")
Back to Journals » International Journal of Nanomedicine » Volume 12
Heterogeneous dimer peptide-conjugated polylysine dendrimer-Fe3O4 composite as a novel nanoscale molecular probe for early diagnosis and therapy in hepatocellular carcinoma
Authors Shen JM, Li XX, Fan LL, Zhou X, Han JM, Jia MK, Wu LF, Zhang XX, Chen J
Received 7 November 2016
Accepted for publication 4 January 2017
Published 10 February 2017 Volume 2017:12 Pages 1183—1200
DOI https://doi.org/10.2147/IJN.S126887
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Linlin Sun
Jian-Min Shen,1 Xin-Xin Li,1 Lin-Lan Fan,2 Xing Zhou,3 Ji-Min Han,1 Ming-Kang Jia,1 Liang-Fan Wu,1 Xiao-Xue Zhang,1 Jing Chen1
1School of Life Sciences, 2School of Basic Medical Sciences, Lanzhou University, 3The People’s Hospital of Gansu Province, Lanzhou, Gansu, China
Abstract: A novel nanoscale molecular probe is formulated in order to reduce toxicity and side effects of antitumor drug doxorubicin (DOX) in normal tissues and to enhance the detection sensitivity during early imaging diagnosis. The mechanism involves a specific targeting of Arg-Gly-Asp peptide (RGD)-GX1 heterogeneous dimer peptide-conjugated dendrigraft poly-l-lysine (DGL)–magnetic nanoparticle (MNP) composite by αvβ3-integrin/vasculature endothelium receptor-mediated synergetic effect. The physicochemical properties of the nanoprobe were characterized by using transmission electron microscope, Fourier transform infrared spectroscopy, X-ray diffraction, dynamic light scattering (DLS), and vibrating sample magnetometer. The average diameter of the resulting MNP–DGL–RGD-GX1–DOX nanoparticles (NPs) was ~150-160 nm by DLS under simulate physiological medium. In the present experimental system, the loading amount of DOX on NPs accounted for 414.4 mg/g for MNP–DGL–RGD-GX1–DOX. The results of cytotoxicity, flow cytometry, and cellular uptake consistently indicated that the MNP–DGL–RGD-GX1–DOX NPs were inclined to target HepG2 cells in selected three kinds of cells. In vitro exploration of molecular mechanism revealed that cell apoptosis was associated with the overexpression of Fas protein and the significant activation of caspase-3. In vivo magnetic resonance imaging and biodistribution study showed that the MNP–DGL–RGD-GX1–DOX formulation had high affinity to the tumor tissue, leading to more aggregation of NPs in the tumor. In vivo antitumor efficacy research verified that MNP–DGL–RGD-GX1–DOX NPs possessed significant antitumor activity and the tumor inhibitory rate reached 78.5%. These results suggested that NPs could be promising in application to early diagnosis and therapy in hepatocellular carcinoma as a specific nanoprobe.
Keywords: heterogeneous dimer peptide (HDP), molecular probe, magnetic nanoparticles (MNPs), targeting, hepatocellular carcinoma (HCC)
Introduction
Hepatocellular carcinoma (HCC) is mostly diagnosed at the middle or advanced stage, which causes difficulties to chemotherapy and surgery. In addition, doxorubicin (DOX) is regarded as an excellent broad-spectrum anticancer drug, but its administration is often limited because of the enormous cardiovascular side effects.1,2 Nanoscale molecular probe is promising in the comprehensive application of early imaging diagnosis and reducing toxicity and side effects of antitumor drug.3,4 However, nowadays, molecular probes that have been synthesized possess defects such as weak specificity and low sensitivity to some extent, which is unable to meet the requirements of clinical applications.5,6
Magnetic resonance imaging (MRI) has become one of the most powerful detection means in the contemporary clinical diagnosis.7,8 Magnetic resonance molecular imaging is an emerging technique for cancer diagnosis at the molecular level.9 It usually needs an intrinsic or extrinsic molecular probe which comprises a targeting ligand and a signaling element that can be detected by MRI. The former is usually highly specific to a certain type of tumor or its microenvironment. The latter can change the alignment of magnetic dipoles, which is used for the establishment of corresponding relationship between signal contrast and tumor molecular process, especially for the diagnosis of solid tumor.10,11 Therefore, there are two issues that need to be urgently addressed in the process of developing desired MRI molecular probes: how to improve the targeting specificity and detection sensitivity.
It is known that cell adhesion molecules αvβ3 integrins and vascular endothelial growth factor receptors (VEGFRs) are highly expressed in many tumor tissues,12–14 which are almost undetectable in normal tissues,15,16 which provides a theoretical basis for synthesizing probes that can specifically bind to the tumor. Several peptide-based probes have been fabricated and have demonstrated promising applications in animal studies,17,18 and some have even been successfully studied in human clinical trials.17 The previous studies in vitro showed that the GX1 peptide is a tumor vasculature endothelium-specific ligand.19 The study has also manifested the potential applications of Arg-Gly-Asp (RGD) peptide for integrin-targeted tumor treatment by specific identification and drug delivery.20 In these applications, the peptides were used as ligand components, and the resulting probes usually bound to a single target by receptor mediation. If a probe contains two ligands that can target VEGFRs and αvβ3 integrins at the same time, the targeting efficiency should be greatly improved under the dual-receptor mediation. This dual-targeting strategy is undoubtedly a good approach for developing molecular probes.21 Hence, fabrication and application of the probe decorating dual ligands is a main task of this study.
At present, the MRI contrast agents most broadly used in cancer imaging are chelated gadolinium (Gd) compounds.22 However, clinically, there are many adverse factors, including short biological half-lives (a few hours) because of hydrophilic nature and weak MRI signal due to low relaxation rate.23 By contrast, superparamagnetic iron oxide nanoparticles (SPIONPs) have been used as a high-relaxivity imaging agent since 1990s for molecular and cellular MRI applications in vitro.24 Unfortunately, unmodified SPIONPs are unfit for accurate diagnosis of tumor in vivo and tend to the large interception and accumulation in reticuloendothelial system.25 Therefore, developing a tailored SPIONP MRI agent that does not compromise basic characteristics is highly desirable. Dendrigraft of lysine (DGL) scaffolds have excellent biological functionality, compatibility, non-immunogenicity, biodegradability, and good water solubility. It has been extensively applied because of its versatile properties,26 such as 3-dimensional spherical shape, highly branched internal cavity, and abundant active amino groups, which are easier for both the modification of different imaging agents and targeting ligands and the entrapment of anticancer drugs on dendrimer surface. It has been feasible to design and synthesize hybrid SPIONP–DGL nanoparticles (NPs) as an MRI contrast for good blood circulation.27 More importantly, the MRI detection signal may be greatly improved when a straight substitution of the hybrid SPIONP–DGL NPs for individual Gd ion is done, because the signal generated by individual Gd ion is very weak.28 However, if DGL dendrimers are used as the skeleton ornamented magnetic nanoparticles (MNPs), the active amino groups on its surface will be largely blocked, which will hinder both the conjugation with specific targeting ligands such as peptides and uploading of drug. In order to ensure that the amino groups on the DGL surface are free, in present study, the synthesis protocol of combination of layer-by-layer assembly with sol-gel processes, covalent conjugation, and physical absorption was adopted. In this, Fe3O4 MNPs with diameter of ~140 nm were selected as magnetic nucleus, which was then orderly coated with DGL, conjugated with peptides, and finally uploaded as anti-cancer drug DOX (Figure 1).
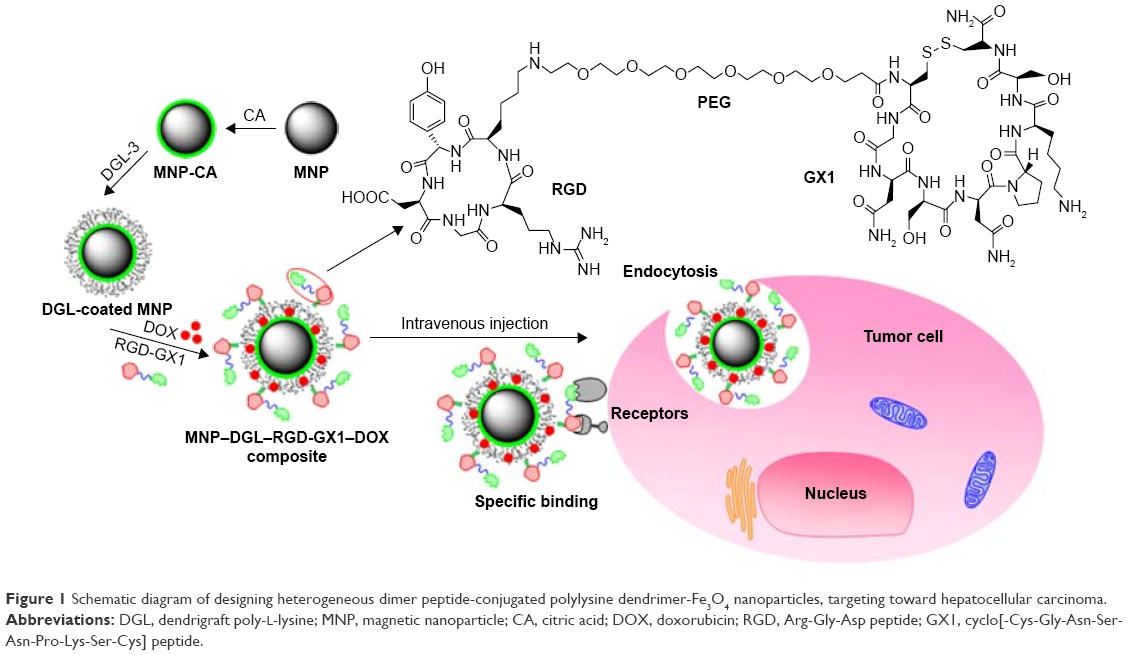 | Figure 1 Schematic diagram of designing heterogeneous dimer peptide-conjugated polylysine dendrimer-Fe3O4 nanoparticles, targeting toward hepatocellular carcinoma. |
Based on our previous research,29 the integration of multiple properties and magnetic function into a single network endows the nanoprobe with multiple performances, such as dual-targeting capability and diagnosis–therapeutical synchronization, which would be vastly beneficial for cancer nanomedicine. In view of this, the study is important for promoting both molecular imaging technology and tumor-targeted therapy.
Materials and methods
Materials
Ferric trichloride hexahydrate, ethanediol, sodium acetate, polyethylene glycol-2000 (PEG), citric acid, N-hydroxysuccinimide (NHS), 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDAC), dendrigraft poly-L-lysine (DGL-G3) containing 123 primary amino groups, and DOX were purchased from Sigma Aldrich (Shanghai, China). RGD pentapeptide (cyclo[-Arg-Gly-Asp-Phe-Lys]) (MW 603.7 Da), GX1 nonapeptide (cyclo[-Cys-Gly-Asn-Ser-Asn-Pro-Lys-Ser-Cys]) (MW 907 Da), and RGD-GX1 heterogeneous dimer peptide (MW 1927.8 Da) were purchased from ChinaPeptides Co., Ltd. (Shanghai, China). RPMI-1640 medium was obtained from Thermo Fisher Scientific (Waltham, MA, USA). Analytical grade dimethyl sulfoxide (DMSO) was obtained from Gansu Yinguang Chemical Industry Co. (China). Calf serum was acquired from Sijiqing Biological Engineering Materials Co., Ltd (Hangzhou, China). Human hepatocytes cancer HepG2 cells, lung cancer A549 cells, and normal human hepatocytes L02 cells lines were obtained from the cell bank of Shanghai Science Academe (China). All the other chemicals were of analytical grade. Water used in the study was prepared using a Milli-Q Water Purification System (Milli-Pore, Bedford, MA, USA). Milli-Q water purged with nitrogen (N2) gas was used in all the steps involved in the synthesis and formulation of MNPs.
Synthesis of citric acid (CA)-modified MNPs (MNPs-CA)
Fe3O4 MNPs were prepared as described in a previous paper.30 The magnetite surface was further modified with CA. In a typical reaction, 2.025 g FeCl3·6H2O, 5.4 g NaAc, and 1.5 g PEG-2000 were thoroughly dissolved in 60 mL ethylene glycol under ultrasonication for 40 min. Later, the solution was transferred into an autoclave and reacted in a vacuum oven at 200°C for 5 h. Then, the MNP precipitation was collected by centrifugation and was washed 6 times with deionized water. MNPs were adjusted to 2.0% (w/v) with 0.3 mol/L trisodium citrate (Na3CA) solution. The MNP suspension was uniformly dispersed by ultrasonication for 30 min and being vibrated for 3 h to prepare MNP–CA. Later, MNP–CA was readjusted to 50 mg/mL with deionized water and kept at 4°C refrigerator. Approximately 10 μL of MNP–CA suspension was properly diluted with deionized water and then uniformly dispersed by ultrasonication. A drop of uniform MNP–CA suspension was spotted on the brass carbon membrane and dried to measure TEM.
Synthesis of MNP–DGL composite
DGL-G3 was used as a fluffy shell material for further modification of the peptide and drug loading. First, in order to acquire activated DGL, 30 mg of DGL was dissolved in 500 μL of phosphate buffer solution (PBS) (50 mM) containing 0.9% NaCl in a 25-mL round-bottom flask. Subsequently, 15 mg of EDAC and 15 mg of NHS that act as a catalyzer was added to the flask followed by ultrasonication for 10 s and shaking for 2 h. Second, the as-prepared MNP–CA suspension (50 mg/mL) was uniformly dispersed through ultrasound. Later, 500 μL of MNP–CA suspension (50 mg/mL) was mixed with the activated DGL solution and sonicated for 30 s so that it was well dispersed and then shaking for another 4 h. Four samples of such solution were prepared. Third, after the reaction, the mixture was transferred into a 50 mL beaker and washed 6 times with the PBS to remove the unreacted DGL. The end product, MNP–DGL, was adjusted to the concentration of 50 mg/mL by adding 500 μL of PBS. Then the suspension was transferred into a 25 mL round-bottom flask and kept at 4°C. Approximately 10 μL of MNP–DGL suspension was diluted properly with deionized water and then uniformly dispersed through ultrasound in order to measure TEM.
Synthesis of MNP–DGL–RGD-GX1 composite
For the preparation of RGD-GX1 peptide solution (1 mg/mL), 5 mg of RGD-GX1 heterogeneous dimer peptide was mixed with 5 mL Buffer A (50 mM, pH 7.4 PBS containing 0.1 M NaCl) in a 25 mL round-bottom flask. Two identical samples were prepared. At the same time, 5 mL of RGD (1 mg/mL) and GX1 (1 mg/mL) peptide solution were also prepared, respectively. Next, four copies of catalysts (18 mg of EDAC and 10 mg of NHS) were added to the above peptide solutions and vibrated for 5 h at room temperature in order to activate the carboxyl groups on the peptide molecules. The four copies of MNP–DGL suspension (50 mg/mL) kept in the refrigerator were dispersed by ultrasonication for 10 min. Later, the two identical activated RGD-GX1, RGD, and GX1 peptide solution was added dropwise to the four copies of MNP–DGL suspension, respectively. The suspensions were shaken for 3 h on a level swing bed at room temperature and then kept at 4°C overnight. On the second day, after shaking for 0.5 h, the precipitation was separated by magnetic decantation and washed thrice with Buffer A to remove the excessive nonreactive peptide. The end products were denoted as MNP–DGL–RGD-GX1, MNP–DGL–RGD, and MNP–DGL–GX1 composites.
Drug loading and release in vitro
In order to synthesize the drug-loaded composites, DOX was selected as anticancer drug. First, 30 mg of MNP–DGL–RGD-GX1, MNP–DGL–RGD, MNP–DGL–GX1, and MNP–DGL composites were dispersed in 30 mL of DOX solution (1 mg/mL in PBS) by ultrasonication for 30 s, respectively, and shaken for 24 h at room temperature until DOX concentration in the solution was stabilized. Then the mixtures were centrifuged at 10,000 rpm for 10 min. To remove redundant DOX, the composites were further rinsed 3 times with PBS. All the supernatants in every system were collected, and the contents of non-encapsulated DOX in eluant were measured by UV-visible spectrophotometer at 479 nm. Then, the loading efficiency and loading amount of DOX were determined according to Equations 1 and 2. All the experiments were carried out in triplicate.
The loading efficiency of DOX
|
|
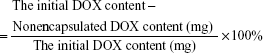
The loading amount of DOX
|
|
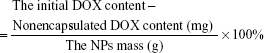
The release mechanisms of DOX at different pH were researched by the dialysis method. Albumin, the most abundant protein in plasma proteins, generally has a concentration of 0.6 mM.31 Hence, the release profiles of DOX were obtained in a simulated normal body fluid (pH 7.4, 50 mM PBS containing 0.6 mM human serum albumin [HSA] and 0.1 M NaCl) and an acidic environment (pH 5.3 50 mM PBS containing 0.6 mM HSA and 0.1 M NaCl) at 37°C±1°C. First, 30 mg of the DOX-loaded MNP–DGL–RGD-GX1, MNP–DGL–RGD, and MNP–DGL–GX1 composites were dispersed in 5 mL of medium and placed in a dialysis bag (molecular weight cut-off of 14 kDa), respectively. The release pattern of free DOX was also researched to testify that DOX molecules were not trapped inside dialysis bag. Then the dialysis bags were sunk in 45 mL of the release medium in a water bath with gentle shaking. Approximately 0.5 mL of dialysate was removed at certain time interval and assayed by UV spectrophotometer at 479 nm. An amount of 0.5 mL of fresh-buffered medium was supplemented and the amount of drug released was calculated as described.32 Each series of experiments was conducted in triplicate and the results were presented as mean ± standard deviation.
Physico-chemical characterization
The inner microstructure of formulated NPs was observed with a transmission electron microscopy (TEM, TecnaiG2 F30; FEI, USA) with an acceleration voltage of 200 kV. The particle size suspended in PBS media at pH 7.4 was measured in triplicate using a Malvern dynamic light scattering (DLS) (Zetasizer Nano 3600; UK). The crystallographic structure was characterized by large-angle powder X-ray diffraction (XRD) patterns from 10° to 80° using Cu Kα radiation at X’Pert PRO (PANalytical, Holland). The composition of the NPs was assayed by a Fourier transform infrared (FTIR) spectrometer (NEXUS 670 FT-IR; Nicolet, USA). The FTIR spectrum was collected between the wave number of 400 and 4,000 cm−1. The magnetic performance was recorded by a vibrating sample magnetometer (VSM) (Lake Shore, USA).
In vitro cytotoxicity, flow cytometry (FCM), and cellular imaging
In vitro cytotoxicities of empty NPs (MNP–DGL–RGD-GX1, MNP–DGL–RGD, and MNP–DGL–GX1) and their corresponding DOX-loaded NPs were assessed by MTT method and FITC-labeled Annexin-V and propidium iodide (PI) staining. HepG2, A549, and L02 cells were seeded in a 96-well plate (1×104 cells/well) in RPMI-1640 medium containing calf serum (10%) and 1% penicillin-streptomycin, respectively. The cells were incubated in a fully humidified incubator at 37°C with 5% CO2 until 80% confluence with normal morphology. The empty or DOX-loaded NPs with different concentration (12.5~800 μg/mL, in PBS) were added to the cell wells, respectively. The cells added with equal amount of PBS without NPs or DOX were used as control. After incubation at 37°C for another 48 h, these cells were washed with PBS thrice to remove the free NPs adhered on the outer surface of cell membrane. Then, the experiment was divided into two systems. In the first system, MTT reagent diluted in culture medium (0.5 mg/mL, 20 μL) replace the culture medium, followed by incubation for another 2 h. The MTT/medium was cleaned carefully and 150 μL of DMSO was added to each well to dissolve the formazan crystals. Absorbance was determined on a microplate spectrophotometer at 570 nm (iMark™; Bio-Rad Laboratories Inc., Hercules, CA, USA). In the second system, the cells treated with the empty or DOX-loaded NPs with a concentration of 400 μg/mL were stained with Annexin-V/PI and assayed by flow cytometer (BD LSRFortessa, USA) to quantificationally detect inducing cell apoptosis. The FCM of the control group was also performed with HepG2 cells incubated with PBS without NPs or DOX. There were 20,000 cells counted in each event. The cells labeled by fluorescence were further observed on laser scanning confocal microscopy (LSCM, Olympus, FV-300, IX71). All the experiments were conducted in triplicate.
Fas protein expression and caspase activation assay
HepG2 cells were collected after 48 h of treatment with free DOX or MNP–DGL–RGD-GX1–DOX NPs and were washed with PBS thrice to remove the free drug or NPs. Later, the cells were stained with a FITC-labeled mouse anti-human CD95 and FITC rabbit anti-active caspase-3 apoptosis Kit (BD Pharmingen™ reagent; Shiga, USA), according to the manufacturer’s instructions, respectively. Then, the cells were assayed by flow cytometer (BD LSRFortessa) to detect Fas protein expression and caspase-3 activation of HepG2 cells, respectively.
Biodistribution studies
Establishment of tumor-bearing model and treatment in vivo
In order to analyze the distribution of DOX in vivo, 60 healthy male Balb/c mice (body weight 18–22 g) were obtained from the Experimental Animal Center of Lanzhou University (Lanzhou, China). The mice were housed as 6 per cage at 22°C±1°C and 50%–60% relative humidity with free access to food and water under a standard 12 h light/dark circadian cycle condition (light on from 8:30 am to 8:30 pm). Animal welfare and experimental procedures were carried out in accordance with protocols approved by the Ethics Committee of Animal Experiments of Lanzhou University. The HepG2 cells (2×106 cells/0.2 mL) were inoculated to the right front oxter of mice. The mice were randomly divided into 4 groups at day 14 after inoculation (in general, the primary tumor grew to 1.0~1.2 cm3). 0.2 milliliter of the MNP–DGL–RGD-GX1–DOX (group 1), MNP–DGL–RGD–DOX (group 2), and MNP–DGL–GX1–DOX NPs (group 3) in sterile PBS were injected by tail vein at a dose of 2 mg/kg body weight after the mice were anesthetized by intraperitoneal injection of 20% chloral hydrate, respectively. DOX group (group 4) was injected with 0.2 mL of free DOX solution (in sterile PBS) corresponding to the amount of DOX in injection dose of NP group. The amount of DOX was determined according to the DOX loading amount on NPs and release amount from NPs.
DOX quantification
The blood samples were collected by excising the eyeball of the mice at different time intervals (1, 2, and 3 h, 5 mice at each time point) after injection. The total blood samples obtained were centrifuged at 1,500 rpm for 10 min to collect plasma for DOX distribution analysis. Then all mice were sacrificed. The tissues, including heart, liver, spleen, lung, kidney, and tumor, were taken out quickly and rinsed thoroughly in order to remove the blood stains on them with PBS (50 mM, pH 7.4) and weighed. The tissues (1 g) were homogenized with 5 mL of chloroform-methanol mixture (4:1, v/v). The tissue homogenates were vortexed for 5 min for extracting DOX and centrifuged at 3,000 rpm for 10 min. The lower chloroform layer was extracted and evaporated to dryness under a stream of nitrogen in water bath at 37°C. The residues were redissolved in 60 μL of deionized water for further DOX analysis. The DOX in plasma (1 mL) were extracted with 5 mL of chloroform–methanol mixture (4:1, v/v) according to the aforementioned method omitting the homogenization process.
The quantifications of DOX in tissue extracts were carried out through RP-HPLC on a Diamonsil ODS column (250×4.6 mm i.d., 5 μm; Dikma) at 37°C±0.1°C. HPLC analysis was conducted on a Skyray LC-UV 310 HPLC system (Tianrui, China) equipped with a Skyray 310 UV–vis detector. The injection volume was 20 μL. The mobile phase consisted of (A) 0.01 mol/L sodium acetate-acetic acid buffer and (B) methanol. An isocratic elution (A:B =35:65, v/v) was achieved at a flow rate of 1.0 mL/min. All the analytes were detected at a wavelength of 254 nm. Each experiment was conducted in triplicate and results were presented as mean ± standard deviation.
In vivo MRI
As tumor-bearing model was established as the aforementioned mode, tumors were allowed to develop without treatment. Twenty HepG2 tumor-bearing mice were randomly divided into 4 groups (n=5/group): 1) MNP–DGL–DOX, 2) MNP–DGL–RGD–DOX, 3) MNP–DGL–GX1–DOX, and 4) MNP–DGL–RGD-GX1–DOX. The MRI study was performed using a 3.0 T system (Signa Excite HDx; GE Healthcare, Milwaukee, WI, USA), horizontal bore MRI scanner equipped with a dedicated small animal coil (15 channels). Tumor-bearing mice from the each group were first imaged at 2 weeks after tumor implant. Then the mice were given 0.2 mL of the different NPs irradiated by 60Co in sterile PBS through tail vein at a dose of 2 mg/kg body weight. Three hours later, the mice were anesthetized with 20% urethane one by one. The turbo spin echo technique was applied with acquisitions in the T2-weighted sequence in the transverse plane. The indicator “signal intensity (SI)” was determined at the same region of interest of tumor using T2-weighted sequence. T2-weighted image parameters were as follows: TR 3,000 ms, TE 100 ms, NEX 4, matrix size 192×192, FOV 190×190 mm, slice thickness 2 mm. The T2 relative signal intensity (SIR) was calculated by the following equation: SIR = SIEN/SIPL×100%,33 where SIEN and SIPL indicated the signal intensity of contrast-enhanced imaging (after injection of NPs) and plain imaging (before injection of NPs), respectively. The time–SIR curves were conducted to compare the enhancement effect of the NPs in tumor.
In vivo antitumor efficacy
On day 7 after inoculation, another 144 mice were randomly divided into 6 groups (n=24 per group): 1) control, 2) free DOX, 3) MNP–DGL–RGD-GX1, 4) MNP–DGL–RGD–DOX, 5) MNP–DGL–GX1–DOX, and 6) MNP–DGL–RGD-GX1–DOX. Each group included three treatment periods (for 10, 14, 18 days, respectively). For receiving NP mice, 0.2 mL of the NPs was injected at the aforementioned dose and mode on alternate days for 10, 14, 18 days, respectively. In control groups, each mouse was given 0.2 mL of sterile PBS in the same way. In free DOX groups, each mouse was injected with 0.2 mL of sterile DOX solutions (3 μg/mL) corresponding to the DOX content loaded on NPs. After this treatment, all the mice were sacrificed. The tumor tissues were quickly excised and weighed accurately. Tumor inhibition rate was computed according to the following formula: TIR = (the average tumor weight of the control group - the average tumor weight of treatment group)/the average tumor weight of the control group ×100%.34
Statistical analysis
All the data were presented as mean ± standard deviation (SD). Statistical analyses between the samples and the controls were conducted using ANOVA. P<0.05 was considered statistically significant.
Results and discussion
Synthesis of the MNP–DGL–RGD-GX1–DOX NPs
Synthesis of the MNP–DGL–RGD-GX1–DOX NPs comprises four steps. First, it is necessary to cover the MNPs with CA to obtain dispersive hydrophilic MNP–CA NPs and provide plenty of carboxyl groups on the surface. Second, MNP–DGL NPs were synthesized by covalent bonding of CA carboxyl groups with DGL amino groups. Third, the MNP–DGL–RGD-GX1 NPs were fabricated by the formation of amide bond between free carboxyl groups from heterogeneous dimer peptide RGD-GX1 and amino groups of DGL. In the posterior two reactions, NHS and EDAC served as catalyst, and PBS buffer (50 mM) containing 0.9% NaCl was used as solvent. Finally, anticancer drug DOX was uploaded by physical absorption in water phase.
MNPs are increasingly being considered for a number of biomedical applications because of their biocompatibility.35 DGL-G3 is a three-dimensional polyvalent nanomolecule (2–16 nm) with high number of NH2 surface groups (123). DGL are non-immunogenic and biocompatible dendritic polypeptides.36 However, the cationic dendrimers have a certain disadvantages such as high toxicity. The toxicity is mainly caused by cations that can produce electrostatic attraction with anions on the surface of the cell membrane.37 However, in the present study, this is not the case. The cationic amino groups have been blocked by covalent bonding of carboxyl groups on MNP–CA with amino groups on DGL. Furthermore, the MNP–DGL–RGD-GX1 NPs were fabricated by the formation of amide bond between free carboxyl groups from heterogeneous dimer peptide RGD-GX1 and amino groups of DGL. Finally, high DOX payload in NPs also covered exposed polylysine residues. After the aforementioned 3 steps, the cationic amino groups on DGL were blocked. So, ingredients used for the present NP formulation demonstrate high biocompatibility.
Characterization of the MNP–DGL–RGD-GX1–DOX NPs
Figure 2 shows the morphology of the resulting NPs taken by TEM. The hydroxyl groups on the dispersive Fe3O4 MNP–CA surface provide the adequate condition for dendri-graft poly-L-lysine (DGL) grafting (Figure 2A). From Figure 2, it is observed that the surface of MNP–DGL particles became smoother than that of MNP–CA, and there was a thin layer of dense and uniform covering (~10 nm). The TEM images reveal that the covering layer of MNP–DGL–RGD-GX1–DOX NPs is denser and thicker than that of MNP–DGL (Figure 2C). It can be seen that the average diameter of NPs was ~150 nm.
 | Figure 2 TEM images of (A) MNP–CA, (B) MNP–DGL, and (C) MNP–DGL–RGD-GX1–DOX composite. |
Figure 3A shows the FTIR spectra of the resulting NPs and uniformly presents a peak at 589 cm−1 related to the Fe-O vibration, indicating the existence of Fe3O4 MNPs in the products synthesized step by step. As shown in Figure 3A(b), the peak at 3,450 cm−1 corresponding to the carboxyl groups indicated CA coated on the MNPs, showing MNP–CA has been obtained. Compared with the spectrum of MNP–CA, the peak at 3,450 cm−1 in MNP–DGL (Figure 3A(c)) became stronger, which is due to the introduction of amino groups from DGL. Two new characteristic peaks occurred at 1,700 cm−1 and 1,660 cm−1, confirming that the CONH2 groups have been formed between the carboxyl groups of CA and amino groups of DGL, indicating the successful synthesis of MNP–DGL. The spectrum of MNP–DGL–RGD (Figure 3A(d)) displayed the absorption peaks at 3,000 cm−1 and 2,940 cm−1 related to the stretching vibration of =C-H from phenyl group of phenylalanine (Phe) existing in the cyclopeptide cRGDfk, two peaks at 1,600 cm−1 and 1,500 cm−1 resulted from the stretching vibration of C=C and two peaks at 1,380 cm−1 and 1,200 cm−1 corresponding to C-H and -O-, respectively. All of these results show that the RGD successfully combined with DGL. Likewise, in the MNP–DGL–GX1 (Figure 3A(e)), the peak at 3,450 cm−1 became stronger related to the superposition of amino group and carboxyl group peaks, the peaks at 1,700 cm−1 and 1,660 cm−1 resulted from the carboxyl group, the peak at 2,520 cm−1 corresponding to the vibration of sulfydryl groups existing in GX1, powerfully suggesting that MNP–DGL–GX1 has been prepared through the formation of amide bond between GX1 and DGL. According to the spectrum of MNP–DGL–RGD-GX1 (Figure 3A(f)), a big overlapping peak corresponding to hydroxyl group, amino group, and carboxyl group clearly shifted to higher wave numbers (3,600~3,400 cm−1) and became wider. The peaks of the stretching vibration of =C-H from phenyl group and C=C from aromatic ring skeleton appeared at 3,000, 2,940, 1,600, and 1,500 cm−1 as previously mentioned. Furthermore, a special peak at 1,140 cm−1 related to -O- deriving from the introduction of PEG linking RGD to GX1, fully demonstrating that heterogeneous dimer peptide RGD-GX1 successfully conjugated with DGL. In the Figure 3A(g), the peak occurred between 3,600 and 3,400 cm−1 became stronger compared with that of MNP–DGL–RGD-GX1, which is attributed to the large number of hydroxyl groups from DOX. Two new characteristic peaks at 2,692 cm−1 and 1,660 cm−1 corresponding to -CH3 and C=O from DOX, respectively, further verify the DOX loading.
 | Figure 3 (A) Fourier transform infrared spectroscopy spectra of MNP (a), MNP-CA (b), MNP-DGL (c), MNP-DGL-RGD (d), MNP-DGL-GX1 (e), MNP-DGL-RGD-GX1 (f), and MNP-DGL-RGD-GX1-DOX (g) NPs. (B) XRD patterns. (C) The size distribution of the MNP–DGL–RGD-GX1–DOX measured by DLS. (D) VSM magnetization curves. |
On the basis of XRD mode (Figure 3B), the peaks at (220), (311), (400), (422), and (440) were the characteristic peaks of Fe3O4 nanocrystal. Their peak values have a little decline although the intensities were still strong, which is because MNP nucleus was covered step by step. However, no peaks related to other crystallographic structure were observed, demonstrating the purity of the NPs.
The average diameter of the MNP–DGL–RGD-GX1–DOX NPs was measured to be ~150-160 nm by DLS (Figure 3C). Compared with the mean diameters based on TEM (Figure 1C), those of the MNP–DGL–RGD-GX1–DOX NPs only increased by ~10 nm, which is attributed to wet sample in DLS assay.
Figure 3D shows VSM magnetization curves of MNP, MNP–CA, MNP–DGL, and MNP–DGL–RGD-GX1–DOX NPs. With step-by-step synthesis, the saturation magnetization is gradually reduced due to the appearance and thickening of the covering. The saturation magnetization of end product MNP–DGL–RGD-GX1–DOX (49.0 emu/g) was ~56.3% and 72.9% of that of MNP–CA (87.0 emu/g) and MNP–DGL (63.4 emu/g), suggesting a sufficient magnetic responsiveness in the applications of MRI as a contrast agent.
Drug loading and release
In the present experimental system, the loading efficiencies of DOX were 83.8 wt%, 82.4 wt%, and 82.6 wt% for MNP–DGL–RGD, MNP–DGL–GX1, and MNP–DGL–RGD-GX1; in other words, the actual loading amount of DOX on 3 NPs accounted for ~418.4 mg/g, 410.8 mg/g, and 414.4 mg/g, respectively. These values were basically consistent with one another, and the mean value was ~414.5 mg/g. It is not difficult to speculate that DOX bound similarly to 3 carriers. DOX molecules possess both acidic phenolic and hydroxyl groups and alkaline amino groups (Figure 4, inset) that are able to interact with the amino groups of DGL and hydroxyl groups of peptides to form intermolecular complexes by hydrogen bonding on the resulting carriers. At the same time, there were both hydrophobic force and aromatic-ring stack force between DOX and phenylalanine in RGDfk pentapeptide, resulting in a slightly higher DOX loading efficiencies in MNP–DGL–RGD and MNP–DGL–RGD-GX1 than MNP–DGL–GX1. The introduction of polylysine dendrimer in NPs provided ample space for high DOX payload, which was significantly higher than that of amphiphilic copolymer micelles in the previous report.38
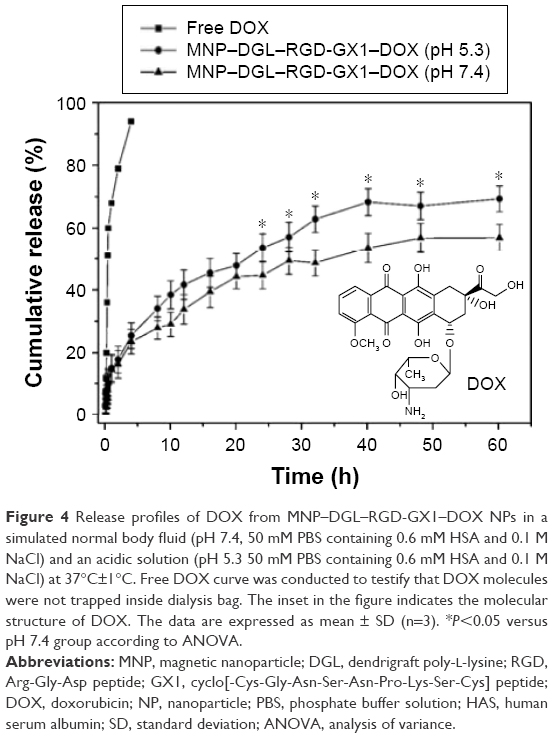 | Figure 4 Release profiles of DOX from MNP–DGL–RGD-GX1–DOX NPs in a simulated normal body fluid (pH 7.4, 50 mM PBS containing 0.6 mM HSA and 0.1 M NaCl) and an acidic solution (pH 5.3 50 mM PBS containing 0.6 mM HSA and 0.1 M NaCl) at 37°C±1°C. Free DOX curve was conducted to testify that DOX molecules were not trapped inside dialysis bag. The inset in the figure indicates the molecular structure of DOX. The data are expressed as mean ± SD (n=3). *P<0.05 versus pH 7.4 group according to ANOVA. |
DOX release profiles in vitro were conducted under dialysis condition at 37°C in a simulative normal body fluid and an acidic environment. As expected, free DOX in dialysis bag was rapidly dialyzed from the medium. In this study, the release results of the only end product MNP–DGL–RGD-GX1–DOX are presented (Figure 4) because there was no significant difference among MNP–DGL–RGD–DOX, MNP–DGL–GX1–DOX, and MNP–DGL–RGD-GX1–DOX. First, the initial burst releases of DOX were clearly found in two types of medium. For example, 34% and 28% of DOX from MNP–DGL–RGD-GX1–DOX NPs can be released at pH 5.3 and pH 7.4 at the initial 8 h, respectively, occupying approximately half of release amount within 60 h. This release profiles may attribute to the loading pattern of DOX. The combination maintained by non-bonding interaction may be unstable, such as hydrophobic force and aromatic-ring stack force. Hence, DOX molecules readily fell off when NPs were shaken in water bath. Subsequently, more DOX were released from 8 to 40 h, suggesting that DOX loading by hydrogen bond began to separate from NPs. The release reached a plateau after 40 h, indicating that the DOX release has been in a balance state. Second, the release efficiency from MNP–DGL–RGD-GX1–DOX was higher at pH 5.3 than at pH 7.4. A possible reason may be DGL structural response to environmental pH. Under acidic condition (pH 5.3), the side-chain amino groups of DGL within NPs obtain proton and form NH3+; hence, Lys chains are in stretched conformation in order to reduce internal repulsion.39
Accordingly, the DOX would be more liable to dissolution because of exposure. When at pH 7.4, the side-chain amino groups of DGL gradually shift to weak nucleophilic reagent, so that DGL form stable coiled helical structure by hydrogen bonds within the main chain,39 and accordingly the DOX would have low solubility because of compact structure, which is the expected result, namely the antitumor drug DOX could largely be delivered to acidic tumor microenvironment rather than distributed around normal tissues.
In vitro cytotoxicity
In order to assess the in vitro cytotoxicity of the resulting NPs and to compare the differences of targeting between single and double ligand-modified NPs to different cancer cells, in the present study, human hepatocyte cancer HepG2 cells, lung cancer A549 cells, and control L02 hepatic cells were selected. The cells were treated with a concentration range of 12.5~800 μg/mL of the empty or DOX-loaded NPs. As shown in Figure 5, empty MNP–DGL–RGD-GX1 NPs always showed high cell viability for HepG2 cells (72%), A549 cells (79%), and L02 cells (80%) even at a high concentration of 800 μg/mL, suggesting that the carrier had relatively good biocompatibility. By contrast, the DOX-loaded NPs revealed apparent growth inhibition activity for cancer and normal cells. The rising cytotoxicity resulted mainly from the DOX bound on different NPs. Especially, MNP–DGL–RGD-GX1–DOX and MNP–DGL–RGD–DOX showed lower cell viability for HepG2 cells than MNP–DGL–GX1–DOX at all tested concentrations, reflecting that the previous two NPs possessed better targeting capacity to tumor. According to previous research studies, NPs modified with RGD could more efficiently bind with HepG2 cells by the mediation of ανβ3 integrin overexpressed on tumor cells,40 whereas the binding effect of GX1 peptide with cultured HepG2 cells was not significant.41 Furthermore, according to the results, it was found that the targeting effects of the corresponding NPs to hepatocellular carcinoma were slightly better than lung cancer. A further comparison indicated that a lower but not negligible cytotoxic effect has also been observed on normal hepatocytes. In the case of free DOX, two types of cancer cells and L02 cells demonstrated similar cell viabilities, showing nonspecific effect of free drug.
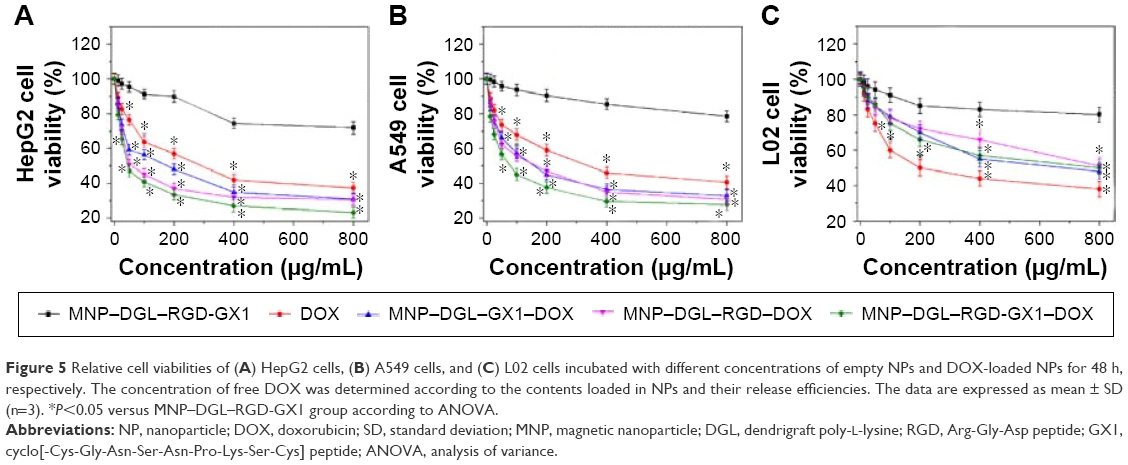 | Figure 5 Relative cell viabilities of (A) HepG2 cells, (B) A549 cells, and (C) L02 cells incubated with different concentrations of empty NPs and DOX-loaded NPs for 48 h, respectively. The concentration of free DOX was determined according to the contents loaded in NPs and their release efficiencies. The data are expressed as mean ± SD (n=3). *P<0.05 versus MNP–DGL–RGD-GX1 group according to ANOVA. |
Apoptosis assay assessed by FCM
To further quantitatively analyze the apoptosis induced by free DOX or different NPs, above HepG2 cells treated with NPs for 48 h were co-stained with Annexin V-FITC/PI. The apoptosis in the cells from various groups was assayed by FCM. The gate of positive cells was uniformly set in 79.8% of the living cells (Q4) derived from the control group. If the percentage of positive cells in the given gate compared with the control was >10%, the event was considered as significant (P<0.05). As shown in the control group (Figure 6A), untreated HepG2 cells occupied 20.18% of apoptotic cells (Q2 + Q3) and few dead cells (Q1). Similarly, HepG2 cells were treated with empty MNP–DGL–RGD-GX1 NPs and possessed 73.2% of the living cells (Q4), 26.8% of apoptotic cells (Q2 + Q3), and negligible dead cells (Q1) (Figure 6B), which shows that empty carrier has high biocompatibility. However, free DOX induced 22.5% of dead cells (Q1), 25.9% of late-stage apoptotic cells (Q2), and 3.45% of early stage apoptotic cells (Q3) (Figure 6C), and the number of living cell is reduced to 48.1%. Furthermore, the MNP–DGL–GX1–DOX NPs induced 10.8% of dead cells (Q1), 41.1% of late stage apoptotic cells (Q2), and 6.91% of early stage apoptotic cells (Q3) (Figure 6D), and their sum is similar to the total cell sum (Q1 + Q2 + Q3) induced by free DOX, suggesting that the DOX released from the NPs still possessed similar anticancer activity as free DOX. By contrast, the dead cells (Q1) induced by MNP–DGL–RGD–DOX NPs (Figure 6E) and MNP–DGL–RGD-GX1–DOX NPs (Figure 6F) reached 40.9% and 46.7%, respectively. The results indicated that RGD- or RGD-GX1-modified NPs were easier to induce cell death than GX1-modified NPs, which verified that RGD could enhance integrin-mediated endocytosis in cultured HepG2 cells.42
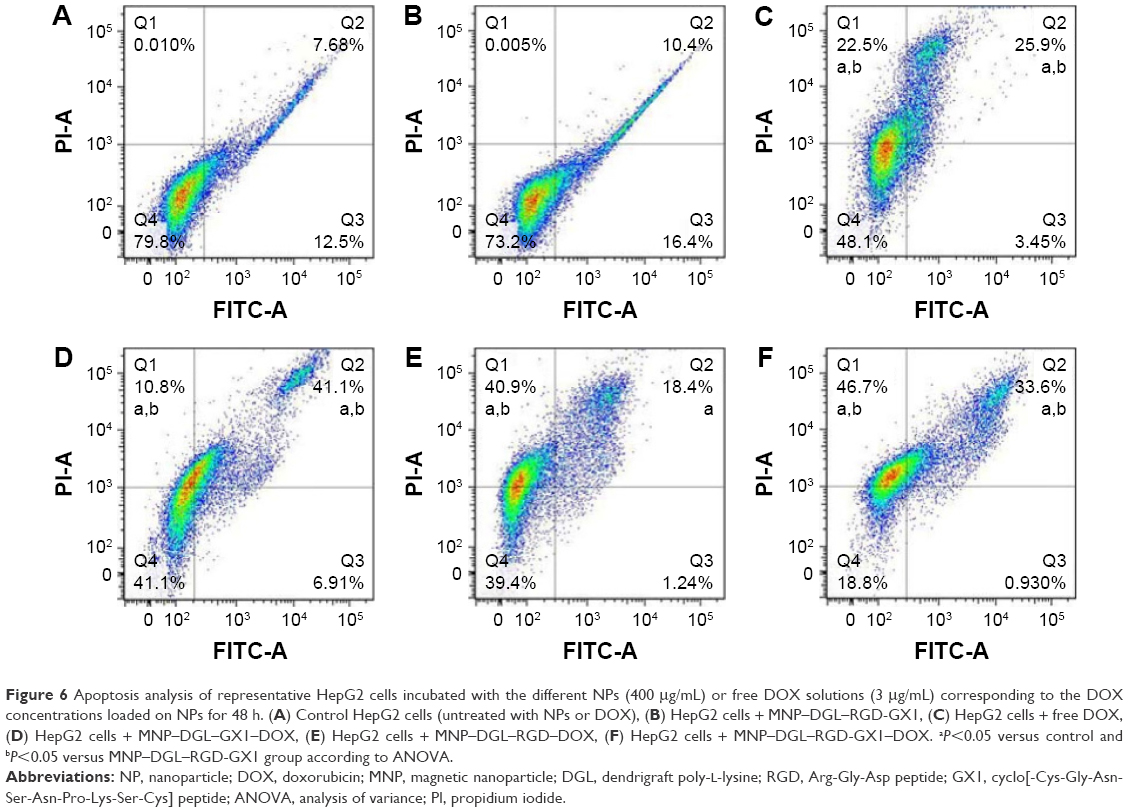 | Figure 6 Apoptosis analysis of representative HepG2 cells incubated with the different NPs (400 μg/mL) or free DOX solutions (3 μg/mL) corresponding to the DOX concentrations loaded on NPs for 48 h. (A) Control HepG2 cells (untreated with NPs or DOX), (B) HepG2 cells + MNP–DGL–RGD-GX1, (C) HepG2 cells + free DOX, (D) HepG2 cells + MNP–DGL–GX1–DOX, (E) HepG2 cells + MNP–DGL–RGD–DOX, (F) HepG2 cells + MNP–DGL–RGD-GX1–DOX. aP<0.05 versus control and bP<0.05 versus MNP–DGL–RGD-GX1 group according to ANOVA. |
In vitro cellular imaging on LSCM
In order to intuitively compare the differences of single or double ligand-modified NPs on cellular uptake, the morphologies of representative apoptotic HepG2 cells from each group were imaged on LSCM. As seen in Figure 7A, there was no fluorescence material within the control HepG2 cells, showing that cells came out unharmed. It can be clearly seen that the HepG2 cells treated with MNP–DGL–RGD-GX1 have green fluorescence from Annexin V-FITC that can only react with phosphatide acyl serine inside membrane, indicating that the membrane has been broken and cells began apoptosis (Figure 7B). However, the morphologies of the cells were still normal. In fact, Figure 7B is presented as specific field of vision with apoptosis. MNP–DGL–RGD-GX1 can also induce apoptosis of the HepG2 cells because of cellular uptake of MNP–DGL–RGD-GX1 NPs by the mediation of RGD-GX1. Although MNP–DGL–RGD-GX1 NPs did not contain DOX, there was more or less some toxicity, which was consistent with the previous results of in vitro cytotoxicity. In the case of the HepG2 cells treated with free DOX or three types of DOX-loaded NPs, the cytoplasm severely collapsed and deformed, the nuclei had bright red fluorescence besides the cellular membrane, and the matrix possessed green fluorescence (Figure 7C–F), confirming that the cells have been at later stages of apoptosis, even died because PI can only stain dead cells. These results showed that DOX released from the NPs still has the same antitumor activity as free DOX. Particularly, the HepG2 cells treated with MNP–DGL–RGD–DOX or MNP–DGL–RGD-GX1–DOX NPs were completely destroyed (Figure 7E and F), for example, the cell membrane disappeared, the matrix flowed out, and nucleuses severely deformed, verifying that the apoptosis can be enhanced by the specificity of RGD peptide conjugated on NPs to αvβ3 integrin on tumor cells. The mechanism involves the specific targeting of peptide-conjugated nanoprobe. The drug was internalized by the mediation of peptide, which in turn caused higher cytotoxicity. Figure 7E and F illustrates the morphology of apoptosis that were included in the cases caused by MNP–DGL–RGD–DOX or MNP–DGL–RGD-GX1–DOX NPs.
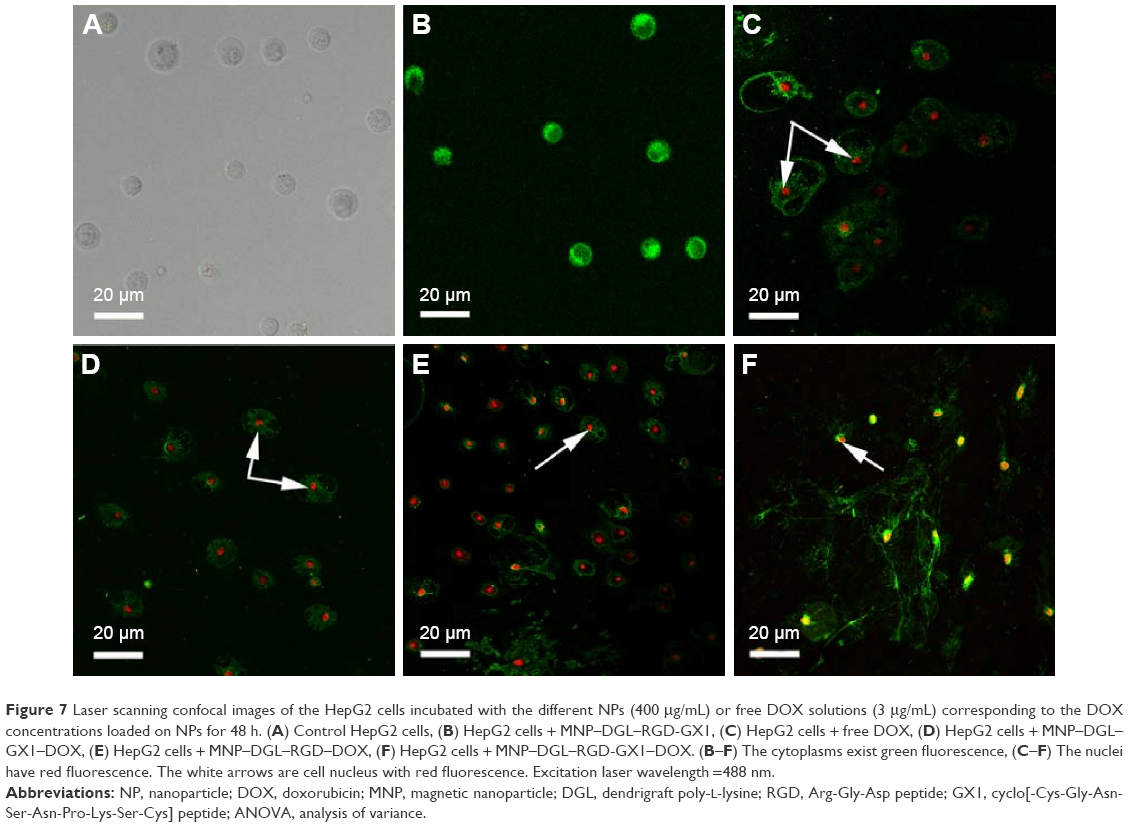 | Figure 7 Laser scanning confocal images of the HepG2 cells incubated with the different NPs (400 μg/mL) or free DOX solutions (3 μg/mL) corresponding to the DOX concentrations loaded on NPs for 48 h. (A) Control HepG2 cells, (B) HepG2 cells + MNP–DGL–RGD-GX1, (C) HepG2 cells + free DOX, (D) HepG2 cells + MNP–DGL–GX1–DOX, (E) HepG2 cells + MNP–DGL–RGD–DOX, (F) HepG2 cells + MNP–DGL–RGD-GX1–DOX. (B–F) The cytoplasms exist green fluorescence, (C–F) The nuclei have red fluorescence. The white arrows are cell nucleus with red fluorescence. Excitation laser wavelength =488 nm. |
Exploration of the Fas-mediated apoptotic mechanism
In order to explore the potential signaling pathway of MNP–DGL–RGD-GX1–DOX NP-induced apoptosis, the expression level of Fas protein and the activation of caspase-3 were examined by FCM analysis by using monoclonal antibodies. The results showed that Fas proteins were upregulated in a dose-dependent mode in HepG2 cells in the presence of MNP–DGL–RGD-GX1–DOX or free DOX (Figure 8A and B) compared with control. The activities of caspase-3 were also significantly enhanced in a dose-dependent pattern after the treatment with the NPs (Figure 8C and D).
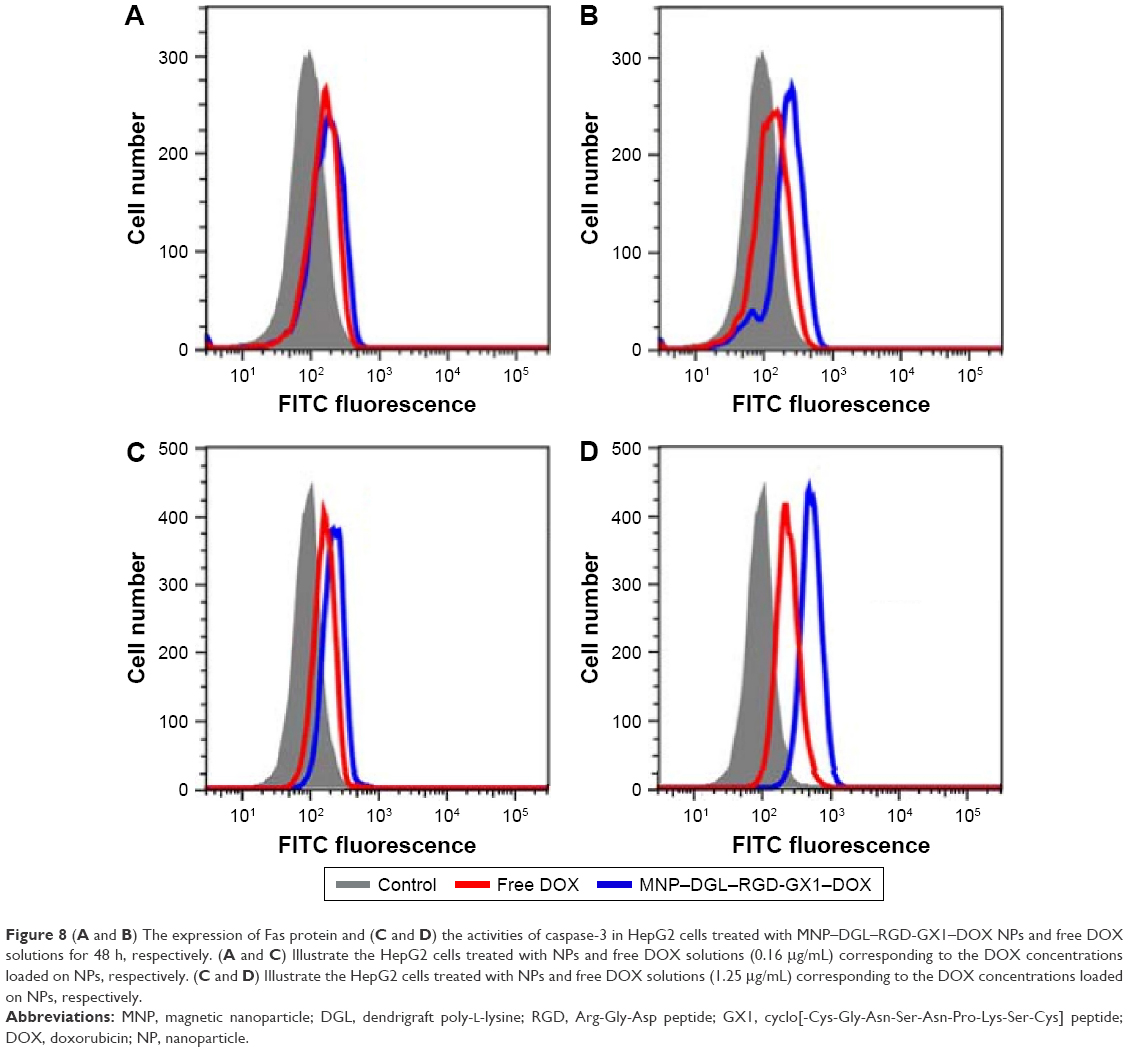 | Figure 8 (A and B) The expression of Fas protein and (C and D) the activities of caspase-3 in HepG2 cells treated with MNP–DGL–RGD-GX1–DOX NPs and free DOX solutions for 48 h, respectively. (A and C) Illustrate the HepG2 cells treated with NPs and free DOX solutions (0.16 μg/mL) corresponding to the DOX concentrations loaded on NPs, respectively. (C and D) Illustrate the HepG2 cells treated with NPs and free DOX solutions (1.25 μg/mL) corresponding to the DOX concentrations loaded on NPs, respectively. |
According to a previous report,43 DOX-induced apoptosis in HepG2 cells involved Fas-mediated pathway. Fas is a member of death receptors that belong to the tumor necrosis factor receptor family and play a critical role in cell apoptosis by transmitting apoptosis signals and activating caspases.44 Caspase-3 is a pivotal downstream caspase on the apoptotic pathway. After being activated, it can induce cell death.45,46 Hence, it could be inferred that MNP–DGL–RGD-GX1–DOX NPs induced the cell death in the same way as free DOX, namely the apoptosis initiated through triggering the expression of Fas protein and the activities of caspase-3. A further comparison showed that the values of Fas protein and caspase-3 in NPs group were 1.68 times and 2.58 times at low dose, 3.66 times and 1.70 times at high dose of those in free DOX group, respectively (Figure 8). Huang et al declared that RGD-modified polymeric NPs could enhance the internalization of the NPs in cultured HepG2 cells through integrin-mediated endocytosis with rapid DOX release intracellularly.42 In addition, a weak binding of GX1 to HepG2 cells was also observed in specific phage-displayed peptide binding experiment by Zhi et al, although GX1 mainly selectively targeted to vascular endothelium of tumor.41 Therefore, there was more or less synergistic effect of the targeting to HepG2 cells in vitro through the mediation of RGD-GX1 heterogeneous dimer peptide. This high accumulation of NPs in HepG2 cells enhanced the intracellular release of DOX.41,47 Correspondingly, the apoptosis of HepG2 cells induced by DOX-loaded RGD-GX1-modified NPs was more effective than free DOX.21,48
Distribution results
The distribution patterns of DOX in various tissues from free DOX, MNP–DGL–RGD–DOX, MNP–DGL–GX1–DOX, and MNP–DGL–RGD-GX1–DOX NPs-treated mice were shown in Figure 9. In free DOX-treated group (Figure 9A), DOX was widely and rapidly distributed into various tissues by intravenous administration of free DOX solution in the first 1 h, particularly in blood, liver, and kidney. When the time was extended to 3 h, the distribution of DOX was homogeneous in all the tissues. This is mainly because the small molecule DOX tends to diffuse distribution to various tissues, which causes toxic side effects on normal tissues. In the MNP–DGL–RGD–DOX (Figure 9B) or MNP–DGL–GX1–DOX (Figure 9C) NPs-treated mice, DOX abundances in tumor tended to very significant increase at every time point compared with free DOX group. The results indicate that the RGD and GX1 peptides play an active role in targeting the tumor. A large number of literatures reported that RGD and GX1 peptides could specifically target to αvβ3-integrin2,49,50 and vasculature endothelium receptors,51–54 respectively, which were excessively expressed on plasma membrane of tumor cells. In addition, the distribution mode of double ligand-modified NPs was significantly different from those of the free DOX and single ligand-modified NPs. As can be clearly seen in Figure 9D, the values observed for MNP–DGL–RGD-GX1–DOX formulation were higher than those found for MNP–DGL–RGD–DOX and MNP–DGL–GX1–DOX NPs in the tumor at 1 h. The highest DOX concentration was found in tumor > liver > kidney >lung > spleen > blood > heart. Importantly, at 3 h time point, DOX abundance significantly (P<0.05) increased in tumor and markedly decreased in the other tissues, especially in liver, compared with the two single ligand-modified NP groups. This result testified that RGD-GX1 heterogeneous dimer peptide-modified NPs enhanced the delivery of DOX to tumor tissue. This is the synergistic activity of targeting αvβ3-integrin receptors on cancer cells and GX1 receptors on tumor vasculature.48,55
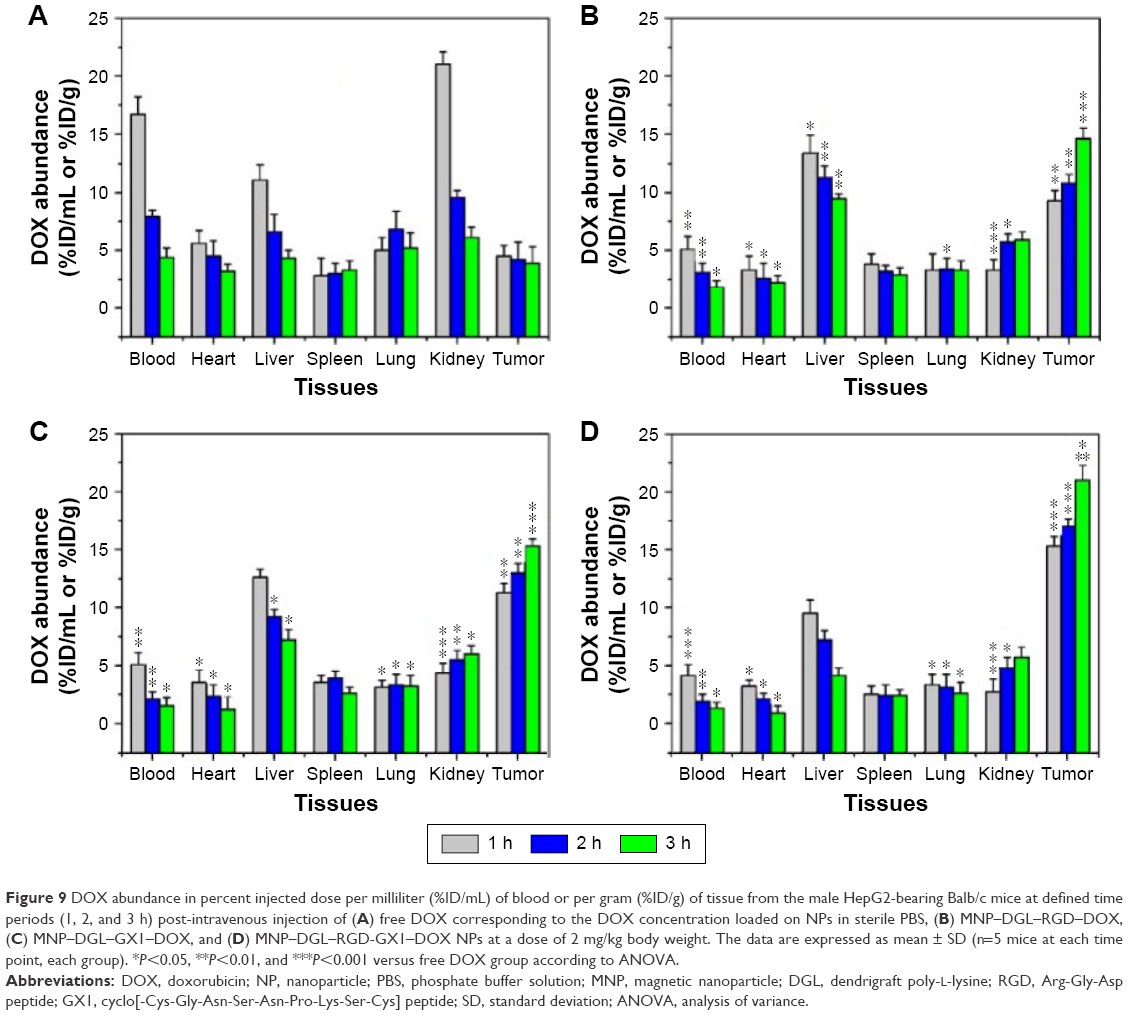 | Figure 9 DOX abundance in percent injected dose per milliliter (%ID/mL) of blood or per gram (%ID/g) of tissue from the male HepG2-bearing Balb/c mice at defined time periods (1, 2, and 3 h) post-intravenous injection of (A) free DOX corresponding to the DOX concentration loaded on NPs in sterile PBS, (B) MNP–DGL–RGD–DOX, (C) MNP–DGL–GX1–DOX, and (D) MNP–DGL–RGD-GX1–DOX NPs at a dose of 2 mg/kg body weight. The data are expressed as mean ± SD (n=5 mice at each time point, each group). *P<0.05, **P<0.01, and ***P<0.001 versus free DOX group according to ANOVA. |
MRI in vivo
In order to compare the targeting effect of different peptide-modified NPs in vivo, T2-weighted MR images of HepG2 tumor-bearing mice transverse plane were acquired before and after intravenous administration of NPs. Figure 10A and B showed representative MR images of tumor in mice untreated (control) and treated with MNP–DGL–DOX NPs, respectively. The signal intensity (image color) of tumor had hardly any difference, suggesting that ligand-unmodified NPs had negligible targeting capability to tumor. On the contrary, the periphery of tumors treated with MNP–DGL–RGD–DOX NPs (Figure 10C) or MNP–DGL–GX1–DOX NPs (Figure 10D) Is darkened, reflecting the aggregation of the contrast agent due to the uptake of NPs in tumor. Especially, the regional signal intensity (Figure 10D) of tumor for mice treated with MNP–DGL–GX1–DOX NPs was apparently lower than one treated with MNP–DGL–RGD–DOX NPs (Figure 10C). For mice that received MNP–DGL–RGD-GX1–DOX NPs, the MR signal further declined compared with mice that received single-ligand-modified NPs. The most lesion zone turned homogenously into black area (Figure 10E). As shown in the time–SIR curves (Figure 10F), the tumor SIR reduced quickly and reached a negative peak value (maximal negative enhancement) after MNP–DGL–RGD-GX1–DOX NPs were injected. Then, the value gradually rose to 68%±2.1% of the baseline after 3 h. A negligible enhancement was shown in tumor after MNP–DGL–DOX.
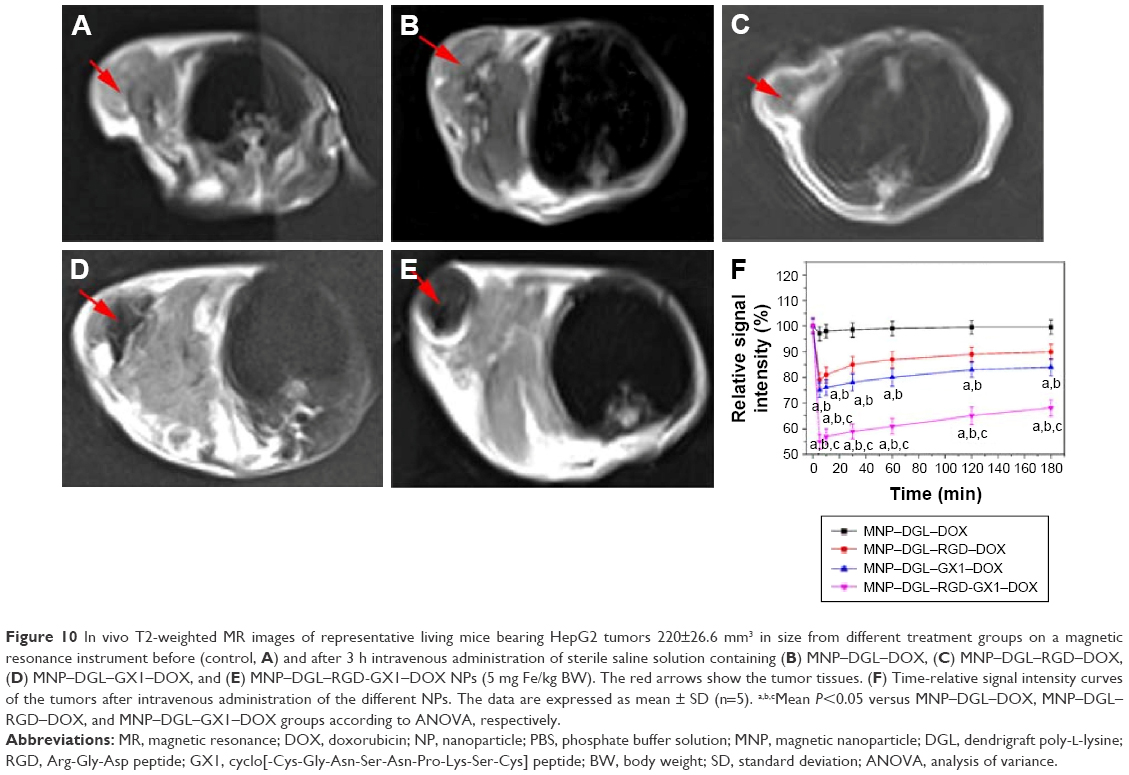 | Figure 10 In vivo T2-weighted MR images of representative living mice bearing HepG2 tumors 220±26.6 mm3 in size from different treatment groups on a magnetic resonance instrument before (control, A) and after 3 h intravenous administration of sterile saline solution containing (B) MNP–DGL–DOX, (C) MNP–DGL–RGD–DOX, (D) MNP–DGL–GX1–DOX, and (E) MNP–DGL–RGD-GX1–DOX NPs (5 mg Fe/kg BW). The red arrows show the tumor tissues. (F) Time-relative signal intensity curves of the tumors after intravenous administration of the different NPs. The data are expressed as mean ± SD (n=5). a,b,cMean P<0.05 versus MNP–DGL–DOX, MNP–DGL–RGD–DOX, and MNP–DGL–GX1–DOX groups according to ANOVA, respectively. |
NPs were injected, where the SIR always kept at the baseline level. For mice that received MNP–DGL–RGD–DOX NPs or MNP–DGL–GX1–DOX NPs, the tumor SIR fell between the above two sides. From these results, the MNP–DGL–RGD-GX1–DOX formulation seemed to have a higher affinity to the tumor tissue, leading to more accumulation of NPs (contrast agent) in the tumor. This finding can be explained by the fact that the RGD-GX1 heterogeneous dimer peptide could play a synergetic role in targeting to tumor tissue in vivo because of their specific receptors overexpressed in tumor cells and tumor blood vessels.
In vivo antitumor efficacy
The antitumor activities of free DOX and different NPs were evaluated in HepG2 tumor-bearing Balb/c mice. There was an indication that mice treated with free DOX showed systemic toxicity giving rise to ~2.1% weight loss for 10 days, 3.8% for 14 days, and 11.2% for 18 days (Table 1), respectively. By contrast, the body weight of mice in DOX-loaded NP groups was increased in various degrees, which showed that the ligand-conjugated DOX-loaded NPs could decrease the toxicity of DOX and improve the anticancer drug security. For tumor-bearing mice, when NPs play a role of tumor suppression, the mice will grow better, and weight will be increased.
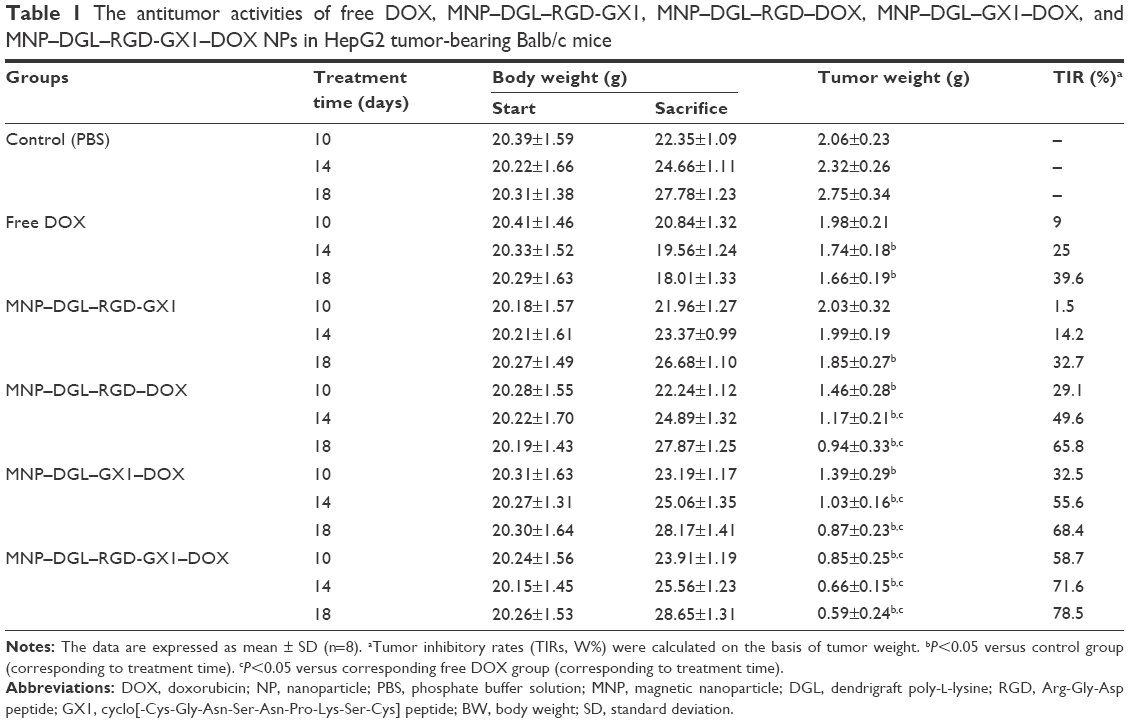 | Table 1 The antitumor activities of free DOX, MNP–DGL–RGD-GX1, MNP–DGL–RGD–DOX, MNP–DGL–GX1–DOX, and MNP–DGL–RGD-GX1–DOX NPs in HepG2 tumor-bearing Balb/c mice |
The changes in tumor weight were measured as shown in Table 1. The mean weight of tumors increased from 2.06±0.23 g at 10 days to 2.75±0.34 g at 18 days for the control groups, whereas the values in all treatment groups decreased with time. Particularly, at the end of the treatment (day 18), the mean weight of tumors was 1.66±0.19 g for free DOX group, 1.85±0.27 g for MNP–DGL–RGD-GX1 group, 0.94±0.33 g for MNP–DGL–RGD–DOX group, 0.87±0.23 g for MNP–DGL–GX1–DOX group, and 0.59±0.24 g for MNP–DGL–RGD-GX1–DOX group, respectively. It is clear that the tumor mass of mice in MNP–DGL–RGD-GX1–DOX group was the smallest in all the groups. Although free DOX and empty MNP–DGL–RGD-GX1 NPs could inhibit the growth of tumor to some extent, the tumor inhibitory rates (TIRs) of 39.6% and 32.7% at 18 days were relatively low, respectively (Table 1). This can be attributed to the deficiency of targeting efficacy because of the diffuse administration of free DOX group. Another possible reason may be that free DOX molecules are apt to lose its anticancer activity in body fluid. In terms of the empty NPs, the absence of DOX is the main cause. In the case of MNP–DGL–RGD–DOX and MNP–DGL–GX1–DOX groups, their TIRs were higher compared with two previous groups, reaching ~1.66 times and 1.73 times as high as those of free DOX group, respectively. The differences were significant in contrast to the control group (P<0.05). In particular, the TIR in the MNP–DGL–RGD-GX1–DOX group was markedly the highest in all the groups, and this value accounted for 78.5% (Table 1), occupying ~1.19 times and 1.15 times as high as those of MNP–DGL–RGD–DOX and MNP–DGL–GX1–DOX groups, respectively. The results showed that the TIR obtained upon treatment with MNP–DGL–RGD-GX1–DOX NPs was significantly better than those in single-ligand-modified groups. According to the previous literatures, moreover, the TIR of RGD-modified DOX-loaded NPs in Bel-7402-bearing mice was 56.64%,49 and the TIR of RGD-conjugated EPB-loaded NPs in H22 tumor-bearing ICR mice was 59.48%.56 The present TIR of 78.5% powerfully testified that the specific integrin/vasculature endothelium receptor-mediated chemotherapy significantly promoted the tumor growth inhibition.
Conclusion
The novel heterogeneous dimer peptide-conjugated polylysine dendrimer-Fe3O4 nanoscale probe was prepared and characterized for early diagnosis and therapy in hepatocellular carcinoma. The introduction of polylysine dendrimer in NPs provided ample space for high DOX payload. In vitro cytotoxicity, apoptosis assessment, and cellular imaging consistently indicated the preferential enrichment capability of the resulting probe to HepG2 cells in selected three types of cells. The molecular mechanism research demonstrated that MNP–DGL–RGD-GX1–DOX NPs accelerated the apoptosis through the death receptor way. In vivo distribution data and MRI verified that the nanoprobe bound effectively to tumor by specific mediation of αvβ3-integrin/vasculature endothelium dual receptors. A high tumor inhibition rate has powerfully testified its antitumor efficacy. In short, the synthesized nanoprobe possesses the potential of a sensitive contrast agent and specific nanovehicle.
Acknowledgments
This study was supported by the National Natural Science Foundation of China Fund (No 81541060). The authors are grateful to Core Facility of School of Life Sciences, Lanzhou University for the assistance during some experiments.
Disclosure
The authors report no conflicts of interest in this work.
References
Elberry AA, Abdel-Naim AB, Abdel-Sattar EA, et al. Cranberry (Vaccinium macrocarpon) protects against doxorubicin-induced cardiotoxicity in rats. Food Chem Toxicol. 2010;48(5):1178–1184. | ||
Shen JM, Gao FY, Yin T, et al. cRGD-functionalized polymeric magnetic nanoparticles as a dual-drug delivery system for safe targeted cancer therapy. Pharmacol Res. 2013;70(1):102–115. | ||
Xu C, Shi SX, Feng LZ, et al. Long circulating reduced graphene oxide-iron oxide nanoparticles for efficient tumor targeting and multimodality imaging. Nanoscale. 2016;8(25):12683–12692. | ||
Zhao Q, Yi X, Li MF, Zhong XY, Shi QL, Yang K. High near-infrared absorbing Cu5FeS4 nanoparticles for dual-modal imaging and photothermal therapy. Nanoscale. 2016;8(27):13368–13376. | ||
Adegoke O, Kato T, Park EY. An ultrasensitive alloyed near-infrared quinternary quantum dot-molecular beacon nanodiagnostic bioprobe for influenza virus RNA. Biosens Bioelectron. 2016;80:483–490. | ||
He XW, Zeng T, Li Z, Wang GL, Ma N. Catalytic molecular imaging of microRNA in living cells by DNA-programmed nanoparticle disassembly. Angew Chem Int Edit. 2016;55(9):3073–3076. | ||
Hussain T, Nguyen QT. Molecular imaging for cancer diagnosis and surgery. Adv Drug Deliv Rev. 2014;66:90–100. | ||
Li XM, Wei JR, Aifantis KE, et al. Current investigations into magnetic nanoparticles for biomedical applications. J Biomed Mater Res A. 2016;104(5):1285–1296. | ||
Li KA, Wen SH, Larson AC, et al. Multifunctional dendrimer-based nanoparticles for in vivo MR/CT dual-modal molecular imaging of breast cancer. Int J Nanomedicine. 2013;8:2589–2600. | ||
Massoud TF, Gambhir SS. Molecular imaging in living subjects: seeing fundamental biological processes in a new light. Genes Dev. 2003;17(5):545–580. | ||
Jacobs RE, Cherry SR. Complementary emerging techniques: high-resolution PET and MRI. Curr Opin Neurobiol. 2001;11(5):621–629. | ||
Takahashi S. Vascular endothelial growth factor (VEGF), VEGF receptors and their inhibitors for antiangiogenic tumor therapy. Biol Pharm Bull. 2011;34(12):1785–1788. | ||
Massaguer A, Gonzalez-Canto A, Escribano E, et al. Integrin-targeted delivery into cancer cells of a Pt(IV) pro-drug through conjugation to RGD-containing peptides. Dalton T. 2015;44(1):202–212. | ||
Zheng Y, Ji S, Czerwinski A, Valenzuela F, Pennington M, Liu S. FITC-conjugated cyclic RGD peptides as fluorescent probes for staining integrin αvβ3/αvβ5 in tumor tissues. Bioconjug Chem. 2014;25(11):1925–1941. | ||
Eliceiri BP, Cheresh DA. The role of αv integrins during angiogenesis: insights into potential mechanisms of action and clinical development. J Clin Invest. 1999;103(9):1227–1230. | ||
Holzer TR, Fulford AD, Reising LO, et al. Profiling of vascular endothelial growth factor receptor heterogeneity identifies protein expression-defined subclasses of human non-small cell lung carcinoma. Anticancer Res. 2016;36(7):3277–3288. | ||
Sun X, Li Y, Liu T, Li Z, Zhang X, Chen X. Peptide-based imaging agents for cancer detection. Adv Drug Deliv Rev. 2016 Epub Jun 18. | ||
Hui XL, Han Y, Liang SH, et al. Specific targeting of the vasculature of gastric cancer by a new tumor-homing peptide CGNSNPKSC. J Control Release. 2008;131(2):86–93. | ||
Chen K, Yap LP, Park R, et al. A Cy5.5-labeled phage-displayed peptide probe for near-infrared fluorescence imaging of tumor vasculature in living mice. Amino Acids. 2012;42(4):1329–1337. | ||
Liu Z, Niu G, Wang F, Chen X. 68Ga-labeled NOTA-RGD-BBN peptide for dual integrin and GRPR-targeted tumor imaging. Eur J Nucl Med Mol Imaging. 2009;36(9):1483–1494. | ||
Liu ZF, Wang F. Dual-targeted molecular probes for cancer imaging. Curr Pharm Biotechnol. 2010;11(6):610–619. | ||
Bellin MF. MR contrast agents, the old and the new. Eur J Radiol. 2006;60(3):314–323. | ||
Gilbert FJ, Ahearn TS. Dynamic contrast-enhanced MRI in cancer. Imaging Med. 2009;1:173–186. | ||
Mahmoudi M, Sant S, Wang B, Laurent S, Sen T. Superparamagnetic iron oxide nanoparticles (SPIONs): development, surface modification and applications in chemotherapy. Adv Drug Deliv Rev. 2011;63(1–2):24–46. | ||
Sitthichai S, Pilapong C, Thongtem T, Thongtem S. CMC-coated Fe3O4 nanoparticles as new MRI probes for hepatocellular carcinoma. Appl Surf Sci. 2015;356:972–977. | ||
Ibrahim A, Koval D, Kašička V, Faye C, Cottet H. Effective charge determination of dendrigraft poly-L-lysine by capillary isotachophoresis. Macromolecules. 2013;46(2):533–540. | ||
Qiao Z, Shi XY. Dendrimer-based molecular imaging contrast agents. Prog Polym Sci. 2015;44:1–27. | ||
Rohrer M, Bauer H, Mintorovitch J, Requardt M, Weinmann HJ. Comparison of magnetic properties of MRI contrast media solutions at different magnetic field strengths. Invest Radiol. 2005;40(11):715–724. | ||
Shen JM, Yin T, Tian XZ, Gao FY, Xu S. Surface charge-switchable polymeric magnetic nanoparticles for the controlled release of anticancer drug. ACS Appl Mater Interfaces. 2013;5(15):7014–7024. | ||
Shen JM, Huang G, Zhou X, et al. Safety evaluation of graphene oxide-based magnetic nanocomposites as MRI contrast agents and drug delivery vehicles. RSC Adv. 2014;4(92):50464–50477. | ||
Guo M, Su XY, Kong L, Li X, Zou HF. Characterization of interaction property of multicomponents in Chinese Herb with protein by microdialysis combined with HPLC. Anal Chim Acta. 2006;556(1):183–188. | ||
Fisher KA, Huddersman KD, Taylor MJ. Comparison of micro- and mesoporous inorganic materials in the uptake and release of the drug model fluorescein and its analogues. Chemistry. 2003;9(23):5873–5878. | ||
Zhang S, Gong MF, Zhang D, Yang H, Gao FB, Zou LG. Thiol-PEG-carboxyl-stabilized Fe2O3/Au nanoparticles targeted to CD105: Synthesis, characterization and application in MR imaging of tumor angiogenesis. Eur J Radiol. 2014;83(7):1190–1198. | ||
Liu PF, Wang HZ, Li YG, Duan YR. Preparation of DHAQ-loaded PLA-PLL-RGD nanoparticles and comparison of antitumor efficacy to hepatoma and breast carcinoma. J Macromol Sci A. 2009;46(10):1024–1029. | ||
Yallapu MM, Othman SF, Curtis ET, Gupta BK, Jaggi M, Chauhan SC. Multi-functional magnetic nanoparticles for magnetic resonance imaging and cancer therapy. Biomaterials. 2011;32(7):1890–1905. | ||
Sisavath N, Leclercq L, Le Saux T, Oukacine F, Cottet H. Study of interactions between oppositely charged dendrigraft poly-L-lysine and human serum albumin by continuous frontal analysis capillary electrophoresis and fluorescence spectroscopy. J Chromatogr A. 2013;1289:127–132. | ||
Kodama Y, Nakamura T, Kurosaki T, et al. Biodegradable nanoparticles composed of dendrigraft poly-L-lysine for gene delivery. Eur J Pharm Biopharm. 2014;87(3):472–479. | ||
Jia X, Tian K, Zhao XB, Zhou TT, Pei ML, Liu P. Fluorescent amphiphilic copolymer-based tumor theranostics for facile DOX-loading and tumor microenvironment-triggered release. Mater Des. 2016;105:333–340. | ||
Wu SR, Geng X, Chen C, Cai YK, Han GZ. Synthesis of poly-L-lysine and its structural response to environmental pH. Chinese J Org Chem. 2013;33:1769–1773. | ||
Gao W, Ji L, Li L, et al. Bifunctional combined Au-Fe2O3 nanoparticles for induction of cancer cell-specific apoptosis and real-time imaging. Biomaterials. 2012;33(14):3710–3718. | ||
Zhi M, Wu KC, Dong L, et al. Characterization of a specific phage-displayed peptide binding to vasculature of human gastric cancer. Cancer Biol Ther. 2004;3(12):1232–1235. | ||
Huang PS, Song HJ, Wang WW, et al. Integrin-targeted zwitterionic polymeric nanoparticles with acid-induced disassembly property for enhanced drug accumulation and release in tumor. Biomacromolecules. 2014;15(8):3128–3138. | ||
Seitz SJ, Schleithoff ES, Koch A, et al. Chemotherapy-induced apoptosis in hepatocellular carcinoma involves the p53 family and is mediated via the extrinsic and the intrinsic pathway. Int J Cancer. 2010;126(9):2049–2066. | ||
Ashkenazi A, Dixit VM. Death receptors: signaling and modulation. Science. 1998;281(5381):1305–1308. | ||
Grütter MG. Caspases: key players in programmed cell death. Curr Opin Struct Biol. 2000;10(6):649–655. | ||
Balamurugan K, Rajaram R, Ramasami T. Caspase-3: its potential involvement in Cr(III)-induced apoptosis of lymphocytes. Mol Cell Biochem. 2004;259(1–2):43–51. | ||
Humphries JD, Byron A, Humphries MJ. Integrin ligands at a glance. J Cell Sci. 2006;119(19):3901–3903. | ||
Oliveira EA, Faintuch BL. Radiolabeling and biological evaluation of the GX1 and RGD-GX1 peptide sequence for angiogenesis targeting. Nucl Med Biol. 2015;42(2):123–130. | ||
Wang C, Li Y, Chen BB, Zou MJ. In vivo pharmacokinetics, biodistribution and the anti-tumor effect of cyclic RGD-modified doxorubicin-loaded polymers in tumor-bearing mice. Colloids Surf B Biointerfaces. 2016;146:31–38. | ||
Zhang LH, Zhu SJ, Qian LL, Pei YY, Qiu YM, Jiang YY. RGD-modified PEG-PAMAM-DOX conjugates: In vitro and in vivo studies for glioma. Eur J Pharm Biopharm. 2011;79(2):232–240. | ||
Chen B, Cao S, Zhang Y, et al. A novel peptide (GX1) homing to gastric cancer vasculature inhibits angiogenesis and cooperates with TNF alpha in anti-tumor therapy. BMC Cell Biol. 2009;10:63. | ||
Chen K, Sun XL, Niu G, et al. Evaluation of 64Cu labeled GX1: a phage display peptide probe for PET imaging of tumor vasculature. Mol Imaging Biol. 2012;14:96–105. | ||
Xin J, Zhang X, Liang J, et al. In vivo gastric cancer targeting and imaging using novel symmetric cyanine dye-conjugated GX1 peptide probes. Bioconjug Chem. 2013;24(7):1134–1143. | ||
Hu H, Yin J, Wang M, et al. GX1 targeting delivery of rmhTNFα evaluated using multimodality imaging. Int J Pharm. 2014;461(1–2):181–191. | ||
de Oliveira EA, Faintuch BL, Targino RC, et al. Evaluation of GX1 and RGD-GX1 peptides as new radiotracers for angiogenesis evaluation in experimental glioma models. Amino Acids. 2016;48(3):821–831. | ||
Zhang L, Li G, Gao M, et al. RGD-peptide conjugated inulin-ibuprofen nanoparticles for targeted delivery of Epirubicin. Colloids Surf B Biointerfaces. 2016;144:81–89. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.