")
Back to Journals » International Journal of Nanomedicine » Volume 16
Heptamethine Cyanine Dye MHI-148-Mediated Drug Delivery System to Enhance the Anticancer Efficiency of Paclitaxel
Authors Raveendran A, Poilil Surendran S, Ser J , Alam K , Cho H , Jeong YY
Received 17 June 2021
Accepted for publication 20 September 2021
Published 21 October 2021 Volume 2021:16 Pages 7169—7180
DOI https://doi.org/10.2147/IJN.S325322
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Farooq A. Shiekh
Athira Raveendran,1,* Suchithra Poilil Surendran,2,* Jinhui Ser,1 Khurshed Alam,1 Hoonsung Cho,1 Yong Yeon Jeong3
1Department of Materials Science & Engineering, Chonnam National University, Gwangju, 61186, Republic of Korea; 2Department of Biomedical Sciences, Chonnam National University Medical School, Hwasun, 58128, Republic of Korea; 3Department of Radiology, Chonnam National University Hwasun Hospital, Hwasun, 58128, Republic of Korea
*These authors contributed equally to this work
Correspondence: Hoonsung Cho
Department of Materials Science & Engineering, Chonnam National University, Gwangju, 61186, Republic of Korea
Tel +82-62-530-1717
Fax +82-62-530-1699
Email [email protected]
Yong Yeon Jeong
Department of Radiology, Chonnam National University Hwasun Hospital, Hwasun, 58128, Republic of Korea
Email [email protected]
Introduction: Paclitaxel (PTX) is a conventional chemotherapeutic drug that effectively treats various cancers. The cellular uptake and therapeutic potential of PTX are limited by its slow penetration and low solubility in water. The development of cancer chemotherapy methods is currently facing considerable challenges with respect to the delivery of the drugs, particularly in targeting the tumor site without exerting detrimental effects on the healthy surrounding cells. One possibility for improving the therapeutic potential is through the development of tumor-targeted delivery methods.
Methods: We successfully synthesized paclitaxel-MHI-148 conjugates (PTX-MHI) by coupling PTX with the tumor-targeting heptamethine cyanine dye MHI-148. Synthesis and purification were characterized using the absorbance spectrum and the results of time-of-flight mass spectrometry. Cellular uptake and cytotoxicity studies were conducted in vitro and in vivo.
Results: PTX-MHI accumulates in tumor cells but not in normal cells, as observed by in vitro near-infrared fluorescent (NIRF) imaging along with in vivo NIRF imaging and organ biodistribution studies. We observed that MHI-148-conjugated PTX shows greater efficiency in cancer cells than PTX alone, even in the absence of light treatment. PTX-MHI could also be used for specific drug delivery to intracellular compartments, such as the mitochondria and lysosomes of cancer cells, to improve the outcomes of tumor-targeting therapy.
Conclusion: The results indicated that PTX-MHI-mediated cancer therapy exerts an excellent inhibitory effect on colon carcinoma (HT-29) cell growth with low toxicity in normal fibroblasts (NIH3T3).
Keywords: cancer therapy, colon cancer, bioconjugation, fluorescent dye, paclitaxel
Introduction
The therapeutic potential of several drugs used for cancer therapy is restricted owing to their poor targeting and low penetration in tumors.1 The inability of anticancer drugs to selectively target tumors increases the adverse effects on healthy tissues and surrounding organs, thus limiting the dosage of the drugs that can be safely administered in patients with cancer and ultimately reducing the treatment efficiency.2 Targeted cancer therapy is a useful approach for overcoming this issue and is currently the prime focus of anticancer drug advancement research. Targeted drug delivery involves the conjugation of a tumor-targeting carrier that can deliver anticancer drugs to tumors. For instance, antibodies, peptides, and nanoparticles have been identified as suitable delivery vehicles.Targeted carrier-mediated cancer treatment enhances the accumulation of anticancer drugs in tumor cells with minimal damage to normal tissues, thereby reducing the side effects and considerably improving the therapeutic potential of anticancer drugs.3
Paclitaxel (PTX) is a chemotherapeutic drug4 that has been widely used for many years to treat breast cancer, head cancer, lung cancer, ovarian cancer, Kaposi’s sarcoma,5 and colon cancer.6 The mechanism underlying the anticancer activity of PTX is well-recognized and has been reported in several studies; the activity of this drug involves the breakdown of microtubules during cell division and cell cycle arrest in the G2/M phase. Consequently, the cells are unable to proceed with the regular cell cycle.7,8 Microtubule-stabilizing agents such as PTX can act on the mitochondria to trigger apoptosis9–11 and also via cathepsin B-dependent lysosomal pathways, thereby leading to cancer cell death.12 Therefore, PTX may exhibit a cytotoxic effect via the induction of apoptosis, apart from exerting effects on microtubules that lead to cell cycle arrest. However, PTX application in anticancer therapy faces challenges because of its low solubility, hydrophobic nature, and the development of drug resistance.13 These factors substantially limit the bioavailability of PTX in tumor cells because of the low tumor targeting potential of the drug. Additionally, PTX can exhibit some side effects, including myelosuppression, peripheral neuropathy, and hypersensitivity, because of non-specific drug distribution.14,15 Considering the side effects caused by PTX, it is necessary to design novel methods for the effective target-specific delivery of PTX to tumors while limiting its impact on healthy tissues. In recent years, several approaches have been reported to improve the delivery of paclitaxel to the tumor site.16–18 Findings from studies indicate that drugs delivered via nanoparticle, peptide, and fluorescent dye carriers are retained for extended periods in the tumors, causing a higher suppression rate of the tumor.19 This type of a drug delivery system can efficiently deliver the drug to the targeted tissue without blocking the capillaries while preserving the bioactivity and stability of the drug.20
MHI-148 is a near-infrared heptamethine cyanine dye that possesses tumor-targeting properties.21 Its dual imaging and dual-targeting characteristics make this dye a unique and efficient agent for cancer detection, diagnosis, and therapy. MHI-148 is immediately taken up and accumulated in the lysosomes and mitochondria of tumor cells, but not in those of normal cells.22–25 Considering these qualities, in this study, we selected MHI-148 as a promising tumor-targeting candidate and conjugated it with the anticancer drug PTX for successfully synthesizing PTX-MHI-148 conjugates (PTX-MHI). A group of cell membrane-bound solute carriers called organic anion-transporting polypeptides (OATPs) play a vital role in facilitating the tumor-specific delivery of near-infrared fluorescent (NIRF) dyes.26,27 The tumor-specific accumulation of MHI-148 is associated with the upregulated expression of OATPs in various types of tumors, including lung, brain, bladder,28 and colon carcinomas,29 and in the hypoxic microenvironment of cancer cells.30
We aimed to couple PTX with the tumor-specific dye MHI-148 and enhance the bioavailability of PTX inside tumors, thereby improving the therapeutic effect of PTX while decreasing toxicity to normal cells. This study demonstrated the conjugation of PTX with the heptamethine cyanine dye MHI-148 in the synthesis of PTX-MHI and the anticancer effects of PTX-MHI in colon cancer cells and normal fibroblasts. The fluorescent dye MHI-148 used here did not cause noticeable cytotoxicity. PTX has already been studied for its anticancer effects. Nevertheless, PTX exhibits disadvantages such as low tumor specificity, thereby causing side effects in healthy tissues,31 which are commonly accountable for failures in cancer therapy. Because of these reasons, novel drug delivery systems with better targeting ability are required for cancer treatment. Here, we focused on minimizing the side effects of PTX in healthy tissues by tumor-targeted delivery with PTX-MHI conjugates. We used the tumor-targeting property of MHI-148 along with the anticancer property of paclitaxel to destroy colon cancer cells selectively. The method did not involve light irradiation treatments, but the efficiency of the treatment was high, and the treatment may be of significance in the field of cancer-targeting therapies. Our results show that PTX-MHI effectively suppresses the proliferation of colon cancer cells, while exerting minimal effects on normal fibroblasts, even in the absence of light treatment.
Materials and Methods
HT-29 colorectal adenocarcinoma cells and NIH3T3 mouse embryonic fibroblasts were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA). The following kits and reagents were used in the experimental protocols: McCoy’s 5A Medium Modified (Welgene Co., No. LM 005–02), RPMI1640 (Welgene), MHI-148 (2-[2-[2-chloro-3-[2-[1,3-dihydro-3,3-dimethyl-1-(5-carboxypentyl)-2H-indol-2-ylidene]-ethylidene]-1-cyclohexene-1-yl]-ethyl]-3,3-dimethyl-1-(5-carboxypentyl)-3H-indolium bromide) [purchased from Bioacts (DKC corporation, Incheon, South Korea)], PTX (Taxol) (purchased from Sigma-Aldrich, South Korea), fetal bovine serum (Thermo Fisher Scientific, No. 16000044), DMSO anhydrous (Life Technologies Co, No. LOT. 0305C186), penicillin-streptomycin (Life Technologies, No. 15140122), trypsin-EDTA (Welgene Co, No. LS 015–10), Dulbecco’s PBS (DPBS) and LysoTracker Green (Thermo Fisher Scientific, No. L7526), N,N’-diisopropyl carbodiimide (DIC), 4-dimethylaminopyridine (DMAP), and rhodamine 123 mitochondria specific dye (Sigma-Aldrich, St Louis, USA). For the cell cytotoxicity assay, MTT was obtained from Life Technologies (Carlsbad, CA, USA). To purify the conjugates, a Mini Dialysis Membrane (MWCO cutoff 1k) was obtained from Thermo Scientific. Female BALB/c-Nu/Nu mice (six weeks old) were obtained from Orient Bio (Seongnam, South Korea) and maintained under specific pathogen-free conditions.
Synthesis and Purification of PTX-MHI
PTX-MHI was synthesized by the ester formation reaction between PTX and MHI-148 using a previously published method.32 The 2′-OH group of PTX was coupled with the carboxylic group of MHI-148 in the presence of DIC and DMAP as activating agents. Briefly, 500 µL of PTX (5 mM in DMSO) was first mixed with an equal concentration of MHI-148 (5 mM in DMSO) and an additional 500 µL of 1X PBS in the presence of DIC and DMAP. The ratio of PTX-MHI was 1:1. The pH was adjusted to 9 using sodium hydroxide buffer with magnetic stirring. The reaction was carried out in a vial at 27°C for 4 h. For the purification and effective removal of unconjugated MHI-148 and the byproducts, dialysis was performed using the Mini Dialysis membrane (MWCO cutoff 1k) in triple-distilled water for 2 days. After the purification process, the conjugation and purification of PTX-MHI were confirmed via time-of-flight mass spectrometry (MALDI-TOF-MS).
Cellular Uptake Studies of MHI-148 and PTX-MHI
To compare the intracellular uptake of MHI-148 in cancer and normal cells, cellular uptake assays of MHI-148 and PTX-MHI were conducted using fluorescence microscopy. HT-29 colon carcinoma cells and NIH3T3 fibroblasts were seeded in a confocal dish and incubated at 37°C for 1 day before treating with MHI-148 and PTX-MHI. Next, 10 µM PTX-MHI and MHI-148 were added to the culture dish and culture medium; after 1 h, the cells were washed multiple times with DPBS to remove excess dye and were resuspended in fresh media. Images were captured using a Nikon inverted microscope eclipse DSRi2, Qi2 (Ex/Em, 633/780 nm: exposure time, 30 s).
Intracellular Localization Studies in the Lysosomes and Mitochondria
To determine the intracellular distribution of PTX-MHI in HT-29 and NIH3T3 cells, a co-staining experiment was conducted using commercially available LysoTracker (a green fluorescent dye that stains lysosomes) and rhodamine 123 (a cationic fluorescent dye that labels mitochondria). To conduct localization imaging with LysoTracker and MitoTracker, HT-29 colon carcinoma cells and NIH3T3 fibroblasts were seeded in a confocal dish and incubated at 37°C for 1 day before treating with PTX-MHI. Next, 10 µM PTX-MHI was added to the culture dish and the culture medium and incubated for 1 h. Next, cells were stained with rhodamine 123 (2 µM) and LysoTracker Green (50 nM) at 37°C. After incubation, the cells were washed multiple times with DPBS and resuspended in fresh media. Images were captured using the Nikon inverted microscope eclipse DSRi2, Qi2.
In vitro Cytotoxicity Assay of MHI-148, PTX-MHI, and PTX
To compare the cytotoxicity of PTX-MHI in cancer cells and normal cells, the HT-29 and NIH3T3 cells were harvested by trypsinization and resuspended in 96-well plates at a concentration of 10,000 cells/well in a fresh culture medium and incubated for 1 day at 37°C under 5% CO2. After 24 h of incubation, both cell types were treated with MHI-148, PTX-MHI, and PTX at concentrations of 0.01, 0.05, 0.1, 0.5, and 1.5 µM, along with the controls. The cell death activities after MHI-148, PTX-MHI, and PTX treatment were evaluated using MTT assays for 3 consecutive days. Cell viability was measured after an interval of 24 h. The MTT assay was carried out by adding 10 μL of 5 mg/mL MTT to each well and incubating for 2 h. Afterward, culture media containing the drugs were removed, and the resulting purple-colored formazan crystals were dissolved in DMSO. The plates were shaken well using a digital orbital shaker for 5 min (Scilab Instruments Co, No. SSO-2D) at 200 rpm before measuring the relative color intensity. The absorbance of individual wells was estimated at 590 nm on a microplate reader (Thermo Scientific, Varioskan™ LUX multimode).
Animal Tumor Model
All animal experiments were carried out according to the regulations of the National Institutes of Health guide for the care and use of Laboratory Animals, which is approved by the Chonnam National University Medical School Research Institutional Animal Care and Use Committee (CNUHH 2014–148). For the biodistribution studies and in vivo tumor reduction study, female BALB/c nude mice (18–20 g, 5 weeks old) were acquired from Jungang Lab Animal Inc., Korea. HT-29 colon carcinoma tumors in mice were established by injecting 3×106 HT-29 cells subcutaneously near the hind limb of mice. The mice were kept in filter-topped cages and fed sterile food and water in a specific pathogen-free facility. We used the following formula to calculate tumor volume:
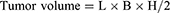
where L is the tumor length (major axis), B is the tumor breadth (minor axis), and H is the height or thickness of the tumor.
In vivo Biodistribution of PTX-MHI
When HT-29 tumors reached approximately 5–7 mm in size (after 15 days), PTX-MHI (in PBS, 150 μL) was intravenously administered at 2 µg/mouse through the tail vein of tumor-bearing mice. In vivo and ex vivo imaging was also performed in HT-29 tumor-induced BALB/c nude mice administered 2 µg/kg PTX-MHI at different time points. Imaging was conducted using the FOBI in vivo imaging system (Neuroscience, Korea), with 825-nm emission filters at an exposure time of 1 s to analyze the biodistribution.
In vivo Tumor Growth Inhibition Study
For the PTX-MHI-mediated tumor reduction study, an HT-29 colon cancer tumor model was developed, and when the tumor reached a size of 200 mm3, the mice were separated into four groups (n = 5). The mice in the different groups were intravenously injected with 200 μL of PBS, 200 μL of PTX-MHI (2 mg/kg PTX), 200 μL of PTX (2 mg/kg), or 200 μL of MHI-148 (2 mg/kg) at three intervals (day 0, 7, and 14). The tumor size and mouse whole body weights were measured every alternate day.
Results
Conjugation and Characterization of PTX-MHI
The conjugation of MHI-148 and PTX is based on a solution-phase reaction carried out between the 2′-OH group of PTX and the carboxylic group of MHI-148, in the presence of DIC and DMAP as activating agents, forming a stable33,34 ester linkage (Figure 1A). The purification and effectual removal of unconjugated MHI-148 and PTX were performed via dialysis using the Mini Dialysis membrane (MWCO cut off 1k) in triple-distilled water for 2 days. PTX-MHI, which had the highest molecular mass, was retained in the dialysis tube, whereas PTX (MW: 853.90614 g/mol) and MHI-148 (MW: 684.33 g/mol) were eluted from the dialysis membrane. Figure 1B shows the absorbance spectra of PTX-MHI, MHI-148, and PTX, using which the conjugation and purification of PTX-MHI was confirmed. The absorbance peaks of PTX-MHI represent a combination of the absorbance peaks of pure PTX and pure MHI-148. MALDI-TOF-MS confirmed the efficient conjugation of MHI-148 and PTX and indicated the successful synthesis of PTX-MHI (MW ≈ 1538.2 Da; Figure 1C).
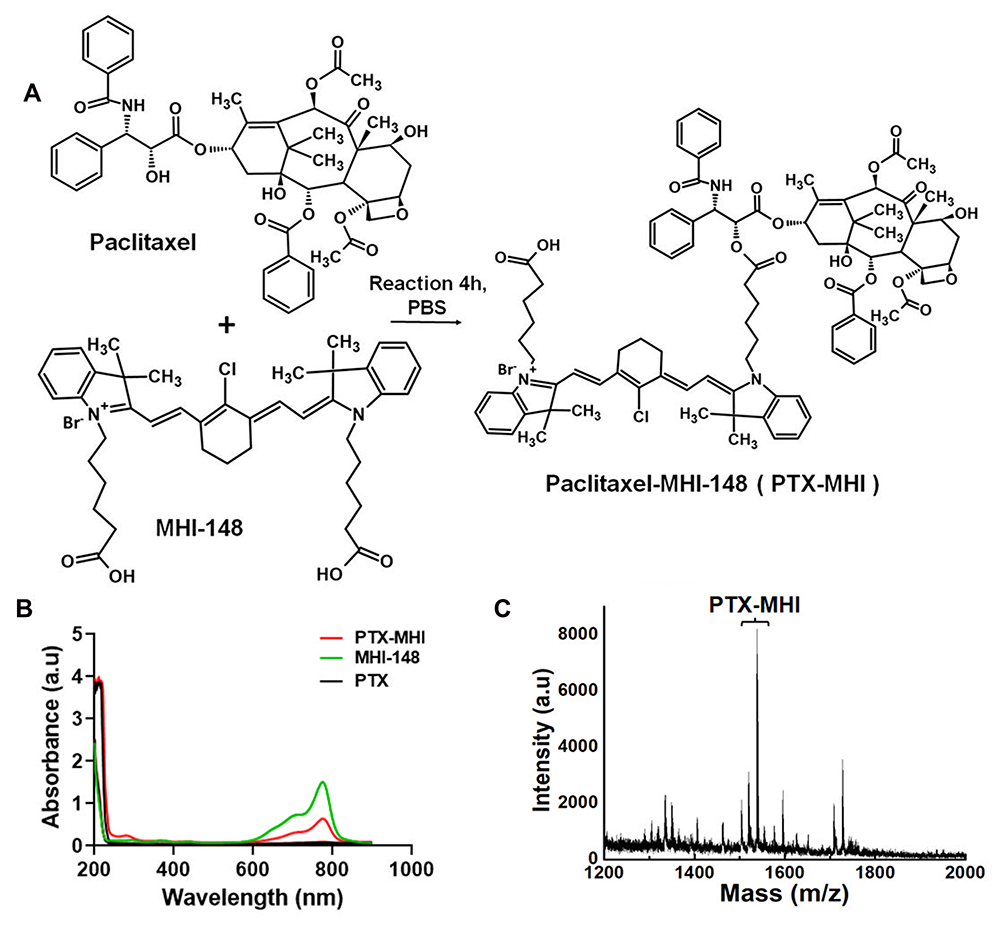 |
Figure 1 Synthesis and purification of PTX-MHI. (A) The hydroxyl group of PTX reacted with the carboxylic group of MHI-148 to yield PTX-MHI. (B) Absorption spectra of PTX, MHI-148, and purified PTX-MHI. (C) MALDI-TOF-MS data confirmed conjugation, as indicated by the peak of PTX-MHI after purification. |
Assessment of PTX-MHI and MHI-148 Uptake in HT-29 and NIH3T3 Cell Lines
In vitro cellular uptake experiments were performed to compare the uptake of MHI-148 and PTX-MHI in colon carcinoma (HT-29) cells and fibroblasts (NIH3T3). The NIRF signal was exclusively observed in HT-29 cells (Figure 2A), and low uptake was observed in NIH3T3 cells (Figure 2B) in both MHI-148- and PTX-MHI-treated groups. This experiment showed that the uptake of MHI-148 and PTX-MHI in colon cancer cells is markedly greater than that in normal cells. Using human colon cancer cells and normal fibroblasts to study dye uptake, we confirmed that MHI-148 and PTX-MHI possess tumor imaging and targeting properties. The fluorescent spots observed in the cancer cells suggested that the dye accumulated in the mitochondria and lysosomes of the tumor cells.35
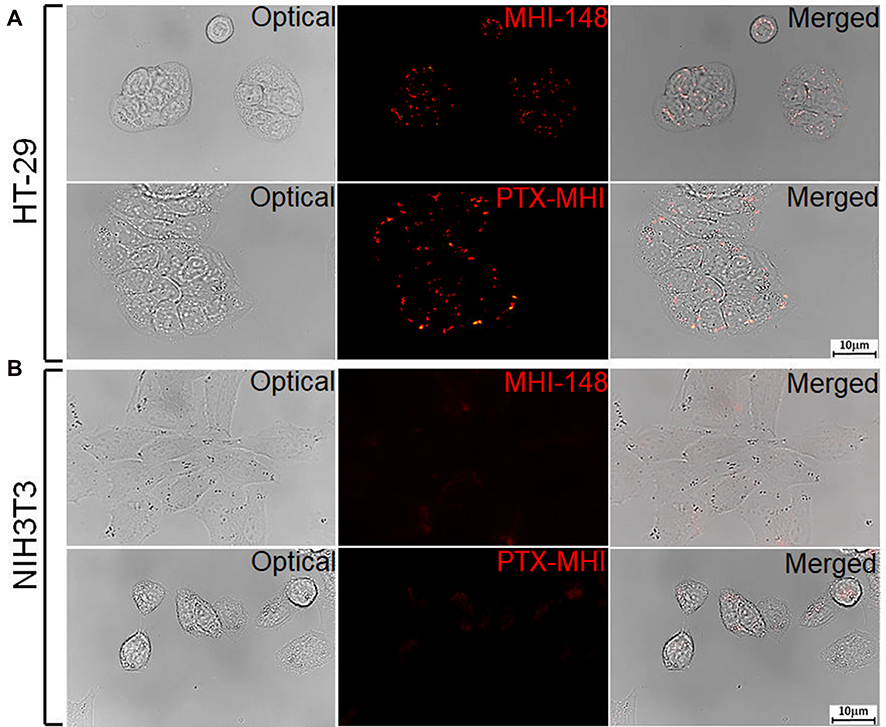 |
Figure 2 Uptake and intracellular endosomal localization of MHI-148 and PTX-MHI in HT-29 and NIH3T3 cells. (A) HT-29 cells treated with MHI-148 and PTX-MHI. (B) NIH3T3 cells treated with MHI-148 and PTX-MHI. MHI-148 and PTX-MHI appear to be internalized in HT-29 cells, but in NIH3T3 cells, the dye’s internalization is significantly lower. All images were acquired via fluorescence microscopy (exposure time of 10 s). |
Intracellular Localization Studies in Lysosomes and Mitochondria
We evaluated the targeting property of PTX-MHI in HT-29 and NIH3T3 cells through co-localization experiments using lysosomal and mitochondrial markers. The NIR signal appeared to condense on the mitochondria and lysosomes of HT-29 cells only, as observed by fluorescent imaging (Figure 3A). Merged images show a considerable overlap of the PTX-MHI signal in the lysosomes and mitochondria of cancer cells, indicating that PTX-MHI selectively targets cancer cells and accumulates in colon cancer cell lysosomes and mitochondria. However, only a weak NIRF signal was detected in NIH3T3 fibroblasts (Figure 3B) owing to the non-specific staining of the dye.
 |
Figure 3 Co-localization study using LysoTracker Green and rhodamine 123 as the standard lysosomal and mitochondrial markers, respectively. HT-29 (A) and NIH3T3 (B) cells were treated with PTX-MHI and stained with LysoTracker Green and rhodamine 123 for 30 min. PTX-MHI was localized to the lysosomes and mitochondria of HT-29 cells, whereas only a weak fluorescent signal was observed in NIH3T3 fibroblasts. Images were acquired via fluorescence microscopy (exposure time of 10 s). |
In vitro Cytotoxic Effects of PTX-MHI, MHI-148, and PTX
A cell viability test was carried out for up to 3 days after the HT-29 cells and NIH3T3 cells were treated with PTX-MHI, MHI-148, and PTX. We evaluated the effects of PTX-MHI, MHI-148, and PTX administered at different concentrations in HT-29 colon cancer cells and NIH3T3 normal fibroblasts to compare the cytotoxicity of the conjugates in these cells. Figure 4A–C show the cytotoxic effect of PTX-MHI in HT-29 and NIH3T3 cells from day 1, day 2, and day 3. Figure 4D–F show the cytotoxic effect of PTX in HT-29 and NIH3T3 cells from day 1, day 2, and day 3. Figure 4G–I show the effect of MHI-148 in HT-29 and NIH3T3 cells after day 1, day 2, and day 3 of treatment.
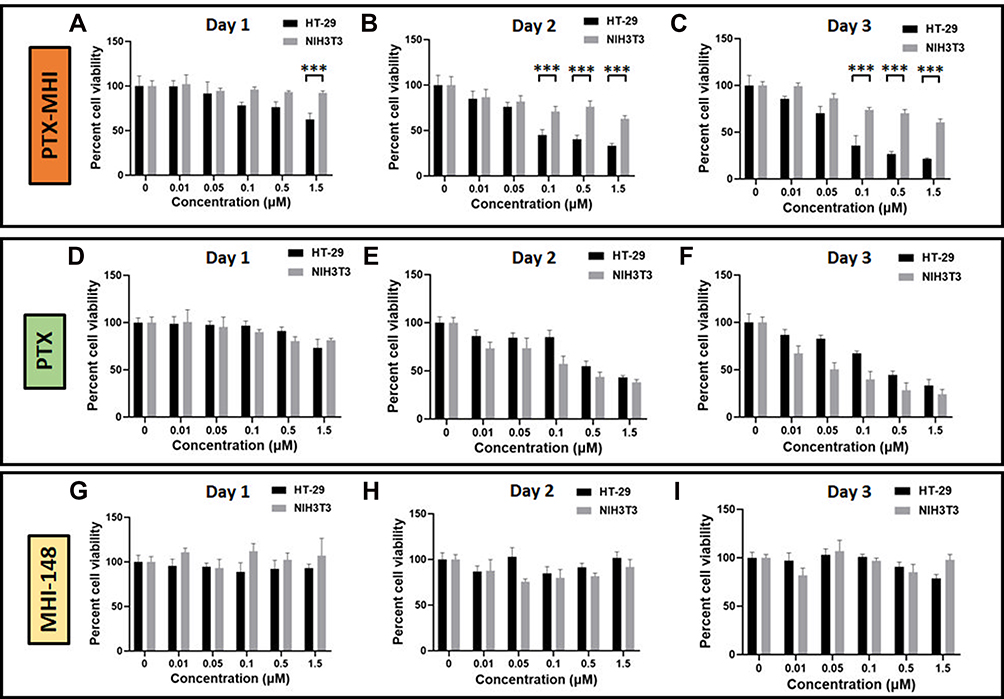 |
Figure 4 Cytotoxicity analysis of PTX-MHI, MHI-148, and PTX (n = 5). (A–C) HT-29 and NIH3T3 cells were treated with various concentrations of PTX-MHI, and cell viability was assessed after day 1 (A), 2 (B), and 3 (C). (D–F) HT-29 and NIH3T3 cells were treated with various concentrations of PTX, and cell viability was assessed after day 1 (D), 2 (E), and 3 (F). (G–I) HT-29 and NIH3T3 cells were treated with various concentrations of MHI-148, and cell viability was assessed after day 1 (G), 2 (H), and 3 (I). Cells were treated with 0, 0.01, 0.05, 0.1, 0.5, and 1.5 µM PTX-MHI, MHI-148, or PTX, and then incubated for 3 days. Cell viability assays were conducted using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT). Significant differences are shown as ***P < 0.001. |
The PTX-MHI treatment groups exhibited a greater reduction in the number of HT-29 cancer cells than in that of NIH3T3 normal fibroblasts. The PTX-MHI treatment group on day 1 (Figure 4A), day 2 (Figure 4B), and day 3 (Figure 4C) showed a gradual and significant reduction in the HT-29 cancer cell population. However, NIH3T3 cells treated with PTX-MHI showed no toxicity on day 1, and only showed toxicity on days 2 and 3. The tumor reduction rate in HT-29 cells increased upon treatment with 0.1 to 1.5 µM of PTX-MHI. Treatment after days 2 and 3 caused a greater decline in the tumor cell population compared to that after day 1 in all concentration groups. After 3 days of treatment of HT-29 cells, the cell viability decreased considerably. The cytotoxicity of PTX-MHI in HT-29 cells increased from 37.5% to 78.4% between the first and third day of treatment, but the cytotoxicity observed in the NIH3T3 fibroblasts treated with PTX-MHI led to an increase in cell death from 8% to only 39.4%.
When comparing the cytotoxicities in the PTX and PTX-MHI treatment groups, HT-29 cells from the PTX-MHI treatment group showed a greater reduction in cell population than those from the PTX treatment group. The PTX-MHI group showed toxicity toward cancer cells at 0.1, 0.5, and 1.5 µM concentrations from day 1, but only 1.5 µM PTX showed slight toxicity in cancer cells on day 1. Additionally, on day 2, 0.1 µM PTX-MHI showed a further reduction of approximately 40%; on the third day, 32% additional reduction was observed in the cancer cell population compared to that in the PTX treatment group. In HT-29 cells, the cytotoxicity observed after treatment was greater in the PTX-MHI-treated group than in the PTX groups at all concentrations.
In NIH3T3 fibroblasts, PTX-MHI treatment showed lower cytotoxicity compared to PTX treatment. On day 1, PTX treatment showed cytotoxicity, but PTX-MHI treatment did not show considerable cytotoxicity in normal cells. On day 2, the NIH3T3 cell population in the PTX-treated group decreased by 61.9% compared to that in the control group. Concurrently, there was only 37.1% reduction in the PTX-MHI group. On day 3, the NIH3T3 cell population in the PTX treatment group decreased to 75.9% compared to that in the control group, but the cell death rate in the PTX-MHI treatment group was only 39.4% compared to that in the control group. The results showed that the PTX treatment group exhibited greater NIH3T3 cell death than the PTX-MHI groups at all concentrations. When comparing the cytotoxicity of PTX-MHI and MHI-148, MHI-148 caused only negligible toxicity in HT-29 and NIH3T3 cells.
Biodistribution of PTX-MHI in the HT-29 Tumor Model
To evaluate the tumor accumulation and biodistribution of PTX-MHI, we administered PTX-MHI to HT-29 tumor-induced BALB/c nude mice. Whole-body imaging was performed after 2, 4, 6, 12, 24, and 48 h of injection (Figure 5A). Maximum accumulation was found 12 h after injection, and the signal was sustained until day 3. The ex vivo imaging results also showed maximum accumulation of PTX-MHI in the tumor at 12 h (Figure 5B). The quantification of fluorescence signals in organs showed that the dye uptake in tumor tissues was significantly higher than that in other vital organs (Figure 5C). The biodistribution results indicated the ability of PTX-MHI to precisely target tumors as well as to be retained in tumor tissues rather than in other organs.
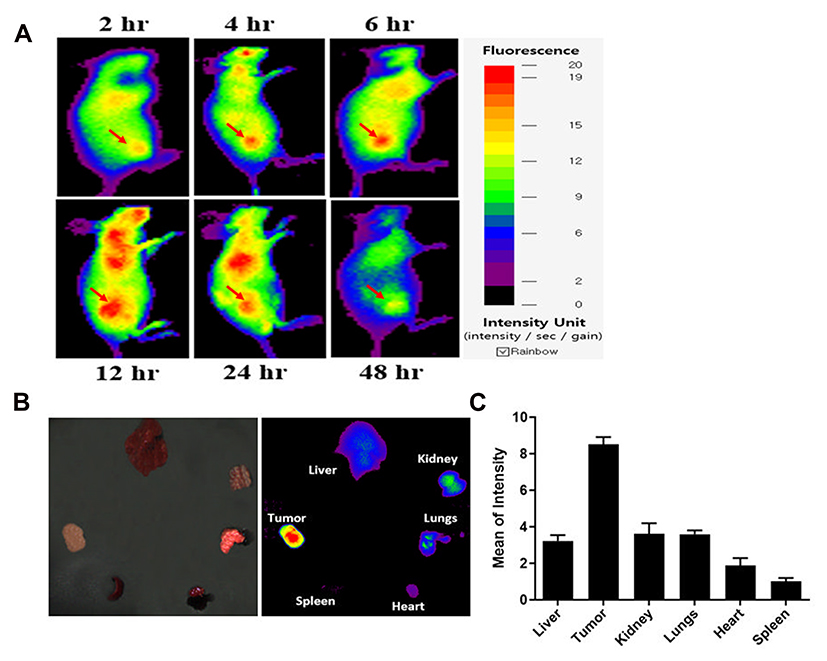 |
Figure 5 In vivo and ex vivo biodistribution study. (A) Biodistribution of PTX-MHI in HT-29 tumor-induced BALB/c nude mice (n = 3). In vivo fluorescence analysis of PTX-MHI in HT-29 cells was carried out after the intravenous administration of PTX-MHI (2 µg/mouse). Maximum tumor accumulation was achieved after 12 h of administration. (B) Ex vivo imaging of organs (heart, spleen, tumor, liver, kidneys, and lungs) harvested after euthanizing the mice after 12 h of intravenous PTX-MHI injection. (C) The fluorescence intensities in the organs were quantified and plotted as mean intensity values ± SD (n=3). |
Tumor Growth Inhibition Study in the HT-29 Tumor Model
To confirm our hypothesis that PTX-MHI exerts a stronger therapeutic effect than PTX, we tested the in vivo antitumor efficacy of PTX-MHI, MHI-148, and PTX. For the antitumor study, an HT-29 colon cancer tumor model was established using female BALB/c nude mice. The mice were intravenously injected with 200 μL of PBS, 200 μL of PTX-MHI (2 mg/kg PTX), 200 μL of PTX (2 mg/kg), or 200 μL of MHI-148 (2 mg/kg). The intravenous treatment was conducted in three intervals (day 0, 7, and 14) to a total dose of 6 mg/kg. All mice were weighed during the experimental period, and tumor volumes were measured every alternate day for up to 30 days. A significant rate of tumor growth inhibition was observed in mice treated with PTX-MHI compared with that in mice treated with PTX, MHI-148, and PBS (Figure 6A). The PTX-MHI-treated group showed relatively slower tumor growth after 15 days of treatment than the other groups. To further confirm that PTX-MHI did not induce weight changes, the body weights of mice were measured every other day for 30 days. No weight loss was observed in the PTX-MHI-treated group compared to that in the control (Figure 6B). The body weights of the mice remained almost the same in all treatment groups.
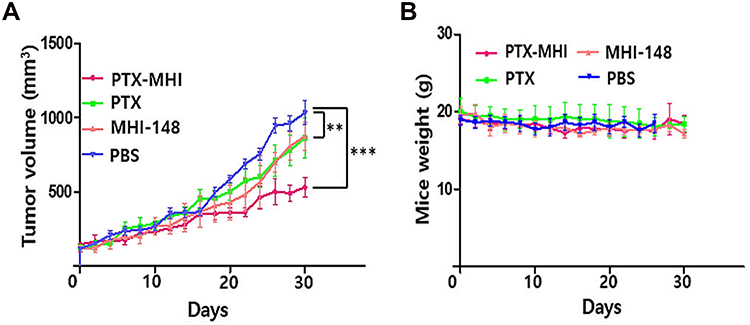 |
Figure 6 In vivo tumor growth inhibition and weight studies in HT-29 subcutaneous tumor-induced BALB/c nude mice treated with PTX-MHI-148. Tumor volume measurement (A) and body weight change (B) in different groups. Data are presented as mean ± SD (n = 5, ***P < 0.001, **P < 0.01). |
Discussion
In this study, we synthesized PTX-MHI by coupling the anticancer drug PTX and heptamethine cyanine dye MHI-148. We significantly improved the solubility of PTX and increased the bioavailability of PTX inside tumor cells. The tumor-specific targeting property of MHI-148 assisted in the effective delivery of PTX into the tumor without accumulation in normal cells. The higher accumulation of MHI-148 in cancer cells than in normal cells occurs because of the hypoxic tumor microenvironment and OATP signaling.30,36,37 Previous studies have reported that HT-29 colon carcinoma cells overexpress OATPs.29 OATPs are a superfamily of solute carrier transporters that facilitate the transport of multiple substrates, including drugs, organic acids, and hormones, to specific organelles.38 The uptake of some NIR dyes, including MHI-148, is mediated by OATP channel proteins whose expression is regulated by HIF-1α, which is regulated by hypoxia, a standard condition that accompanies tumor growth.36 Hypoxia is frequently observed in many solid tumors and affects gene expression and cell behavior, primarily through HIF-1α.39 In the presence of oxygen, the HIF-1α protein undergoes degradation, which may contribute to the relatively low level of OATPs in normal cells, leading to the reduced uptake of MHI-148.30 Considering the characteristics of MHI-148, such as tumor hypoxia and OATP-mediated cancer cell targeting, we suggest that PTX-MHI uses a similar mechanism for cancer-specific uptake signaling. MHI-148 can selectively accumulate in the lysosomes and mitochondria of tumor cells by OATP signaling, making it a suitable candidate for dual imaging and targeted therapy.22,23 Thus, this delivery system could help overcome low PTX bioavailability in cancer cells and improve PTX-mediated cancer therapy without causing considerable side effects in adjacent healthy cells. The results of the in vitro fluorescent imaging of PTX-MHI and co-localization experiments with LysoTracker and MitoTracker showed that PTX-MHI efficiently accumulates in the lysosomes and mitochondria of cancer cells.
Several studies have investigated the mechanism underlying PTX-mediated anticancer therapy. Some studies have shown that the primary means of PTX-mediated cytotoxicity is microtubule stabilization, which leads to cell cycle arrest at the G2/M phase and termination of cell proliferation.40,41 Several studies have shown that PTX can cause cytotoxicity via mitochondrial9,41,42 and lysosomal43 cell death pathways. A study on mitochondria-mediated cell death reported that PTX acts on Bcl-2 phosphorylation, accelerating the release of mitochondrial cytochrome c. The association of cytochrome c with pro-caspase-9 and Apaf-1 converts procaspase-9 to its active form, thereby triggering the intrinsic pathway of apoptosis. Pro-caspase-3 is cleaved and forms caspase-9 and caspase-8, eventually leading to apoptosis.41 Some studies have shown that microtubule-stabilizing agents, such as PTX, can induce cell death in cancer cells via cathepsin B-mediated lysosomal pathways. Briefly, PTX triggers lysosome disruption, followed by cathepsin B release and activation; this may lead to cathepsin B-dependent cell death.43 The findings from our in vitro imaging studies showed that PTX-MHI specifically accumulates inside the mitochondria and lysosomes of cancer cells. Thus, we suggest that PTX-MHI causes cell death via both lysosomal and mitochondrial pathways, as the dye seems to accumulate in both organelles (Figure 7). The accumulation of PTX-MHI was greater in tumors than in normal organs, including the liver, kidneys, spleen, lungs, and heart, in both in vivo and ex vivo studies. Since PTX alone has no tumor-targeting properties, the conjugation with the tumor-targeting MHI-148 dye could likely provide significant advantages over conventional PTX-mediated cancer therapy.
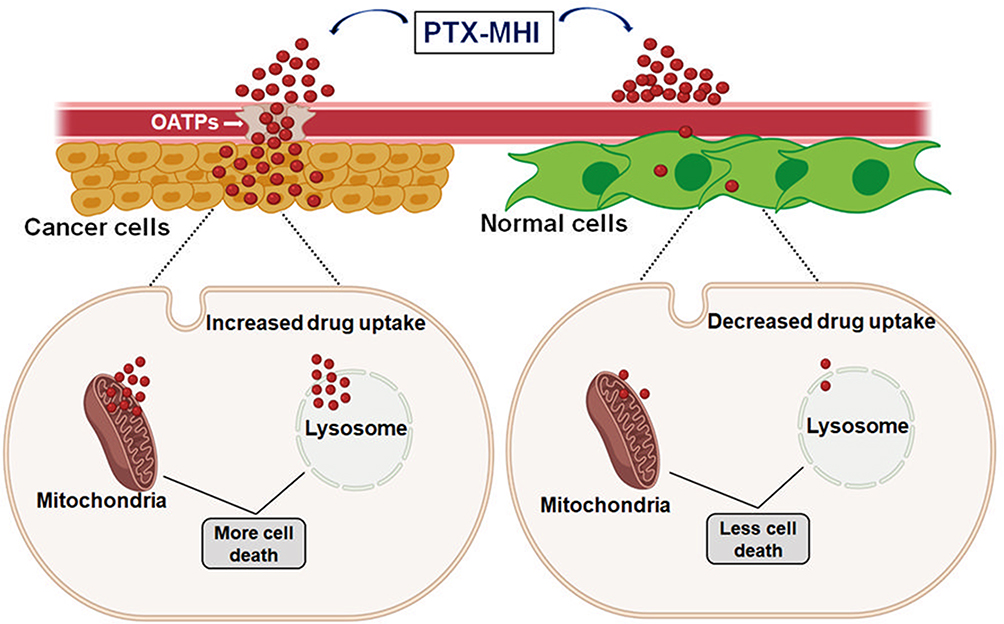 |
Figure 7 Mechanism of action of PTX-MHI in cancer cells and normal cells. PTX-MHI accumulates inside the mitochondria and lysosomes of cancer cells with the help of a group of transport proteins, organic anion-transporting polypeptides (OATPs), which are overexpressed in cancer cells, and promotes cancer cell death. Conversely, in normal cells, owing to a low level or absence of OATPs, PTX-MHI cannot accumulate sufficiently, and causes low levels of cell death. Created with BioRender.com. |
In vitro, NIRF imaging in HT-29 and NIH3T3 cells indicated that PTX-MHI could assist with the tumor-targeted delivery of PTX to cancer cells only. Hence, we successfully increased PTX accumulation in the tumor site by preventing its non-specific accumulation in healthy cells. We believe that the mechanism by which PTX-MHI causes selective cytotoxicity in colon cancer tissues involves the specific accumulation of the drug in the lysosomes and mitochondria, which leads to mitochondria- and lysosome-mediated cell death.
Conclusion
Traditional chemotherapy is frequently inefficient because of the poor targeting capabilities and off-target effects of drugs. It is critical to develop innovative methods to increase the selectivity of anticancer medication while minimizing the harmful effects on healthy cells.
In this study, we show that PTX-MHI can be employed to treat cancer successfully, with considerable tumor shrinkage in colon cancer cells and minimal cytotoxicity in normal fibroblasts. Our modification of PTX-MHI demonstrated the efficacy of the heptamethine dye MHI-148 as a tumor-targeting and optical imaging agent for successful cancer therapy without the compulsion for light irradiation treatments. This approach can possibly serve as an innovative and cost-effective method of cancer treatment in the absence of light assistance and can help advance research in the field of non-irradiation treatments.
PTX-MHI can be used as an efficient delivery system for the effective tumor-targeted delivery of the anticancer agent PTX and holds great promise in cancer imaging and therapy.
Acknowledgment
This work was supported by the Technology Innovation Program (or Industrial Strategic Technology Development Program-Development of Core Industrial Technology) (20003822, Development of Navigation System Technologies of Micro Nano Robots with Drug for Brain Disease Therapy) funded By the Ministry of Trade, Industry & Energy (MOTIE, Korea) and by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (2018R1D1A1B07049867, 2021R1F1A1064062).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Jin Z, Lv Y, Cao H, et al. Core-shell nanocarriers with high paclitaxel loading for passive and active targeting. Sci Rep. 2016;6(1):1–10.
2. Minchinton AI, Tannock IF. Drug penetration in solid tumours. Nat Rev Cancer. 2006;6(8):583–592. doi:10.1038/nrc1893
3. Ebaston T, Rozovsky A, Zaporozhets A, et al. Peptide‐driven targeted drug‐delivery system comprising turn‐on near‐infrared fluorescent xanthene–cyanine reporter for real‐time monitoring of drug release. Chem Med Chem. 2019;14(19):1727–1734. doi:10.1002/cmdc.201900464
4. Crown J, O’Leary M. The taxanes: an update. Lancet. 2000;355(9210):1176–1178. doi:10.1016/S0140-6736(00)02074-2
5. Jordan MA, Wilson L. Microtubules as a target for anti-cancer drugs. Nat Rev Cancer. 2004;4(4):253–265. doi:10.1038/nrc1317
6. Zou H, Li L, Carcedo IG, Xu ZP, Monteiro M, Gu W. Synergistic inhibition of colon cancer cell growth with nanoemulsion-loaded paclitaxel and PI3K/mTOR dual inhibitor BEZ235 through apoptosis. Int J Nanomedicine. 2016;11:1947.
7. Taxman DJ, MacKeigan JP, Clements C, Bergstralh DT, Ting JP. Transcriptional profiling of targets for combination therapy of lung carcinoma with paclitaxel and mitogen-activated protein/extracellular signal-regulated kinase kinase inhibitor. Cancer Res. 2003;63(16):5095–5104.
8. van Vuuren RJ, Visagie MH, Theron AE, Joubert AM. Antimitotic drugs in the treatment of cancer. Cancer Chemother Pharmacol. 2015;76(6):1101–1112. doi:10.1007/s00280-015-2903-8
9. Maushagen R, Reers S, Pfannerstill A-C, et al. Effects of paclitaxel on permanent head and neck squamous cell carcinoma cell lines and identification of anti-apoptotic caspase 9b. J Cancer Res Clin Oncol. 2016;142(6):1261–1271. doi:10.1007/s00432-016-2150-3
10. André N, Braguer D, Brasseur G, et al. Paclitaxel induces release of cytochrome c from mitochondria isolated from human neuroblastoma cells. Cancer Res. 2000;60(19):5349–5353.
11. André N, Carré M, Brasseur G, et al. Paclitaxel targets mitochondria upstream of caspase activation in intact human neuroblastoma cells. FEBS Lett. 2002;532(1–2):256–260. doi:10.1016/S0014-5793(02)03691-8
12. Mediavilla-Varela M, Pacheco FJ, Almaguel F, et al. Docetaxel-induced prostate cancer cell death involves concomitant activation of caspase and lysosomal pathways and is attenuated by LEDGF/p75. Mol Cancer. 2009;8(1):1–15. doi:10.1186/1476-4598-8-68
13. Tian W, Liu J, Guo Y, Shen Y, Zhou D, Guo S. Self-assembled micelles of amphiphilic PEGylated rapamycin for loading paclitaxel and resisting multidrug-resistant cancer cells. J Mater Chem B. 2015;3(7):1204–1207. doi:10.1039/C4TB01633E
14. Gupta N, Hatoum H, Dy GK. First line treatment of advanced non-small-cell lung cancer–specific focus on albumin bound paclitaxel. Int J Nanomedicine. 2014;9:209.
15. Abu Samaan TM, Samec M, Liskova A, Kubatka P, Büsselberg D. Paclitaxel’s mechanistic and clinical effects on breast cancer. Biomolecules. 2019;9(12):789. doi:10.3390/biom9120789
16. Ou K, Xu X, Guan S, et al. Nanodrug carrier based on Poly(Ursolic Acid) with self-anticancer activity against colorectal cancer. Adv Funct Mater. 2020;30:1907857. doi:10.1002/adfm.201907857
17. Zheng Y, You X, Guan S, et al. Poly(Ferulic Acid) with an anticancer effect as a drug nanocarrier for enhanced colon cancer therapy. Adv Funct Mater. 2019;29:1808646. doi:10.1002/adfm.201808646
18. Zhang X, Zhang R, Huang J, et al. Albumin enhances PTX delivery ability of dextran NPs and therapeutic efficacy of PTX for colorectal cancer. J Mater Chem B. 2019;7(22):3537. doi:10.1039/C9TB00181F
19. Ayalew L, Acuna J, Urfano SF, et al. Conjugation of paclitaxel to hybrid peptide carrier and biological evaluation in Jurkat and A549 cancer cell lines. ACS Med Chem Lett. 2017;8(8):814–819. doi:10.1021/acsmedchemlett.7b00117
20. Surapaneni MS, Das SK, Das NG. Designing paclitaxel drug delivery systems aimed at improved patient outcomes: current status and challenges. ISRN Pharmacol. 2012;2012:623139.
21. Thomas RG, Moon MJ, Surendran SP, et al. cyanine dye conjugated chitosan nanomicelle with NIR light-trigger release property as cancer-targeting theranostic agent. Mol Imaging Biol. 2018;20(4):533–543. doi:10.1007/s11307-018-1169-z
22. Yang X, Shi C, Tong R, et al. Near IR heptamethine cyanine dye–mediated cancer imaging. Clin Cancer Res. 2010;16(10):2833–2844. doi:10.1158/1078-0432.CCR-10-0059
23. Choi PJ, Park TIH, Cooper E, Dragunow M, Denny WA, Jose J. Heptamethine cyanine dye mediated drug delivery: hype or hope. Bioconjug Chem. 2020;31(7):1724–1739. doi:10.1021/acs.bioconjchem.0c00302
24. Yuan J, Yi X, Yan F, et al. Near‑infrared fluorescence imaging of prostate cancer using heptamethine carbocyanine dyes. Mol Med Rep. 2015;11(2):821–828. doi:10.3892/mmr.2014.2815
25. Shi C, Wu JB, Pan D. Review on near-infrared heptamethine cyanine dyes as theranostic agents for tumor imaging, targeting, and photodynamic therapy. J Biomed Opt. 2016;21(5):050901. doi:10.1117/1.JBO.21.5.050901
26. Yi X, Yan F, Wang F, et al. IR-780 dye for near-infrared fluorescence imaging in prostate cancer. Med Sci Monitor. 2015;21:511. doi:10.12659/MSM.892437
27. Zhang E, Luo S, Tan X, Shi C. Mechanistic study of IR-780 dye as a potential tumor targeting and drug delivery agent. Biomaterials. 2014;35(2):771–778. doi:10.1016/j.biomaterials.2013.10.033
28. Buxhofer-Ausch V, Secky L, Wlcek K, et al. Tumor-specific expression of organic anion-transporting polypeptides: transporters as novel targets for cancer therapy. J Drug Deliv. 2013;2013:863539.
29. Lee W, Belkhiri A, Lockhart AC, et al. Overexpression of OATP1B3confers apoptotic resistance in colon cancer. Cancer Res. 2008;68(24):10315–10323. doi:10.1158/0008-5472.CAN-08-1984
30. Wu JB, Shao C, Li X, et al. Near-infrared fluorescence imaging of cancer mediated by tumor hypoxia and HIF1α/OATPs signaling axis. Biomaterials. 2014;35(28):8175–8185. doi:10.1016/j.biomaterials.2014.05.073
31. Ndungu JM, Lu YJ, Zhu S, et al. Targeted delivery of paclitaxel to tumor cells: synthesis and in vitro evaluation. J Med Chem. 2010;53(8):3127–3132. doi:10.1021/jm901763f
32. Machulkin AE, Skvortsov DA, Ivanenkov YA, et al. Synthesis and biological evaluation of PSMA-targeting paclitaxel conjugates. Bioorg Med Chem Letters. 2019;29(16):2229–2235. doi:10.1016/j.bmcl.2019.06.035
33. Chou PL, Huang YP, Cheng MH, Rau KM, Fang YP. Improvement of paclitaxel-associated Adverse Reactions (ADRs) via the use of nano-based drug delivery systems: a systematic review and network meta-analysis. Int J Nanomedicine. 2020;15:1731–1743. doi:10.2147/IJN.S231407
34. Leonelli F, Bella AL, Migneco LM, Bettolo RM. Design, synthesis and applications of hyaluronic acid-paclitaxel bioconjugates†. Molecules. 2008;13:360–378. doi:10.3390/molecules13020360
35. Zhang C, Zhao Y, Zhao N, et al. NIRF optical/PET dual-modal imaging of hepatocellular carcinoma using heptamethine carbocyanine dye. Contrast Media Mol Imaging. 2018;2018:1–12.
36. Shi C, Wu JB, Chu GC, et al. Heptamethine carbocyanine dye-mediated near-infrared imaging of canine and human cancers through the HIF-1α/OATPs signaling axis. Oncotarget. 2014;5(20):10114. doi:10.18632/oncotarget.2464
37. Zhao N, Zhang C, Zhao Y, et al. Optical imaging of gastric cancer with near-infrared heptamethine carbocyanine fluorescence dyes. Oncotarget. 2016;7(35):57277. doi:10.18632/oncotarget.10031
38. Giacomini KM, Huang S-M, Tweedie DJ, et al. Membrane transporters in drug development. Nat Rev Drug Discov. 2010;9(3):215.
39. Lu X, Kang Y. Hypoxia and hypoxia-inducible factors: master regulators of metastasis. Clin Cancer Res. 2010;16(24):5928–5935. doi:10.1158/1078-0432.CCR-10-1360
40. Fan W. Possible mechanisms of paclitaxel-induced apoptosis. Biochem Pharmacol. 1999;57(11):1215–1221.
41. George J, Banik NL, Ray SK. Molecular mechanisms of taxol for induction of cell death in glioblastomas. In: Ray SK, editor, Glioblastoma. Springer; 2010:283–298.
42. Esfandyari-Manesh M, Mohammadi A, Atyabi F, et al. Enhancement mitochondrial apoptosis in breast cancer cells by paclitaxel-triphenylphosphonium conjugate in DNA aptamer modified nanoparticles. J Drug Deliv Sci Technol. 2019;54:101228. doi:10.1016/j.jddst.2019.101228
43. Bröker LE, Huisman C, Span SW, Rodriguez JA, Kruyt FA, Giaccone G. Cathepsin B mediates caspase-independent cell death induced by microtubule stabilizing agents in non-small cell lung cancer cells. Cancer Res. 2004;64(1):27–30. doi:10.1158/0008-5472.CAN-03-3060
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.