")
Back to Journals » International Journal of Nanomedicine » Volume 11
Heterogeneous response of different tumor cell lines to methotrexate-coupled nanoparticles in presence of hyperthermia
Authors Stapf M, Pömpner N, Teichgräber U, Hilger I
Received 13 August 2015
Accepted for publication 21 October 2015
Published 4 February 2016 Volume 2016:11 Pages 485—500
DOI https://doi.org/10.2147/IJN.S94384
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 6
Editor who approved publication: Dr Thomas Webster
Marcus Stapf,* Nadine Pömpner,* Ulf Teichgräber, Ingrid Hilger
Institute of Diagnostic and Interventional Radiology, Department of Experimental Radiology, Jena University Hospital, Friedrich-Schiller University, Jena, Germany
*These authors contributed equally to this work
Abstract: Today, the therapeutic efficacy of cancer is restricted by the heterogeneity of the response of tumor cells to chemotherapeutic drugs. Since those therapies are also associated with severe side effects in nontarget organs, the application of drugs in combination with nanocarriers for targeted therapy has been suggested. Here, we sought to assess whether the coupling of methotrexate (MTX) to magnetic nanoparticles (MNP) could serve as a valuable tool to circumvent the heterogeneity of tumor cell response to MTX by the combined treatment with hyperthermia. To this end, we investigated five breast cancer cell lines of different origin and with different mutational statuses, as well as a bladder cancer cell line in terms of their response to exposure to MTX as a free drug or after its coupling to MNP as well as in presence/absence of hyperthermia. We also assessed whether the effects could be connected to the cell line-specific expression of proteins related to the uptake and efflux of MTX and MNP. Our results revealed a very heterogeneous and cell line-dependent response to an exposure with MTX-coupled MNP (MTX–MNP), which was almost comparable to the efficacy of free MTX in the same cell line. Moreover, a cell line-specific and preferential uptake of MTX–MNP compared with MNP alone was found (probably by receptor-mediated endocytosis), agreeing with the observed cytotoxic effects. Opposed to this, the expression pattern of several cell membrane transport proteins noted for MTX uptake and efflux was only by tendency in agreement with the cellular toxicity of MTX–MNP in different cell lines. Higher cytotoxic effects were achieved by exposing cells to a combination of MTX–MNP and hyperthermal treatment, compared with MTX or thermotherapy alone. However, the heterogeneity in the response of the tumor cell lines to MTX could not be completely abolished – even after its combination with MNP and/or hyperthermia – and the application of higher thermal dosages might be necessary.
Keywords: magnetic nanoparticles, SPION, in vitro, methotrexate, hyperthermia, breast cancer, bladder cancer
Introduction
The heterogeneity of tumors dramatically impacts a patient’s survival due to a selective response of differently dedifferentiated cell populations to the respective cancer treatment.1 Based on this circumstance, the limited efficacy of a single treatment, for example, a single chemotherapeutic drug, is not surprising. For this reason, several chemotherapeutic drugs are usually combined in the clinics in order to target multiple cellular signaling pathways and increase the antitumor effect.2 Nevertheless, their dosage in cancer treatment is restricted due to severe side effects affecting the whole body, as they were mostly applied intravenously and do not exert their effects solely at the tumor region. As a consequence, drug-based treatments were often applied in several cycles and used in combination with other therapies like radiation. In spite of several advantages to increase therapeutic efficacy, the problems related to the occurrence of side effects still remain.
To overcome these drawbacks, a combination of localized antitumor therapies is preferential. In this regard, magnetic nanoparticles (MNP) working as drug carriers after being coupled to (eg, chemotherapeutic) drugs can provide a handy alternative. In particular, systemically applied MNP can be specifically enriched in the tumor region by magnetic forces (magnetic targeting). Hereto, MNP will be able to deposit their cargo (eg, a coupled chemotherapeutic drug) at the target site whereby unwanted side effects can be reduced.3–7 Moreover, MNP can be heated in an alternating magnetic field, allowing a localized sensitization or destruction of tumor cells or tumor tissue by hyperthermal or even thermoablative temperatures.8–11 For magnetic heating purposes, iron oxide MNP with a clustered magnetite or maghemite core and an appropriate coating (polyethylene glycol [PEG], dextran, dimercaptosuccinic acid [DMSA], etc) have been shown to exhibit good heating capabilities and biocompatibility.10,12–16
One chemotherapeutic drug that can effectively be coupled to MNP is methotrexate (MTX). By this approach, combinatory treatments consisting of MTX-coupled MNP (MTX–MNP) and magnetic hyperthermia have the capability of interfering with multiple phases of the cell cycle, as MTX is known to act, for example, at the G1/S transition (eg, probably by restoring p53 pathways), whereas hyperthermia treatments are reported to act mostly in later phases like S or M phase.17–21
MTX is a structural analog of folate (antifolate) that inhibits key enzymes of the purine and pyrimidine synthesis by targeting dihydrofolate reductase and thymidylate synthetase. The inhibition of specific steps of the folate metabolism leads to a depletion of intracellular folates, which finally provoke cytotoxic effects, particularly by impairing DNA synthesis, methylation, and repair.22–24 However, MTX shows heterogeneous toxicity and resistances among different tumor entities, which result from several mechanisms including alterations in various transport mechanisms responsible for the uptake and/or efflux of MTX from the cell.25
The two main transport mechanisms responsible for the uptake of MTX by cells are the folate receptor (FR) and the reduced folate carrier (RFC). Among them, the unidirectional FRα is overexpressed in the majority of cancer tissues. As such, it has been considered as target in several therapeutic strategies.26–33 The bidirectional RFC, which is ubiquitously expressed in mammalian cells and reported to be overexpressed in cancer, is also an important uptake mechanism of folate and antifolates.34–41 An impairment of RFC synthesis or mutations in the RFC protein can lead to cellular dysfunctions and therefore to resistances against chemotherapeutic drugs like MTX.26,36 Besides the RFC, which also participates in the efflux of MTX from the cells due to its bidirectionality, the efflux of MTX from the cells is additionally dictated by several cell membrane transport proteins, including the breast cancer resistance protein (BCRP), and different members of the multidrug resistance protein (MRP) family, including MRP1 and MRP5. Interestingly, short-time resistances to MTX seem to be related at least to the latter mentioned proteins.26,42–48
Although fundamental knowledge about the mechanisms that are involved in mediating MTX resistance is available, up to now, there is almost no understanding about the feasibility of hyperthermia for overcoming the heterogenic and unfavorable variation in MTX response in tumor cells. Moreover, despite the aforementioned advantages of using MNP for cancer treatment, the potential of MTX–MNP in overcoming the heterogenic cytotoxicity observed for free MTX, especially when combined with hyperthermia, is mostly unknown. MTX–MNP offer a promising tool for a localized tumor treatment with low systemic effects.
Here, we studied if the large variability of MTX cytotoxicity to tumor cells could be restricted by the combination with MNP and hyperthermia. To this end, we coupled MTX to MNP to model the feasibility of the used MNP as drug carriers. Moreover, the cellular uptake of MTX–MNP as well as the release kinetic of MNP-bound MTX was analyzed to confirm the suitability of MTX as targeting agent, estimate the potential of MTX–MNP in generating drug depots after cellular internalization, and determine the feasibility of MTX–MNP for multimodal treatments. The presence of hyperthermia was modeled via the utilization of hot air hyperthermia, since hot air hyperthermia offers the exposure of cells to homogeneous temperatures in vitro independently of the internalized amount of MNP. In this regard, a combinatory treatment consisting of MTX–MNP and hot air hyperthermia (1 hour, 44°C) was performed to study the influence of hyperthermia on MTX–MNP-related cytotoxicity of selected cancer cell lines and assess the potential of MTX–MNP and hyperthermia in later in vivo applications when used as single or combinatory/multimodal treatment. With consideration of tumors to be prospectively treated, we focused our investigations on breast and bladder cell lines, as these tumor entities will exhibit a good accessibility for magnetic heating in later applications (eg, for magnetic field inductors) due to their localization outside the body or in a hollow organ. Moreover, multiple cell lines with a different mutational status (including p53, H-ras, wnt), grade of differentiation, and origin were chosen to address the heterogeneity of tumors. To correlate the uptake of MTX–MNP with cytotoxic effects, the expression of the FR, RFC, and various ATP-binding cassette transporters was analyzed on mRNA and protein level.
Materials and methods
Cells and cell culture conditions
All breast cancer cell lines (T47D, MDA-MB-231, MX1, AU-565, and SK-BR-3) as well as the bladder cancer T24 and the nontumor cell line HMEC-1, were cultured as recommended by the vendor (CLS Cell Lines Service, Eppelheim, Germany; or American Type Culture Collection [ATCC], Wesel, Germany) supplemented with 10% (T47D, MX1, AU-565, SK-BR-3, HMEC-1) or 5% (MDA-MB-231, T24) fetal bovine serum (Life Technologies, Grand Island, NE, USA). Ethical approval was deemed not necessary as neither human tissues nor primary human cell lines were used.
Preparation of MTX-coupled MNP
The PEG-coated and amine-functionalized superparamagnetic iron oxide nanoparticles (hydrodynamic diameter: 130 nm Nanomag®-D PEG-NH2) were obtained from micromod (micromod Partikeltechnologie GmbH, Rostock, Germany). They consisted of a multicore, which was composed of several smaller single core particles (~12 nm), giving a total core size of ~115–125 nm.10 MNP were covalently coupled to the carboxyl groups of MTX (AppliChem, Darmstadt, Germany) using carbodiimide method by utilizing the coupling agent 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC, Sigma-Aldrich, St Louis, MO, USA). In brief, equal amounts of EDC and MTX were dissolved in 0.1 M MES (pH 5) and carefully mixed at ambient temperature to allow activation of the carboxyl groups of MTX. PEG-NH2 MNP were added to the solution at the same amount as MTX and incubated for 2 hours under steady mixing. MTX-conjugated MNP (MTX–MNP) were separated by a permanent magnet and washed with phosphate buffered saline (PBS) until no MTX in supernatant could be detected. For the validation of the coupling efficiency, the obtained supernatants were collected and the free MTX was quantitatively measured by ultraviolet/visible (UV/Vis) spectrometry using an Ultrospec 4300 pro UV/Visible Spectrophotometer (Amersham Pharmacia Biotech, Freiburg, Germany). In this context, the MTX–MNP content was measured indirectly by quantifying the amount of uncoupled MTX in the supernatant of the washing steps at a wavelength of 302 nm.
MTX release kinetics
The release of MTX from the particle’s surface was analyzed via UV/Vis spectrometry at 302 nm as described before. In this regard, washed MTX–MNP were incubated for another 2, 24, 48, or 72 hours with PBS under physiological conditions (pH 7.4), with a subsequent collection of the related supernatants. Moreover, acidic conditions, as present in endosomes (pH 6–6.5) and lysosomes (pH 4.5–5) after an uptake of MTX–MNP into cells were simulated by using PBS pH 5 and the same points in time.49
Particle characterization
The hydrodynamic diameter of the used MNP formulations was measured in quintuplicates at a concentration of 50 μg/mL in bidistilled water by dynamic light scattering using a Zetasizer Nano ZS and DTS1061 disposable capillary cells (both Malvern Instruments GmbH, Herrenberg, Germany) with a measurement angle of 173° backscatter. Within this publication, the hydrodynamic diameter refers to the z-average, the most proper value for size determination provided by dynamic light scattering for small polydispersity indices (<0.3). Coincidently, the ζ-potential, which represents the particle’s surface charge, was measured in triplicates. The standard error of the mean was calculated by using the mean values of at least five independent experiments. Used particles showed a sufficient heating capability (specific absorption rate) for later in vivo applications as determined in earlier experiments.10
In vitro cytotoxicity assay
At 24 hours after cell seeding, MTX–MNP or MNP in equal concentrations (100 μg/mL) were mixed with fresh culture medium and added to the cells. In the free MTX group, the MTX concentration (10 μg/mL) was adapted to the coupling efficiency of MTX–MNP (100 μgMTX/mgMNP). As an additional control (“none”), cells in culture medium without further additives were used. After an incubation time of either 48 or 72 hours with MTX–MNP, MTX, MNP, or “none”, the cells were washed with Hank’s balanced salt solution (HBSS, Biochrom GmbH, Berlin, Germany) and fresh medium was added. Cellular viability was assessed based on the mitochondrial dehydrogenase activity using AlamarBlue®Cell Proliferation Reagent (1:10; Life Technologies, Thermo Fischer Scientific, Langenselbold, Germany).50 Fluorescence measurements (excitation/emission: 545/590 nm) were performed in a fluorescence spectrometer (Tecan Infinite M1000 Pro, Tecan Trading AG, Männedorf, Switzerland) according to the manufacturer’s instructions.
For calculation of median effective dose (ED50) values, the cells were treated with MTX at concentrations ranging from 0.22 pM to 220 μM in the culture medium. ED50 values with the corresponding standard errors were calculated from sextuplicates using Origin (Origin 9.0.0G, OriginLab Corp., Northampton, MA, USA).
Cellular uptake of nanoparticles
To investigate uptake of MNP in dependency of coupled MTX in the different cell lines, Prussian blue staining of intracellular iron was performed. In particular, three exemplary cell lines exhibiting a high (T24), medium (AU-565), or low (SK-BR-3) sensitivity after an incubation of 48 hours with either MTX–MNP or MTX alone were chosen. At 24 hours after seeding, the cells were washed and equal particle concentrations (100 μg/mL) of either MNP or MTX–MNP were added. After further 24, 48, or 72 hours of incubation, the cells were washed again to remove free nanoparticles and fixed with 4% formaldehyde (Carl Roth GmbH & Co. KG, Karlsruhe, Germany). Afterward, slides were treated with 20% HCl (Carl Roth GmbH & Co.) and 10% potassium hexacyanoferrate (II) trihydrate (Sigma-Aldrich GmbH, Steinheim, Germany) solution in order to stain the iron of intrinsic nanoparticles. Cell nuclei were stained with nuclear fast red (Carl Roth GmbH & Co.). Slides were dehydrated using ascending alcohol concentration and embedded in Pertex® (Medite GmbH, Burgdorf, Germany).
For semiquantitative analyses of cellular MNP uptake, the cells in three randomly chosen fields of view per cell line (T24, AU-565, and SK-BR-3) were counted and categorized by three independent observers according to the incorporated MNP amount into “high uptake”, “low uptake”, and “no uptake”. The number of cells in each category was normalized to the total number of cells of the specific field of view to calculate the relative values for the particle uptake in each category. Statistical calculations concerning the differences in the particle uptake in dependency of MNP-coupled MTX were performed with SPSS (V21, IBM Corporation, Armonk, NY, USA) using a linear mixed model and the compound symmetry with correlation parameterization method.
mRNA expression of selected genes involved in MTX metabolization
Quantitative real-time reverse transcription polymerase chain reaction was performed for mRNA expression analysis of FRα, RFC, BCRP, MRP1, and MRP5. In this context, total RNA was isolated from cell pellets using the High Pure RNA Isolation Kit (Roche, Mannheim, Germany) and transcribed into cDNA by a peqSTAR 96 Universal thermocycler (PEQLAB Biotechnologie GmBH, Erlangen, Germany) according to the manufacturer’s manual. All primers (Table 1) were synthesized by Jena Bioscience GmbH (Jena, Germany) except for FRα, which was purchased from BIOMOL GmbH (Hamburg, Germany). β-2-microglobulin was used as reference gene. Negative controls consisted of RNAse-free water instead of cDNA. Experiments were performed in triplicates within each of the three independent experiments. Product amplification was measured via the LightCycler® 480 SYBR Green I Master (Roche, Germany) and the Mastercycler® ep realplex4 S device (Eppendorf AG, Hamburg, Germany). The purity of amplified products was verified by melting curve analysis. Primer efficiency was determined by REST software (V2.0.13, Qiagen GmbH, Hilden, Germany) after measuring dilution series of a mixture of all used cDNA samples. The expression of genes was calculated with REST software by using the nontumor cell line HMEC-1 as control group and the tumor cell lines as treatment group.
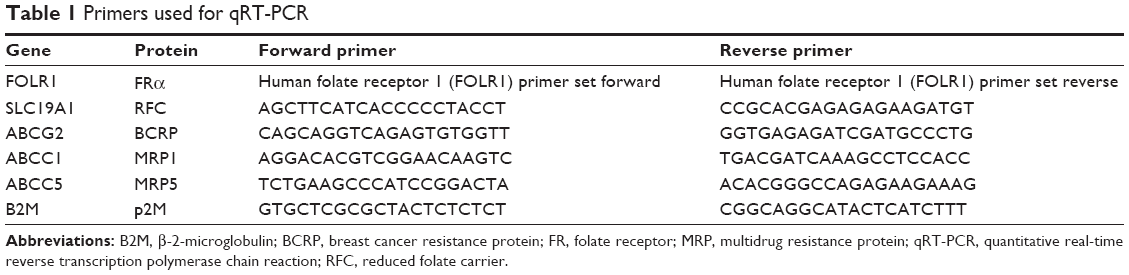 | Table 1 Primers used for qRT-PCR |
Expression of selected proteins involved in MTX metabolization
Total protein of cell pellets was extracted by using the radioimmunoprecipitation assay lysis buffer supplemented with protease inhibitors (complete, mini, ethylenediaminetetraacetic acid-free, Hoffman-La Roche Ltd., Basel, Switzerland). For sodium dodecyl sulfate polyacrylamide gel electrophoresis and electroblotting to a polyvinylidene difluoride membrane (Immobilon®-P, EMD Millipore Corporation, Billerica, MA, USA), either 30 μg (FRα, BCRP) or 50 μg (RFC, MRP1, MRP5) of protein of each sample was mixed with sodium dodecyl sulfate sample buffer and loaded onto a 8% (for MRP5), 10% (for MRP1, RFC, and BCRP), or 12% (for FRα) polyacrylamide gel.51 Afterward, the blotted polyvinylidene difluoride membrane was incubated with blocking buffer (1× PBS, 0.2% Tween, 5% nonfat dry milk, or bovine serum albumin). Membranes were incubated with primary antibodies against FRα (anti-folate binding protein antibody [EPR4708(2)], rabbit antihuman), MRP1 (anti-MRP1 antibody [EPR4658(2)] – C-terminal, rabbit antihuman), MRP5 (anti-MRP5 antibody, rabbit antihuman), or BCRP (anti-BCRP/ABCG2 antibody [ERP2099(2)], rabbit antihuman) purchased from Abcam Inc. (Cambridge, UK). The used RFC (anti-SLC19A1 antibody, rabbit antihuman) antibody was acquired from BIOZOL Diagnostica Vertrieb GmbH (Eching, Germany). Antibody against beta actin (anti-beta actin antibody, mouse antihuman) was purchased from Abcam Inc. and served as loading control.
Afterward, membranes were incubated with the appropriate secondary horseradish peroxidase-conjugated antibodies (goat anti-rabbit IgG-horseradish peroxidase, Santa Cruz Biotechnology, Inc., Heidelberg, Germany). For protein detection, Western blot chemiluminescent HRP substrate (EMD Millipore Corporation) and a digital imaging system (ImageQuant LAS 4000, GE Healthcare Europe GmbH, Freiburg, Germany) were used.
Hyperthermia treatment
For hyperthermal treatment, the cells were seeded and cultured as described before. To allow the cellular internalization of either MTX–MNP or MNP, at 24 hours after the addition of MTX, MNP, or MTX–MNP, the cells were exposed to 44°C (Figure 1). By using an external heat source (hot air), homogeneous temperatures and comparability among different treatment groups independent of the presence of MNP was achieved. The target temperature was maintained for 60 minutes. Subsequently, the cells were post-incubated at 37°C until the assessment of cellular viability after a total incubation time of either 48 or 72 hours with MTX–MNP, MTX, MNP, or “none”. The thermal isoeffect dose, expressed as cumulative equivalent minutes at a reference temperature of 43°C, was calculated to ensure comparability between different experiments.52
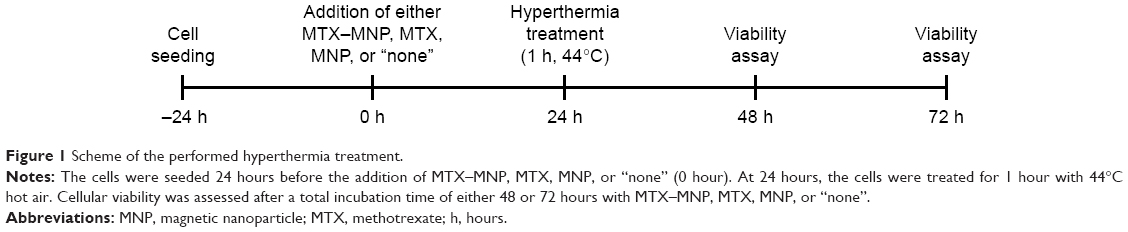 | Figure 1 Scheme of the performed hyperthermia treatment. |
Results
Particle characteristics
Dynamic light scattering measurements revealed a slight increase in the hydrodynamic diameter from 163 to 173 nm of the MNP after coupling of MTX (Table 2). Additionally, MTX coupling resulted in a slightly more negative ζ-potential (−21 vs −23 mV). In all cases, the polydispersity indices were smaller than 0.3. The coupling efficiency for the used MTX–MNP was calculated to be 100 μgMTX/mgMNP. MTX–MNP showed a very stable MTX binding with almost no MTX release (<0.1%) under physiological (pH 7.4) and acidic (pH 5) conditions over 72 hours (data not shown).
 | Table 2 Hydrodynamic diameter and ζ-potential of MNP is slightly increased after MTX coupling (MTX–MNP) |
Time- and cell line-dependent cytotoxicity of free MTX and MTX–MNP
As depicted in Table 3, the lowest ED50 values were observed for the bladder cancer cell line T24 (16.7 nM). Selected breast cancer cell lines showed a high variability of ED50 values ranging from 52.6 nM to above 22 μM. Nevertheless, no correlation between cellular toxicity and cellular doubling times were found (data not shown).
 | Table 3 Heterogenic and cell line-dependent ED50 values after MTX incubation |
In regard to the dependency of the incubation time either with MTX–MNP, MTX, or MNP on the cellular dehydrogenase level as a marker for cellular viability, distinct relationships were found (Figure 2). In particular, after 48 hours of incubation, the lowest viability (dehydrogenase activity normalized to nontreated controls) was found for MTX–MNP regarding T24 cells (18%) whereas a medium viability could be observed for AU-565 (46%) and MDA-MB-231 (40%) cells. MTX–MNP exhibited the lowest impact on cellular viability for SK-BR-3 (75%), MX1 (88%), and T47D (80%) cells. Concerning the impact of free MTX on cells, the MTX control group revealed almost the same cytotoxicity profile for T24 (16%), AU-565 (41%), and MDA-231 (38%) in comparison to the effects after binding of MTX to MNP. A higher cytotoxicity in the MTX control group was found for SK-BR-3 (65%) and T47D (70%) as opposed to the MTX–MNP group. The largest impact of free MTX on cellular viability was measured for MX1 cells (54% of nontreated cells; reduction of ~34% compared with MTX–MNP). Native nanoparticles (MNP control group) showed rather low effects after 48 hours of incubation for all cell lines (>90%) except for MDA-MB-231 (68%).
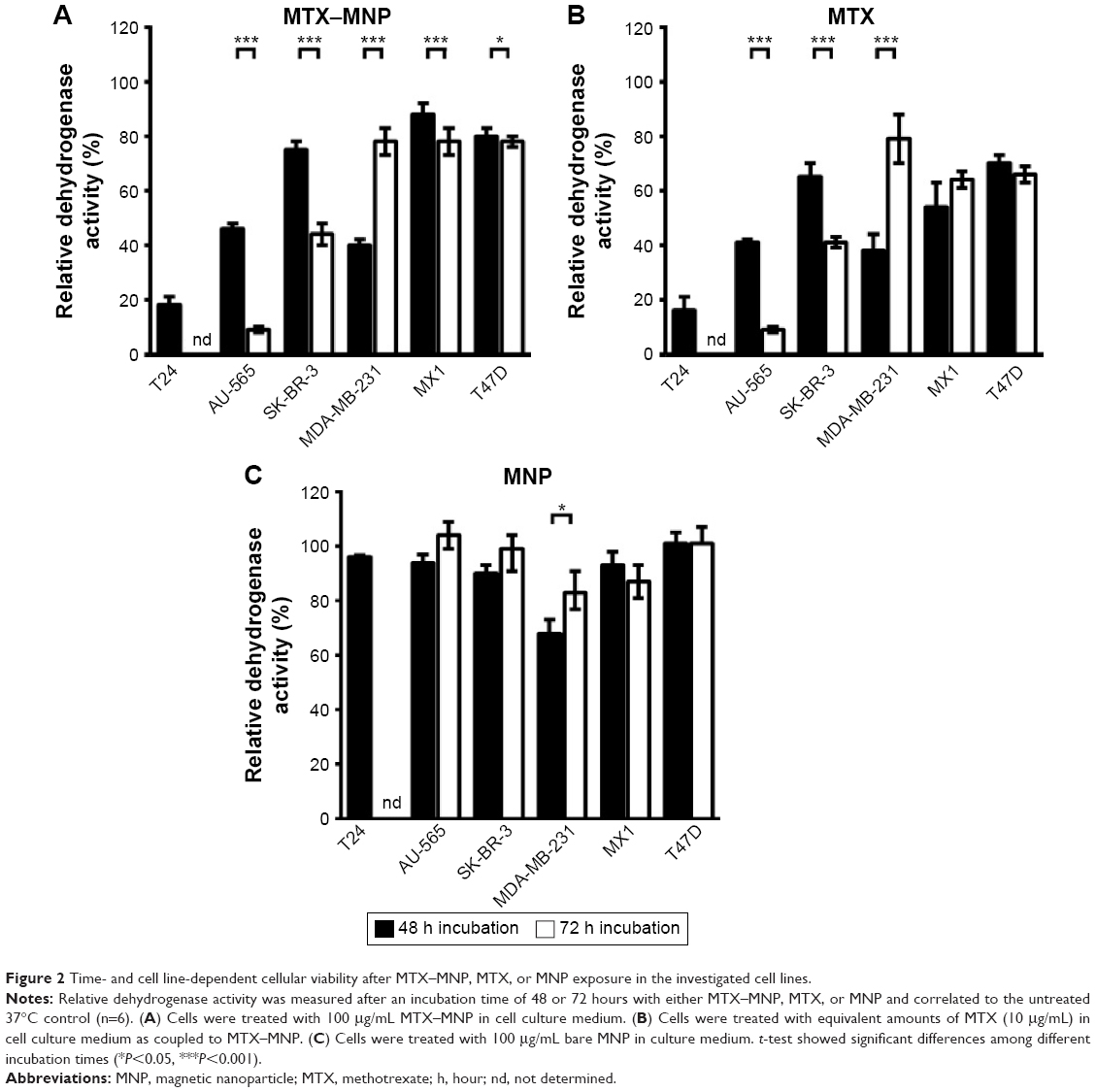 | Figure 2 Time- and cell line-dependent cellular viability after MTX–MNP, MTX, or MNP exposure in the investigated cell lines. |
The extension of the incubation time with MTX–MNP, MTX, or MNP from 48 to 72 hours did further change the response pattern of the different cell lines compared with a 48 hours incubation period. In the MTX–MNP group, the cellular viability was remarkably reduced for AU-565 (to 9%; −37%), SK-BR-3 (to 44%; −31%), and MX1 (to 78%; −10%) cells, whereas it remained almost unchanged in T47D (78%; −2%) cells. Interestingly, a dramatic recovery of viability was found for MDA-MB-231 (to 78%; +38%) cells after 72 hours. At the same time, free MTX (MTX control group) revealed a comparable regulation of the cellular viability as MTX bound to MNP (MTX–MNP group). In this regard, the viability of AU-565 (9%), SK-BR-3 (41%), and T47D (66%) was reduced after extended incubation times, whereas it was dramatically increased for MDA-MB-231 (79%) and slightly increased for MX1 (64%) cells. Again, MNP alone (MNP control group) showed almost no cytotoxic effects (>87%) even for MDA-MB-231 (83%) cells.
Taken together, MTX–MNP exhibited a comparable and cell line-dependent cytotoxicity as observed for free MTX. Concurrently, MNP alone showed almost no cytotoxic effects indicating the MTX-mediated cytotoxicity of MTX–MNP.
MTX influences the uptake of MNP in a cell line-dependent manner
Representative microscopy images of three exemplarily chosen cell lines (T24, AU-565, and SK-BR-3) which exhibed a varying MTX sensitivity towards MTX–MNP or MTX alone, revealed a preferential uptake of MTX–MNP in comparison to bare MNP in dependence of the cell line and incubation time (Figure 3, left panel). As opposed to the treatment with bare MNP, a reduced cell number and an altered cellular morphology was found for the MTX–MNP group, indicating a higher cytotoxicity of MTX–MNP. These results were in good agreement with the cellular viability as measured by dehydrogenase level.
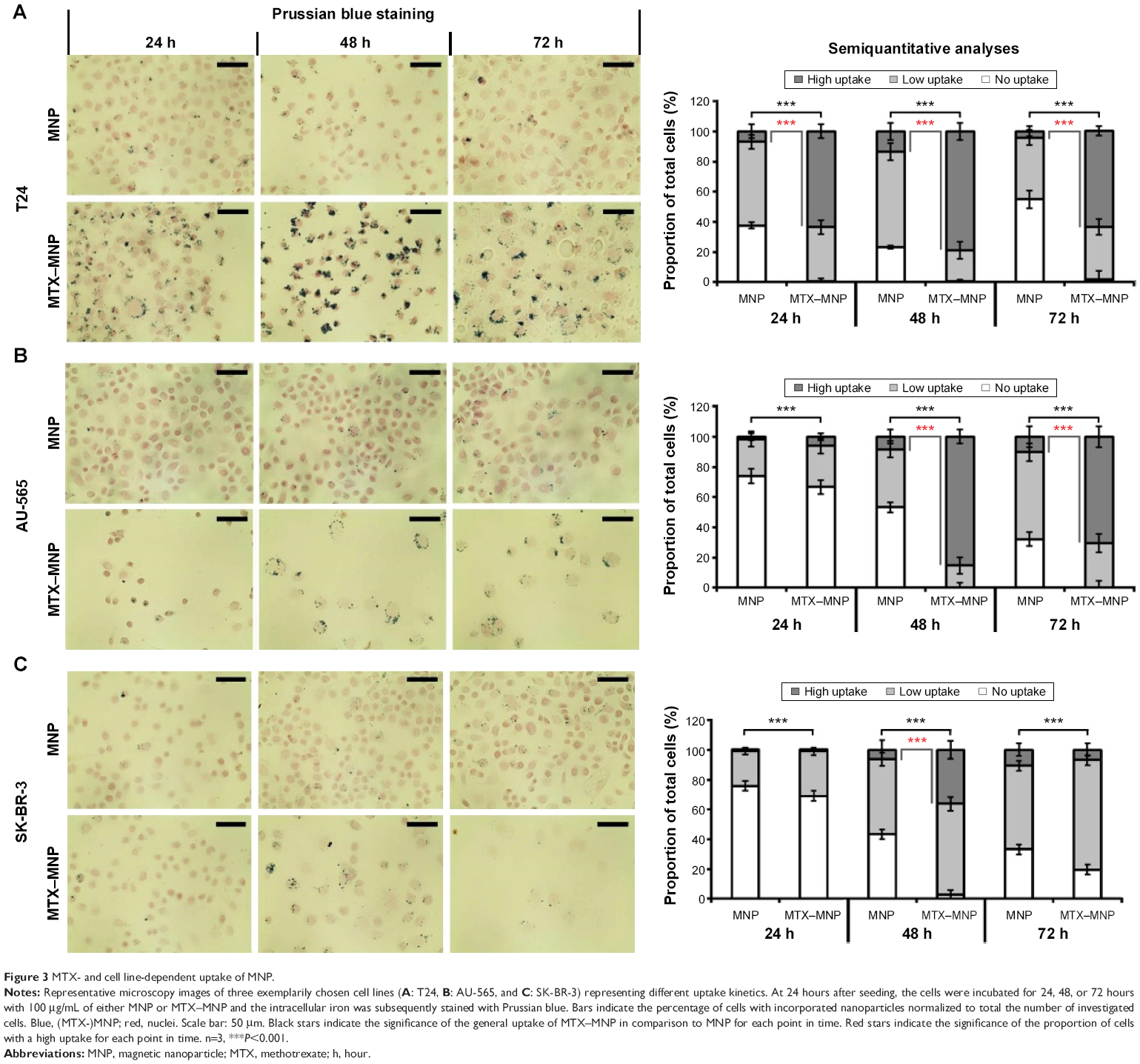 | Figure 3 MTX- and cell line-dependent uptake of MNP. |
Semiquantitative analyses of intracellular iron staining confirmed the microscopy images (Figure 3, right panel). By tendency, a significantly (P<0.001) increased uptake of MTX–MNP in contrast to MNP alone was found for all investigated cell lines and points in time (Figure 3, light gray and dark gray bars). Moreover, a significantly (P<0.001) higher proportion of cells exhibiting a remarkable uptake of MTX–MNP was found for all points in time (24–72 hours) in the case of T24 cells. On the contrary, AU-565 cells showed only a significantly (P<0.001) higher proportion of cells with a high uptake of MTX–MNP at longer incubation times (48 and 72 hours). SK-BR-3 cells revealed only at one point in time (48 hours) a significantly (P<0.001) higher proportion of cells with a high uptake of MTX–MNP. The fastest and strongest uptake of MTX–MNP was found for T24 cells, followed by AU-565 and SK-BR-3. In most cell lines, the uptake of bare MNP increased with longer incubation time (except for T24, 72 hours) but not as strong as found for MTX–MNP.
Heterogeneous mRNA and protein levels of FRα, RFC, BCRP, MRP1, and MRP5 among different cell lines
The expression pattern of mRNA (normalized to HMEC-1) and proteins responsible for MTX–MNP uptake (FRα), efflux (BCRP, MRP1, MRP5), or both (RFC) were found to be heterogeneous among the investigated cell lines (Figure 4).
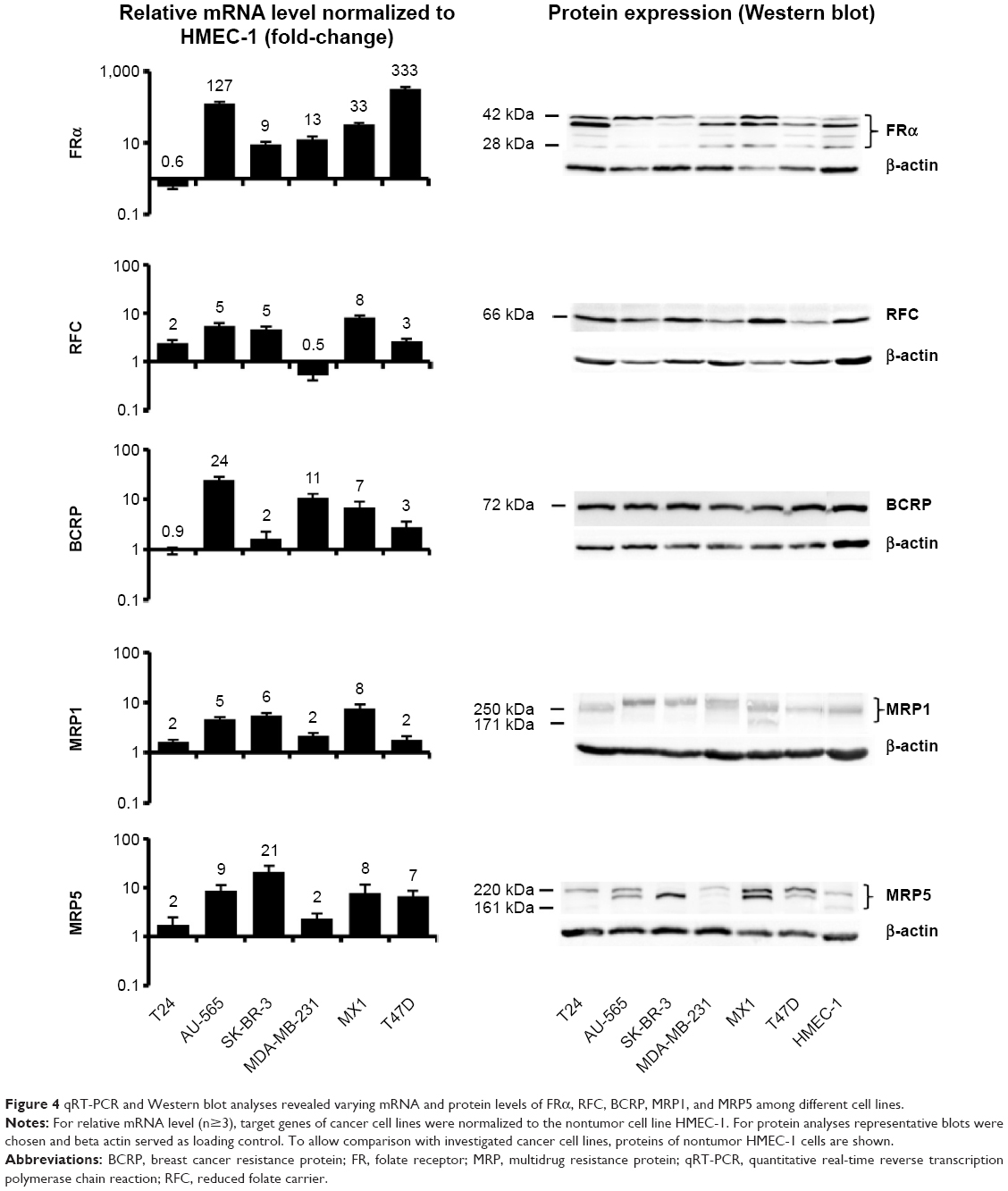 | Figure 4 qRT-PCR and Western blot analyses revealed varying mRNA and protein levels of FRα, RFC, BCRP, MRP1, and MRP5 among different cell lines. |
In the case of the FRα, the heterogeneity on the mRNA level was visible by a high expression, especially in AU-565 and T47D cells, whereas a low expression was observed in T24 cells. On the protein level, expression heterogeneity was visible not only by the degree of expression but also by the presence of several protein forms (between 28 and 42 kDa).
A similar situation was observed in relation to the RFC. On the mRNA level, the highest expression was visible in MX1 cells, whereas the lowest expression was observed in MDA-MB-231 cells. On the protein level, the RFC was expressed to a different degree among the cell lines but only in one protein variant (66 kDa).
BCRP showed an upregulation on the mRNA level for almost all cell lines except for T24 cells. On the protein level, the expression was nearly homogeneous among all investigated cell lines (comparable degree of expression, one protein form at 72 kDa).
MRP1 mRNA was highly overexpressed in all cell lines between two- and eightfold compared with the nontumor cell line HMEC-1. A comparable protein expression pattern was detected in all investigated cell lines, even though the single proteins exhibited diverging molecular weights (171–300 kDa).
At the same time, the MRP5 mRNA expression was elevated in SK-BR-3 and lowest in T24 and MDA-MB-231 cells. On protein level, the degree of expression was heterogeneous among the cell lines, and several protein forms with molecular weights ranging from 161 to 220 kDa were present.
Mostly higher cytotoxic effects of MTX–MNP after hyperthermal treatment
In all heating experiments, the target temperature of 44°C was reached within the first 10 minutes and maintained for further 60 minutes. As a result, comparable cumulative equivalent minutes at a reference temperature of 43°C values (~130 minutes) were achieved among different hyperthermia treatments. Furthermore, the cellular viability after 48 and 72 hours was in accordance to microscopic observations. In each control (“none”, MNP, MTX) and the treatment (MTX–MNP) group, significant (P≤0.05) additive cytotoxic effects could be observed in combination with hyperthermia for both points in time and most investigated cell lines (Figure 5, white bars) compared with the treatment without hyperthermia (Figure 5, black bars).
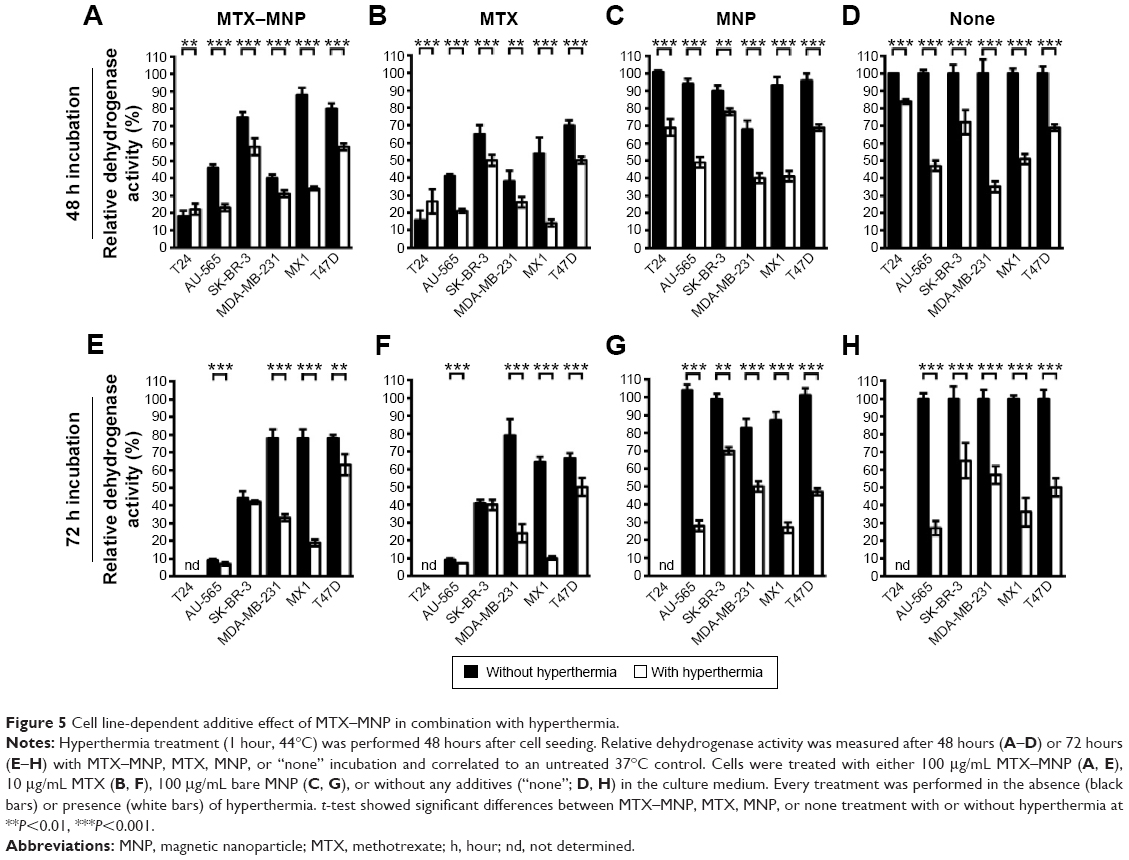 | Figure 5 Cell line-dependent additive effect of MTX–MNP in combination with hyperthermia. |
In particular, after 48 hours of incubation with MTX–MNP, the combination with hyperthermia resulted in a significant (P<0.001) decrease of the cellular viability in most cell lines compared with MTX–MNP treatment alone (Figure 5A). In this regard, the most dramatic impact was found for MX1 (34% vs 88%) and AU-565 (23% vs 46%) cells, whereas a medium impact for SK-BR-3 (58% vs 75%) and T47D (58% vs 80%) cells was observed. MDA-MB-231 showed only a small reduction of cellular viability (31% vs 40%) and T24 cells remained almost unaltered (22% vs 18%). The effect of free MTX was occasionally higher than that of MTX–MNP, regardless of the presence of hyperthermia (Figure 5B). In this regard, especially MX1 cells showed an increased sensitivity of free MTX in contrast to MNP-bound MTX (MTX–MNP group). Interestingly, T24 cells showed a slightly decreased sensitivity after the combination of MTX with hyperthermia (27% vs 16%). Bare MNP alone (without hyperthermia; Figure 5C) had no effect on the different cell lines. The addition of a hyperthermia treatment in presence of MNP led to a cell viability pattern which was comparable to that of hyperthermia alone (Figure 5D).
Increasing the incubation time of the treatment (MTX–MNP) or control (MTX, MNP, “none”) groups from 48 to 72 hours influenced the cellular viability in a heterogenic and cell line-dependent manner (Figure 5E–H). In particular, in combination with hyperthermia, the MTX–MNP group showed after 72 hours of MTX–MNP incubation, a reduced viability for AU-565 (−16%), SK-BR-3 (−16%), and MX1 (−15%) cells, whereas slight increases were observed for MDA-MB-231 (+2%) and T47 (+5%) cells compared to 48 hours (Figure 5E). Interestingly, in the case of MDA-MB-231 without hyperthermia, a dramatic recovery of cellular viability (+38%) was observed after 72 hours compared with 48 hours. In this regard, especially, the eradication of AU-565, SK-BR3, and MX1 cells benefited from an increasing incubation time with MTX–MNP in combination with hyperthermia. After 72 hours, free MTX in combination with hyperthermia (Figure 5F) revealed lower cellular viabilities for most cell lines compared with the MTX–MNP group. Nevertheless, those viabilities were almost comparable to those after 48 hours of MTX incubation. In comparison with MTX–MNP, free MTX in the absence of hyperthermia revealed on average a reduced cellular viability after 72 hours. Without hyperthermia, only a marginal cytotoxicity after 72 hours of incubation with bare MNP (which was comparable to 48 hours) was observed (Figure 5G). Moreover, bare MNP in combination hyperthermia showed similar cellular viabilities as observed after hyperthermia treatment alone (Figure 5H). In tendency, hyperthermia treatment alone had higher cytotoxic effects at 72 hours compared with 48 hours.
Taken together, these results showed a very heterogenic and cell line-dependent reaction to the different treatments and incubation times and highlight the necessity of the combination of MTX–MNP and hyperthermia in order to neglect the possibility of cellular recovery.
Discussion
By investigating the impact of free MTX on the viability of different cancer cell lines, a very heterogeneous and cell line-dependent MTX cytotoxicity with ED50 values ranging from 16.7 nM to over 22 μM was found. Moreover, no correlation between the ED50 values and the cellular doubling times was observed (data not shown), although MTX is involved in nucleotide metabolism and therefore permits its cytotoxic effects, for example, by impairing DNA synthesis and cell cycle progression.22–24 The results clearly illustrate the insufficiency of using MTX as a mono-modal strategy in cancer therapy as the MTX sensitivity will strongly vary among different cell populations in a heterogenic tumor. At the same time, the used bare iron oxide MNP caused almost no cytotoxic effects in all cell lines, demonstrating their high biocompatibility. In this regard, the combination of MTX, MNP, and hyperthermia provides a promising tool for overcoming the drawbacks of a mono-modal MTX therapy.
A cell line-specific and preferential uptake of MTX–MNP vs bare MNP was revealed by Prussian blue staining of intracellular iron, which was in agreement with the observed cytotoxicity. In this regard, T24 and AU-565 cells showed an increased MTX–MNP uptake and concurrently were more sensitive to MTX, whereas for SK-BR-3 cells a lesser uptake of MTX–MNP and a lesser sensitivity to MTX were observed. As the cytotoxicity of MTX–MNP and free MTX was almost comparable (whereas bare MNP showed only a marginal cytotoxicity), these results demonstrate that both formulations were taken up in a comparable manner by the target cells, since MTX was shown to exert its action only after internalization into cells.23,25 In this regard, the observed slightly higher cytotoxicity of MTX was addressed to a faster incorporation of the smaller free MTX in comparison to the larger MTX–MNP, as free MTX is taken up mainly by FR and RFC, whereas an increased uptake of MTX–MNP was thought to be mainly caused by receptor-mediated endocytosis after binding to FR. The involvement of receptor-mediated endocytosis is additionally confirmed by the performed cellular uptake studies, revealing a preferential uptake of MTX–MNP opposing to MNP without MTX, independent of the particle’s hydrodynamic diameter and ζ-potential.
No dissociation of the MTX from the MTX–MNP was found under physiological (pH 7.4) as well as acidic (pH 5) conditions, indicating a strong binding of MTX and cytotoxic activity of MNP–MTX. In this regard, MTX–MNP have the capability of being used as an efficient and localized tumor treatment (eg, as drug depot) since a delocalization of MTX from the tumor region is minimized, resulting in a reduction of severe systemic side effects. Interestingly, a recovery of cellular viability was found for some cell lines with an increasing time of MTX–MNP and MTX incubation. This observation may indicate a release of free MTX from the cells or effective cellular damage-repair mechanisms. An exocytosis of MTX–MNP is more unlikely, since the larger MNP (here 130 nm MNP were used) are known to be hardly released by exocytosis.53,54
Our data show that the utilization of MTX–MNP is preferential over the usage of the free drug, since MTX will remain localized in the tumor region for a longer period of time (depot effect) in comparison to systemically applied MTX as given by intravenous injection. The localized application favors the induction of local effects at the tumor area, with a concomitant reduction of side effects. Furthermore, MTX–MNP also offer the possibility of magnetic targeting, allowing the “guidance” of MTX–MNP to the tumor region by magnetic forces and, hence, facilitating a localized accumulation of MTX–MNP in the tumor region.3–7 In this regard, the low release of MTX from the MTX–MNP will additionally lead to a reduction of systemic side effects during the targeting process.
Interestingly, the mRNA and protein levels of the FRα, the RFC, and the membrane transport proteins are only by tendency in agreement with the cellular toxicity of MTX–MNP in different cell lines. In general, these findings can be attributed to the fact that the translational processing from mRNA into a functional protein is a complex process involving multiple steps that determine protein functionality.55–57 The observed mRNA expression of FRα is in good agreement with reports revealing elevated FR levels (approximately 50%) in breast tumors and nonelevated (or even slightly reduced) levels in bladder tumors.28,58,59 Opposed to this, the protein level of FRα showed a cell line-specific glycosylation state that was almost in line with the observed cytotoxicity of MTX–MNP.55 In this context, high levels of immature and inactive FRα protein in the MTX-resistant cell lines could explain the low MTX cytotoxicity in relation to the reduced MTX uptake. At the same time, at least for the investigated T24, AU-565, and SK-BR-3 cells, the FR expression correlated with the MTX–MNP uptake, confirming the hypothesis of a FR-mediated preferential uptake of MTX–MNP and supports utilization of MTX for drug targeting purposes. Concerning the RFC, the expression on mRNA level was present to a different extent. These differences could be attributed to cell-specific promoter methylations.60,61 The fact that the RFC on mRNA and protein level correlated only by tendency with the cellular viability might implicate that specific mechanism, other than the expression by itself, are responsible for the observed cell line-specific cytotoxicity (eg, a cell line-specific posttranslational modification of protein functionality). In relation to BCRP, the mRNA expression level was heterogeneous among the investigated cell lines in contrast to the protein level. In consequence, BCRP-based MTX efflux, was expectedly similar in all cell lines, but not the primary reason for the cell line-specific impact of MTX.62–64 Considering the transmembrane transport proteins MRP1 and MRP5, the different posttranslational phosphorylation and glycosylation statuses might be related to the impact of MTX on cytotoxicity. For example, it was found that a strong MRP1 glycosylation is associated with high intracellular accumulation and cytotoxicity of chemotherapeutic drugs.56,57
Although we could basically demonstrate a sufficient magnetic heating capability of the used MNP in previous investigations, the usage of an external heat source (hot air) ensured the elucidation of the tumor cell response at very defined treatment temperatures, since variations in the MNP uptake (eg, depending cell line and MNP functionalization) would have led to different temperature dosages when performing magnetic hyperthermia (see MNP uptake experiments in Figure 3).10 Moreover, the utilization of hot air hyperthermia allowed an estimation of the impact of the hyperthermia treatment alone (control groups without any MNP), which would not be possible in the case of magnetic heating.
On the downside, the usage of external heating methods (including hot air) instead of magnetic heating results in a more indirect and slower heating of the target structures (eg, tumor cells), prolonging the time needed for reaching the target temperature. In this regard, a higher efficacy in cases of magnetic heating can be assumed, as the heat will be generated directly at the site of interest reducing the ability of tumor cells to adapt to the applied heat stress. Although the efficiency of external heating may be reduced compared with magnetic heating approaches, the applied thermal dose was shown to be sufficient to reduce cellular viability.65
We could achieve higher cytotoxic effects by MTX–MNP exposure in combination with an additional hyperthermal treatment compared with MTX or thermotherapy alone in nearly all cell lines. As it is known, MTX and hyperthermia treatments will result in an impairment of different phases of the cell cycle.17–21 In this regard, the combination of both treatments was hypothesized to be, at least partly, responsible for the increased cytotoxicity of the combinatory treatment. Moreover, these additive effects of the combination therapy might well be connected to the altered presence of efflux proteins in the cell membrane.66 Another explanation is related to a heating-based impairment of protein synthesis affecting the activity of the dihydrofolate reductase, a critical enzyme in folate metabolism.67 Interestingly, for T24 bladder cancer cells, a slightly higher cellular viability was found after combining either MTX–MNP or MTX with hyperthermia, as opposed to MTX–MNP or MTX treatment alone. Based on this finding, we hypothesized that not all cancer cell lines fully benefit from the performed therapy regime (with additional hyperthermia) after MTX–MNP or MTX treatment, although MTX–MNP in combination with hyperthermia were found to be preferential over free MTX and heat. It remains unclear, if these circumstances are based on the fast cellular doubling time of T24 cells or the different cancer entities (bladder instead of breast cancer). Further studies need to clarify if, for example, a faster applied hyperthermia treatment (within the cellular doubling time) is needed when treating this cancer cell line (or tumor entity). With consideration of the incubation time, a very cell line-specific and heterogeneous response to the treatment with MTX–MNP and hyperthermia was observed, indicating a cell line-specific onset of survival mechanisms. This finding supports the importance of a long term post-therapy observation in the clinical situation. Our results show that the treatment of cancers with a combinatory treatment consisting of MTX–MNP and hyperthermia is very effective. Nevertheless, the heterogeneous cellular viability between different types of tumor cells still persists.68,69 In this regard, the survival of treatment-resistant tumor cells could be further decreased by the utilization of MNP with higher dosages of immobilized drugs as well as higher temperature dosages.70–74
In later clinical applications, applied MNP will stay at the site of injection up to weeks, allowing the generation of drug depots for a prolonged and/or repeated tumor treatment.75 By this, the utilization of MTX–MNP as a combinatory therapy is capable of achieving higher cytotoxic results in later clinical applications, with less systemic side effects than systemically applied free MTX.
Taken together, MTX bound to MNP was comparably effective as its free counterpart, allowing the utilization of the advantages of MNP without scarifying the cytotoxicity of MTX. Considering the dramatic recovery of cellular viability of some cell lines after 72 hours of incubation with either free MTX or MTX–MNP without hyperthermia, these results underline the necessity of combining both modalities with an additional cellular stressor like hyperthermia in order to prohibit a possible cellular recovery. In this regard, the addition of a hyperthermia treatment resulted in an additive cytotoxic effect in most cell lines. Nevertheless, the heterogenic cell line response even after a combination with hyperthermia was not completely negligible and could not solely be explained by a cell line-specific expression of proteins involved in uptake and efflux of MTX. It was shown that MTX can be used for drug targeting purposes and that MTX–MNP were preferentially taken up in a cell line-dependent manner compared with bare MNP. Moreover, almost no desorption of MTX from the MTX–MNP could be found within 72 hours under physiological and acidic conditions, suggesting a low release of MTX from the tumor region resulting in a higher patient compliance by reduced systemic effects compared with free MTX. Additionally, MTX–MNP can be used for magnetic targeting applications, allowing a “guidance” of MTX–MNP to the target area by magnetic forces and, therefore, minimize the systemic burden after intravenous application.
Conclusion
In conclusion, the combination of MTX–MNP and hyperthermia allows a more efficient and localized eradication of tumor cells and is capable of reducing the recovery of cellular viability. Nevertheless, the heterogenic efficacy of MTX or MTX–MNP among the different cell lines cannot only be solved by an additional hyperthermia treatment. Further, MTX–MNP exhibit important features to minimize the release of MTX from the tumor region and consequently allow additional treatments like magnetic targeting and/or magnetic hyperthermia, helping to increase therapeutic efficiency and reduce systemic side effects.
Acknowledgments
The present investigation was carried out within the projects “PAK-151” (HI-689/7-4), which is funded by the Deutsche Forschungsgemeinschaft, and “Multifunctional Nanoparticles for the Selective Detection and Treatment of Cancer” (MultiFun), which is funded by the European Seventh Framework Program (FP7/2007-2013) under grant agreement no 262943. M Stapf was also supported by the Graduate Academy of the Friedrich Schiller University, Jena. We acknowledge R Ludwig for the design of the B2M primer and Dr L Leistritz for his advice in statistical calculations. Moreover, we acknowledge Dr J Clement for the support concerning the performed qRT-PCR experiments.
Disclosure
Prof Dr Hilger is holding several patents related to the treatment of tumors via magnetic hyperthermia. The authors report no other conflicts of interest in this work.
References
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. doi:10.1016/j.cell.2011.02.013. | ||
Von der Maase H, Hansen SW, Roberts JT, et al. Gemcitabine and cisplatin versus methotrexate, vinblastine, doxorubicin, and cisplatin in advanced or metastatic bladder cancer: results of a large, randomized, multinational, multicenter, phase III study. J Clin Oncol. 2000;18(17):3068–3077. | ||
Tietze R, Lyer S, Dürr S, et al. Efficient drug-delivery using magnetic nanoparticles – biodistribution and therapeutic effects in tumour bearing rabbits. Nanomedicine. 2013;9(7):961–971. doi:10.1016/j.nano.2013.05.001. | ||
Janko C, Dürr S, Munoz LE, et al. Magnetic drug targeting reduces the chemotherapeutic burden on circulating leukocytes. Int J Mol Sci. 2013;14(4):7341–7355. doi:10.3390/ijms14047341. | ||
Fortin-Ripoche J-P, Martina MS, Gazeau F, et al. Magnetic targeting of magnetoliposomes to solid tumors with MR imaging monitoring in mice: feasibility. Radiology. 2006;239(2):415–424. doi:10.1148/radiol.2392042110. | ||
Yang Y, Jiang J-S, Du B, Gan Z-F, Qian M, Zhang P. Preparation and properties of a novel drug delivery system with both magnetic and biomolecular targeting. J Mater Sci Mater Med. 2009;20(1):301–307. doi:10.1007/s10856-008-3577-0. | ||
Huang HS, Hainfield JF. Intravenous magnetic nanoparticle cancer hyperthermia. Int J Nanomedicine. 2013;8:2521–2532. doi:10.2147/IJN.S43770. | ||
Stapf M, Pömpner N, Kettering M, Hilger I. Magnetic thermoablation stimuli alter BCL2 and FGF-R1 but not HSP70 expression profiles in BT474 breast tumors. Int J Nanomedicine. 2015;10(10):1931–1939. doi:10.2147/IJN.S77372. | ||
Kossatz S, Ludwig R, Dähring H, et al. High therapeutic efficiency of magnetic hyperthermia in xenograft models achieved with moderate temperature dosages in the tumor area. Pharm Res. 2014;31(12):3274–3288. doi:10.1007/s11095-014-1417-0. | ||
Ludwig R, Stapf M, Dutz S, Müller R, Teichgräber U, Hilger I. Structural properties of magnetic nanoparticles determine their heating behavior – an estimation of the in vivo heating potential. Nanoscale Res Lett. 2014;9(1):602. doi:10.1186/1556-276X-9-602. | ||
Zaloga J, Stapf M, Nowak J, et al. Tangential flow ultrafiltration allows purification and concentration of lauric acid-/albumin-coated particles for improved magnetic treatment. Int J Mol Sci. 2015;16(8):19291–19307. doi:10.3390/ijms160819291. | ||
Friedrich RP, Janko C, Poettler M, et al. Flow cytometry for intracellular SPION quantification: specificity and sensitivity in comparison with spectroscopic methods. Int J Nanomedicine. 2015;10:4185–4201. doi:10.2147/IJN.S82714. | ||
Weissleder R, Bogdanov A, Neuwelt EA, Papisov M. Long-circulating iron oxides for MR imaging. Adv Drug Deliv Rev. 1995;16(2–3):321–334. doi:10.1016/0169-409X(95)00033-4. | ||
Okon E, Pouliquen D, Okon P, et al. Biodegradation of magnetite dextran nanoparticles in the rat. A histologic and biophysical study. Lab Invest. 1994;71(6):895–903. | ||
Lãbbe AS, Bergemann C, Huhnt W, Fricke T, Riess H. Predinical experiences drug targeting: tolerance and efficacy. Statistics (Ber). 1996:4694–4701. | ||
Hergt R, Dutz S, Müller R, Zeisberger M. Magnetic particle hyperthermia: nanoparticle magnetism and materials development for cancer therapy. J Phys Condens Matter. 2006;18(38):S2919–S2934. doi:10.1088/0953-8984/18/38/S26. | ||
Spurlock CF, Tossberg JT, Fuchs HA, Olsen NJ, Aune TM. Methotrexate increases expression of cell cycle checkpoint genes via JNK activation. Arthritis Rheum. 2012;64(6):1780–1789. doi:10.1002/art.34342. | ||
Goncharova SA, Frankfurt OS. Effect of methotrexate on the cell cycle of L1210 leukemia. Cell Tissue Kinet. 1976;9(4):333–340. | ||
Lim C-U, Zhang Y, Fox MH. Cell cycle dependent apoptosis and cell cycle blocks induced by hyperthermia in HL-60 cells. Int J Hyperthermia. 2006;22(1):77–91. doi:10.1080/02656730500430538. | ||
Coss RA, Dewey C, Bamburg JR. Effects of hyperthermia on dividing Chinese hamster Ovary cells and on microtubules in vitro. Cancer Res. 1982;42(3):1059–1107. | ||
Bhuyan BK. Kinetics of cell kill by hyperthermia. Cancer Res. 1979;39(6 II):2277–2284. | ||
Zwicke G, Mansoori G, Jeffery C. Utilizing the folate receptor for active targeting of cancer nanotherapeutics. Nano Rev. 2012;1:1–11. http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3521101/. Accessed September 26, 2014. | ||
Chan ESL, Cronstein BN. Mechanisms of action of methotrexate. Bull NYU Hosp Jt Dis. 2013;71(suppl 1):5–8. doi:10.1016/S0162-3109(00)00189-2. | ||
Cheok MH, Evans WE. Acute lymphoblastic leukaemia: a model for the pharmacogenomics of cancer therapy. Nat Rev Cancer. 2006;6(2):117–129. doi:10.1038/nrc1800. | ||
Genestier L, Paillot R, Quemeneur L, Izeradjene K, Revillard J-P. Mechanisms of action of methotrexate. Immunopharmacology. 2000;47(2–3):247–257. doi:10.1016/S0162-3109(00)00189-2. | ||
Brzezinska A, Winska P, Balinska M. Cellular aspects of folate and antifolate membrane transport. Acta Biochim Pol. 2000;47(3):735–749. http://www.ncbi.nlm.nih.gov/pubmed/11310973 | ||
Yoo HS, Park TG. Folate-receptor-targeted delivery of doxorubicin nano-aggregates stabilized by doxorubicin-PEG-folate conjugate. J Control Release. 2004;100(2):247–256. doi:10.1016/j.jconrel.2004.08.017. | ||
Parker N, Turk MJ, Westrick E, Lewis JD, Low PS, Leamon CP. Folate receptor expression in carcinomas and normal tissues determined by a quantitative radioligand binding assay. Anal Biochem. 2005;338(2):284–293. doi:10.1016/j.ab.2004.12.026. | ||
Werner ME, Karve S, Sukumar R, et al. Folate-targeted nanoparticle delivery of chemo- and radiotherapeutics for the treatment of ovarian cancer peritoneal metastasis. Biomaterials. 2011;32(33):8548–8554. doi:10.1016/j.biomaterials.2011.07.067. | ||
Saul JM, Annapragada A, Natarajan JV, Bellamkonda RV. Controlled targeting of liposomal doxorubicin via the folate receptor in vitro. J Control Release. 2003;92(1–2):49–67. http://www.ncbi.nlm.nih.gov/pubmed/14499185 | ||
Ali ASG, Reza MA, Eshghi H, Sazgarnia A, Montazerabadi AR. Cancerous cells targeting and destruction using folate conjugated gold nanoparticles. Dyn Biochem Process Biotechnol Mol Biol. 2010;4:6–12. | ||
Dhawan D, Ramos-Vara JA, Naughton JF, et al. Targeting folate receptors to treat invasive urinary bladder cancer. Cancer Res. 2013;73(2):875–884. doi:10.1158/0008-5472.CAN-12-2101. | ||
Campbell IG, Jones TA, Foulkes WD, Trowsdale J. Folate binding protein is a marker for ovarian cancer. Cancer Res. 1991;51(33):5329–5338. http://www.biomednet.com/db/medline/92005454 | ||
Matherly LH, Goldman DI. Membrane transport of folates. Vitam Horm. 2003;66:403–456. | ||
Matherly LH, Hou Z, Deng Y. Human reduced folate carrier: translation of basic biology to cancer etiology and therapy. Cancer Metastasis Rev. 2007;26:111–128. doi:10.1007/s10555-007-9046-2. | ||
Matherly LH, Hou Z. Chapter 5 structure and function of the reduced folate carrier. A paradigm of a major facilitator superfamily mammalian nutrient transporter. Vitam Horm. 2008;79(08):145–184. doi:10.1016/S0083-6729(08)00405-6. | ||
Whetstine JR, Flatley RM, Matherly LH. The human reduced folate carrier gene is ubiquitously and differentially expressed in normal human tissues: identification of seven non-coding exons and characterization of a novel promoter. Biochem J. 2002;367:629–640. doi:10.1042/BJ20020512. | ||
Gong M, Cowan KH, Gudas J, Moscow JA. Isolation and characterization of genomic sequences involved in the regulation of the human reduced folate carrier gene (RFC1). Gene. 1999;233(1–2):21–31. doi:10.1016/S0378-1119(99)00166-3. | ||
Liu M, Ge Y, Cabelof DC, et al. Structure and regulation of the murine reduced folate carrier gene: identification of four noncoding exons and promoters and regulation by dietary folates. J Biol Chem. 2005;280(7):5588–5597. doi:10.1074/jbc.M412662200. | ||
Wang Y, Zhao R, Russell RG, Goldman ID. Localization of the murine reduced folate carrier as assessed by immunohistochemical analysis. Biochim Biophys Acta – Biomembr. 2001;1513(1):49–54. doi:10.1016/S0005-2736(01)00340-6. | ||
Spinella MJ, Brigle KE, Sierra EE, Goldman ID. Distinguishing between folate receptor-α-mediated transport and reduced folate carrier-mediated transport in L1210 leukemia cells. J Biol Chem. 1995;270(14):7842–7849. doi:10.1074/jbc.270.14.7842. | ||
Hooijberg JH, Broxterman HJ, Kool M, et al. Antifolate resistance mediated by the multidrug resistance proteins MRP1 and MRP2 advances in brief antifolate resistance mediated by the multidrug resistance proteins MRP1. Cancer Res. 1999;59(11):2532–2535. | ||
Kool M, van der Linden M, de Haas M, et al. MRP3, an organic anion transporter able to transport anti-cancer drugs. Proc Natl Acad Sci U S A. 1999;96(June):6914–6919. doi:10.1073/pnas.96.12.6914. | ||
Assaraf YG, Goldman ID. Loss of folic acid exporter function with markedly augmented folate accumulation in lipophilic antifolate-resistant mammalian cells. J Biol Chem. 1997;272(28):17460–17466. doi:10.1074/jbc.272.28.17460. | ||
Stark M, Rothem L, Jansen G, Scheffer GL, Goldman ID, Assaraf YG. Antifolate resistance associated with loss of MRP1 expression and function in Chinese hamster ovary cells with markedly impaired export of folate and cholate. Mol Pharmacol. 2003;64(2):220–227. doi:10.1124/mol.64.2.220. | ||
Assaraf YG, Rothem L, Hooijberg JH, et al. Loss of multidrug resistance protein 1 expression and folate efflux activity results in a highly concentrative folate transport in human leukemia cells. J Biol Chem. 2003;278(9):6680–6686. doi:10.1074/jbc.M209186200. | ||
Ifergan I, Shafran A, Jansen G, Hooijberg JH, Scheffer GL, Assaraf YG. Folate deprivation results in the loss of breast cancer resistance protein (BCRP/ABCG2) expression: a role for BCRP in cellular folate homeostasis. J Biol Chem. 2004;279(24):25527–25534. doi:10.1074/jbc.M401725200. | ||
Wielinga P, Hooijberg JH, Gunnarsdottir S, et al. The human multidrug resistance protein MRP5 transports folates and can mediate cellular resistance against antifolates. Cancer Res. 2005;65(10):4425–4430. doi:10.1158/0008-5472.CAN-04-2810. | ||
Mindell JA. Lysosomal acidification mechanisms. Annu Rev Physiol. 2012;74(1):69–86. doi:10.1146/annurev-physiol-012110-142317. | ||
Spencer JPE, Wong J, Jenner A, Aruoma OI, Cross CE, Halliwell B. Base modification and strand breakage in isolated calf thymus DNA and in DNA from human skin epidermal keratinocytes exposed to peroxynitrite or 3-morpholinosydnonimine. Chem Res Toxicol. 1996;9(7):1152–1158. doi:10.1021/tx960084i. | ||
Towbin H, Staehelin T, Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci U S A. 1979;76(9):4350–4354. doi:10.1073/pnas.76.9.4350. | ||
Sapareto SA, Dewey WC. Thermal dose determination in cancer therapy. Int J Radiat Oncol Biol Phys. 1984;10(6):787–800. doi:10.1016/0360-3016(84)90379-1. | ||
Jin H, Heller DA, Sharma R, Strano MS. Size-dependent cellular uptake and expulsion of single-walled carbon nanotubes: single particle tracking and nanoparticles. ACS Nano. 2009;3(1):149–158. | ||
Chithrani BD, Chan WCW. Elucidating the mechanism of cellular uptake and removal of protein-coated gold nanoparticles of different sizes and shapes. Nano Lett. 2007;7(6):1542–1550. doi:10.1021/nl070363y. | ||
Roberts SJ, Petropavlovskaja M, Chung KN, Knight CB, Elwood PC. Role of individual N-linked glycosylation sites in the function and intracellular transport of the human alpha folate receptor. Arch Biochem Biophys. 1998;351(2):227–235. doi:10.1006/abbi.1997.0551. | ||
Liang X-J, Shen D-W, Garfield S, Gottesman MM. Mislocalization of membrane proteins associated with multidrug resistance in cisplatin-resistant cancer cell lines. Cancer Res. 2003;63(18):5909–5916. | ||
Beretta GL, Benedetti V, Cossa G, et al. Increased levels and defective glycosylation of MRPs in ovarian carcinoma cells resistant to oxaliplatin. Biochem Pharmacol. 2010;79(8):1108–1117. doi:10.1016/j.bcp.2009.12.002. | ||
Hartmann LC, Keeney GL, Lingle WL, et al. Folate receptor overexpression is associated with poor outcome in breast cancer. Int J Cancer. 2007;121(5):938–942. doi:10.1002/ijc.22811. | ||
Jhaveri MS, Rait AS, Chung K-N, Trepel JB, Chang EH. Antisense oligonucleotides targeted to the human alpha folate receptor inhibit breast cancer cell growth and sensitize the cells to doxorubicin treatment. Mol Cancer Ther. 2004;3(12):1505–1512. | ||
Worm J, Kirkin AF, Dzhandzhugazyan KN, Guldberg P. Methylation-dependent silencing of the reduced folate carrier gene in inherently methotrexate-resistant human breast cancer cells. J Biol Chem. 2001;276(43):39990–40000. doi:10.1074/jbc.M103181200. | ||
Yang R, Li W-W, Hoang BH, et al. Quantitative correlation between promoter methylation and messenger RNA levels of the reduced folate carrier. BMC Cancer. 2008;8:124. doi:10.1186/1471-2407-8-124. | ||
Volk EL, Farley KM, Wu Y, Li F, Robey RW, Schneider E. Overexpression of wild-type breast cancer resistance protein mediates methotrexate resistance. Cancer Res. 2002;62(17):5035–5040. | ||
Chen ZS, Robey RW, Belinsky MG, et al. Transport of methotrexate, methotrexate polyglutamates, and 17β-estradiol 17-(β-D-glucuronide) by ABCG2: effects of acquired mutations at R482 on methotrexate transport. Cancer Res. 2003;63(14):4048–4054. | ||
Rhee MS, Schneider E. Lack of an effect of breast cancer resistance protein (BCRP/ABCG2) overexpression on methotrexate polyglutamate export and folate accumulation in a human breast cancer cell line. Biochem Pharmacol. 2005;69(1):123–132. doi:10.1016/j.bcp.2004.09.007. | ||
Rampersaud EN, Vujaskovic Z, Inman BA. Hyperthermia as a treatment for bladder cancer. Oncology (Williston Park). 2010;24(12):1149–1155. | ||
Franke K, Kettering M, Lange K, Kaiser WA, Hilger I. The exposure of cancer cells to hyperthermia, iron oxide nanoparticles, and mitomycin C influences membrane multidrug resistance protein expression levels. Int J Nanomedicine. 2013;8:351–363. doi:10.2147/IJN.S37465. | ||
Herman TS, Cress AE, Sweets C, Gerner EW. Reversal of resistance to methotrexate by hyperthermia in Chinese hamster ovary cells. Cancer Res. 1981;41(10):3840–3843. | ||
Goss P, Parsons PG. The effect of hyperthermia and melphalan on survival of human fibroblast strains and melanoma cell lines the effect of hyperthermia and melphalan on survival of human fibroblast strains and melanoma cell lines. Cancer Res. 1977;37(1):152–156. | ||
Van der Heijden AG, Verhaegh G, Jansen CFJ, Schalken JA, Witjes JA. Effect of hyperthermia on the cytotoxicity of 4 chemotherapeutic agents currently used for the treatment of transitional cell carcinoma of the bladder: an in vitro study. J Urol. 2005;173(4):1375–1380. doi:10.1097/01.ju.0000146274.85012.e1. | ||
Hauck TS, Jennings TL, Yatsenko T, Kumaradas JC, Chan WCW. Enhancing the toxicity of cancer chemotherapeutics with gold nanorod hyperthermia. Adv Mater. 2008;20(20):3832–3838. doi:10.1002/adma.200800921. | ||
Murray TG, Cicciarelli N, McCabe CM, et al. In vitro efficacy of carboplatin and hyperthermia in a murine retinoblastoma cell line. Investig Ophthalmol Vis Sci. 1997;38(12):2516–2522. | ||
Teicher BA, Kowal CD, Kennedy KA, Sartorelli AC. Enhancement by hyperthermia of the in vitro cytotoxicity of mitomycin C toward hypoxic tumor cells. Cancer Res. 1981;41(3):1096–1099. | ||
Wallner KE, Banda M, Li GC. Hyperthermic enhancement of cell killing by mitomycin C in mitomycin C-resistant Chinese hamster ovary cells. Cancer Res. 1987;47(5):1308–1312. | ||
Wallner KE, DeGregorio MW, Li GC. Hyperthermic potentiation of cis-diamminedichloroplatinum(II) cytotoxicity in Chinese hamster ovary cells resistant to the drug. Cancer Res. 1986;46(12 I):6242–6245. | ||
Kettering M, Richter H, Wiekhorst F, et al. Minimal-invasive magnetic heating of tumors does not alter intra-tumoral nanoparticle accumulation, allowing for repeated therapy sessions: an in vivo study in mice. Nanotechnology. 2011;22(50):505102. doi:10.1088/0957-4484/22/50/505102. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.