")
Back to Journals » International Journal of Nanomedicine » Volume 14
Heterozygous Disruption of Beclin 1 Alleviates Zinc Oxide Nanoparticles-Induced Disturbance of Cholesterol Biosynthesis in Mouse Liver
Authors Liu X, Wang B , Jiang X, Zhang J , Tang Q, Zhang Y, Qin X, Chen C , Zou Z
Received 22 July 2019
Accepted for publication 21 November 2019
Published 12 December 2019 Volume 2019:14 Pages 9865—9875
DOI https://doi.org/10.2147/IJN.S224179
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Mian Wang
Xuemei Liu,1,* Bin Wang,2,* Xuejun Jiang,3 Jun Zhang,2 Qianghu Tang,1 Yujia Zhang,1 Xia Qin,4 Chengzhi Chen,1,5 Zhen Zou2,5
1Department of Occupational and Environmental Health, School of Public Health and Management, Chongqing Medical University, Chongqing 400016, People’s Republic of China; 2Institute of Life Sciences, Chongqing Medical University, Chongqing 400016, People’s Republic of China; 3Center of Experimental Teaching for Public Health, Experimental Teaching and Management Center, Chongqing Medical University, Chongqing 400016, People’s Republic of China; 4Department of Pharmacy, The First Affiliated Hospital of Chongqing Medical University, Chongqing 400016, People’s Republic of China; 5Dongsheng Lung-Brain Disease Joint Lab, Chongqing Medical University, Chongqing 400016, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Zhen Zou Email [email protected]
Chengzhi Chen Email [email protected]
Purpose: Liver is regarded as one of the primary target organs for zinc oxide nanoparticles (ZnONPs) toxicity. Since liver represents the leading site for de novo cholesterol biosynthesis in mammals, the injuries of liver could result in the disruption of cholesterol biosynthesis. In this study, we aimed to investigate whether pulmonary ZnONPs exposure induces disturbance of cholesterol biosynthesis in mouse liver.
Methods and results: Our data demonstrated intratracheally instilled with a single dose of 3, 6, and 12 μg/animal ZnONPs could induce histopathological deterioration in mouse liver in a dose-related manner at 3 days, but remission was observed at 7 days after treatment. Moreover, ZnONPs caused the disturbance of cholesterol biosynthesis by increasing both 3-hydroxy-3-methylglutaryl-CoA (HMG-CoA) reductase and sterol regulatory element-binding protein 2 (SREBP2) protein expressions. To further reveal the underlying toxic mechanisms, we detected the biomarkers of autophagy and found that pulmonary ZnONPs exposure led to the elevation of LC3B-II and Beclin 1, suggesting ZnONPs might trigger autophagy in liver tissues. By using both beclin 1+/+ and beclin 1+/- mice, we demonstrated that inhibition of autophagy by heterozygous disruption of beclin 1 attenuated the disturbance of cholesterol biosynthesis induced by ZnONPs in liver.
Conclusion: Pulmonary exposure of ZnONPs would induce the cholesterol biosynthesis disturbance in mouse liver through Beclin-1-dependent autophagy activation, suggesting that inhibition of autophagy may contribute to preventing the cholesterol biosynthesis disturbance and its associated pathologies induced by ZnONPs in liver.
Keywords: zinc oxide nanoparticles, cholesterol biosynthesis, autophagy, beclin 1, liver
Introduction
Zinc oxide nanoparticles (ZnONPs) are the primary nanoscale materials and widely used in industrial products, including cosmetics, rubber, textile, imaging, food packaging, and additive.1 The annual production volume of ZnONPs grows exponentially and ranks third-highest in worldwide.2 Release of ZnONPs from industrial and household products into the environment may pose a severe threat to human health.
The bio-distribution research has revealed that ZnONPs can be translocated from entry portals into circulatory systems, then ultimately to various body organs, such as brain, liver, heart, and kidney.3,4 Liver has been considered as one of the primary target organs for ZnONPs toxicity. Cholesterol is an essential biochemical molecule that serves as a vital part of membrane structure and as a precursor for the synthesis of steroid hormones, bile acids and vitamin D.5 Nearly half of the cholesterol in the body derives from biosynthesis de novo. Moreover, biosynthesis and utilization of cholesterol must be tightly regulated within the body to avoid over-accumulation.5 Of particular clinical importance is the over-accumulation of cholesterol that may eventually result in the various artery disorders, such as atherosclerosis.6 Since biosynthesis of cholesterol in liver accounts for most of the amount produced each day,5 the injuries in liver induced by exogenous substances, such as ZnONPs, may undoubtedly affect the biosynthesis of cholesterol.
Beclin 1, the mammalian orthologue of yeast autophagy-related gene 6 (Atg6), is a central regulator of autophagy.7 Therefore, detection of beclin 1 is used to monitor autophagosome formation and autophagic flux. Importantly, increased beclin 1 expression and elevated autophagy were also observed in sphingolipid storage diseases characterized by disrupted cholesterol and sphingolipid trafficking.8,9 A recent study has found the novel evidence that autophagy can promote lipid droplet formation in a beclin 1-dependent manner.10
In the present study, we aimed to investigate whether pulmonary ZnONPs' exposure induced disturbance of cholesterol biosynthesis in liver. To reveal the toxic mechanisms involved, by using both beclin 1+/+ and beclin 1+/- mice, our results further demonstrated that inhibition of autophagy by heterozygous disruption of the beclin 1 relived the disturbance of cholesterol biosynthesis induced by ZnONPs in mouse liver. These findings together indicate therapeutic strategies to inhibit autophagy may provide a new approach to prevent the cholesterol biosynthesis disturbance and its associated pathologies in liver induced by ZnONPs.
Materials and Methods
Chemicals and Reagents
Zinc oxide nanoparticles (ZnONPs), less than 50 nm particle size, were purchased from Sigma Aldrich Chemical Co. (MO, USA). Cy3 AffiniPure Goat anti-Rabbit IgG (H + L) was from EarthOx Life Sciences (CA, USA). Immobilon Western Chemiluminescent HRP Substrate, RIPA lysis buffer, phenylmethanesulfonylfluoride (PMSF) and bicinchoninic acid (BCA) assay kit were all purchased from Beyotime Institute of Biotechnology (Shanghai, China). β-actin antibody was obtained from ABclonal Biotechnology (MA, USA). Antibodies against HMG-CoA, LC3B, and p62 were all from Abcam Co. (Cambridge, UK). Beclin 1 antibody was purchased from Cell Signaling Technology (Beverly, MA, USA). GAPDH antibody was obtained from Bioss Biotechnology Co., Ltd. (Beijing, China). SREBP2 antibody was from Novus Biologicals Inc. (Littleton, CO, USA).
Animal Husbandry
All animal experiment procedures were approved by the Institutional Animal Care and Use Committee of Chongqing Medical University. All procedures were conducted following the guidelines contained in the guide for the care and use of laboratory animals. All the treatments were performed gently and all efforts were made to minimize animal suffering. Healthy-specific pathogen-free adult male C57BL/6J mice, aged 8–10 weeks and weighed 22–25 g, were purchased from Experimental Animal Center of Chongqing Medical University [Chongqing, China, license numbers: SCXK(Yu)2012–001]. Mice were housed in standard polycarbonate animal cages with five animals per cage in a controlled-specific pathogen-free environment. The animals’ room was maintained a 12:12 hrs light–dark cycle, at an ambient temperature of 23 ± 1°C and 55 ± 10% humidity. The animals were free to access to standard mouse chow and tap water provided. Beclin 1+/− mice on a C57BL/6J background were obtained from the laboratory of Beth Levine11 and their corresponding age and weight-matched wild-type littermates were used as controls. Both the wild-type (beclin 1+/+) and beclin 1+/− mice were sacrificed at the same time points for the designed experiments.
Characterization of ZnONPs and Preparation
The characteristics of ZnONPs, such as level of agglomeration, chemical elemental composition, size, and zeta potential, were all detected and described in detail in our previous work.12 To prepare the suspended solution, ZnONPs were diluted in 2% heat-inactivated sibling mouse serum in MilliQ water and sonicated with an ultrasonic cleaner set at 20% of the maximum amplitude (SB-5200DT, Ningbo Scientz Biotechnology Co., Ltd, Ningbo, China) for 20 mins in an ice water bath. To minimize agglomeration and ensure their homogeneity, ZnONPs were freshly prepared each time before use. Vehicle solution was also prepared by sonication of 2% heat-inactivated sibling mouse serum in MilliQ water in the same ways.
Intratracheal Instillation of ZnONPs
Animals were intratracheally instilled with a single dose of 3, 6 and 12 μg/animal ZnONPs, respectively. The concentrations of ZnONPs used in the present study were calculated according to the current Chinese occupational exposure limit of ZnONPs at 3 mg/m3. The calculation process was shown in our previous study.12 After anesthetizing with 1% pentobarbital sodium, the animals were placed on their back on a 40-degree slope. The trachea of mice was intubated by an insyte catheter (Becton & Dickinson Co., NJ, USA) with a shortened needle. Suspended ZnONPs solution (50 μL) was instilled followed by air (150 μL) with a syringe. Mice were gently placed on the 37°C heating plate and kept their head up until proper breathing and full recovery from anesthesia. After ZnONPs treatment, animals were sacrificed after 1, 3 or 7 days under anesthesia. The liver tissues were quickly collected, and one part of tissue was immersed in the 4% paraformaldehyde used for hematoxylin-eosin staining and immunofluorescence assay, the other part of tissue was used for Western blot assay. The beclin 1+/− mice and wild-type littermates (beclin 1+/+ mice, age and weight matched) were treated with ZnONPs at the concentration of 12 μg/animal and the liver tissues were collected at day 3 after intratracheal instillation.
Hematoxylin-Eosin Staining
Hematoxylin-eosin (H&E) staining on the liver tissues was performed as described previously.13 Briefly, the tissues were dissected immediately and fixed in the 4% paraformaldehyde. The sections were prepared by using standard pathology slide preparation procedures. Sections were deparaffinized by xylene and dehydrated in the gradient concentrations of ethanol. Then, sections were stained with hematoxylin for 5 mins and rinsed with distilled water. After staining with eosin for 1 min, sections were washed and subjected to dehydration in ethanol, trans-parented in xylene and mounted with neutral balsam. The sections were observed under a light microscope (Leica Application Suite, 4.9.0, Germany).
Immunofluorescence Assay
Immunofluorescence assay was carried out as described previously.14 In brief, the sections were rinsed with PBS and subsequently incubated in the blocking solution containing normal serum for 30 mins at 37°C. After incubation of primary anti-SREBP2 (1:50) antibody at 4°C overnight, the sections were incubated with the Cy3-conjugated secondary antibody at room temperature for 1 hr on the next day. Finally, sections were sealed with an anti-fluorescent quencher and observed under a fluorescence microscope (Olympus, IX53, Tokyo, Japan).
Western Blot Analysis
Western blot analysis was conducted according to the procedures described previously.15 In brief, the liver tissues were lysed with ice-cold RIPA lysis buffer containing PMSF and protease inhibitors (Thermo Fisher Scientific, MA, USA). The protein concentrations of tissues were determined by the BCA assay kit. Proteins were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis and then transferred to polyvinylidene difluoride (PVDF) membrane (Millipore Corp., Billerica, MA). After blocking with 5% non-fat milk for 2 hrs, the PVDF membrane was incubated with the primary antibodies (HMG-CoA, 1:2000; LC3B, 1:2000; p62, 1:2000; Beclin 1, 1:1000; β-actin, 1:5000; GAPDH, 1:5000) overnight at 4°C. The next day, the membrane was incubated with horse radish peroxidase-conjugated secondary antibodies for 1 hr at room temperature. Enhanced chemiluminescence reagents were used to visualize the brands by a Molecular Imager Gel Doc XR System (Bio-Rad, USA). The intensities of the bands were obtained and analyzed using Image J software (NIH, Bethesda, MD, USA).
Cholesterol Content Determination
The concentrations of total cholesterol in the liver of mouse were determined by the total cholesterol assay kit (Nanjing Jiancheng Bioengineering Institute, Jiangsu, China) according to the manufacture’s instruction.
Statistical Analysis
Results were obtained from at least three independent experiments and were reported as the mean ± standard deviation. Data were analyzed and plotted using GraphPad Prism 6.0 (GraphPad Software, La Jolla, CA). Statistical significance was determined using one-way analysis of variance (ANOVA) followed by Tukey’s honestly significant difference post hoc test, unless otherwise indicated. Statistical significance level was set at P<0.05.
Results
Pulmonary ZnONPs Exposure Caused the Pathological Changes in Mouse Liver
Unintended exposure to ZnONPs may mainly occur via inhalation, and lung is an undoubted target organ for ZnONPs.16 Moreover, the long length of airways and a large number of alveoli can provide a large surface area for presenting ultrafine nanoparticle in the lung. Indeed, adverse systemic effects have been observed after inhalation of ZnONPs. Moreover, inhalation of ZnONPs has been found not only to reduce pulmonary function, but also lead to damage of other organs because of the systemic translocation of nanoparticles via blood circulation.12,17 However, there are very few studies to demonstrate the toxicity of ZnONPs on liver via inhalation exposure. Thus, in this study, the animals were administrated intratracheally with low (3 μg/animal), medium (6 μg/animal) and high (12 μg/animal) doses of ZnONPs. The liver tissues were collected on days 1, 3 and 7. H&E staining assay was used to observe the histopathological changes in the liver after airway exposure of ZnONPs. Interestingly, our results found that ZnONPs could induce histopathology deterioration in the mouse liver, manifested by hepatocyte ballooning, abundance of micro and macro vesicles, multi necrotic foci filled with edema and hemorrhage in the central vein (Figure 1A). The pathological damages of liver were observed in a dose-related manner for treatment of ZnONPs for 3 days (Figure 1A). But after treating of ZnONPs for 7 days, the H&E staining results showed that these pathological changes could partially recover with time after a single dose of ZnONPs treatment (Figure 1B). Based on these observations, we chose the time point on day 3 for the following designed experiments.
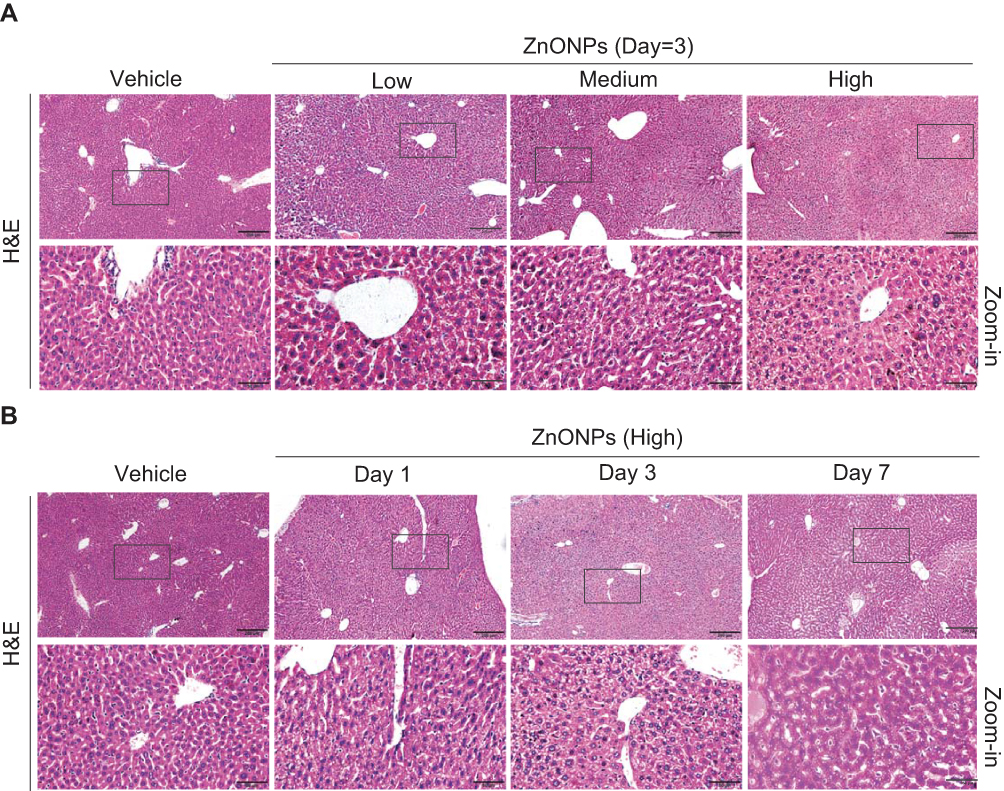 |
Figure 1 Pulmonary ZnONPs exposure caused the pathological changes in mouse liver. (A) After a single treatment of 3, 6, 12 μg/animal ZnONPs via tracheal instillation, H&E staining on liver tissues of mice showed a dose-related manner on pathological changes.(B) The mice were treated with 12 μg/animal ZnONPs via tracheal instillation, the liver tissues were collected at post-exposure day 1, day 3 and day 7. H&E staining was used to observe pathological changes. Scale bar = 200 μm in original figures or 50 μm in Zoom-in figures. |
Pulmonary ZnONPs Exposure Affected the Cholesterol Biosynthesis in Mouse Liver
Liver is the primary site of cholesterol biosynthesis in mammals;5 injuries in liver might result in the disruption of cholesterol biosynthesis. Biosynthesis of cholesterol begins with the transport of acetyl-CoA from mitochondria to cytosol. The rate-limiting step in cholesterol biosynthesis is catalyzed by the 3-hydroxy-3-methylglutaryl-CoA (HMG-CoA) reductase.18,19 To test whether airway exposure of ZnONPs shows a potential impact on the cholesterol biosynthesis in mouse liver, the protein expression level of HMG-CoA was assessed by Western blot analysis. Our results demonstrated that the protein expression of HMG-CoA was significantly enhanced by medium and high doses of ZnONPs (Figure 2A and B). Herein, this increment of HMG-CoA indicated that ZnONPs treatment is capable of provoking the biosynthesis of cholesterol, and further trigger cholesterol accumulation in liver.
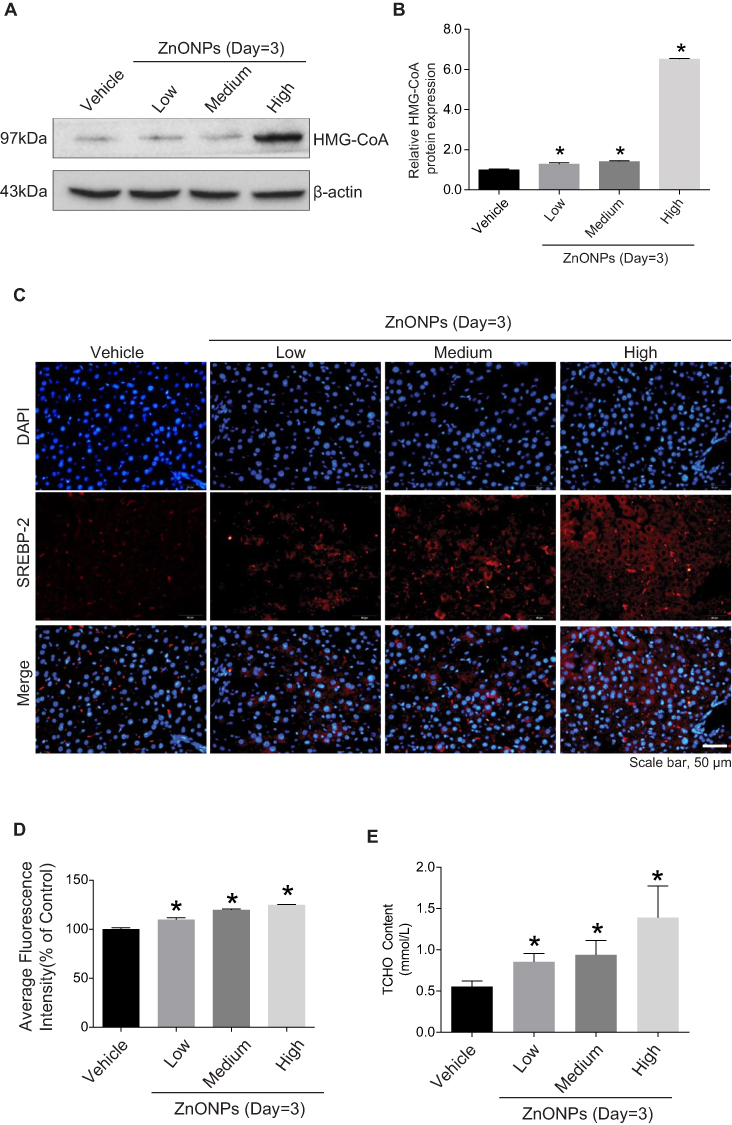 |
Figure 2 Pulmonary ZnONPs exposure affected the cholesterol biosynthesis in mouse liver. After a single treatment of 3, 6, 12 μg/animal ZnONPs, liver tissues were collected for Western blot assay at post-exposure day 3. (A) Representative Western blot reflecting HMG-CoA protein levels in mouse liver. (B) The protein expression of HMG-CoA obtained from at least three independent experiments. β-actin was served as loading control. (C) Representative images obtained from immunofluorescence reflecting SREBP-2 expression in mouse liver. Scale bar = 50 μm. (D) Fluorescence intensities of SREBP-2 analyzed by Image-Pro Plus image analysis. (E) The total cholesterol in the liver of mouse was determined. Data were derived from at least three independent experiments and were reported as mean ± S.D. “*” denoted P<0.05, compared with the vehicle control. |
Sterol regulatory element-binding proteins (SREBPs) are membrane-bound transcription factors that play as the master regulators of cholesterol homeostasis in mammals.20 Activated SREBPs can bind to specific sterol regulatory element DNA sequences, thus upregulating the synthesis of HMG-CoA and many other enzymes involved in cholesterol biosynthesis.21 SREBP-2 is the predominant form of SREBPs in liver and it exhibits preference at controlling the expression of genes involved in cholesterol biosynthetic enzymes.20,22 Thus, to further test our hypothesis, we detected the expressions of SREBP-2 by using immunofluorescence assay. Our data revealed that the fluorescence intensities of SREBP-2 in ZnONPs-treated mice were much higher than those in the control mice (Figure 2C and D). Furthermore, we also found that the total cholesterol was elevated in the liver of mouse upon ZnONPs treatment (Figure 2E). These findings suggest that, in response to ZnONPs pulmonary exposure, the activation of SREBP-2 may contribute to the enhancement of HMG-CoA, and thus further leading to the disturbance of cholesterol biosynthesis.
Pulmonary ZnONPs Exposure Induced Autophagy in Mouse Liver
Previous investigation has demonstrated that pharmacological inhibition of autophagy with 3-methyladenine remarkably increased hepatocyte triglyceride content in the absence or presence of exogenous lipid supplementation, indicating that autophagy may play a vital role in the regulation of cholesterol homeostasis.23 To reveal the potential mechanism underlying how ZnONPs disturbed the biosynthesis of cholesterol, the protein expressions of classical autophagy biomarkers LC3B, p62 and Beclin 1 were detected. Our results showed that ZnONPs markedly increased the levels of LC3B-II (Figure 3A and B), p62 (Figure 3C and D) and Beclin 1 (Figure 3E and F) in particular dose-dependent manner at post-exposure days 3. The above results indicate that pulmonary ZnONPs exposure is capable of triggering autophagy in the mouse liver tissues, and autophagy may be involved in the ZnONPs-induced disturbance of cholesterol biosynthesis.
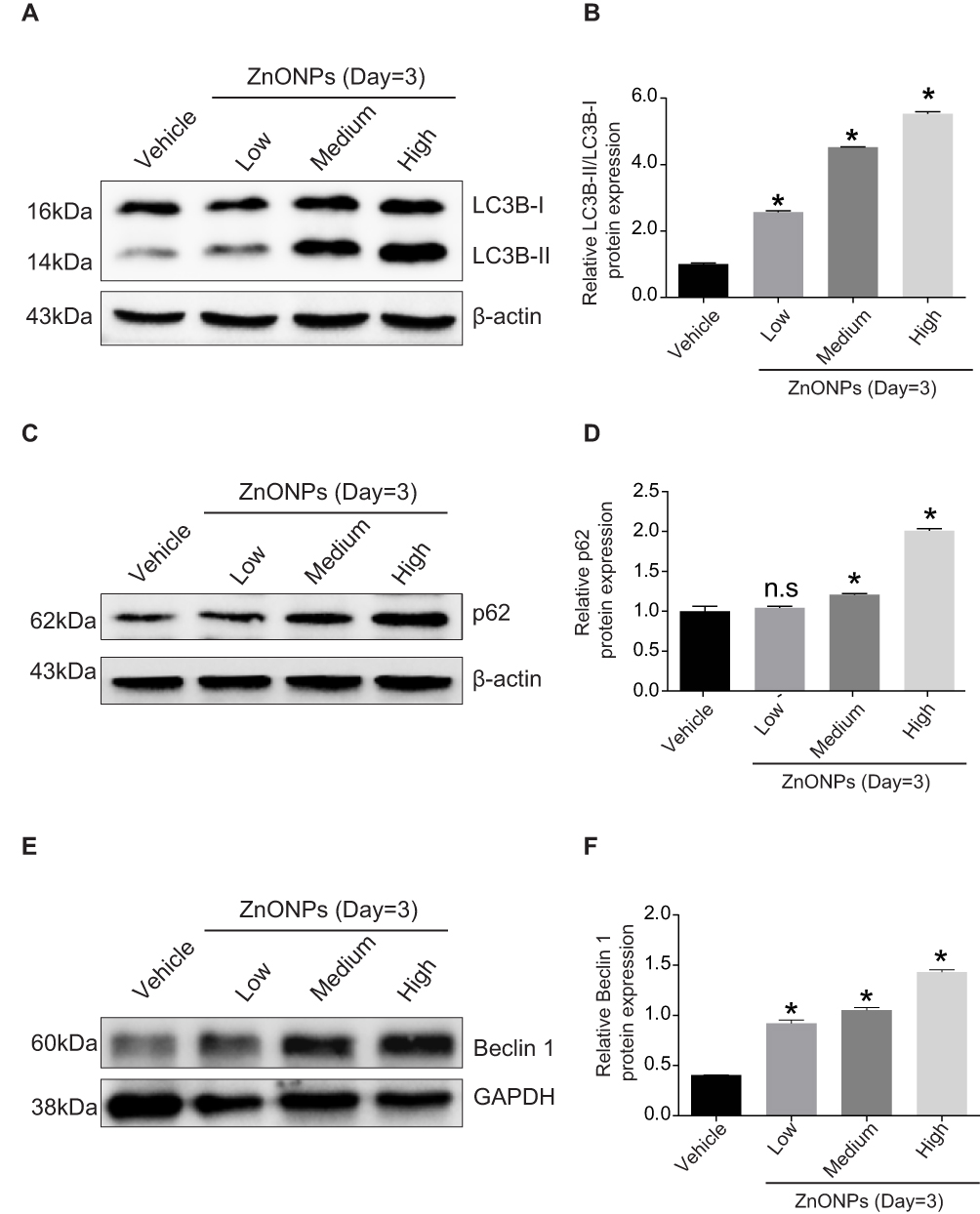 |
Figure 3 Pulmonary ZnONPs exposure induced autophagy in mouse liver. (A) Representative Western blot reflecting LC3B protein levels in mouse liver. (B) The ratio of LC3B-II/LC3B-I obtained from at least three independent experiments. (C) Representative Western blot reflecting p62 protein levels in mouse liver. (D) Protein expression of p62 obtained from at least three independent experiments. (E) Representative Western blot reflecting Beclin 1 protein levels in mouse liver. (F) Protein expression of Beclin 1 obtained from at least three independent experiments. β-actin and GAPDH were served as loading control, respectively. Data were derived from three independent experiments and were reported as mean ± S.D. “*” denoted P<0.05, compared with the vehicle control. |
Heterozygous Disruption of the Beclin 1 Alleviated the Disturbance of Cholesterol Biosynthesis and Injuries Induced by ZnONPs in Mouse Liver
To further verify whether autophagy regulates the ZnONPs-induced disruption of cholesterol biosynthesis and liver injuries, both the beclin 1+/+ and beclin 1 +/-mice were exposed to ZnONPs via tracheal instillation. After the collection of liver tissues, H&E staining assay firstly observed that heterozygous disruption of the beclin 1 significantly alleviated the pathological damage in mouse liver induced by airway ZnONPs exposure (Figure 4A). In the beclin 1+/- mice liver tissues, the elevated protein expression of HMG-CoA triggered by ZnONPs treatment was lower than that in beclin 1+/+ mice liver (Figure 4B and C). Additionally, the down-regulated effect on Beclin 1 protein expression of beclin 1+/- mice was confirmed by using Western blot assay (Figure 4B and D). Together, these findings suggest heterozygous disruption of the beclin 1 can show a protective effect against liver injuries and cholesterol biosynthesis disturbance induced by ZnONPs airway exposure. Moreover, these results may indicate inhibition of autophagy by reduction of its essential regulated gene contributes to the beneficial effects for the prevention of ZnONPs liver toxicity.
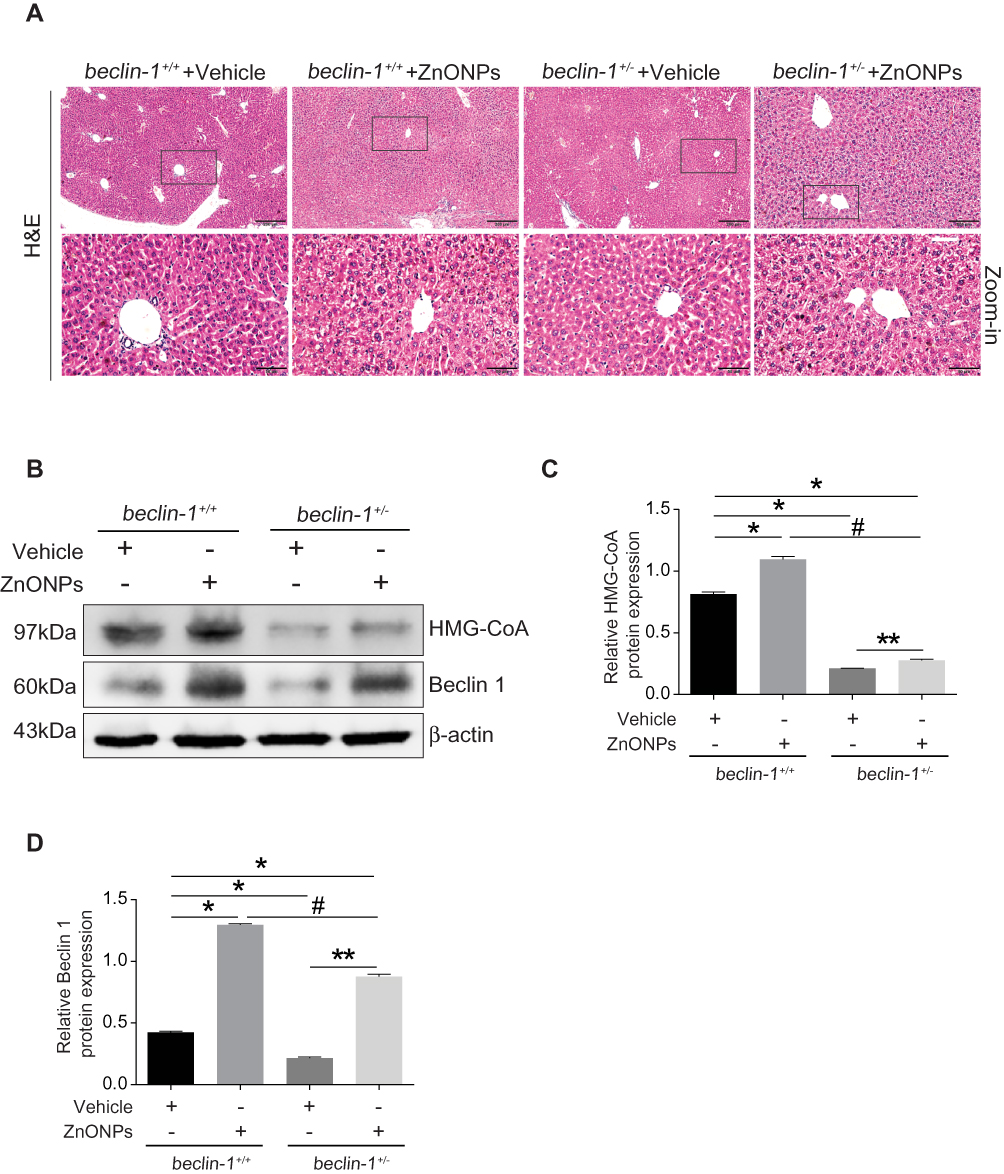 |
Figure 4 Heterozygous disruption of the beclin 1 alleviated the disturbance of cholesterol biosynthesis and injuries induced by ZnONPs in mouse liver. (A) The beclin 1+/+ and beclin 1+/- mice were treated with 12 μg/animal ZnONPs via tracheal instillation, the liver tissues were collected at post-exposure day 3. H&E staining was used to observe pathological changes. 100 ×, scale bar = 200 μm; 400 ×, scale bar = 50 μm. (B) Representative Western blot reflecting HMG-CoA and Beclin 1 protein levels in beclin 1+/+ and beclin 1+/-mice liver. (C and D) Protein expressions of Beclin 1 and HMG-CoA obtained from at least three independent experiments in beclin 1+/+ and beclin 1+/- mice liver. Data were derived from three independent experiments and were reported as mean ± S.D. “*” denoted P<0.05, compared with the vehicle control in beclin 1+/+ mice. “**” denoted P<0.05, compared with vehicle control in beclin 1+/- mice. “#” denoted P<0.05, compared with ZnONPs-treated beclin 1+/+ mice. |
Discussion
In our previous work, we found that pulmonary ZnONPs' exposure could induce acute lung injury, characterized by histopathological changes, infiltration of inflammatory cells in lung tissues and elevation of total protein and cytokine interleukin-6 in bronchoalveolar lavage fluid in time- and dose-dependent manners.12 Because of the small size, inhaled ZnONPs are thought to cross the blood-air barrier, and therefore transporting to extrapulmonary organs or tissues through systemic circulation by blood. Indeed, in the tissue biodistribution study on ZnONPs, their data showed that intraperitoneally injected ZnONPs (2.5 g/kg bw) were absorbed into circulation within 30 mins post-dosing, and mainly distributed to the liver, spleen and kidney.24 Similar results were observed by airway exposure to ZnONPs. For instance, Wang et al revealed that inhalation of 20 nm ZnONPs (2.5 mg/kg bw) by rats for 3 days caused an elevated zinc content in the liver after 12 hrs and in the kidneys after 36 hrs.25 Consistent with these previous findings,25,26 the results obtained in this study also provided evidence that pulmonary exposure of ZnONPs could result in liver pathological damage.
The present study aimed to investigate the detail of ZnONP-induced toxic response with a view to better understanding the mechanisms in mouse liver following a single intratracheal instillation. Herein, we chose the intratracheal instillation for the animal model. Inhalation is the primary route for human exposure to ZnONPs in the occupational environment, and exposure to ZnONPs via respiratory tract is known to cause metal fume fever in workers.16,26 The exposure dose of nanoparticles by tracheal instillation is uniform and accurate. It is easy for us to ensure the consistent amounts of inhaled nanoparticles per mouse by tracheal instillation. Also, many previous studies used this administration route to reveal the toxicological mechanisms of ZnONPs.27,28 By using this way, administered ZnONPs can be prepared as an aerosol and spray directly into the airway tract of mice. Moreover, this administration is an effective way to deliver ZnONPs and facilitate its absorption.
In this study, the concentrations of ZnONPs were calculated based on the permissible concentration-time weighted average, the current Chinese occupational exposure limit of ZnONPs at 3 mg/m3. The calculation process was described in detail in our previous study.12 Interestingly, we observed that the pathological changes in mouse liver tissue at day 7 were not more severe than that at day 3 after treatment of ZnONPs. Similar trends were found in the lung tissues, manifested by the significantly reduced bronchoalveolar lavage fluid protein concentrations and lactate dehydrogenase activities to the normal level at day 7.12 This phenomenon can be partially explained for the metabolism or excretion of nanoparticles via urine or feces with increasing time.
Mounting evidence suggests that exposure to ZnONPs can induce atherosclerotic alterations via direct and indirect actions. This process may involve oxidative stress, inflammation, cholesterol metabolism disorders, etc.29–31 Among these potential mechanisms, cholesterol metabolism disorders may be the central part of pathophysiological pathways, linking ZnONPs exposure with atherosclerosis. A recent investigation has shown the evidence that ZnONPs' exposure influences cholesterol metabolism by increasing total cholesterol, low-density lipoprotein and visceral fat while decreasing high-density lipoprotein in blood.29 Since liver is a significant organ for controlling cholesterol homeostasis, the damage in liver can undoubtedly affect the cholesterol biosynthesis and metabolism.5 Cholesterol biosynthesis is an enormously intricate and precisely regulated process mainly mediated by rate-limiting enzyme HMG-CoA.5 In the present study, we observed that the HMG-CoA protein expression was significantly elevated by ZnONPs exposure in mouse liver. Moreover, the master transcriptional regulator of HMG-CoA, SREBP-2 expression level showed a corresponding increase in response to ZnONPs treatment in a dose-dependent manner. These findings suggest that the enhancement of cholesterol in blood induced by ZnONPs possibly result from the over-activated cholesterol biosynthesis in liver.
There are two main potential mechanisms to explain how pulmonary ZnONPs exposure can initiate disturbance in cholesterol biosynthesis. On the one hand, ZnONPs deposited in the lung elicit local inflammatory responses that may further develop into systemic inflammation in the whole body organs, including liver.26 This inflammation reaction in liver can further regulate cholesterol biosynthesis, and therefore promote the excessive production of cholesterol.32 On the other hand, ZnONPs deposited in the lung can translocate into liver by the systemic circulation in blood and directly interact with liver tissues to induce injury or cholesterol biosynthesis disturbance.
The detailed mechanism underlying how ZnONPs cause cholesterol biosynthesis disturbance in liver remain largely unknown. Autophagy is regarded primarily as cell survival or death response to exogenous substances. It also plays a vital role in various pathologies, including liver diseases.33 In 2009, for the first time, Singh et al. showed a previously unknown function for autophagy in regulating intracellular lipid stores. They also revealed that inhibition of autophagy in cultured hepatocytes and mouse liver increased triglyceride storage in lipid droplets, suggesting that autophagy could regulate lipid metabolism.23 Since then, autophagy is considered as a new alternative lipid metabolic pathway and serves as a potential therapeutic target for diseases related to lipid metabolism disorders.
Given that autophagy has been shown to regulate cholesterol metabolism, we assumed that autophagy was involved in the regulation of cholesterol biosynthesis in liver. Based on this assumption, the typical biomarkers of autophagy were detected in response to ZnONPs treatment. Our results found that airway exposure to ZnONPs could trigger autophagy in the liver tissues, manifested by increasing protein expressions of LC3B, p62 and Beclin 1. We further used the beclin 1+/+ and beclin 1+/- mice to verify our hypothesis. Intriguingly, our data showed inhibition of autophagy by heterozygous disruption of beclin 1 not only extenuated the liver injuries but also relieved the disruption of cholesterol biosynthesis caused by ZnONPs. These findings demonstrate the inter-relationship between autophagy and cholesterol biosynthesis in ZnONPs liver toxicity. Autophagy regulates cholesterol biosynthesis because the loss of autophagy significantly rescues the ZnONPs-triggered HMG-CoA overexpression. Similar results were observed in beclin 1 knockout luteal cells in which fewer lipid droplets were found as compared with control cells.34 Their findings also revealed the significant differences in the cholesterol biosynthesis genes between wild type and beclin conditional knockout mice, and a significant reduction was found for HMG-CoA in the beclin 1 knockout group.34 More interestingly, previous studies have proposed the notion that inhibition of HMG-CoA can induce autophagy flux, manifested by LC3-II accumulation.35 The existing evidence indicates that there is a feedback network between autophagy and cholesterol biosynthesis.
The limitation of the current study is that we do not provide a detailed mechanism of how autophagy (or beclin 1) can regulate cholesterol biosynthesis. Another interesting question is whether other autophagy genes (besides beclin 1) would affect cholesterol biosynthesis. The future work is still required to address the underlying mechanisms on the interactions between autophagy and cholesterol biosynthesis, and also discover more key autophagic genes which can regulate cholesterol biosynthesis. It is expected that the modulation of autophagy might contribute to the prevention and treatment of ZnONPs-induced liver dysfunction.
In summary, we herein demonstrated for the first time that pulmonary exposure of ZnONPs caused the disturbance of cholesterol biosynthesis in mouse liver, and inhibition of autophagy by heterozygous disruption of the beclin 1 was capable of attenuating the disturbance of cholesterol biosynthesis and liver injuries induced by ZnONPs. These findings will provide the novel evidence that inhibition of autophagy by specific strategies will provide us a novel approach for the prevention or therapy of cholesterol biosynthesis disturbance and its related pathologies in liver induced by ZnONPs airway exposure.
Acknowledgments
This research was partially supported by National Science Foundation of China (81602820, 81703187, 81903358), Foundation and Frontier Research Program of Chongqing Municipal Science and Technology Commission (cstc2017jcyjAX0162), and the Science and Technology Research Program of Chongqing Municipal Education Commission (KJQN201800434, KJQN201900418, KJQN201900419, KJQN201900421).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Jiang J, Pi J, Cai J. The advancing of zinc oxide nanoparticles for biomedical applications. Bioinorg Chem Appl. 2018;2018:1–18. doi:10.1155/2018/1062562
2. Bondarenko O, Juganson K, Ivask A, Kasemets K, Mortimer M, Kahru A. Toxicity of Ag, CuO and ZnO nanoparticles to selected environmentally relevant test organisms and mammalian cells in vitro: a critical review. Arch Toxicol. 2013;87:1181–1200. doi:10.1007/s00204-013-1079-4
3. Rajput VD, Minkina TM, Behal A, et al. Effects of zinc-oxide nanoparticles on soil, plants, animals and soil organisms: a review. Environ Nanotechnol Monit Manag. 2018;9:76–84. doi:10.1016/j.enmm.2017.12.006
4. Liu J, Feng X, Wei L, Chen L, Song B, Shao L. The toxicology of ion-shedding zinc oxide nanoparticles. Crit Rev Toxicol. 2016;46:348–384. doi:10.3109/10408444.2015.1137864
5. Cerqueira NM, Oliveira EF, Gesto DS, et al. Cholesterol biosynthesis: a mechanistic overview. Biochemistry. 2016;55:5483–5506. doi:10.1021/acs.biochem.6b00342
6. Chistiakov DA, Bobryshev YV, Orekhov AN. Macrophage‐mediated cholesterol handling in atherosclerosis. J Cell Mol Med. 2016;20:17–28. doi:10.1111/jcmm.12689
7. Kang R, Zeh H, Lotze M, Tang D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011;18:571. doi:10.1038/cdd.2010.191
8. Pacheco CD, Lieberman AP. Lipid trafficking defects increase Beclin-1 and activate autophagy in Niemann-Pick type C disease. Autophagy. 2007;3:487–489. doi:10.4161/auto.4586
9. Pacheco CD, Kunkel R, Lieberman AP. Autophagy in Niemann–Pick C disease is dependent upon Beclin-1 and responsive to lipid trafficking defects. Hum Mol Genet. 2007;16:1495–1503. doi:10.1093/hmg/ddm100
10. Yan Q, Song Y, Zhang L, et al. Autophagy activation contributes to lipid accumulation in tubular epithelial cells during kidney fibrosis. Cell Death Discov. 2018;5:2.
11. Qu X, Yu J, Bhagat G, et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest. 2003;112:1809–1820. doi:10.1172/JCI20039
12. Jiang X, Tang Q, Zhang J, et al. Autophagy-dependent release of zinc ions is critical for acute lung injury triggered by zinc oxide nanoparticles. Nanotoxicology. 2018;12:1068–1091. doi:10.1080/17435390.2018.1513094
13. Zhang Y, Tu B, Jiang X, et al. Exposure to carbon black nanoparticles during pregnancy persistently damages the cerebrovascular function in female mice. Toxicology. 2019;422:44–52. doi:10.1016/j.tox.2019.04.014
14. Tang Q, Bai L, Zou Z, et al. Ferroptosis is newly characterized form of neuronal cell death in response to arsenite exposure. Neurotoxicology. 2018;67:27–36. doi:10.1016/j.neuro.2018.04.012
15. Bai L, Tang Q, Zou Z, et al. m6A demethylase FTO regulates dopaminergic neurotransmission deficits caused by arsenite. Toxicol Sci. 2018;165:431–446. doi:10.1093/toxsci/kfy172
16. Adamcakova-Dodd A, Stebounova LV, Kim JS, et al. Toxicity assessment of zinc oxide nanoparticles using sub-acute and sub-chronic murine inhalation models. Part Fibre Toxicol. 2014;11:15. doi:10.1186/1743-8977-11-15
17. Chen J-K, Ho -C-C, Chang H, et al. Particulate nature of inhaled zinc oxide nanoparticles determines systemic effects and mechanisms of pulmonary inflammation in mice. Nanotoxicology. 2015;9:43–53. doi:10.3109/17435390.2014.886740
18. Ness GC. Physiological feedback regulation of cholesterol biosynthesis: role of translational control of hepatic HMG-CoA reductase and possible involvement of oxylanosterols. Biochim Biophys Acta Mol Cell Biol Lipids. 2015;1851:667–673.
19. DeBose-Boyd RA. Feedback regulation of cholesterol synthesis: sterol-accelerated ubiquitination and degradation of HMG CoA reductase. Cell Res. 2008;18:609. doi:10.1038/cr.2008.61
20. Horton JD, Goldstein JL, Brown MS. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest. 2002;109:1125–1131. doi:10.1172/JCI0215593
21. Bengoechea-Alonso MT, Ericsson J. SREBP in signal transduction: cholesterol metabolism and beyond. Curr Opin Cell Biol. 2007;19:215–222. doi:10.1016/j.ceb.2007.02.004
22. Sato R. Sterol metabolism and SREBP activation. Arch Biochem Biophys. 2010;501:177–181. doi:10.1016/j.abb.2010.06.004
23. Singh R, Kaushik S, Wang Y, et al. Autophagy regulates lipid metabolism. Nature. 2009;458:1131. doi:10.1038/nature07976
24. Li C-H, Shen C-C, Cheng Y-W, et al. Organ biodistribution, clearance, and genotoxicity of orally administered zinc oxide nanoparticles in mice. Nanotoxicology. 2012;6:746–756. doi:10.3109/17435390.2011.620717
25. Wang L, Wang L, Ding W, Zhang F. Acute toxicity of ferric oxide and zinc oxide nanoparticles in rats. J Nanosci Nanotechnol. 2010;10:8617–8624. doi:10.1166/jnn.2010.2483
26. Vandebriel RJ, De Jong WH. A review of mammalian toxicity of ZnO nanoparticles. Nanotechnol Sci Appl. 2012;5:61. doi:10.2147/NSA
27. Jacobsen NR, Stoeger T, Van Den Brûle S, et al. Acute and subacute pulmonary toxicity and mortality in mice after intratracheal instillation of ZnO nanoparticles in three laboratories. Food and Chem Toxicol. 2015;85:84–95. doi:10.1016/j.fct.2015.08.008
28. Fukui H, Horie M, Endoh S, et al. Association of zinc ion release and oxidative stress induced by intratracheal instillation of ZnO nanoparticles to rat lung. Chem Biol Interact. 2012;198:29–37. doi:10.1016/j.cbi.2012.04.007
29. Yan Z, Wang W, Wu Y, et al. Zinc oxide nanoparticle-induced atherosclerotic alterations in vitro and in vivo. Int J Nanomedicine. 2017;12:4433. doi:10.2147/IJN.S134897
30. Wang M, Yang Q, Long J, et al. A comparative study of toxicity of TiO2, ZnO, and Ag nanoparticles to human aortic smooth-muscle cells. Int J Nanomedicine. 2018;13:8037. doi:10.2147/IJN.S188175
31. Chuang K-J, Lee K-Y, Pan C-H, et al. Effects of zinc oxide nanoparticles on human coronary artery endothelial cells. Food and Chem Toxicol. 2016;93:138–144. doi:10.1016/j.fct.2016.05.008
32. Hajjar DP, Hajjar KA. Alterations of cholesterol metabolism in inflammation-induced atherogenesis. J Enzymol Metab. 2016;1(1).
33. Rautou P-E, Mansouri A, Lebrec D, Durand F, Valla D, Moreau R. Autophagy in liver diseases. J Hepatol. 2010;53:1123–1134. doi:10.1016/j.jhep.2010.07.006
34. Gawriluk TR, Ko C, Hong X, Christenson LK, Rucker EB. Beclin-1 deficiency in the murine ovary results in the reduction of progesterone production to promote preterm labor. Proc Natl Acad Sci. 2014;111:E4194–E203. doi:10.1073/pnas.1409323111
35. Ghavami S, Yeganeh B, Stelmack G, et al. Apoptosis, autophagy and ER stress in mevalonate cascade inhibition-induced cell death of human atrial fibroblasts. Cell Death Dis. 2012;3:e330. doi:10.1038/cddis.2012.61
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.