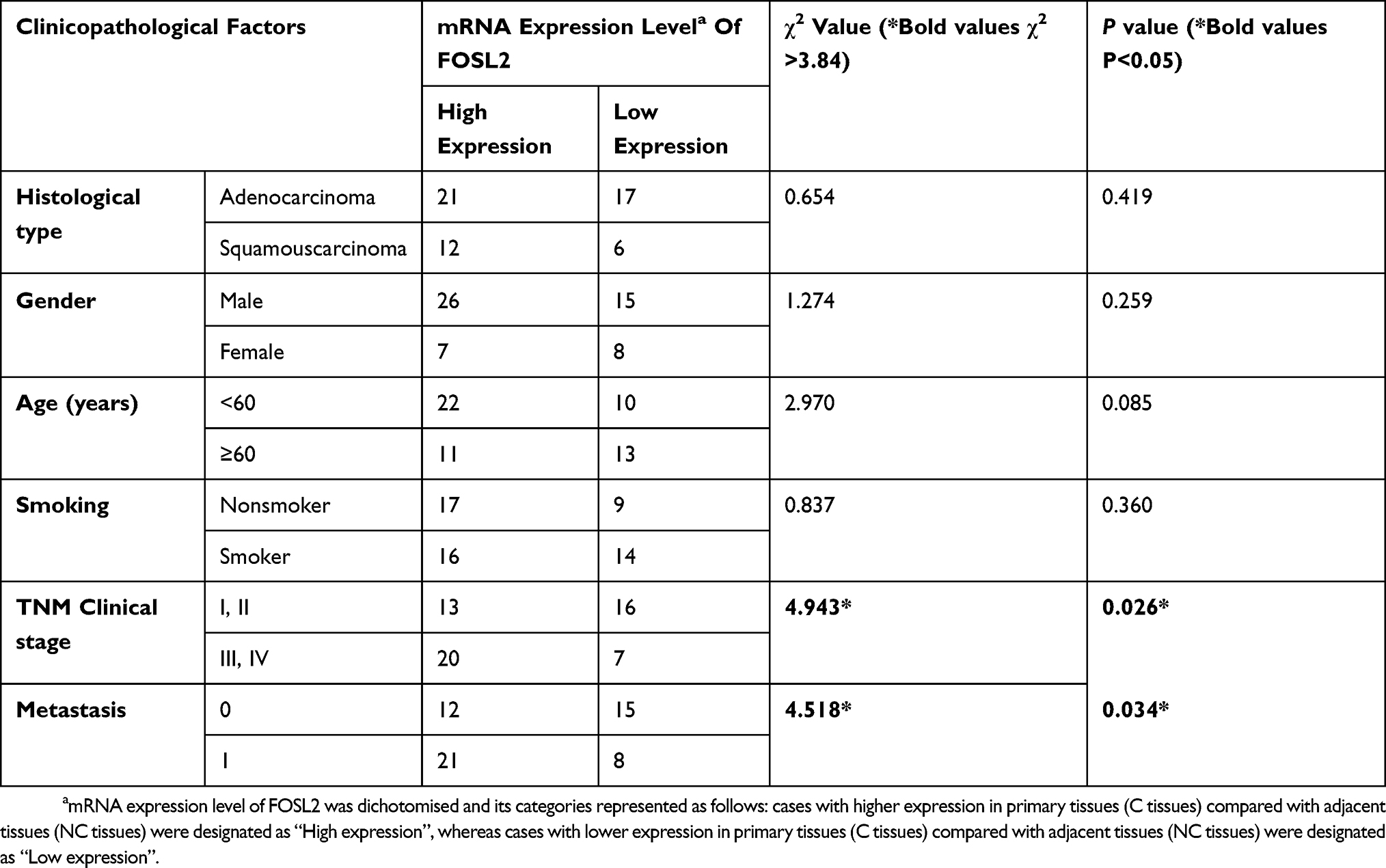 |
Table 1 Correlation Between mRNA Expression Level Of FOSL2 And The Clinicopathological Features Of NSCLC Patients. |
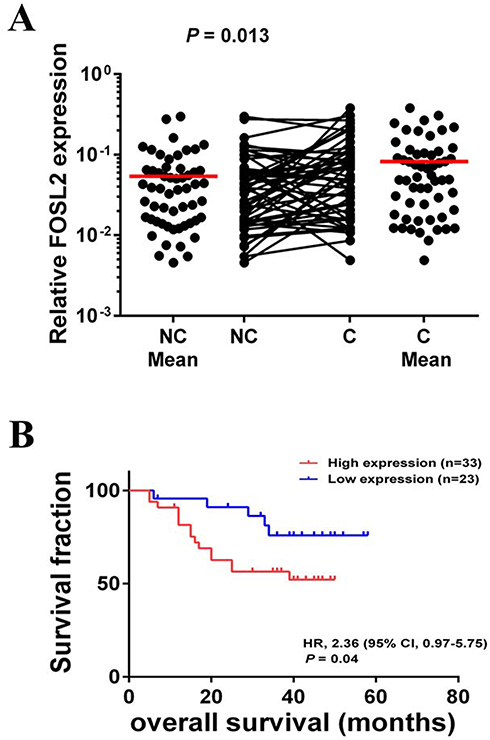 |
Figure 6 FOSL2 expression levels were associated with prognosis of NSCLC. (A) Scatter plots of expression levels of FOSL2 in NC and C tissues. (B) Kaplan-Meier analysis of overall survival in NSCLC patients with primary tumors assessed for expression levels of FOSL2. The P values correspond to hazard ratios (HR). NC, adjacent carcinoma tissues; C, primary carcinoma tissues. |
Discussion
HGF is a cell growth factor that can bind to its MET receptor, causing MET dimerization and autophosphorylation, and subsequently activating intracellular downstream signal transduction pathways that play important roles in development, organ regeneration and cancer.17 In normal tissues, HGF/MET is involved in a wide range of biological functions, such embryogenesis, organogenesis, adult tissue regeneration and carcinogenesis, and several lines of in vitro studies have revealed that HGF has regenerative effects on the epithelium in kidney, lung and other tissues.18,19 Recently, it has been found that HGF/MET can promote EMT of NSCLC cells and is associated with NSCLC metastasis.20–22 In this study, we found that HGF/MET can promote the expression of SNAI2 in both the lung adenocarcinoma cell line (A549) and lung squamous carcinoma cell line (SK-MES-1). Knockdown of SNAI2 terminated the promotion effects of HGF/MET on EMT, invasion and migration. This suggested that HGF/MET can promote the EMT, invasion and migration of NSCLS cells by inducing the expression of SNAI2.
FOSL2 is one of the FOS family transcription factors that binds to JUN family transcription factors to form the AP1 complexes, which are activated in response to extracellular signals, such as cytokines, growth factors, inflammation and stress, The central role of such complexes in transcriptional regulation places AP1 complexes at a functional epicenter for almost all areas of eukaryotic cellular behavior, such as stress response, proliferation, apoptosis and development.23 Moreover, many human cancer studies found that AP1 complexes are often deregulated during cancer progression and metastasis.24 It has been found that the ERK2/FOSL1/ZEB pathway can induce EMT.25 In this study, we found that HGF/MET can induce phosphorylation and upregulation of FOSL2 by ERK1/2 kinase and FOSL2 can promote the transcription of SNAI2 by binding with the SNAI2 promoter. We also found that FOSL2 overexpression can promote SNAI2 expression, EMT, invasion and migration, while knockdown of FOSL2 terminated the regulatory effects of HGF/MET on SNAI2 expression, EMT, invasion and migration. Therefore, these results indicated that the HGF/MET-ERK1/2-FOSL2-SNAI2 pathway was involved in the regulation of EMT and metastasis of NSCLC.
Generally speaking, cell growth factor stimulation requires signal transductions to control cell functions.26 Signal transduction pathways begin from extracellular signaling ligands binding to cell surface receptors, and subsequently activating the intracytoplasmic kinase cascade and the immediate early genes. The immediate early genes further activate the delayed response genes, which finally regulate cell functions.27 FOS family transcription factors are immediate early genes and are responsive to signals or stimuli, suggesting that FOSL2 can act as an immediate early transcription factor of HGF/MET signal to regulate the delayed response transcription factor, SNAI2, which promotes the EMT, invasion and migration of NSCLC cells. Interestingly, it has been reported that phosphorylated FOSL2 can induce the transcription of FOSL2.28 Therefore, we speculated that during the process of HGF/MET inducing EMT of NSCLC cells, HGF/MET first activates the phosphorylation of FOSL2 by the ERK1/2 kinase. Phosphorylated FOSL2 may then upregulate the transcriptional expression of itself; thus, forming a positive feedback loop to enhance the transcriptional expression of SNAI2 and promote the EMT, invasion and migration of NSCLC cells. Therefore, FOSL2 is considered to be the key factor for HGF/MET inducing EMT and metastasis.
In recent years, FOSL2 also has been found to have abnormal expression in cancer, which was closely related to tumor occurrence and progression.29–31 In NSCLC, it has been found that FOSL2 was overexpressed in lung squamous carcinoma.32 Additionally, it has been reported that FOSL2 is involved in the regulation of TGF-β1-induced migration of NSCLC cells and the FOSL2 expression in lung cancer tissues correlate with postoperative relapse and survival of lung cancer patients.33 In this study, we detected the expression levels of FOSL2 in 56 NSCLC samples and found that the expression levels of FOSL2 were higher in the primary tissues than in those of adjacent tissues. The high expression levels of FOXF1 were associated with metastasis and shortened survival times of NSCLC patients.
Conclusion
This study revealed that the roles of HGF/MET in inducing the EMT and metastasis of NSCLC cells were achieved through the phosphorylation and upregulation of FOSL2, which regulates the transcription of SNAI2. Furthermore, an understanding of the molecular mechanisms of FOSL2-mediated HGF/MET pathway may provide insights and approaches for the treatment of NSCLC.
Ethics Approval And Consent To Participate
All patients provided written informed consent, and the collection of NSCLC tissues for research purposes was approved by relevant human research ethics committees of the Cancer Center of Guangzhou Medical University (Approval no. (2014) 100).
Acknowledgments
This study was supported by grants from the National Natural Science Foundation of China (81501969, 81572258) and Guangzhou Science and Technology Program (201804010077), and funded by the Guangzhou Key Medical Discipline Construction Project fund.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2015;68:7–30. doi:10.3322/caac.21442
2. Chen W, Zheng R, Baade PD, et al. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66:115–132. doi:10.3322/caac.21338
3. Thomas A, Liu SV, Subramaniam DS, Giaccone G. Refining the treatment of NSCLC according to histological and molecular subtypes. Nat Rev Clin Oncol. 2015;12:511–526. doi:10.1038/nrclinonc.2015.90
4. Gridelli C, Rossi A, Carbone DP, et al. Non-small-cell lung cancer. Nat Rev Dis Primers. 2015;1:15009. doi:10.1038/nrdp.2015.9
5. Steeg PS. Targeting metastasis. Nat Rev Cancer. 2016;16:201–218. doi:10.1038/nrc.2016.25
6. Brabletz T, Kalluri R, Nieto MA, Weinberg RA. EMT in cancer. Nat Rev Cancer. 2018;18:128–134. doi:10.1038/nrc.2017.118
7. Davis FM, Stewart TA, Thompson EW, Monteith GR. Targeting EMT in cancer: opportunities for pharmacological intervention. Trends Pharmacol Sci. 2014;35:479–488. doi:10.1016/j.tips.2014.06.006
8. Li Q, Hutchins AP, Chen Y, et al. A sequential EMT-MET mechanism drives the differentiation of human embryonic stem cells towards hepatocytes. Nat Commun. 2017;8:15166. doi:10.1038/ncomms15166
9. Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol. 2014;15:178–196. doi:10.1038/nrm3758
10. Tsai JH, Donaher JL, Murphy DA, Chau S, Yang J. Spatiotemporal regulation of epithelial–mesenchymal transition is essential for squamous cell carcinoma metastasis. Cancer Cell. 2012;22:725–736. doi:10.1016/j.ccr.2012.09.022
11. Skrypek N, Goossens S, De Smedt E, Vandamme N, Berx G. Epithelial-to-mesenchymal transition: epigenetic reprogramming driving cellular plasticity. Trends Genet. 2017;33:943–959. doi:10.1016/j.tig.2017.08.004
12. Ye X, Weinberg RA. Epithelial-mesenchymal plasticity: a central regulator of cancer progression. Trends Cell Biol. 2015;25:675–686. doi:10.1016/j.tcb.2015.07.012
13. Puisieux A, Brabletz T, Caramel J. Oncogenic roles of EMT-inducing transcription factors. Nat Cell Biol. 2014;16:488–494. doi:10.1038/ncb2976
14. Miranda O, Farooqui M, Siegfried JM. Status of agents targeting the HGF/c-Met axis in lung cancer. Cancers. 2018;10:280. doi:10.3390/cancers10110400
15. Scagliotti GV, Novello S, von Pawel J. The emerging role of MET/HGF inhibitors in oncology. Cancer Treat Rev. 2013;39:793–801. doi:10.1016/j.ctrv.2013.02.001
16. Pérez-Ramírez C, Cañadas-Garre M, Molina MÁ, et al. MET/HGF targeted drugs as potential therapeutic strategies in non-small cell lung cancer. Pharmacol Res. 2015;102:90–106. doi:10.1016/j.phrs.2015.09.016
17. Trusolino L, Bertotti A, Comoglio PM. MET signalling: principles and functions in development, organ regeneration and cancer. Nat Rev Mol Cell Biol. 2010;11:834–848. doi:10.1038/nrm3012
18. Imamura R, Matsumoto K. Hepatocyte growth factor in physiology and infectious diseases. Cytokine. 2017;98:97–106. doi:10.1016/j.cyto.2016.12.025
19. Gallo S, Sala V, Gatti S, Crepaldi T. Cellular and molecular mechanisms of HGF/Met in the cardiovascular system. Clin Sci. 2015;129:1173–1193. doi:10.1042/CS20150502
20. Xiang C, Chen J, Fu P. HGF/Met signaling in cancer invasion: the impact on cytoskeleton remodeling. Cancers. 2017;9:44. doi:10.3390/cancers9050044
21. Sadiq AA, Salgia R. MET as a possible target for non-small-cell lung cancer. J Clin Oncol. 2013;31:1089–1096. doi:10.1200/JCO.2012.43.9422
22. Landi L, Minuti G, D’Incecco A, Cappuzzo F. Targeting c-MET in the battle against advanced nonsmall-cell lung cancer. Curr Opin Oncol. 2013;25:130–136. doi:10.1097/CCO.0b013e32835daf37
23. Lopez-Bergami P, Lau E, Ronai Z. Emerging roles of ATF2 and the dynamic AP1 network in cancer. Nat Rev Cancer. 2010;10:65–76. doi:10.1038/nrc2681
24. Trop-Steinberg S, Azar Y. AP-1 expression and its clinical relevance in immune disorders and cancer. Am J Med Sci. 2017;353:474–483. doi:10.1016/j.amjms.2017.01.019
25. Shin S, Blenis J. ERK2/Fra1/ZEB pathway induces epithelial-to-mesenchymal transition. Cell Cycle. 2010;9:2483–2484. doi:10.4161/cc.9.13.12270
26. Hynes NE, Ingham PW, Lim WA, Marshall CJ, Massagué J, Pawson T. Signalling change: signal transduction through the decades. Nat Rev Mol Cell Biol. 2013;14:393–398. doi:10.1038/nrm3581
27. Kolch W, Halasz M, Granovskaya M, Kholodenko BN. The dynamic control of signal transduction networks in cancer cells. Nat Rev Cancer. 2015;15:515–527. doi:10.1038/nrc3983
28. Murakami M, Ui M, Iba H. Fra-2-positive autoregulatory loop triggered by mitogen-activated protein kinase (MAPK) and Fra-2 phosphorylation sites by MAPK. Cell Growth Differ. 1999;10:333–342.
29. Li S, Fang XD, Wang XY, Fei BY. Fos-like antigen 2 (FOSL2) promotes metastasis in colon cancer. Exp Cell Res. 2018;373:57–61. doi:10.1016/j.yexcr.2018.08.016
30. Sun L, Guo Z, Sun J, et al. MiR-133a acts as an anti-oncogene in hepatocellular carcinoma by inhibiting FOSL2 through TGF-β/Smad3 signaling pathway. Biomed Pharmacother. 2018;107:168–176. doi:10.1016/j.biopha.2018.07.151
31. Sun X, Dai G, Yu L, Hu Q, Chen J, Guo W. miR-143-3p inhibits the proliferation, migration and invasion in osteosarcoma by targeting FOSL2. Sci Rep. 2018;8:606. doi:10.1038/s41598-017-18739-3
32. Gao L, Guo YN, Zeng JH, et al. The expression, significance and function of cancer susceptibility candidate 9 in lung squamous cell carcinoma: a bioinformatics and in vitro investigation. Int J Oncol. 2019;54:1651–1664. doi:10.3892/ijo.2019.4758
33. Wang J, Sun D, Wang Y, et al. FOSL2 positively regulates TGF-β1 signalling in non-small cell lung cancer. PLoS One. 2014;9:e112150. doi:10.1371/journal.pone.0112150
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.