")
Back to Journals » OncoTargets and Therapy » Volume 12
Helicobacter pylori upregulates TRPC6 via Wnt/β-catenin signaling to promote gastric cancer migration and invasion
Authors Song Y, Liu G, Liu S, Chen R, Wang N, Liu Z, Zhang X, Xiao Z, Liu L
Received 10 January 2019
Accepted for publication 20 May 2019
Published 3 July 2019 Volume 2019:12 Pages 5269—5279
DOI https://doi.org/10.2147/OTT.S201025
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr William C. Cho
Yang Song,*,1,2 Gao Liu,*,3,4 Shuang Liu,2 Rong Chen,1 Na Wang,5 Zhaoyu Liu,2 Xiao Zhang,6 Zheng Xiao,2 Lin Liu7,
1Center of Clinical Laboratory, First Medical Center of Chinese PLA General Hospital, Sanya, People’s Republic of China; 2Center of Clinical Laboratory, Hainan Hospital of Chinese PLA General Hospital, Sanya, People’s Republic of China; 3Department of Geriatric Cardiology, Second Medical Center of Chinese PLA General Hospital, Beijing, People’s Republic of China; 4National Centre for Clinical Research on Gerontology, Beijing, People’s Republic of China; 5Outpatient Comprehensive Treatment Area, First Medical Center of Chinese PLA General Hospital, Sanya, People’s Republic of China; 6Central Laboratory, Hainan Hospital of Chinese PLA General Hospital, Sanya, People’s Republic of China; 7Department of General Surgery, First Medical Center of Chinese PLA General Hospital, Sanya, People’s Republic of China
*These authors contributed equally to this work
Background: Helicobacter pylori infection is recognized as a major risk factor for gastric cancer (GC) progression; however, the underlying molecular mechanisms have remained to be fully elucidated.
Methods: qPCR and Western blot were used to detect mRNA level and relative protein expression. Wound healing assay and transwell were used to determine migration and invasion of cells. Calcium imaging was used to determine calcium signaling in cells. Luciferase reporter assay and immunohistochemistry were performed.
Results: In the present study, it was demonstrated that H. pylori infection in GC is closely associated with the depth of tumor invasion, lymph node metastasis, tumor-nodes-metastasis stage, and distant metastasis. Migration and invasion assays indicated that H. pylori infection enhanced the migration and invasion of GC cells in a Ca2+-dependent manner. Calcium imaging was applied to detect intracellular Ca2+ and revealed that H. pylori induced an increase of intracellular Ca2+ in GC cells through release from Ca2+ stores and extracellular Ca2+ influx. Further study indicated that H. pylori infection led to an upregulation of the expression of transient receptor potential cation channel subfamily C member 6 (TRPC6) and induced an increase of Ca2+ through the TRPC6 channel. Furthermore, H. pylori increased TRPC6 transcription through the Wnt/β-catenin pathway, and Wnt/β-catenin/TRPC6 signaling was identified to be at least in part responsible for H. pylori-induced GC migration and invasion. Finally, it was observed that TRPC6 expression was significantly associated with the H. pylori infection status in GC tissues, and H. pylori infection was associated with metastasis and poor prognosis for GC patients.
Conclusion: The present results indicate that H. pylori causes an upregulation of TRPC6 expression through the Wnt/β-catenin pathway to promote GC progression, and this interaction may serve as a promising target for GC therapy.
Keywords: helicobacter pylori, TRPC6, Wnt/β-catenin, calcium signaling, gastric cancer
Introduction
Helicobacter pylori persistently colonizes in the stomach, and has been classified as a type-I carcinogen that induces the development of gastric cancer (GC) by the World Health Organization and the International Agency for Research of Cancer.1,2 The ability of H. pylori to colonize in the human stomach and to promote GC development may be attributed to the production of multiple virulence factors,3,4 which modulate the pathogen–host interaction and determine the degree of bacterial pathogenicity. H. pylori exerts multiple effects on cell proliferation, apoptotic pathways, cytokine secretion, and DNA damage.5–8 Numerous studies have indicated that H. pylori infection drives GC progression,9–11 while the associated molecular mechanisms implicated in GC migration and invasion remain to be fully elucidated.
For the progression of cancer, it is required that tumor cells acquire migratory and invasive abilities, and Ca2+ is a critical regulator of cell migration and invasion.12 Calcium is responsible for the transmission of signals from the microenvironment into cells, and calcium signaling is an important step in cancer cell migration and invasion.13,14 Numerous studies have indicated that Ca2+ channels are essential for the progression of various tumor types.15–18 Among these, transient receptor potential (TRP) channels are non-selective Ca2+ entry channels. Several families of TRP channels exist in humans, among which TRP cation channel subfamily C member 6 (TRPC6) is greatly associated with the progression of GC. TRPC6 is significantly upregulated in human GC, and TRPC6 inhibition was reported to lead to growth suppression of GC cells.19 However, to date, the functions of TRPC6 in H. pylori infection in GC and the underlying and mechanisms have remained largely elusive.
In the present study, H. pylori was identified to upregulate TRPC6 expression through the Wnt/β-catenin pathway, and TRPC6 then mediates the extracellular Ca2+ influx in GC cells to finally promote GC cell migration and invasion in a Ca2+-dependent manner. The present study indicates that the Wnt/β-catenin/TRPC6 signaling cascade is responsible for the mediation of H. pylori-induced human GC progression, providing novel insight that may be utilized to develop strategies for preventing GC development.
Materials and methods
Cell culture
The human GC cell line MKN45 was obtained from the Chinese Academy of Sciences, and the AGS cell line was purchased from the American Type Culture Collection (ATCC). MKN45 and AGS cells were cultured in RPMI 1640 medium (Hyclone, Waltham, MA, USA) supplemented with 10% fetal bovine serum in a humidified incubator containing 5% CO2 at 37°C.
Analysis of TRPC6 expression in GC specimens
Tissue samples were collected from GC patients at the Department of General Surgery, Chinese PLA General Hospital. The written informed consent was obtained from all patients. This study was conducted in accordance with the Declaration of Helsinki. The use of the clinical specimens was approved by the Clinical Research Ethics Committee of Chinese PLA General Hospital. Tissues were obtained from each patient and blocked with 2.5% hydrogen peroxide in methanol. All the GC patients enrolled in our study were tested for H. pylori infection using a rapid urease test before the resection. Patients were regarded as positive for H. pylori infection if they were positive by the rapid urease test. Then, TRPC6 expression in gastric tissues was detected using the immunohistochemical staining. A polyclonal rabbit anti-TRPC6 antibody (Santa Cruz Biotechnology, CA, USA) was used for the staining. Then, the semi-quantitative analysis was performed. Briefly, images of all tissue samples were acquired with a constant set of imaging parameters on the microscope and imaging software. The images were then subjected to optical density (OD) analysis by Image-Pro Plus software. Adjustments to the background and color intensity range were performed. The intensity range selection was based on histograms, with the intensity (I) and saturation (S) set at maximum hue (H) was set at a range in which most of the brown DAB color was selected. These settings were saved and subsequently applied to all images analyzed. After defining the area of interesting, the mean OD of the selected area [integrated optical density/unit area] was determined by the software and represents the immunoreactivity of the candidate protein within tumor tissue. The acquired score of the OD was standard normalized and subjected to ImagePro Plus (Media Cybernetics, MD, USA) for further analysis. Then, patients were evaluated in terms of sex, age, tumor differentiation, depth of invasion, lymph node metastasis, TNM stage, distant metastasis, and survival.
H. pylori culture and infection in GC cells
The CagA- and Vac A- positive H. pylori strain NCTC11637 was obtained from ATCC, which was grown on Columbia agar (Oxoid, Basingstoke Hampshire, UK) plates containing 5% sheep blood and incubated at 37°C under microaerophilic conditions for 48–72 hrs. H. pylori was collected from the plates with PBS and then resuspended in RPMI 1640 medium without antibiotics. The densities of bacteria were measured by the OD at 660 nm [1 OD660=1×108 colony-forming units (CFU)/mL]. Cultured AGS and MKN45 cells were seeded on plates and grown to 80% confluency. Then, the cells were infected with the bacteria at a bacteria-to-cell ratio of 100:1 in the culture media.
Real-time quantitative PCR
Total RNA was extracted by TRIZOL Reagent Kit (Invitrogen, Carlsbad, CA, USA). RNA was reverse transcribed to cDNA using PrimeScript RT-polymerase (Takara, Japan). Quantitative real-time PCR was performed using SYBR Green Mix (Takara, Japan) and primers specific for TRPC6 and GAPDH. GAPDH was used as an internal control. All the experiments were performed in triplicate.
Western blotting
Cells were lysed with a lysis buffer, and protein concentration was measured using a BCA Protein Assay Kit (Beyotime, Beijing, China). Protein expression levels were detected using the Western blotting analysis. Primary antibodies: anti-TRPC6, anti-GAPDH were purchased from Cell Signal Technology (CST, USA). Then, secondary antibodies conjugated with horseradish peroxidase were used. The relative levels were quantified and normalized against GAPDH in the same sample.
Luciferase reporter vectors and Luciferase assays
TRPC6 promoter was amplified from human cDNA, and cloned into the KpnI and BglII sites of a PGL3-basic vector (Promega, USA). Similar plasmids containing mutations in the putative TCF/LEF-binding sites, named Mut, were constructed. AGS and MKN45 cells were co-transfected with the constructed luciferase vectors. Then, H. pylori was added to the cells. After 24 hrs, the cells were harvested in 1×Reporter Lysis Buffer (Catalog no.: E1960, Promega), and luciferase activities were measured using the Dual-Luciferase Reporter Assay System Kit (Promega, WI, USA). The luciferase activity for each lysate was normalized to the renilla luciferase activity.
Scratch wounding assay
GC cells were seeded into 24-well plates as a monolayer at 24 hrs. Cells were scraped with a 10 μL pipette tip. After scratching, the cells were washed to remove the detached cells. H. pylori was added to the cells. The extent of cell migration was observed, and images were captured at 0 and 24 hrs under a microscope. In each group, at least three parallel wells were utilized for testing.
Cell invasion assay
The invasive potential of GC cells treated by H. pylori was assessed using a transwell invasion assay. Matrigel Basement Membrane Matrix (BD Biosciences, San Diego, CA, USA) was added to each well and incubated at 37°C for 4 hrs to assess tumor cell invasion. Cells (approximately 1×104), in 200 μL of serum-free RPMI-1640 medium, were added into the upper compartment of the chamber. A total of 500 μL of RPMI-1640 medium with 10% fetal bovine serum were placed in the bottom compartment of the chamber. Then, H. pylori and the inhibitors were added to the upper chamber. After 24 hrs of incubation at 37°C with 5% CO2, the medium was removed from the upper chamber, and invading cells were fixed and stained with crystal violet. Then, the number of invasive cells were analyzed using a microscope (Olympus, Japan). Triplicate experiments were performed.
Measurement of [Ca2+]cyt by calcium imaging
GC cells were cultured on coverslips and loaded with Fura-2AM (Invitrogen, NY, USA) at 37°C for 45 mins and then washed with PBS for 30 mins. After loading, cells were transferred to a standard perfusion chamber on the stage of a TMD inverted microscope (Nikon, Japan). [Ca2+]i measurements were made at 340/380 nm excitation and 510 nm emission wavelengths from an SLM-Aminco spectrophotometer (Rochester, NY). F340/380 ratios were used to represent [Ca2+]cyt. For the Ca2+-free solution, the CaCl2 was replaced with NaCl. H. pylori and the correspond inhibitors or treatments were applied as described.
TOP/FOP flash reporter assay
A TOP/FOP flash assay was used for Wnt signaling pathway analysis. Briefly, the TCF/LEF-responsive luciferase construct was made under the control of minimal TK promoter and tandem repeats of the TCF/LEF transcriptional response element. GC cells were seeded in a 24-well plate and co-transfected with the pRL-TK plasmid (10 ng/well) and either TOP flash plasmid or FOP flash plasmid (200 ng/well), and then treated by H. pylori and inhibitors as indicated. At 48 hrs later, the cells were analyzed using the Dual-Luciferase Reporter Assay System (Promega, E1910). The ratio of TOP Flash activity and FOP Flash activity represented the results of the activity.
Statistical analysis
Data are expressed as means ± standard error (SE). A two-tailed Student’s t-test or Mann–Whitney test was used to test the difference. The Kaplan–Meier curves and log-rank test were applied to assess survival. The associations between the protein expression and clinicopathological parameters were analyzed with Pearson’s χ2 test. SPSS Statistics software (version 19.0, SPSS Inc., Chicago, IL, USA) was used for statistical analysis, and the graphs were generated using GraphPad Prism 6.0 (Graphpad Software Inc, CA, USA).
Results
H. pylori infection is correlated with the progression of GC. To investigate the correlation between H. pylori infection and the clinicopathological features of GC patients, H. pylori colonization was examined in 360 GC tissues. As presented in Table 1, H. pylori colonization was present in 44.2% (159/360) of GC tissues. Analysis of the potential correlation between H. pylori infection and the clinicopathological features indicated that H. pylori infection in GC was not associated with sex, age, or degree of differentiation, while it was associated to the depth of invasion, TNM stage and lymph node or distant metastasis (Table 1). These results suggest that H. pylori infection has an important role in GC metastasis and is correlated with an advanced stage of GC.
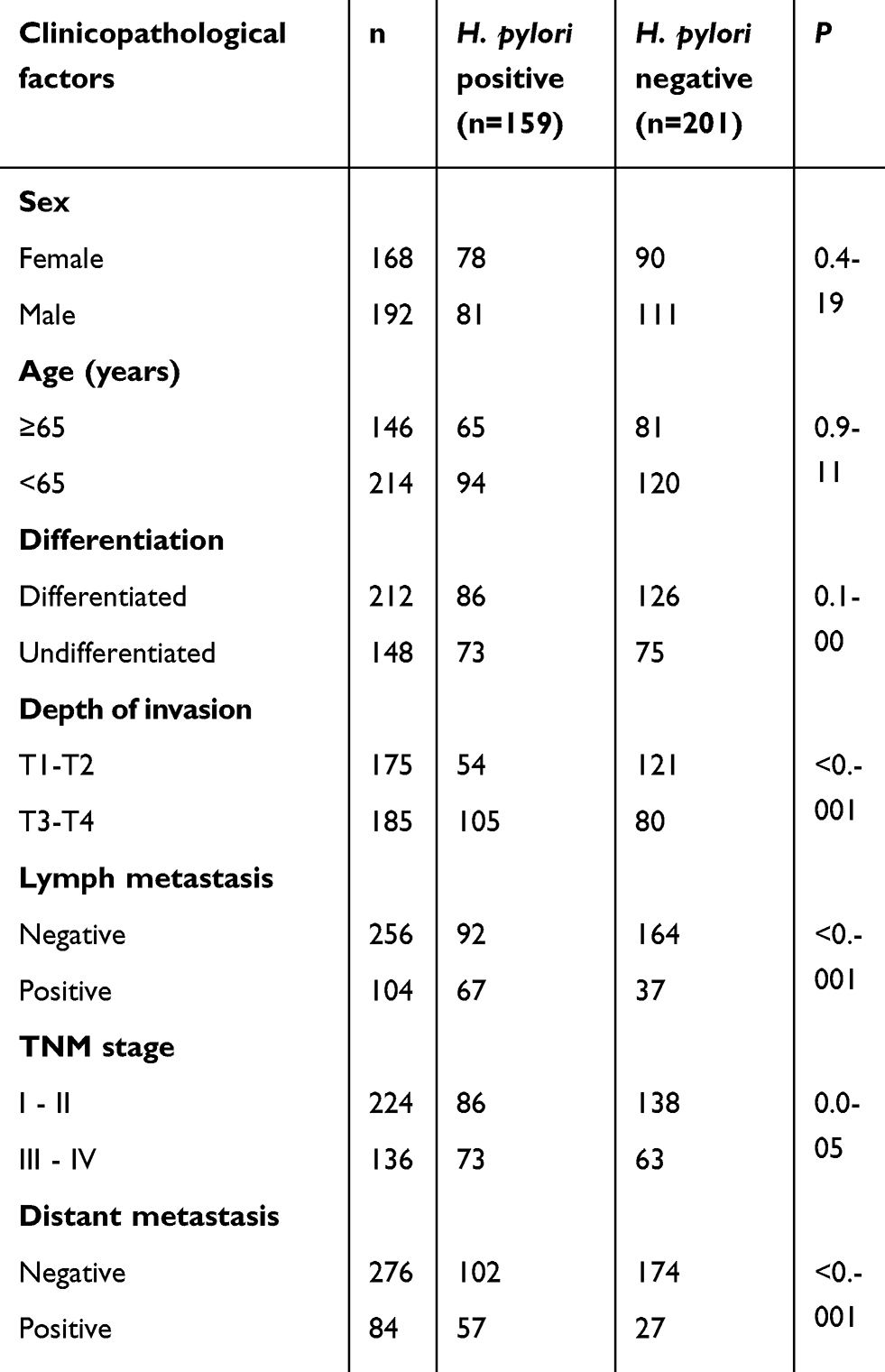 |
Table 1. Correlations of H. pylori infection and clinicopathological features of GC patients |
H. pylori enhances GC cell migration and invasion in a Ca2+-dependent manner. To assess the effect of H. pylori infection on the migratory and invasive capacities of GC cells, AGS and MKN45 cells were infected with H. pylori NCTC 11637. The results indicated that H. pylori infection increased the migration and invasion of GC cells (Figure 1A and B). Since Ca2+ signaling is important in tumor cell migration, it was further explored whether Ca2+ was involved in H. pylori-induced GC cell migration and invasion. The results suggested that pre-treatment of GC cells with the Ca2+ chelator BAPTA-AM significantly attenuated H. pylori-induced GC cell migration and invasion (Figure 1A and B). Taken together, it was demonstrated that H. pylori infection promotes the migration and invasion of GC cells in a Ca2+-dependent manner.
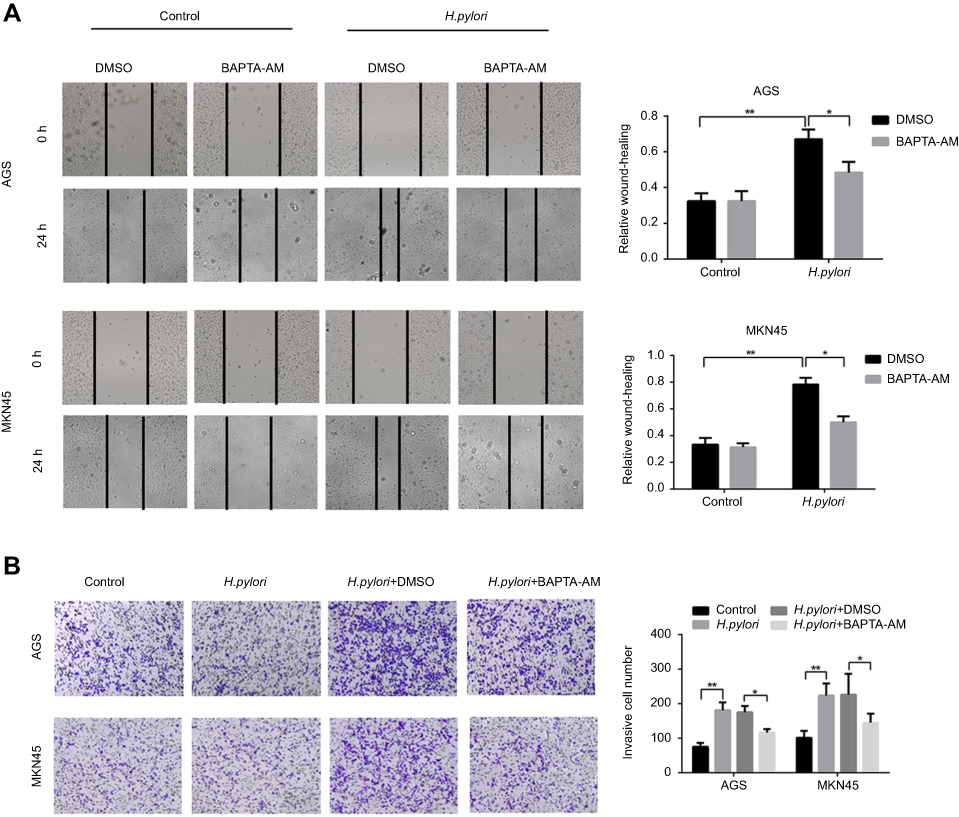 |
Figure 1 H. pylori enhances migration and invasion of gastric cancer cells in Ca2+-dependent manner. AGS and MKN45 cells were preincubated with calcium chelator, BAPTA-AM, and then incubated with H. pylori for 24 hrs. (A) Scratch wounding assay was performed to evaluate the migration of GC cells in response to H. pylori infection and BAPTA-AM treatment, respectively. (B) Representative images and micrographs of the transwell matrix penetration assay showing the H. pylori-induced invasiveness of GC cells pretreated with BAPATA-AM or DMSO. Data shown in the histogram are summary of three independent experiments; *P<0.05 and ** P<0.01. |
H. pylori induces intracellular Ca2+ signaling through intracellular Ca2+ release and Ca2+ influx. The present study further assessed whether H. pylori infection increases the intracellular Ca2+ concentration of GC cells. Various amounts of H. pylori were added to the cell culture and the intracellular Ca2+ concentration, [Ca2+]i, was measured. As presented in Figure 2A, H. pylori induced a dose-dependent increase in [Ca2+]i in GC cells, comprising a biphasic [Ca2+]i increase (“peak” phase), followed by a descent to a [Ca2+]i baseline (“plateau” phase).
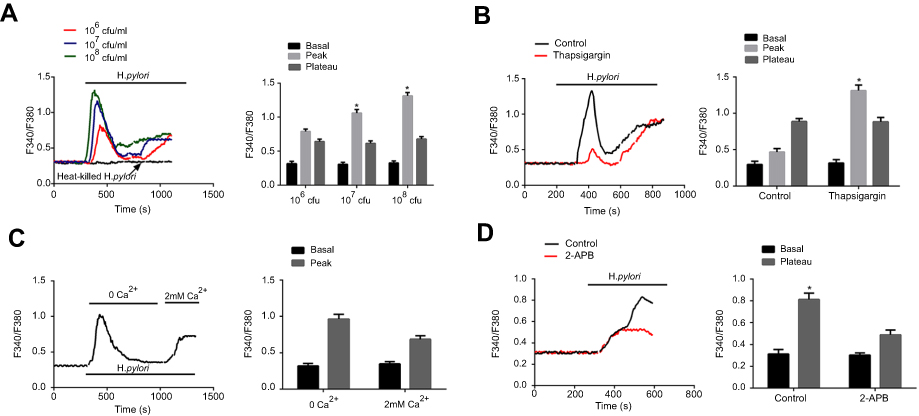 |
Figure 2 H. pylori induces intracellular Ca2+ increase through intracellular Ca2+ release and extracellular Ca2+ influx. (A) Representative tracings of intracellular Ca2+ concentration ([Ca2+]i) changes with varying concentrations [1×106–1×108 colony-forming units (CFU)/mL] of H. pylori on Fura 2-loaded gastric cancer cells. The addition of H. pylori produced a concentration-dependent biphasic change in [Ca2+]i, which was followed by a return to baseline [Ca2+]i levels. Histogram at the right shows the summary data for peak and plateau changes of Fura-2 fluorescence ratio. Heat killed H. pylori was used as a negative control. *P<0.05. (B) Effects of intracellular Ca2+ depletion on H. pylori-induced [Ca2+]i changes in MKN45 cells. Fura 2-loaded human gastric cancer cells were pretreated for 30 mins with 500 nM thapsigargin to release intracellular Ca2+. The H. pylori strains (1×108 CFU/mL) were then added, the peak and plateau changes were recorded. Histogram at the right shows the summary data for peak and plateau changes of Fura-2 fluorescence ratio. *P<0.05. (C) Effects of extracellular Ca2+-free Ringer on H. pylori-induced [Ca2+]i changes in gastric cancer cells. Fura 2-loaded cells were first exposed to extracellular Ca2+-free Ringer for 15 mins, H. pylori strains (1×108 CFU/mL) were added and [Ca2+]i was recorded. Then, 2 mM Ca2+ was readded to the solution and [Ca2+]i was recorded again. Right panel: Summary data for peak changes of Fura-2 fluorescence ratio. (D) Effects of 2-APB on H. pylori-induced [Ca2+]i changes gastric cancer cells. Fura 2-loaded human gastric cancer cells were pretreated for 30 mins with 500 nM thapsigargin for depletion of intracellular Ca2+. Then, time course of [Ca2+]i changes induced by H. pylori in the absence and presence of 100 nM 2-APB was recorded. Histogram at the right shows the summary data for basal and plateau changes of Fura-2 fluorescence ratio. *P<0.05. For all tracings, n=10 independent experiments. |
The Ca2+ source of the H. pylori-evoked increase in [Ca2+]i in GC cells was then determined. One of the major intracellular Ca2+ sources is the sarcoendoplasmic reticular Ca2+ store (SERCA). GC cells were pretreated with thapsigargin, a Ca2+-ATPase inhibitor in the SERCA, followed by addition of H. pylori and measurement of [Ca2+]i. The result indicated that thapsigargin significantly reduced the [Ca2+]i peak levels, while it had no effect on the [Ca2+]i plateau change (Figure 2B).
The role of calcium channels in the cell membrane in the changes in [Ca2+]i was then investigated. When H. pylori was first applied in Ca2+-free solutions and with pre-treatment using thapsigargin, a marked increase in [Ca2+]i was observed in GC cells, and re-addition of extracellular Ca2+ further induced an increase in [Ca2+]i (Figure 2C). Furthermore, the H. pylori-induced rise in [Ca2+]i was inhibited by 2-aminoethoxydiphenyl borate (2-APB, 100 µM; Figure 2D), a non-specific inhibitor of TRP channels. Taken together, these results suggest that the H. pylori-induced peak [Ca2+]i is caused by Ca2+ release from intracellular Ca2+ stores, while the H. pylori-induced plateau [Ca2+]i change is caused by Ca2+ influx.
H. pylori upregulates the TRPC6 channel and induces Ca2+ influx through TRPC6. To further identify the potential calcium channels involved in H. pylori-induced Ca2+ influx, various membrane Ca2+-permeable channels reported in GC cells were screened. It was indicated that SN-6, the Na+/Ca2+ exchanger 1 inhibitor, and RN1734 (10 µM), a selective TRPV4 inhibitor, did not alter the H. pylori-induced increase in [Ca2+]i (Figure 3A and B). However, SKF96365 (10 µM), a blocker for TRPC channels, attenuated the H. pylori-induced rise in [Ca2+]i in GC cells (Figure 3C). Overall, these results suggest that TRPC channels may be the target calcium channels mediating H. pylori-induced Ca2+ entry in GC cells. Since several TRPC channel subtypes are present in the cell membrane, it was attempted to identify the subtype of TRPC channel involved in H. pylori-induced Ca2+ influx in GC cells. GC cells were infected with the H. pylori strain, and the expression of different subtypes of TRPC channel in response to H. pylori treatment was examined. TRPC6 expression was significantly increased in H. pylori-infected GC cells (Figure 3D and E). Therefore, it was indicated that TRPC6 is involved in the H. pylori-induced Ca2+ influx. Furthermore, knockdown of TRPC6 significantly decreased the H. pylori-induced increase in [Ca2+]i in GC cells (Figure 3F). These results strongly indicate that H. pylori increases TRPC6 channel expression to induce Ca2+ entry in GC cells via this channel.
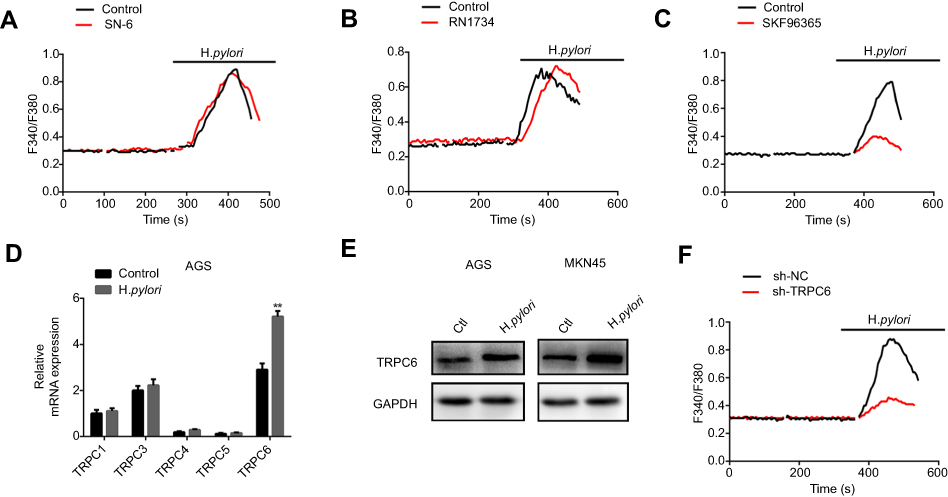 |
Figure 3 H. pylori increases TRPC6 expression and induces extracellular Ca2+ influx through TRPC6 channel. (A–C) Cells were pretreated with 500 nM thapsigargin for 30 mins for the depletion of intracellular Ca2+. Then, SN-6 (10 μM) (A), RN1734 (10 μM) (B), and SKF96365 (10 μM) (C) were added to detect H. pylori-induced [Ca2+]i changes in MKN45 cells. (D) The relative mRNA expression of subtypes of TRPC channels induced by H. pylori in AGS cells was measured by qPCR. (E) H. pylori-induced TRPC6 protein expression was evaluated by Western blot analysis. (F) Effect of TRPC6 knockdown on H. pylori-induced [Ca2+]i changes in MKN45 cells. For all tracings, n=10 independent experiments. |
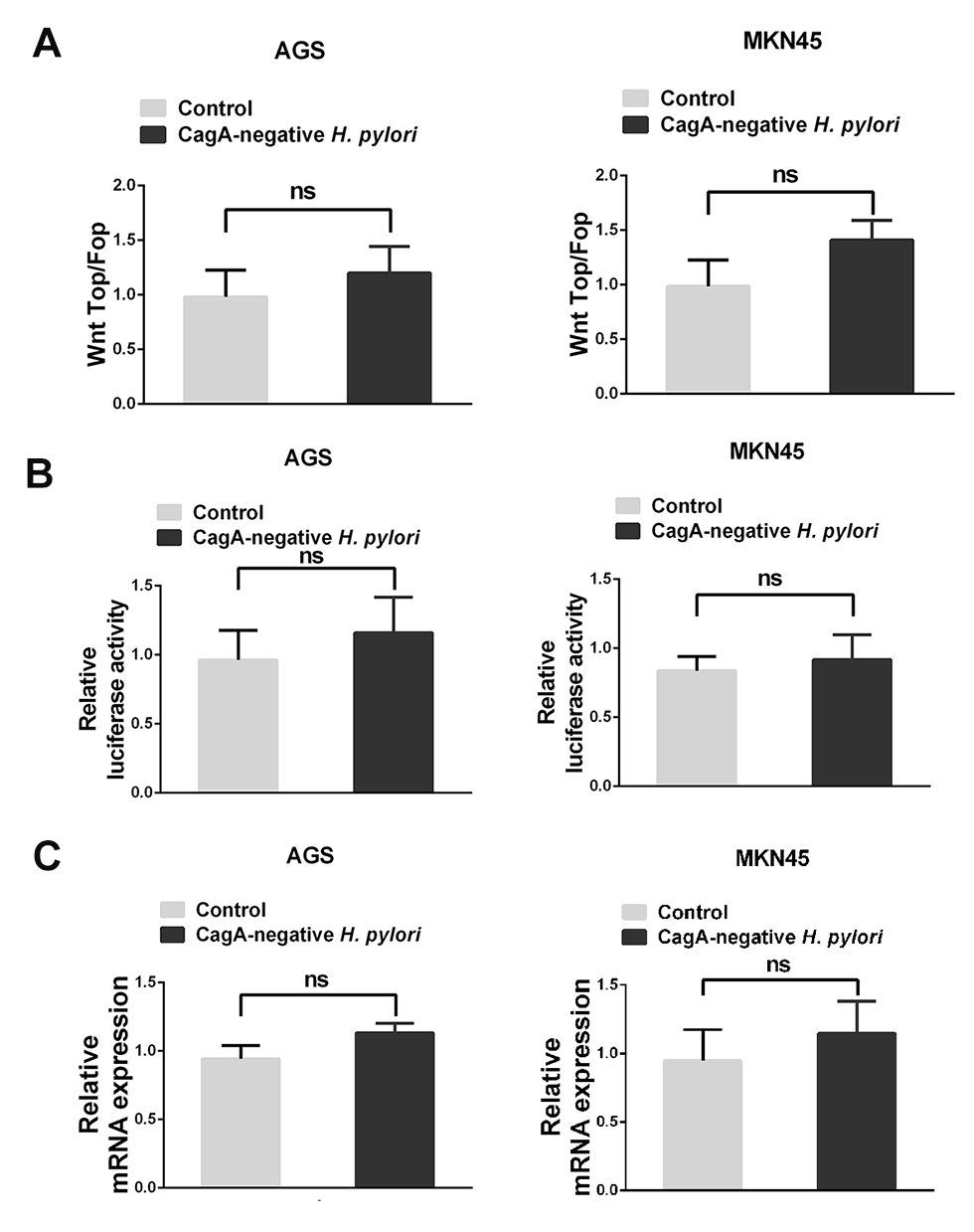
H. pylori upregulates TRPC6 expression via Wnt/β-catenin signaling. Next, it was investigated how H. pylori treatment causes an upregulation of TRPC6 expression in GC cells. It is known that H. pylori infection activates a series of signaling pathways, among which the Wnt/β-catenin pathway is involved in H. pylori-mediated tumor progression.20–22 Therefore, the present study aimed to investigate whether H. pylori promotes TRPC6 expression through the Wnt/β-catenin pathway. The results of the present study also suggested that CagA positive H. pylori enhanced the TOP/FOP activity in GC cells (Figure 4A and B), while CagA negative H. pylori could not enhance TOP/FOP activity (Figure S1A). GC cells were then pretreated with XAV, an inhibitor of Wnt/β-catenin signaling. The results indicated that XAV significantly attenuated CagA positive H. pylori-induced expression of TRPC6 in GC cells (Figure 4C and D).
{kind=link}
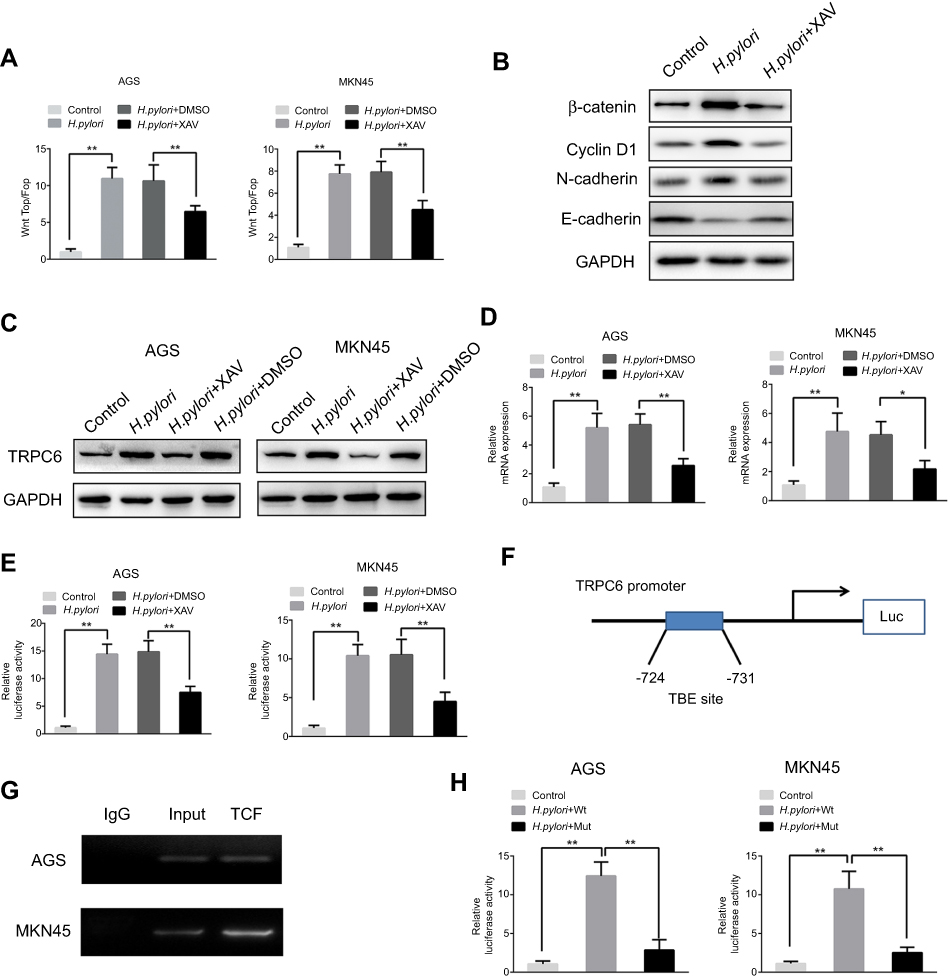 |
Figure 4 Wnt/β-catenin signaling is involved in H. pylori-induced transcriptional expression of TRPC6 in gastric cancer cells. (A) AGS and MKN45 cells were transfected with luciferase reporter constructs containing TCF/LEF-binding motifs (Top-Flash) or mutated TCF/LEF sites (Fop-Flash). Then, H. pylori was added to cells pretreated with XAV(10 μM) or DMSO for 2 hrs, luciferase activity was measured to assess β-catenin transcriptional activity. ** P<0.01. (B) Various markers of Wnt signaling were detected by Western blot in H. pylori treated cells with or without XAV. (C) AGS and MKN45 cells were pretreated with XAV or DMSO, and H. pylori was incubated with cells for 24 hrs. The TRPC6 protein expression was examined by Western blot analysis. (D) AGS and MKN45 cells were pretreated with XAV or DMSO, then H. pylori was incubated with cells for 24 hrs. The relative mRNA expression of TRPC6 was detected by qPCR analysis. ** P<0.01. (E) AGS and MKN45 cells were pre-transfected with luciferase reporter constructs containing the TRPC6 promoter for 4 hrs. Luciferase activity was measured to assess promoter activity in response to H. pylori infection in the absence or presence of XAV. ** P<0.01. (F) The potential TCF binding element (TBE) was identified within the TRPC6 promoter. (G) Chromatin immunoprecipitation (ChIP) assays were conducted in AGS and MKN45 cells using an antibody against TCF or a control IgG antibody. (H) The TBE binding site was mutated in the TRPC6 promoter, the mutated and wild type of TRPC6 promoter activity induced by H. pylori infection was assessed. ** P<0.01. |
Next, it was examined whether Wnt/β-catenin controls the expression of TRPC6 transcription. A luciferase reporter assay revealed that CagA positive H. pylori enhanced the promoter activity of TRPC6, whereas XAV attenuated the H. pylori-induced promoter activity (Figure 4E). Nevertheless, CagA negative H. pylori could not enhance TRPC6 promoter activity and expression (Figure S1B and C). Furthermore, the gene encoding TRPC6 was analyzed using the University of California Santa Cruz (UCSC) genome browser (https://genome.ucsc.edu). One putative transcription factor (TCF) binding site was identified within the TRPC6 promoter (Figure 4F). The chromatin immunoprecipitation assay showed than TCF could directly bind to the promoter of TRPC6 (Figure 4G). Luciferase reporter vectors including the wild type or mutant binding site were then constructed and it was demonstrated that after infection with H. pylori, the luciferase activity of the vector with the mutant binding site was decreased compared with that of the vector with the wild-type promoter (Figure 4H). These results clearly suggest that H. pylori increased the transcription of TRPC6 in GC cells through the Wnt/β-catenin pathway.
Wnt/β-catenin/TRPC6 signaling is responsible for H. pylori-induced GC progression. The role of TRPC6 in H. pylori-induced GC migration and invasion was further examined by performing TRPC6 knockdown, leading to a significant reduction in H. pylori-induced migration and invasion (Figure 5A and B). Taken together, the present results indicate that Wnt/β-catenin/TRPC6 signaling is involved in H. pylori-induced GC progression.
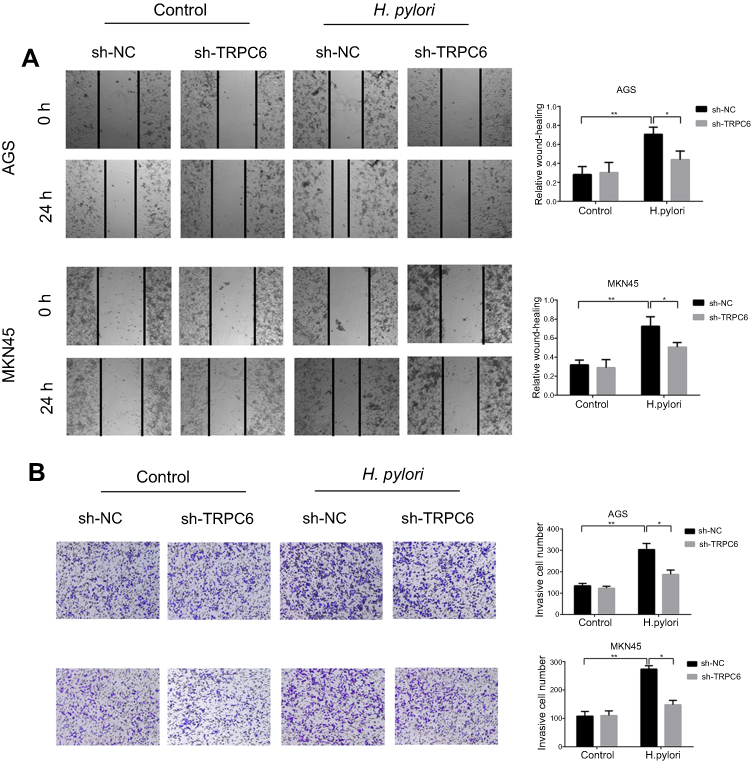 |
Figure 5 Wnt/β-catenin/TRPC6 signaling is involved in H. pylori-induced gastric cancer migration and invasion. (A and B) AGS and MKN45 cells were pre-transfected with sh-NC or sh-TRPC6, and then infected with H. pylori for 24 hrs. Scratch wounding assay (A) and transwell invasion assay (B) were performed to evaluate the migration and invasion of GC cells. *P<0.05; ** P<0.01. |
H. pylori infection increases TRPC6 expression in GC tissues and is associated with poor prognosis of GC patients. Since H. pylori infection caused an increase in TRPC6 expression in GC cells in vitro, TRPC6 expression was further examined in H. pylori-infected GC tissues. qPCR analysis of freshly resected GC tissues indicated that TRPC6 expression in H. pylori-positive GC was markedly higher than that in H. pylori-negative GC (Figure 6A). The expression of TRPC6 was then assessed in 360 GC tissues by using immunohistochemistry. The results suggested an obvious upregulation of TRPC6 expression in H. pylori-positive GC tissues compared with that in H. pylori-negative tissues (Figure 6B, Table S1). Furthermore, survival analysis demonstrated that H. pylori infection was highly associated with poor survival (Figure 6C). Taken together, the present results indicated that H. pylori infection was associated with increased TRPC6 expression and poor prognosis of GC patients.
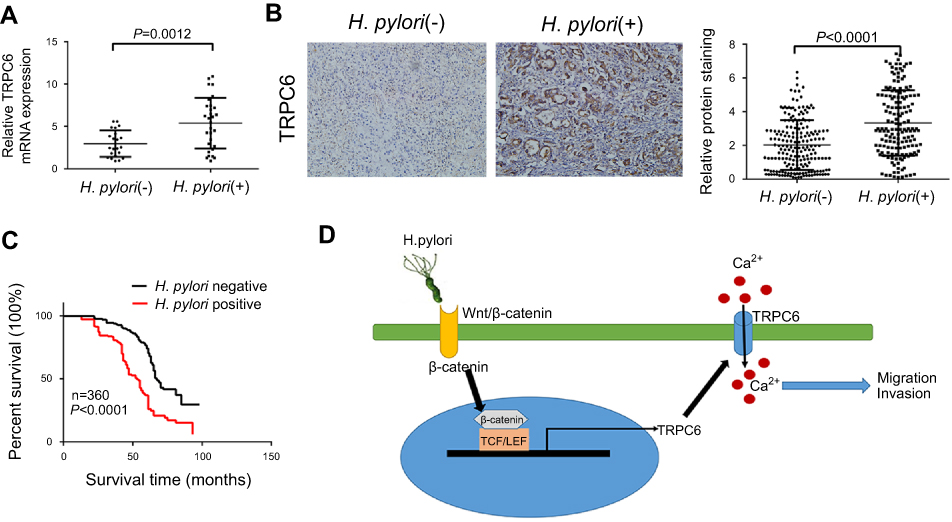 |
Figure 6 H. pylori infection was associated with increased expression of TRPC6 in GC tissues and poor prognosis of GC patients. (A) The relative mRNA expression of TRPC6 in gastric cancer tissues from H. pylori-positive (n=30) and H. pylori-negative (n=22) patients. (B) Representative immunohistochemical staining for TRPC6 in gastric cancer tissues from H. pylori-positive (n=159) and H. pylori-negative (n=201) patients. Right panel: TRPC6 expression levels were scored with semi-quantitative immunohistochemical analysis. (C) Kaplan–Meier curves comparing overall survival in GC patients with positive and negative infection of H. pylori (n=360; P<0.0001). (D) Schematic model of H. pylori-induced TRPC6/Ca2+ signaling through activation of the Wnt/β-catenin signaling pathway, which promotes migration and invasion of human gastric cancer cells. |
Discussion
H. pylori infection-associated chronic inflammation is an important factor for the development of GC.23,24 Several previous studies have also suggested that H. pylori infection is closely associated with GC progression.23–27 The present study indicated that H. pylori infection is associated with invasion and lymph node metastasis of GC in vivo, and it was demonstrated that H. pylori promoted GC cell migration and invasion in vitro. However, the molecular mechanisms of how H. pylori causes GC progression remain largely elusive. The present study revealed a novel mechanism, namely that H. pylori activates Wnt/β-catenin to transcriptionally upregulate TRPC6 expression and eventually induce Ca2+-dependent GC cell migration and invasion.
Calcium signaling is an important regulator for cell migration and invasion.13 In the present study, calcium signaling was demonstrated to be involved in H. pylori-induced GC cell migration and invasion. A previous study revealed that H. pylori promoted a dose-dependent [Ca2+]i increase in human gastric mucous epithelial cells.28 However, the mechanisms and exact dynamics of how H. pylori infection induces calcium mobilization in GC cells remain elusive. The present study indicated that H. pylori induced a biphasic [Ca2+]i increase (transient “peak”) and a [Ca2+]i baseline value (“plateau” phase). The SERCA Ca2+ store is an important intracellular Ca2+ store.29,30 The present study indicated that pre-treatment with the SERCA inhibitor thapsigargin did not affect the [Ca2+]i plateau change. Furthermore, in a Ca2+-free solution, H. pylori induced a [Ca2+]i peak change, while re-addition of extracellular Ca2+ produced a plateau change, suggesting that the H. pylori-induced [Ca2+]i peak change is based on intracellular Ca2+ release, whereas the plateau [Ca2+]i change is dependent on Ca2+ influx.
There are several major classes of plasma membrane Ca2+-permeable channels, which mediate Ca2+ influx in response to various activating stimuli.8 In the present study, a panel of membrane Ca2+-permeable channels was screened, which have been previously reported in GC cells, and it was revealed that only TRPC channel blocker SKF96365 attenuated the H. pylori-induced increase in [Ca2+]i in GC cells. Since the family of TRPC channels has seven members,31,32 the expression of each subtype in response to H. pylori infection was further evaluated by qPCR. The results suggested that H. pylori infection induced TRPC6 expression and that H. pylori may induce the increase in [Ca2+]i by activating the expression of TRPC6. The present study then attempted to investigate the possible mechanisms of how H. pylori induces TRPC6 expression.
Previous studies have indicated that H. pylori activates various intracellular pathways.33–36 The Wnt/β-catenin pathway is critical to tumorigenesis and cancer development,37,38 and is also responsible for H. pylori-induced gastric carcinogenesis.11,39,40 In the present study, it was demonstrated that H. pylori activated Wnt/β-catenin signaling and induced the transcription of TRPC6 in GC cells. A search with the UCSC genome browser indicated that the TRPC6 promoter had a TCF binding site, and mutation of this binding site significantly attenuated H. pylori-induced TRPC6 promoter activity. Furthermore, inhibition of Wnt/β-catenin or TRPC6 markedly inhibited H. pylori-induced migration and invasion of GC cells. In addition, the clinical analysis revealed that TRPC6 levels in GC samples from H. pylori-positive patients were higher compared with those from H. pylori-negative patients.
In conclusion, the present study indicated that H. pylori promotes GC migration and invasion through the Wnt/β-catenin/TRPC6/Ca2+ signaling pathway (Figure 6D). It provides a novel molecular target for developing effective strategies to prevent GC progression.
Acknowledgment
This work was supported by Hainan Provincial Natural Science Foundation of China (grant number 20158275).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Menaker RJ, Sharaf AA, Jones NL. Helicobacter pylori infection and gastric cancer: host, bug, environment, or all three? Curr Gastroenterol Rep. 2004;6(6):429–435.
2. An international association between Helicobacter pylori infection and gastric cancer. The EUROGAST study group. Lancet. 1993;341(8857):1359–1362.
3. Backert S, Neddermann M, Maubach G, Naumann M. Pathogenesis of Helicobacter pylori infection. Helicobacter. 2016;21(Suppl 1):19–25. doi:10.1111/hel.12335
4. Nejati S, Karkhah A, Darvish H, Validi M, Ebrahimpour S, Nouri HR. Influence of Helicobacter pylori virulence factors CagA and VacA on pathogenesis of gastrointestinal disorders. Microb Pathog. 2018;117:43–48. doi:10.1016/j.micpath.2018.02.016
5. Yang F, Xu Y, Liu C, et al. NF-κB/miR-223-3p/ARID1A axis is involved in Helicobacter pylori CagA-induced gastric carcinogenesis and progression. Cell Death Dis. 2018;9(1):12. doi:10.1038/s41419-018-1111-y
6. Yahiro K, Akazawa Y, Nakano M, et al. Helicobacter pylori VacA induces apoptosis by accumulation of connexin 43 in autophagic vesicles via a Rac1/ERK-dependent pathway. Cell Death Discov. 2015;1:15035. doi:10.1038/cddiscovery.2015.35
7. Zhang Y, Sun H, Li J, Rong Q, Ji X, Li B. The leukocyte-associated immunoglobulin (Ig)-like receptor-1 modulating cell apoptosis and inflammatory cytokines secretion in THP-1 cells after Helicobacter pylori infection. Microb Pathog. 2017;109:292–299. doi:10.1016/j.micpath.2017.06.012
8. Sierra JC, Asim M, Verriere TG, et al. Epidermal growth factor receptor inhibition downregulates Helicobacter pylori-induced epithelial inflammatory responses, DNA damage and gastric carcinogenesis. Gut. 2018;67(7):1247–1260. doi:10.1136/gutjnl-2016-312888
9. Wang F, Meng W, Wang B, Qiao L. Helicobacter pylori-induced gastric inflammation and gastric cancer. Cancer Lett. 2014;345(2):196–202. doi:10.1016/j.canlet.2013.08.016
10. Tan X, Tang H, Bi J, Li N, Jia Y. MicroRNA-222-3p associated with Helicobacter pylori targets HIPK2 to promote cell proliferation, invasion, and inhibits apoptosis in gastric cancer. J Cell Biochem. 2018;119(7):5153–5162. doi:10.1002/jcb.26542
11. Yong X, Tang B, Xiao YF, et al. Helicobacter pylori upregulates Nanog and Oct4 via Wnt/β-catenin signaling pathway to promote cancer stem cell-like properties in human gastric cancer. Cancer Lett. 2016;374(2):292–303. doi:10.1016/j.canlet.2016.02.032
12. Monteith GR, McAndrew D, Faddy HM, Roberts-Thomson SJ. Calcium and cancer: targeting Ca2+ transport. Nat Rev Cancer. 2007;7(7):519–530. doi:10.1038/nrc2171
13. Prevarskaya N, Skryma R, Shuba Y. Calcium in tumour metastasis: new roles for known actors. Nat Rev Cancer. 2011;11(8):609–618. doi:10.1038/nrc3105
14. Davis FM, Azimi I, Faville RA, et al. Induction of epithelial-mesenchymal transition (EMT) in breast cancer cells is calcium signal dependent. Oncogene. 2014;33(18):2307–2316. doi:10.1038/onc.2013.187
15. Yang S, Zhang JJ, Huang XY. Orai1 and STIM1 are critical for breast tumor cell migration and metastasis. Cancer Cell. 2009;15(2):124–134. doi:10.1016/j.ccr.2008.12.019
16. Goswamee P, Pounardjian T, Giovannucci DR. Arachidonic acid-induced Ca2+ entry and migration in a neuroendocrine cancer cell line. Cancer Cell Int. 2018;18:30. doi:10.1186/s12935-018-0529-8
17. Gambade A, Zreika S, Guéguinou M, et al. Activation of TRPV2 and BKCa channels by the LL-37 enantiomers stimulates calcium entry and migration of cancer cells. Oncotarget. 2016;7(17):23785–23800. doi:10.18632/oncotarget.8122
18. Tsai FC, Kuo GH, Chang SW, Tsai PJ. Ca2+ signaling in cytoskeletal reorganization, cell migration, and cancer metastasis. Biomed Res Int. 2015;2015:409245. doi:10.1155/2015/409245
19. Cai R, Ding X, Zhou K, et al. Blockade of TRPC6 channels induced G2/M phase arrest and suppressed growth in human gastric cancer cells. Int J Cancer. 2009;125(10):2281–2287. doi:10.1002/ijc.24551
20. Murata-Kamiya N, Kurashima Y, Teishikata Y, et al. Helicobacter pylori CagA interacts with E-cadherin and deregulates the beta-catenin signal that promotes intestinal transdifferentiation in gastric epithelial cells. Oncogene. 2007;26(32):4617–4626. doi:10.1038/sj.onc.1210251
21. Lee DG, Kim HS, Lee YS, et al. Helicobacter pylori CagA promotes Snail-mediated epithelial-mesenchymal transition by reducing GSK-3 activity. Nat Commun. 2014;5:4423. doi:10.1038/ncomms5972
22. Franco AT, Israel DA, Washington MK, et al. Activation of beta-catenin by carcinogenic Helicobacter pylori. Proc Natl Acad Sci USA. 2005;102(30):10646–10651. doi:10.1073/pnas.0504927102
23. Peek RM
24. Venerito M, Vasapolli R, Rokkas T, Delchier JC, Malfertheiner P. Helicobacter pylori, gastric cancer and other gastrointestinal malignancies. Helicobacter. 2007;22(Suppl):1.
25. Kong Y, Ma LQ, Bai PS, et al. Helicobacter pylori promotes invasion and metastasis of gastric cancer cells through activation of AP-1 and up-regulation of CACUL1. Int J Biochem Cell Biol. 2013;45(11):2666–2678. doi:10.1016/j.biocel.2013.08.015
26. Wen J, Wang Y, Gao C, et al. Helicobacter pylori infection promotes Aquaporin 3 expression via the ROS-HIF-1α-AQP3-ROS loop in stomach mucosa: a potential novel mechanism for cancer pathogenesis. Oncogene. 2018;37(26):3549–3561. doi:10.1038/s41388-018-0208-1
27. Luo C, Sun F, Zhu H, et al. Insulin-like growth factor binding protein-1 (IGFBP-1) upregulated by Helicobacter pylori and is associated with gastric cancer cells migration. Pathol Res Pract. 2017;213(9):1029–1036. doi:10.1016/j.prp.2017.08.009
28. Marlink KL, Bacon KD, Sheppard BC, et al. Effects of Helicobacter pylori on intracellular Ca2+ signaling in normal human gastric mucous epithelial cells. Am J Physiol Gastrointest Liver Physiol. 2003;285(1):G163–G176. doi:10.1152/ajpgi.00257.2002
29. Fernandez-Tenorio M, Niggli E. Stabilization of Ca2+ signaling in cardiac muscle by stimulation of SERCA. J Mol Cell Cardiol. 2018;119:87–95. doi:10.1016/j.yjmcc.2018.04.015
30. Smeazzetto S, Armanious GP, Moncelli MR, et al. Conformational memory in the association of the transmembrane protein phospholamban with the sarcoplasmic reticulum calcium pump SERCA. J Biol Chem. 2017;292(52):21330–21339. doi:10.1074/jbc.M117.794453
31. de Souza LB, Ambudkar IS. Trafficking mechanisms and regulation of TRPC channels. Cell Calcium. 2014;56(2):43–50. doi:10.1016/j.ceca.2014.05.001
32. Mulier M, Vriens J, Voets T. TRP channel pores and local calcium signals. Cell Calcium. 2017;66:19–24. doi:10.1016/j.ceca.2017.04.007
33. Allen LA, Allgood JA, Han X, Wittine LM. Phosphoinositide3-kinase regulates actin polymerization during delayed phagocytosis of Helicobacter pylori. J Leukoc Biol. 2005;78(1):220–230. doi:10.1189/jlb.0205091
34. Krueger S, Hundertmark T, Kalinski T, et al. Helicobacter pylori encoding the pathogenicity island activates matrix metalloproteinase 1 in gastric epithelial cells via JNK and ERK. J Biol Chem. 2006;281(5):2868–2875. doi:10.1074/jbc.M511053200
35. Kaebisch R, Mejías-Luque R, Prinz C, Gerhard M. Helicobacter pylori cytotoxin-associated gene A impairs human dendritic cell maturation and function through IL-10-mediated activation of STAT3. J Immunol. 2014;192(1):316–323. doi:10.4049/jimmunol.1302476
36. Buommino E, Donnarumma G, Manente L, et al. The Helicobacter pylori protein HspB interferes with Nrf2/Keap1 pathway altering the antioxidant response of Ags cells. Helicobacter. 2012;17(6):417–425. doi:10.1111/j.1523-5378.2012.00973.x
37. Logan CY, Nusse R. The Wnt signaling pathway in development and disease. Annu Rev Cell Dev Biol. 2004;20:781–810. doi:10.1146/annurev.cellbio.20.010403.113126
38. Sinnberg T, Levesque MP, Krochmann J, et al. Wnt-signaling enhances neural crest migration of melanoma cells and induces an invasive phenotype. Mol Cancer. 2018;17(1):59. doi:10.1186/s12943-018-0773-5
39. Arévalo-Romero H, Meza I, Vallejo-Flores G, Fuentes-Pananá EM. Helicobacter pylori CagA and IL-1β promote the epithelial-to-mesenchymal transition in a nontransformed epithelial cell model. Gastroenterol Res Pract. 2016;2016:4969163. doi:10.1155/2016/4969163
40. Liu N, Zhou N, Chai N, et al. Helicobacter pylori promotes angiogenesis depending on Wnt/beta-catenin-mediated vascular endothelial growth factor via the cyclooxygenase-2 pathway in gastric cancer. BMC Cancer. 2016;16:321. doi:10.1186/s12885-016-2351-9
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.