")
Back to Journals » International Journal of General Medicine » Volume 15
Hemophagocytic Lymphohistiocytosis in Adults: A Retrospective Study in a Belgian Teaching Hospital
Authors Yildiz H , Castanares-Zapatero D, d'Abadie P, Bailly S, Yombi JC
Received 14 September 2022
Accepted for publication 3 November 2022
Published 8 November 2022 Volume 2022:15 Pages 8111—8120
DOI https://doi.org/10.2147/IJGM.S388880
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Scott Fraser
Halil Yildiz,1 Diego Castanares-Zapatero,2 Philippe d’Abadie,3 Sarah Bailly,4 Jean Cyr Yombi1
1Department of Internal Medicine and Infectious Diseases, Cliniques Universitaires Saint-Luc, UClouvain, Bruxelles, Belgium; 2Department of Intensive Care, Cliniques Universitaires Saint-Luc, UClouvain, Bruxelles, Belgium; 3Department of Nuclear Medicine, Cliniques Universitaires Saint-Luc, UClouvain, Bruxelles, Belgium; 4Department of Hematology, Cliniques Universitaires Saint-Luc, UClouvain, Bruxelles, Belgium
Correspondence: Halil Yildiz, Department of Internal Medicine and Infectious Diseases, Cliniques Universitaires Saint-Luc, UClouvain, 10 Av Hippocrate, Bruxelles, 1200, Belgium, Email [email protected]
Introduction: Hemophagocytic lymphohistiocytosis (HLH) is a life-threatening disease, which can be primary (due to genetic mutation) or secondary to malignancy, infection and rheumatologic diseases. Data concerning Belgian patients with adult HLH are lacking.
Methods: This retrospective study was performed in a teaching hospital in Belgium. All cases of adult HLH, from December 2010 to April 2022, were reviewed. Patients with more than five HLH-2004 criteria and/or HScore > 80% were included in the study. The objective of our study was to describe clinical and biological characteristics of patients with HLH and attempt to look for variables associated with mortality.
Results: Fifty-two patients were included in the final analysis. Mean age (SD) of patients was 48 (18) years old, and 29 patients were of male gender (56%). The underlying diseases associated with HLH were malignancy (M-HLH) in 22 patients, infection related HLH in 20 patients, rheumatologic disease related HLH in 7 patients, idiopathic in 2 patients and secondary to pregnancy in 1 patient. Overall mortality, mortality at 30 days and 90 days were 24/52 (46%), 13/52 (25%) and 4/52 (10%), respectively. In univariate analysis, malignancy, male sex, age and disseminated intravascular coagulation (DIC) were associated with mortality (p < 0.05). In multivariate analysis, only age was significantly associated with mortality (odds ratio, 1.053; 95% confidence interval, 1.016– 1.092; p 0.005).
Conclusion: In our study, the most frequent triggers were malignancy and infectious agent followed by rheumatologic disease. Risk factors for mortality were age, male sex, malignancy and DIC, but only age remained significant in multivariate analysis. Treatment guidelines are mainly based on pediatric patients, and it is important for physician to describe adult patients’ outcome to better understand this disease and adapt treatment.
Keywords: hemophagocytic lymphohistiocytosis, macrophage activation syndrome, infection, autoimmune disease, malignancy, corticosteroid, tocilizumab, ruxolitinib, mortality
Introduction
Hemophagocytic lymphohistiocytosis (HLH) is a disease characterized by hyperactivation of macrophages, cytotoxic T lymphocytes and natural killer (NK) cells intense hyperimmune response as well as uncontrolled release of inflammatory cytokines.1,2 It is classically subdivided into primary HLH and secondary HLH.2 Primary HLH refers to pediatric patients with defects in genes implicated in the cytotoxic pathways of T and NK cells or in the eradication of Epstein Barr virus (EBV).3 Secondary or acquired HLH (sHLH) concerns adult’s patients who have the syndrome secondary to infection (viral: EBV and cytomegalovirus [CMV] mainly, but also after bacterial, fungal, or parasitic infections), malignancies (mainly lymphomas), or autoimmune diseases, such as systemic juvenile idiopathic arthritis, Adult Onset Still’s Diseases (AOSD) or systemic lupus erythematosus, pregnancy and even drug induced HLH (lamotrigine, checkpoint inhibitor, CAR-T cells).2,4 However, this classification is not exactly adapted since genetic mutations are also found in adult patients with secondary or acquired HLH and a trigger (such as infection, autoimmune disease) is also found in pediatric patients with HLH.5–10 The North American Consortium for Histiocytosis (NACHO) proposed to categorized according to etiology rather than as primary or secondary:11 familial-HLH (genetic mutation), malignancy-HLH, Rheumatologic-HLH (macrophage activation syndrome in the context of autoimmune disease), Iatrogenic-HLH (drug induced HLH), HLH with immune compromise and non-specific HLH without a clear genetic or infectious trigger (HLH-NOS, idiopathic). This last classification allows to better appreciate the complexity of HLH disease and gives a better information for the treatment decisions. However, this nomenclature of HLH diagnosis is still a matter of discussion/debate and needs to be validated.
Mortality of HLH in adult patients ranges from 26.5% to 74.8% according to studies and etiologies.4 Most of the studies are performed in pediatric patients, and studies in adult patients are scarce and mainly based on case reports and series (mostly in Asian patients). We performed a retrospective study in order to describe clinical and biological characteristics of HLH in a Belgian teaching hospital and attempt to look for variables associated with mortality.
Materials and Methods
Study Design and Patients
This retrospective study was performed at a teaching hospital of 1000 beds in Belgium. All cases of adult HLH, from December 2010 to April 2022, were reviewed. HLH diagnosis was made based on HLH-2004 criteria and Hscore. Patients with more than five HLH-2004 criteria and/or Hscore >80% were included in the study.
Ethical Issues
Our institutional ethics committee (Comité d’Ethique Hospitalo-Facultaire [CEHF], Cliniques Universitaires Saint-Luc, Brussels, Belgium) stated that a written consent is not needed for analyses of anonymized data, retrospective study and give its authorization (N°CEHF 2022/17MAI/211-HLHadult). Our study complies with the Declaration of Helsinki.
Data Collection
Using our institutional database Medical explorer v3r49b7, Epic electronic health record and the database of the department of internal medicine and hematology, we reviewed the medical records of all patients and collected the following data, HScore and HLH-2004 criteria (on admission), demographics, clinical and labs characteristics (on admission), diagnostic investigation (18F-fluorodeoxyglucose positron emission tomography computed tomography [18-F-FDG PET/CT], thoraco-abdominal CT scan), underlying diseases, treatment, mortality (outcome).
Statistical Analysis
All analyses were conducted using SPSS 27 software (IBM SPSS Statistics for Windows, Version 27.0, Armonk, NY: IBM Corp). Discrete variables were reported as numbers and corresponding percentages, while continuous variables were expressed as mean and standard deviations (SD). Categorical variables were compared using Pearson’s chi-square test or exact Fisher test, whereas differences between continuous variables were assessed using unpaired Student’s t-test. An univariate logistic regression was conducted to assess the association between selected variables and mortality. A multivariate logistic regression model, using a forward stepwise method, was performed. Results were expressed as odds ratio with 95% confidence intervals. P-values were two-sided, and <5% was considered as significative.
Results
General Characteristics
Among 56 patients identified with suspected HLH, 52 were included in the final analysis (4 patients were excluded due to diagnosis other than HLH, 1 thrombotic microangiopathy, 1 medullary aplasia due to epidermoid cancer, 1 B-cell acute lymphocytic leukemia and 1 spleen infarction).
Mean age (SD) of patients was 48 (18) years old, and 29 patients were of male gender (56%). Clinical manifestations are shown in Table 1. The most frequent clinical manifestations were fever (98%), bicytopenia (92%), thrombocytopenia (92%). Hemophagocytosis was found in bone marrow in 100% of patients. Splenomegaly and hepatomegaly were found in 67% and 27%, respectively, while peripheral lymphadenopathy was found in 31% of patients. The underlying diseases associated with HLH are presented in Table 1. A malignancy related-HLH was found in 22 patients (42%). A solid cancer was found in 1 patient, while a hematological disease was found in 21 patients (21/22). The hematological diseases associated with HLH were as following, B-cell lymphoma [10/21], T-cell lymphoma [7/21], acute leukemia [3/21] and Castleman disease [1/21]. An infection related HLH was found in 20 patients (38.5%) and was mainly secondary to EBV [8/20]. Other infectious agents are shown in Table 1. A rheumatologic disease related HLH (macrophage activation syndrome) was found in 7 patients (13.5%) and was mainly secondary to AOSD [3/7]. Finally, 2 patients have idiopathic HLH (3.8%) and 1 patient had HLH secondary to pregnancy (1.9%). Table 2 shows clinical and biological characteristics according to etiologies.
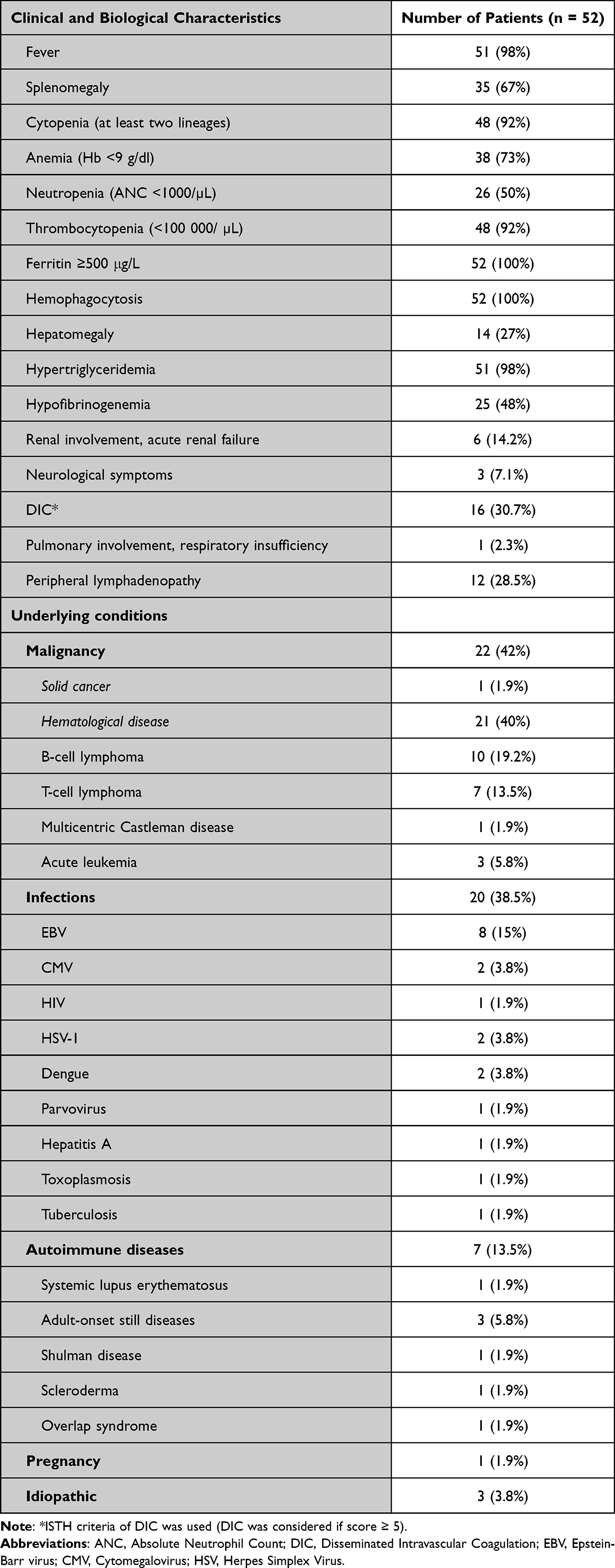 |
Table 1 Clinical, Labs Characteristics and Underlying Conditions of Adult Patients with HLH |
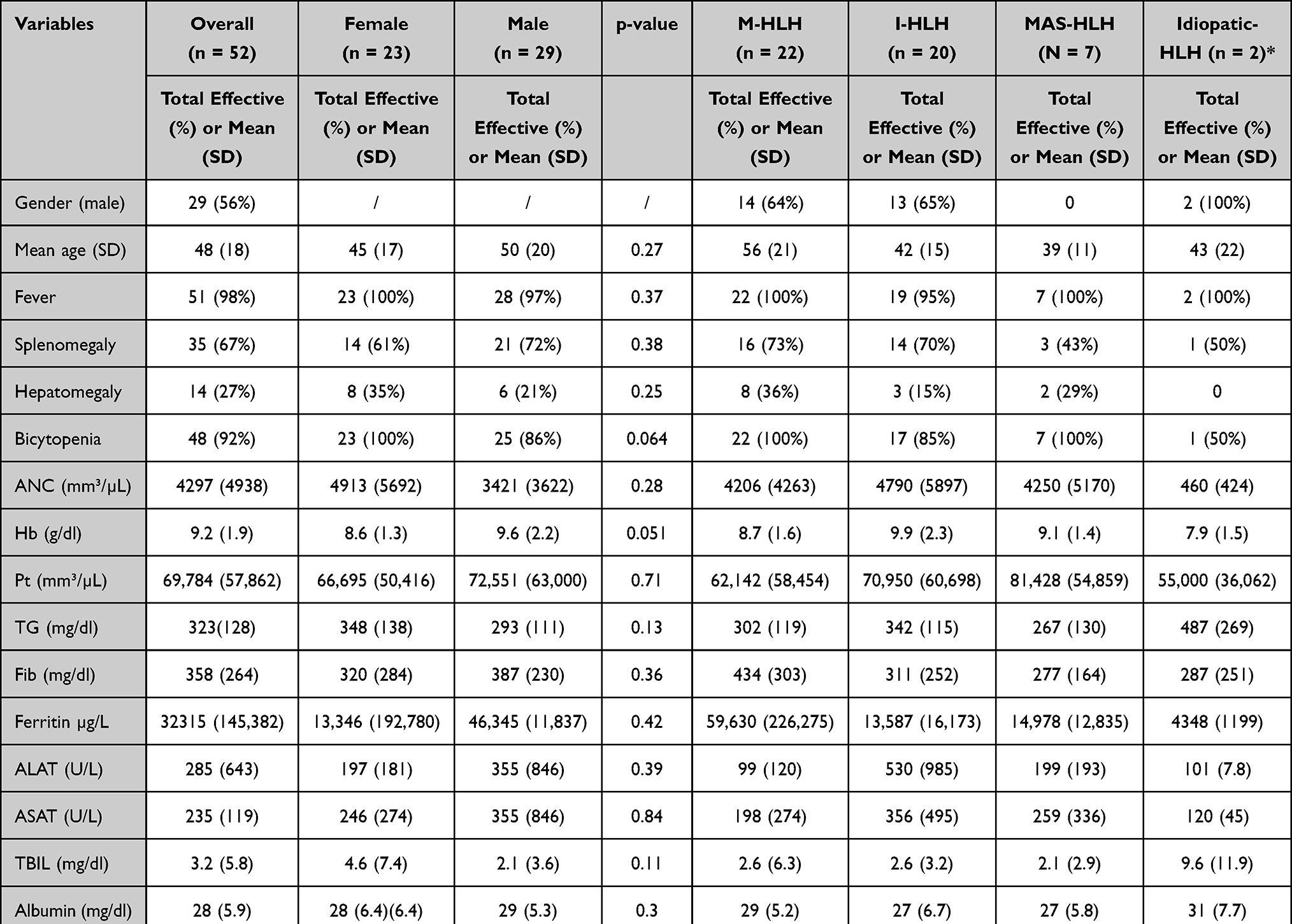 |
Table 2 Clinical and Labs Characteristics of Adult Patients with HLH According to Etiologies |
Overall mortality, mortality at 30 days and 90 days were as following, 24/52 (46%), 13/52 (25%) and 4/52 (10%).
Clinical and biological characteristics were quite similar between women and men despite a slightly male predominance (56%). However, overall mortality was higher in male patients (17/52) compared to female patients (7/52) (p 0.043). There was no difference in mortality at 30 days and 90 days (Table 2).
Comparison of Clinical and Labs Characteristics Between Patients with Malignancy-HLH (M-HLH) and Non Malignancy-HLH (nM-HLH)
Clinical, Labs, evolution and treatment characteristics are presented in Table 3. Mean age (SD) of patients with M-HLH was 56 (21) years old compared to 41 (14) years old in patients with nM-HLH (p 0.003). ALAT and ASAT were more frequently elevated in the nM-HLH subgroup (77%) compared to M-HLH (50%) (p < 0.05). Chemotherapy and etoposide were more frequently given in the M-HLH subgroup (64% and 27% respectively) compared to nM-HLH subgroup (0% and 7% respectively) (p < 0.001 and 0.0042). Overall mortality and mortality at 30 days were higher in M-HLH subgroup (64% and 41%) compared to nM-HLH subgroup (33 and 13%) (p < 0.05).
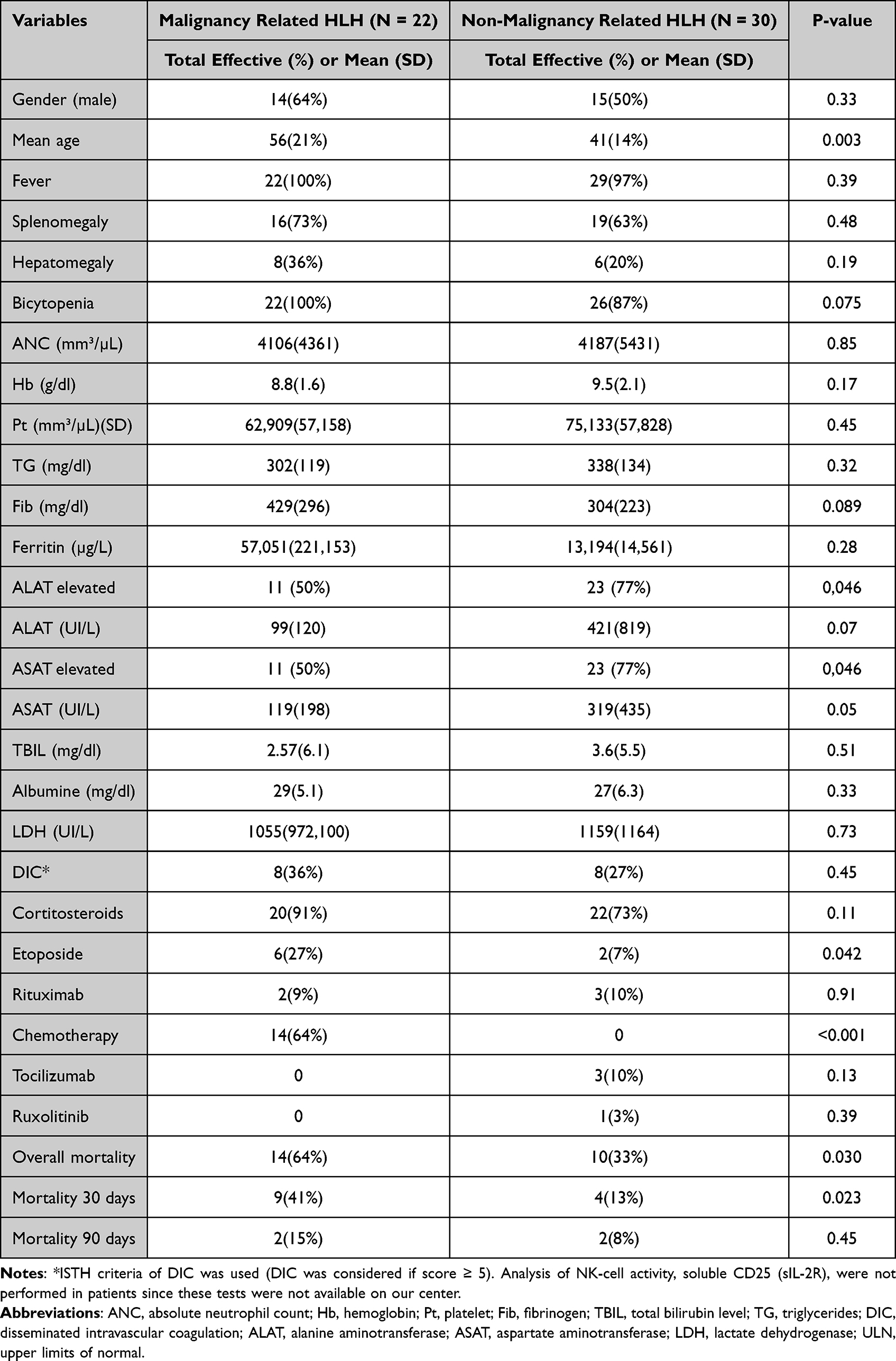 |
Table 3 Comparison of Clinical and Labs Characteristics Among Patients with Malignancy Related HLH and Non-Malignancy Related HLH |
Factors Associated with Mortality in Univariate and Multivariate Analysis
In univariate analysis, malignancy, male sex, age and disseminated intravascular coagulation (DIC) were significantly associated with mortality (p < 0.05). In multivariate analysis, age (odds ratio, 1.053; 95% confidence interval, 1.016–1.092; p 0.005) remains the only factor significantly associated with mortality. The results of univariate and multivariate analysis are available in Table 4.
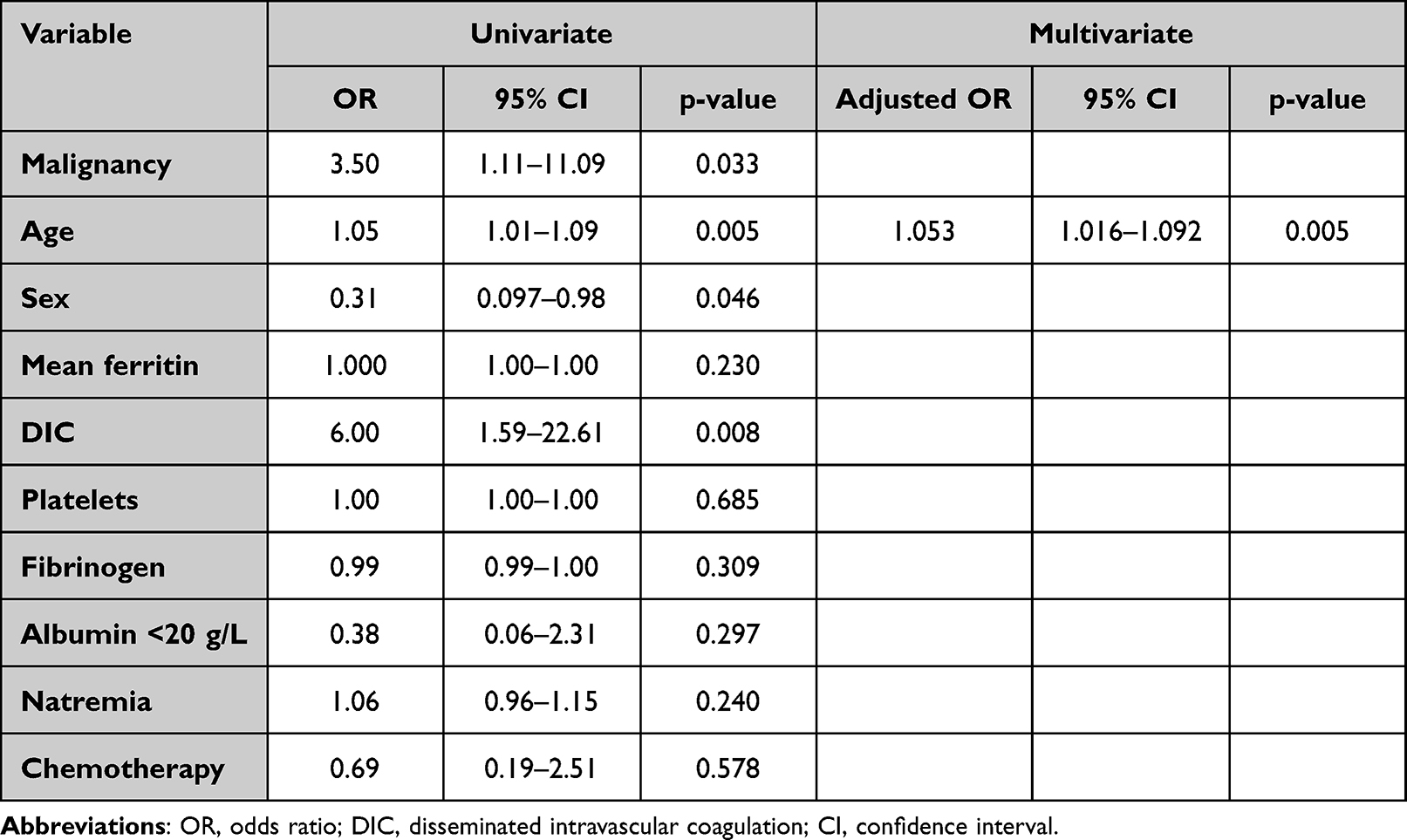 |
Table 4 Factors Associated with Mortality in Adult Patient with HLH (Univariate and Multivariate Analysis) |
Discussion
Data concerning factors associated with mortality in adult patients with HLH are scarce. Our study is, to our knowledge, one of the first concerning Belgian patients. In fact, most of the studies concerned patients in China, only few studies were performed in Europe (Germany and France).11–33 It is important to describe clinical features of adults patients with HLH in different settings to see if there is a difference in the clinical presentation, etiologies and outcome. In our study, we showed that malignancy HLH (M-HLH) is the most common cause of HLH followed by infectious HLH and autoimmune disease associated with HLH such as in previous series.11–33 Factors significantly associated with mortality were malignancy, male sex, age and disseminated intravascular coagulation (DIC) in univariate analysis, while only age remained significant in multivariate analysis. Identifying factors influencing mortality is important since this can lead physician to closer follow-up and adapt treatment.
M-HLH has been showed as being a risk factor for mortality in several studies.13,16,17,21,26 There is only 1 study which showed that non-malignancy HLH (nM-HLH) was associated with higher mortality compared to M-HLH.24 This was explained by 1) delay in the diagnosis in nM-HLH, 2) lack of a clear underlying condition causing HLH in the group of nM-HLh and 3) high number of tuberculosis-associated HLH which is well known to be associated with high mortality rate (>50%).34,35 Since malignancy HLH is associated with poor outcome, some authors suggested that chemotherapy containing etoposide, which is a drug proposed in the HLH-94 and 2004 protocol,36–38 may be associated with better outcome.3,39 Recently, Song et al39 showed that in patients with lymphoma-associated HLH, those treated with chemotherapy containing etoposide have a better survival (p < 0.05).
Another factor associated with mortality in our study was male sex, and this have been described also in other studies.15,25 Several other factors have been described in different studies and are summarized in the Supplemental File (Table S1). Age seems to be a factor of mortality in our study'; however, the age cut-off value varies according to studies ranging from age >50 to ≥60 and needs to be confirmed in prospective studies.11,18,25,33 Disseminated intravascular coagulation (DIC) is also associated with mortality, and it is not surprising since it means that organ damage is ongoing due to the severity of the disease.11–13,26,33 High ferritin and low albumin, hemoglobin and platelet level have also been described as a risk factor of mortality, but this was not confirmed in our study and the best cut-off value still needs to be determined.17,18,27
Another interesting finding in our study was that alanine aminotransferase (ALAT) and aspartate aminotransferase (ASAT) level were more frequently elevated in the nM-HLH subgroup (77%) compared to M-HLH (50%) (p < 0.05). This is probably explained by the infectious agent related HLH in the non-malignancy HLH subgroup (nM-HLH) which were microorganisms with liver tropism (EBV, CMV, Dengue, Herpes Simplex Virus, Tuberculosis).
Our study has several limitations. Firstly, this is a single center and retrospective study. Secondly, the number of patients was low (52 patients) and this may explain why DIC and malignancy failed to be significant factors associated with mortality in multivariate analysis. Despite these limitations, our results depict a valuable picture of the situation in a teaching high-volume center since data concerning Belgian patients are lacking. Thirdly, the diagnosis of DIC was made by using ISTH criteria.40 However, diagnosis of DIC in HLH is a challenge since HLH is associated with hypofibrinogenemia, thrombocytopenia and coagulation abnormalities. Other biological markers are required to better diagnose DIC in patients with HLH. Fourthly, the screening of genes related to primary HLH was not performed which does not exclude the possibility of presence of primary HLH in a minority of our patients.
Conclusions
We described a single center’s experience with adult patients with secondary HLH. The most frequent triggers were malignancy and infectious agent followed by rheumatologic disease. Risk factors for mortality were age, male sex, malignancy and disseminated intravascular coagulation. A prompt diagnosis and early treatment should be performed in order to improve outcome.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Janka GE, Lehmberg K. Hemophagocytic lymphohistiocytosis: pathogenesis and treatment. Hematol Am Soc Hematol Educ Progr. 2013;2013:605–11. doi:10.1182/asheducation-2013.1.605
2. Ramos-Casals M, Brito-Zeron P, Lopez-Guillermo A, Khamashta MA, Bosch X. Adult haemophagocytic syndrome. Lancet. 2014;383:1503–16. doi:10.1016/S0140-6736(13)61048-X
3. La Rosée P, Horne AC, Hines M, et al. Recommendations for the management of hemophagocytic lymphohistiocytosis in adults. Blood. 2019;133:2465–2477. doi:10.1182/blood.2018894618
4. Yildiz H, Van Den Neste E, Defour JP, Danse E, Yombi JC. Adult haemophagocytic lymphohistiocytosis: a review. QJM. 2020;115(4):205–213.
5. Brisse E, Wouters CH, Matthys P. Advances in the pathogenesis of primary and secondary haemophagocytic lymphohistiocytosis: differences and similarities. Br J Haematol. 2016;174(2):203–217. doi:10.1111/bjh.14147
6. Zhang M, Behrens EM, Atkinson TP, Shakoory B, Grom AA, Cron RQ. Genetic defects in cytolysis in macrophage activation syndrome. Curr Rheumatol Rep. 2014;16:439–446. doi:10.1007/s11926-014-0439-2
7. Carvelli J, Piperoglou C, Farnarier C, et al. Functional and genetic testing in adults with HLH reveals an inflammatory profile rather than a cytotoxicity defect. Blood. 2020;136(5):542–552. doi:10.1182/blood.2019003664
8. Miao Y, Zhu HY, Qiao C, et al. Pathogenic gene mutations or variants identified by targeted gene sequencing in adults with hemophagocytic lymphohistiocytosis. Front Immunol. 2019;10:395. doi:10.3389/fimmu.2019.00395
9. Zhang K, Jordan MB, Marsh RA, et al. Hypomorphic mutations in PRF1, MUNC13-4, and STXBP2 are associated with adult onset familial HLH. Blood. 2011;118:5794–8. doi:10.1182/blood-2011-07-370148
10. Chen X, Wang F, Zhang Y, et al. Genetic variant spectrum in 265 Chinese patients with hemophagocytic lymphohistiocytosis: molecular analyses of PRF1, UNC13D, STX11, STXBP2, SH2D1A and XIAP. Clin Genet. 2018;94:200–212. doi:10.1111/cge.13363
11. Tseng YT, Sheng WH, Lin BH, et al. Causes, clinical symptoms, and outcomes of infectious diseases associated with hemophagocytic lymphohistiocytosis in Taiwanese adults. J Microbiol Immunol Infect. 2011;44:191–7. doi:10.1016/j.jmii.2011.01.027
12. Park HS, Kim DY, Lee JH, et al. Clinical features of adult patients with secondary hemophagocytic lymphohistiocytosis from causes other than lymphoma: an analysis of treatment outcome and prognostic factors. Ann Hematol. 2012;91:897–904. doi:10.1007/s00277-011-1380-3
13. Parikh SA, Kapoor P, Letendre L, Kumar S, Wolanskyj AP. Prognostic factors and outcomes of adults with hemophagocytic lymphohistiocytosis. Mayo Clin Proc. 2014;89(4):484–492. doi:10.1016/j.mayocp.2013.12.012
14. Rivière S, Galicier L, Coppo P, et al. Reactive hemophagocytic syndrome in adults: a retrospective analysis of 162 patients. Am J Med. 2014;127:118–25. doi:10.1016/j.amjmed.2014.04.034
15. Li J, Wang Q, Zheng W, et al. Hemophagocytic lymphohistiocytosis, clinical analysis of 103 adult patients. Medicine. 2014;93:100–5. doi:10.1097/MD.0000000000000022
16. Lim SH, Park S, Jang JH, et al. Clinical significance of bone marrow hemophagocytosis in adult patients with malignancy and non malignancy-induced hemophagocytic lymphohistiocytosis. Ann Hematol. 2016;95:325–35. doi:10.1007/s00277-015-2523-8
17. Otrock ZK, Eby CS. Clinical characteristics, prognostic factors, and outcomes of adult patients with hemophagocytic lymphohistiocytosis. Am J Hematol. 2015;90:220–224. doi:10.1002/ajh.23911
18. Dholaria B, Hammond WA, Shreders A, Robinson S, Sher T. Hemophagocytic lymphohistiocytosis: retrospective analysis for prognostic factors. Blood. 2015;126:4615. doi:10.1182/blood.V126.23.4615.4615
19. Schram AM, Comstock P, Campo M, et al. Haemophagocytic lymphohistiocytosis in adults: a multicentre case series over 7 years. Br J Haematol. 2016;172:412–419. doi:10.1111/bjh.13837
20. Shuai X, Xu J, Zhong X, et al. Retrospective treatment analysis of a series of 104 patients with adult onset hemophagocytic lymphohistiocytosis in a single institution of China. Blood. 2016;128:4882. doi:10.1182/blood.V128.22.4882.4882
21. Birndt S, Schenk T, Brunkhorst FM, et al. Hemophagocytic lymphohistiocytosis in adults (aHLH): results from the German HLH Registry. Blood. 2016;128:2523. doi:10.1182/blood.V128.22.2523.2523
22. Yuan L, Kan Y, Meeks JK, Ma D, Yang J. 18F-FDG PET/CT for identifying the potential causes and extent of secondary hemophagocytic lymphohistiocytosis. Diagn Interv Radiol. 2016;22:471–5. doi:10.5152/dir.2016.15226
23. Zheng Y, Hu G, Liu Y, et al. The role of 18F-FDG PET CT in the management of patients with secondary haemophagocytic lymphohistiocytosis. Clin Radiol. 2016;71:1248–1254. doi:10.1016/j.crad.2016.05.011
24. Apodaca E, Rodríguez-Rodríguez S, Tuna-Aguilar EJ, Demichelis-Gómez R. Prognostic factors and outcomes in adults with secondary hemophagocytic lymphohistiocytosis: a single-center experience. Clin Lymphoma Myeloma Leuk. 2018;18(10):e373–e380. doi:10.1016/j.clml.2018.06.014
25. Zhang Q, Li L, Zhu L, et al. Adult onset haemophagocytic lymphohistiocytosis prognosis is affected by underlying disease: analysis of a single-institution series of 174 patients. Swiss Med Wkly. 2018;148:w14641. doi:10.4414/smw.2018.14641
26. Zhou M, Li L, Zhang Q, et al. Clinical features and outcomes in secondary adult hemophagocytic lymphohistiocytosis. QJM. 2018;111:23–31. doi:10.1093/qjmed/hcx183
27. Birndt S, Schenk T, Heinevetter B, et al. Hemophagocytic lymphohistiocytosis in adults: collaborative analysis of 137 cases of a nationwide German registry. J Cancer Res Clin Oncol. 2020;146(4):1065–1077. doi:10.1007/s00432-020-03139-4
28. Diack ND, Kane BS, Fall S, et al. Adult hemophagocytic lymphohistiocytosis in sub-saharan area: a retrospective study of 26 cases. Cureus. 2020;12(3):e7258. doi:10.7759/cureus.7258
29. Bubik RJ, Barth DM, Hook C, et al. Clinical outcomes of adults with hemophagocytic lymphohistiocytosis treated with the HLH-04 protocol: a retrospective analysis. Leuk Lymphoma. 2020;61(7):1592–1600. doi:10.1080/10428194.2020.1737684
30. Zhang FJ, Huang GQ, Li J, Xu J, Li XM, Wang AM. Clinical characteristics of adult hemophagocytic lymphohistiocytosis in the emergency department. Int J Gen Med. 2021;20(14):4687–4694. doi:10.2147/IJGM.S326270
31. Zhou Y, Kong F, Wang S, et al. Increased levels of serum interleukin-10 are associated with poor outcome in adult hemophagocytic lymphohistiocytosis patients. Orphanet J Rare Dis. 2021;16(1):347. doi:10.1186/s13023-021-01973-4
32. Bichon A, Bourenne J, Allardet-Servent J, et al. High mortality of HLH in ICU regardless etiology or treatment. Front Med. 2021;6(8):735796. doi:10.3389/fmed.2021.735796
33. Zhou J, Wu ZQ, Qiao T, Xu HG. Development of laboratory parameters-based formulas in predicting short outcomes for adult hemophagocytic lymphohistiocytosis patients with different underlying diseases. J Clin Immunol. 2022;42:1000–1008. PMID: 35386042. doi:10.1007/s10875-022-01263-z
34. Brastianos PK, Swanson JW, Torbenson M, Sperati J, Karakousis PC. Tuberculosis-associated haemophagocytic syndrome. Lancet Infect Dis. 2006;6(7):447–454. doi:10.1016/S1473-3099(06)70524-2
35. Zhang Y, Liang G, Qin H, Li Y, Zeng X. Tuberculosis-associated hemophagocytic lymphohistiocytosis with initial presentation of fever of unknown origin in a general hospital: an analysis of 8 clinical cases. Medicine. 2017;96(16):e6575. doi:10.1097/MD.0000000000006575
36. Henter J-I, Samuelsson-Horne AC, Arico M, et al. Treatment of hemophagocytic lymphohistiocytosis with HLH-94 immuno-chemotherapy and bone marrow transplantation. Blood. 2002;100:2367–2373. doi:10.1182/blood-2002-01-0172
37. Trottestam H, Horne A, Aricò M, et al. Histiocyte Society. Chemoimmunotherapy for hemophagocytic lymphohistiocytosis: long-term results of the HLH-94 treatment protocol. Blood. 2011;118(17):4577–4584.
38. Bergsten E, Horne A, Aricó M, et al. Confirmed efficacy of etoposide and dexamethasone in HLH treatment: long-term results of the cooperative HLH-2004 study. Blood. 2017;130(25):2728–2738. doi:10.1182/blood-2017-06-788349
39. Song Y, Wang J, Wang Y, Wu L, Wang Z. Requirement for containing etoposide in the initial treatment of lymphoma associated hemophagocytic lymphohistiocytosis. Cancer Biol Ther. 2021;22(10–12):598–606. doi:10.1080/15384047.2021.1996139
40. Taylor FB Jr, Toh CH, Hoots WK, Wada H, Levi M. Scientific subcommittee on Disseminated Intravascular Coagulation (DIC) of the International Society on Thrombosis and Haemostasis (ISTH). Towards definition, clinical and laboratory criteria, and a scoring system for disseminated intravascular coagulation. Thromb Haemost. 2001;86(5):1327–1330. doi:10.1055/s-0037-1616068
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.