")
Back to Journals » Clinical and Experimental Gastroenterology » Volume 9
HCVerso1 and 2: faldaprevir with deleobuvir (BI 207127) and ribavirin for treatment-naïve patients with chronic hepatitis C virus genotype-1b infection
Authors Sarrazin C, Castelli F, Andreone P, Buti M, Colombo M, Pol S, Calinas F, Puoti M, Olveira A, Shiffman M, Stern JO, Kukolj G, Roehrle M, Aslanyan S, Deng Q, Vinisko R, Mensa FJ, Nelson DR
Received 22 April 2016
Accepted for publication 15 August 2016
Published 24 November 2016 Volume 2016:9 Pages 351—363
DOI https://doi.org/10.2147/CEG.S111116
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Andreas M. Kaiser
Christoph Sarrazin,1 Francesco Castelli,2 Pietro Andreone,3 Maria Buti,4 Massimo Colombo,5 Stanislas Pol,6 Filipe Calinas,7 Massimo Puoti,8 Antonio Olveira,9 Mitchell Shiffman,10 Jerry O Stern,11 George Kukolj,12 Michael Roehrle,13 Stella Aslanyan,11 Qiqi Deng,11 Richard Vinisko,11 Federico J Mensa,11 David R Nelson,14
on behalf of the HCVerso1 and 2 study groups
1Department of Internal Medicine 1, JW Goethe University Hospital, Frankfurt, Germany; 2Department of Infectious and Tropical Diseases, University of Brescia, Brescia, 3Department of Medical and Surgical Sciences, Università di Bologna and Azienda Ospedaliero-Universitaria, Policlinico Sant‘Orsola-Malpighi, Bologna, Italy; 4Department of Internal Medicine, Hospital Universitari Vall d’Hebron and CIBERehd del Instituto Carlos III, Barcelona, Spain; 5Division of Gastroenterology and Hepatology, Fondazione IRCCS Cà Granda Ospedale Maggiore Policlinico, Università degli Studi di Milano, Milan, Italy; 6University Paris Descartes, Department of Hepatology, Hospital Cochin, APHP and INSERM UMS-20, Institut Pasteur, Paris, France; 7Department of Gastroenterology, Centro Hospitalar de Lisboa Central, Lisbon, Portugal; 8Department of Infectious Diseases, AO Ospedale Niguarda Cà Granda, Milan, Italy; 9Liver Unit, Hospital Universitario La Paz, CIBERehd, Madrid, Spain; 10Liver Institute of Virginia, Bon Secours Health System, Richmond, VA, USA; 11Boehringer Ingelheim Pharmaceuticals, Inc., Ridgefield, CT, USA; 12Boehringer Ingelheim Ltd/Ltée, Burlington, ON, Canada; 13Boehringer Ingelheim Pharma GmbH & Co. KG, Biberach, Germany; 14Clinical and Translational Science Institute, University of Florida, Gainesville, FL, USA
Abstract: The interferon-free combination of once-daily faldaprevir 120 mg, twice-daily deleobuvir 600 mg, and weight-based ribavirin was evaluated in two Phase III studies (HCVerso1, HCVerso2) in hepatitis C virus genotype-1b-infected, treatment-naïve patients, including those ineligible for peginterferon (HCVerso2). Patients without cirrhosis were randomized to 16 weeks (Arm 1; n=208 HCVerso1, n=213 HCVerso2) or 24 weeks (Arm 2; n=211 in both studies) of faldaprevir + deleobuvir + ribavirin. Patients with compensated cirrhosis received open-label faldaprevir + deleobuvir + ribavirin for 24 weeks (Arm 3; n=51, n=72). Primary endpoints were comparisons of adjusted sustained virologic response (SVR) rates with historical rates: 71% (HCVerso1) and 68% (HCVerso2). Adjusted SVR12 rates were significantly greater than historical controls for Arms 1 and 2 in HCVerso2 (76%, 95% confidence interval [CI] 71–81, P=0.002; 81%, 95% CI 76–86, P<0.0001) and Arm 2 in HCVerso1 (81%, 95% CI 77–86, P<0.0001), but not for Arm 1 of HCVerso1 (72%, 95% CI 66–77, P=0.3989). Unadjusted SVR12 rates in Arms 1, 2, and 3 were 71.6%, 82.5%, and 72.5%, respectively, in HCVerso1 and 75.6%, 82.0%, and 73.6%, respectively, in HCVerso2. Virologic breakthrough and relapse occurred in 24-week arms in 8%–9% and 1% of patients, respectively, and in 16-week arms in 7%–8% and 9%–11% of patients, respectively. The most common adverse events were nausea (46%–61%) and vomiting (29%–35%). Adverse events resulted in discontinuation of all medications in 6%–8% of patients. In treatment-naïve patients with hepatitis C virus genotype-1b infection, with or without cirrhosis, faldaprevir + deleobuvir + ribavirin treatment for 24 weeks resulted in adjusted SVR12 rates significantly higher than historical controls. Both studies were registered in ClinicalTrials.gov (NCT01732796, NCT01728324).
Keywords: chronic hepatitis C, NS3 protease inhibitor, nonnucleoside polymerase inhibitor, cirrhosis, antiviral
Introduction
Chronic infection with hepatitis C virus (HCV) is a major cause of morbidity and mortality worldwide.1–3 Among the numerous genotypes of HCV, genotype-1 (GT-1) is the most prevalent in North America and Europe and has been difficult to treat with peginterferon-α (PegIFN) and ribavirin.2,4,5 There are two major subtypes of HCV GT-1, 1a and 1b, with 1b being the most prevalent subtype outside of North America.5 Although rates of sustained virologic response (SVR) improved markedly with the advent of the first HCV NS3/4A serine protease inhibitors telaprevir and boceprevir, these agents presented new challenges by adding to the already existing substantial toxicity of PegIFN-based treatment regimens.2,6–8 More recently, several new antiviral agents have provided options with simplified and shorter treatment duration for patients with HCV GT-1;9–13 however, at the time of the HCVerso studies, treatment options mainly still required the inclusion of PegIFN/ribavirin in the regimen.9,10 Patients infected with HCV frequently display contraindications to PegIFN treatment (eg, neuropsychiatric disorders, low white blood cell count or platelet count, autoimmune disease). Recognizing the substantial limitations of interferon (IFN)-based regimens, attention is increasingly focused toward combining direct-acting antiviral (DAA) agents in IFN-free treatment strategies.14
Faldaprevir is an HCV NS3/4A protease inhibitor with potent and similar in vitro activity against HCV GT-1a and -1b and a pharmacokinetic profile that supports once-daily dosing.15,16 Deleobuvir (BI 207127) is a nonnucleoside inhibitor of HCV NS5B RNA polymerase that binds reversibly to thumb-pocket I of NS5B, thereby achieving potent and specific antiviral activity.17,18 In vitro and in a Phase I study, deleobuvir was more active against GT-1b than against GT-1a.17,18 The IFN-free combination of once-daily faldaprevir plus either twice-daily or three-times daily deleobuvir, with or without ribavirin, has been investigated in Phase II clinical studies in treatment-naïve patients with chronic HCV GT-1 infection.19–21 In the SOUND-C2 study, 28 weeks of treatment with faldaprevir plus twice-daily deleobuvir and ribavirin was the most effective, driven by a favorable tolerability profile and an SVR 12-week posttreatment (SVR12) of 85% in patients with GT-1b infection.20 SVR12 rates were lower without ribavirin, indicating that ribavirin is an important component of the regimen. Response rates were also lower among patients with HCV GT-1a, particularly those with IL28B nonCC genotypes who experienced a high rate of on-treatment breakthrough. The SOUND-C3 study demonstrated that reduction of the treatment duration of faldaprevir plus twice-daily deleobuvir and ribavirin to 16 weeks was effective in treatment-naïve patients with HCV GT-1b infection, resulting in an SVR12 in 19/20 (95%);21 therefore, Phase III studies assessed 16- and 24-week treatment durations in GT-1b-infected patients only.
Here, we present the results of two Phase III studies (HCVerso1, NCT01732796; HCVerso2, NCT01728324) that aimed to confirm the efficacy and safety of 16 or 24 weeks of treatment with faldaprevir in combination with deleobuvir and ribavirin in treatment-naïve patients with chronic HCV GT-1b infection. Based on the results of these Phase III trials, the development of faldaprevir and deleobuvir has been terminated. Nevertheless, these studies, which include a large number of patients with compensated cirrhosis, provide valuable information about treating chronic hepatitis C with the combination of an NS3 inhibitor and a nonnucleoside NS5B inhibitor that may be relevant to the use of other DAA regimens.
Methods
Study design and patients
HCVerso1 and HCVerso2 were multicentered, randomized, placebo-controlled, partially double-blind Phase III studies. HCVerso1 was carried out at multiple sites in North America and Europe between December 2012 and April 2014, and HCVerso2 was carried out at multiple sites in North America, Australia, New Zealand, and Europe between November 2012 and March 2014. In both studies, eligible patients were aged 18–75 years with a diagnosis of chronic HCV GT-1b infection, as determined by positive antiHCV antibodies and HCV RNA ≥1,000 IU/mL at screening, in addition to at least one of the following: a liver biopsy signifying chronic HCV infection; a positive antiHCV antibody or HCV RNA test; or elevated alanine aminotransferase (ALT) levels at least 6 months prior to the screening visit. Patients with compensated liver cirrhosis were also included. Compensated liver cirrhosis was defined as Ishak Grade ≥5 or METAVIR ≥4 on a liver biopsy within 3 years, or liver stiffness of ≥13 kPa on a FibroScan within 6 months of randomization. All enrolled patients were naïve to IFN, PegIFN, ribavirin treatment, or any DAA agent for HCV infection. Exclusion criteria included mixed-genotype HCV, HCV GT-1a or GT-1 subtype undefined, nonHCV-related liver disease, hepatitis B virus infection, human immunodeficiency virus infection, and decompensated liver disease. In HCVerso1, historical studies used for comparison included only those patients who were eligible for PegIFN/ribavirin. Therefore, patients who would likely be considered ineligible for PegIFN and/or ribavirin were excluded. In HCVerso2, an extended population was included based on modified exclusion criteria allowing inclusion of patients who would have been ineligible for PegIFN; for example, patients with preexisting psychiatric conditions, uncontrolled abnormal thyroid function, active autoimmune-mediated disease known to be exacerbated by PegIFN, or diabetes mellitus (Table S1). PegIFN ineligibility was based on the investigators’ clinical judgment.
Study protocols were approved by the appropriate institutional review board. The study was undertaken in accordance with the Declaration of Helsinki and the International Conference on Harmonization guidelines. Written informed consent was provided by all patients before participation in the study.
Randomization and treatments
In both studies, patients without cirrhosis were randomly assigned in a 1:1 ratio, using an interactive response tool, to treatment Arms 1 or 2. Patients with compensated cirrhosis were directly assigned to Arm 3. Randomization was stratified by PegIFN eligibility in HCVerso2. Patients without cirrhosis in Arms 1 and 2 received oral treatment with faldaprevir 120 mg once daily, deleobuvir 600 mg twice daily, and standard weight-based doses of ribavirin for 16 and 24 weeks, respectively (Figure S1). Blinding of investigators and patients to treatment duration was achieved through the administration of placebo: in HCVerso1, patients in Arm 1 received 16 weeks of active treatment followed by 8 weeks of placebo treatment; in HCVerso2, patients in Arm 1 received an 8-week lead-in with placebo prior to receiving 16 weeks of active treatment. Patients and investigators were unblinded following database lock after the last patient completed assessment of SVR12. In Arm 3, patients with compensated cirrhosis received open-label faldaprevir, deleobuvir, and ribavirin (dosed as in Arms 1 and 2) for 24 weeks. All patients were given a single loading dose of faldaprevir 240 mg on Day 1, followed by 120 mg per day from Day 2 onward. Study treatments were administered with food (a requirement of ribavirin). Dose reductions or temporary discontinuation (with reintroduction at the lowest dose as soon as possible) was permitted only for ribavirin. Concomitant use of immunomodulators, systemic antiviral agents (except for the treatment of herpes simplex infection or influenza A), silibinin (or other extracts from the milk thistle), or medications that could cause photosensitivity reactions was not permitted. Patients were advised to protect any uncovered skin area (including hands, face, and lips) from sun or ultraviolet (UV) light exposure with a daily sunblock cream and a lip balm with sun protection factor ≥50 providing both UVA and UVB protection during the treatment period. Patients were to be followed for up to 96 weeks after the end of treatment (EOT); however, after the termination of deleobuvir development, the extended follow-up period was shortened to 24 weeks after the EOT for responders (with SVR12) and to 48 weeks after the EOT for non-responders, provided they had not started an alternative HCV treatment.
All study treatments were discontinued in the event of a lack of virologic response, defined as virologic breakthrough (an increase in HCV RNA of ≥1 log10 from quantifiable nadir or HCV RNA ≥25 IU/mL following an earlier decrease to <25 IU/mL, confirmed by a second consecutive measurement of ≥25 IU/mL within a 2-week duration) or failure to achieve HCV RNA <25 IU/mL and HCV RNA ≥25 IU/mL at any time after Week 8. Patients with a lack of virologic response could switch to treatment with faldaprevir (12 or 24 weeks) in combination with PegIFN and ribavirin (24 or 48 weeks), with treatment duration determined by response-guided therapy.
Endpoints
The primary objective of the studies was to compare adjusted SVR12 rates for 16- and 24-week regimens, including patients with cirrhosis, with historical SVR rates of 71% for PegIFN-eligible patients and 50% for PegIFN-ineligible patients. The secondary objective was to evaluate whether there is a clinically meaningful difference in efficacy between 16- and 24-week treatment durations. Plasma HCV RNA levels were measured using the quantitative COBAS® TaqMan® HCV/HPS assay (version 2; Roche Molecular Diagnostics, Pleasanton, CA, USA), with a limit of detection between 10 and 20 IU/mL and a linear range of 25–3.91×108 IU/mL. HCV RNA levels are reported as <25 IU/mL (detected or undetected), <25 IU/mL (undetected), or <25 IU/mL (detected).
The primary endpoint in both studies was SVR12, which was defined as HCV RNA <25 IU/mL (detected or undetected) at 12 weeks after the end of active treatment. Key secondary endpoints were SVR at 4 and 24 weeks after completion of active treatment (SVR4 and SVR24, respectively). Further efficacy endpoints included: HCV RNA<25 IU/mL undetected at Week 4; HCV RNA <25 IU/mL detected or undetected at Week 4; HCV RNA <25 IU/mL undetected at Week 12; EOT response (HCV RNA <25 IU/mL undetected at the end of planned treatment); and virologic breakthrough and relapse posttreatment (HCV RNA >25 IU/mL in patients who achieved EOT response).
Safety assessment included the incidence of adverse events (AEs) and changes in laboratory parameters during the course of treatment, which were graded as mild (Grade 1), moderate (Grade 2), severe (Grade 3), or potentially life-threatening (Grade 4) using the Division of AIDS Table for Grading the Severity of Adult and Pediatric Adverse Events.22 Photosensitivity was graded as mild, moderate, or severe according to a dermatologic management plan.
Resistance testing
Virology plasma samples from all visits at which HCV RNA was also measured were collected and stored for monitoring of drug resistance. Population-based sequence analysis of the HCV NS3/4A protease (codons 1–685) and NS5B polymerase (codons 1–592) was performed for baseline plasma samples from all patients and for postbaseline samples, including posttreatment follow-up from patients who did not achieve SVR if HCV RNA rebound was ≥1,000 IU/mL(limit for HCV gene amplification). Treatment-emergent amino acid changes were identified relative to patients’ respective baseline sequence.
Statistical analysis
The primary efficacy and safety analyses were undertaken for all randomized patients who received at least one dose of study medication (intention-to-treat population).
Historical control rates were based on SVR data for GT-1b patients included in Phase III trials of telaprevir and boceprevir: the lowest SVR rate seen in these studies was 71% with boceprevir plus PegIFN/ribavirin response-guided therapy in treatment-naïve patients.23–25 As the historical trials included patients with cirrhosis, whereas the HCVerso trials separated patients with cirrhosis into a nonrandomized treatment arm, for the purposes of the comparison, SVR12 rates in both studies were adjusted using a weighted average (89% times the response rate in noncirrhotic + 11% times the response rate in cirrhotic), where 11% is the highest rate of inclusion of patients with cirrhosis in earlier trials of DAAs + PegIFN/ribavirin.23 For this adjustment, the SVR12 rate from Arm 3 (24 weeks in patients with cirrhosis) was used for both the 16- and 24-week treatment arms. For HCVerso2, the historical response rate was adjusted to take into account the inclusion of PegIFN-ineligible patients. For these patients, a response rate of 50% was used based on several assumptions (see Supplementary materials, including Table S2, for details). The overall historical control rate was calculated as 68%: 86% times the control response rate in PegIFN-eligible patients (71%) + 14% times the control response rate in PegIFN-ineligible patients (50%), where 14% is the proportion of PegIFN-ineligible patients in the study. Statistical comparisons were made using a one-sided z-test with alpha = 0.025. For the comparison of 16- and 24-week treatment in patients without cirrhosis (secondary objective), in HCVerso 2, the Greenland formula with continuity correction was applied in pairwise comparisons of the SVR12 rate (including 95% confidence interval [CI]), with adjustment for stratification. In HCVerso1, a two-sided independent t-test with continuity correction was used. The influence of baseline factors on SVR12 rate was assessed in a subgroup analysis. Logistic regression analysis was also carried out for both studies to explore the potential influence of baseline factors or covariates on response.
For HCVerso1, a sample size of 390 noncirrhotic patients was determined to provide 90% power in order to detect a significant difference in SVR12 compared with historical controls (one-sample z-test with a one-sided alpha of 0.025), assuming an SVR12 rate of 81%, and to provide 80% power in order to detect a significant difference between 16- and 24-week treatment durations, assuming an SVR12 rate of approximately 82.5% in Arm 2 and a clinically relevant difference of 12%. For HCVerso2, a sample size of 420 noncirrhotic patients was determined to provide 83% power in order to detect a significant difference in SVR12 compared with historical controls (one-sample z-test with a one-sided alpha of 0.025), assuming an SVR12 rate of 80% for PegIFN-eligible patients and 60% for PegIFN-ineligible patients, and to provide 80% power in order to detect a significant difference between 16- and 24-week treatment durations, assuming an SVR12 rate of approximately 80% in Arm 2 and a clinically relevant difference of 12%. Sample size for cirrhotic patients was defined as between 50 and 70 patients, based on potential enrollment rate.
Results
Patients
In HCVerso1, 470 patients were randomized into Arms 1 and 2 (noncirrhotic) or allocated to Arm 3 (compensated cirrhosis) and received at least one dose of study medication (Figure 1A). In HCVerso2, 496 patients were randomized into Arms 1 and 2 (noncirrhotic) or allocated to Arm 3 (compensated cirrhosis) and received at least one dose of study medication (Figure 1B). Discontinuation of faldaprevir, deleobuvir, or ribavirin during the active treatment period occurred in 81 (17%), 82 (17%), and 84 (18%) patients, respectively, in HCVerso1, and in 86 (17%), 86 (17%), and 89 (18%) patients, respectively, in HCVerso2. The most frequent reasons for discontinuation of active treatment were AEs and lack of efficacy (Figure 1). In Arm 1, treatment discontinuation during the placebo treatment period occurred in 20 (9.6%) patients in HCVerso1 (placebo given after active treatment), mostly for lack of efficacy, and in two (1%) patients in HCVerso2 (placebo given prior to active treatment), one for an AE and the other one following a protocol violation (Figure 1). Baseline demographics and disease characteristics were generally balanced across the randomized treatment groups in both studies (Table 1). In both studies, among patients with cirrhosis (Arm 3), the percentage of males and the mean age were higher than in Arms 1 and 2. In HCVerso2, most patients were eligible to receive PegIFN; overall, 13% (65/496) were judged to be PegIFN ineligible. The most common reasons for PegIFN ineligibility were psychiatric disease (51% [33/65]) and autoimmune conditions (25% [16/65]).
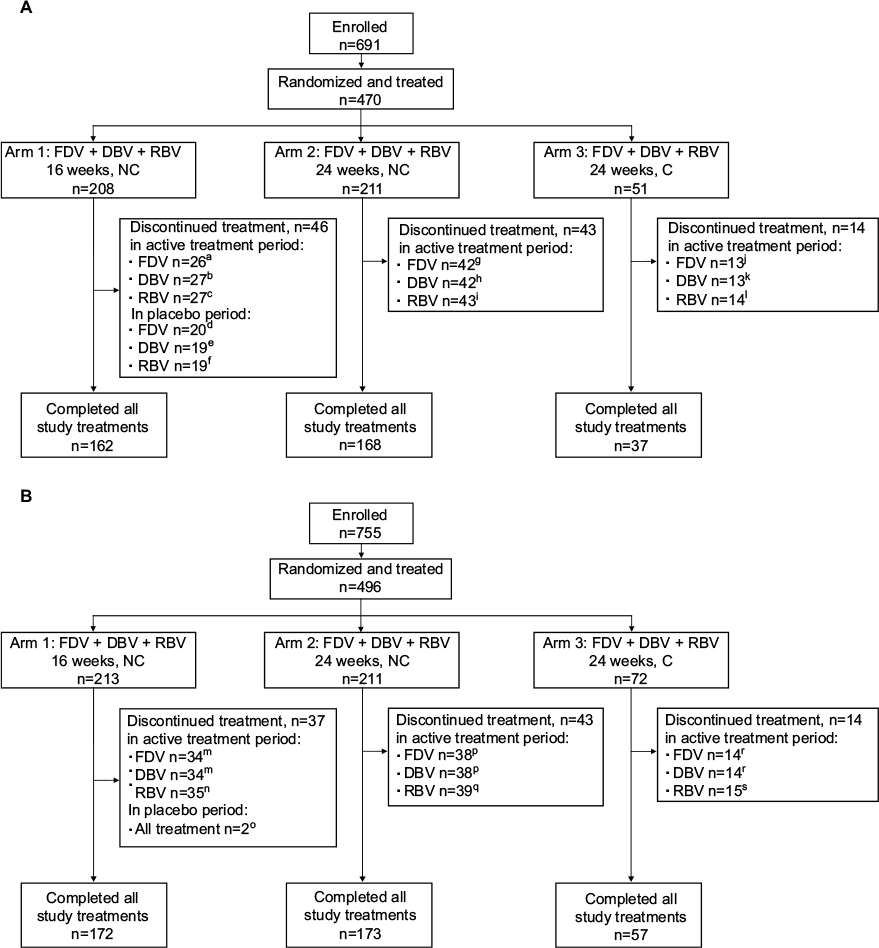 | Figure 1 Patient disposition in HCVerso1 (A) and HCVerso2 (B) Notes: aAdverse event 11, lack of efficacy 7, lost to follow-up 2, withdrawal 4, other 2. bAdverse event 12, lack of efficacy 8, lost to follow-up 2, withdrawal 4, other 1. cAdverse event 12, lack of efficacy 7, lost to follow-up 2, withdrawal 4, other 2. dAdverse event 5, lack of efficacy 13, withdrawal 1, other 1. eAdverse event 4, lack of efficacy 13, withdrawal 2. fAdverse event 4, lack of efficacy 13, withdrawal 1, other 1. gAdverse event 16, lack of efficacy 17, withdrawal 8, other 1. hAdverse event 16, lack of efficacy 18, withdrawal 8. iAdverse event 17, lack of efficacy 17, withdrawal 8, other 1. jAdverse event 4, lack of efficacy 4, withdrawal 2, other 3. kAdverse event 4, lack of efficacy 5, withdrawal 2, other 2. lAdverse event 5, lack of efficacy 4, withdrawal 2, other 3. mAdverse event 13, lack of efficacy 12, withdrawal 6, other 3. nAdverse event 16, lack of efficacy 11, withdrawal 5, other 3. oAdverse event 1, protocol violation 1. pAdverse event 14, lack of efficacy 14, withdrawal 9, other 1. qAdverse event 15, lack of efficacy 14, withdrawal 9, other 1. rAdverse event 4, lack of efficacy 8, withdrawal 2. sAdverse event 5, lack of efficacy 7, withdrawal 2, other 1. Abbreviations: C, compensated cirrhosis; DBV, deleobuvir; DC, discontinuation; FDV, faldaprevir; NC, no cirrhosis; RBV, ribavirin. |
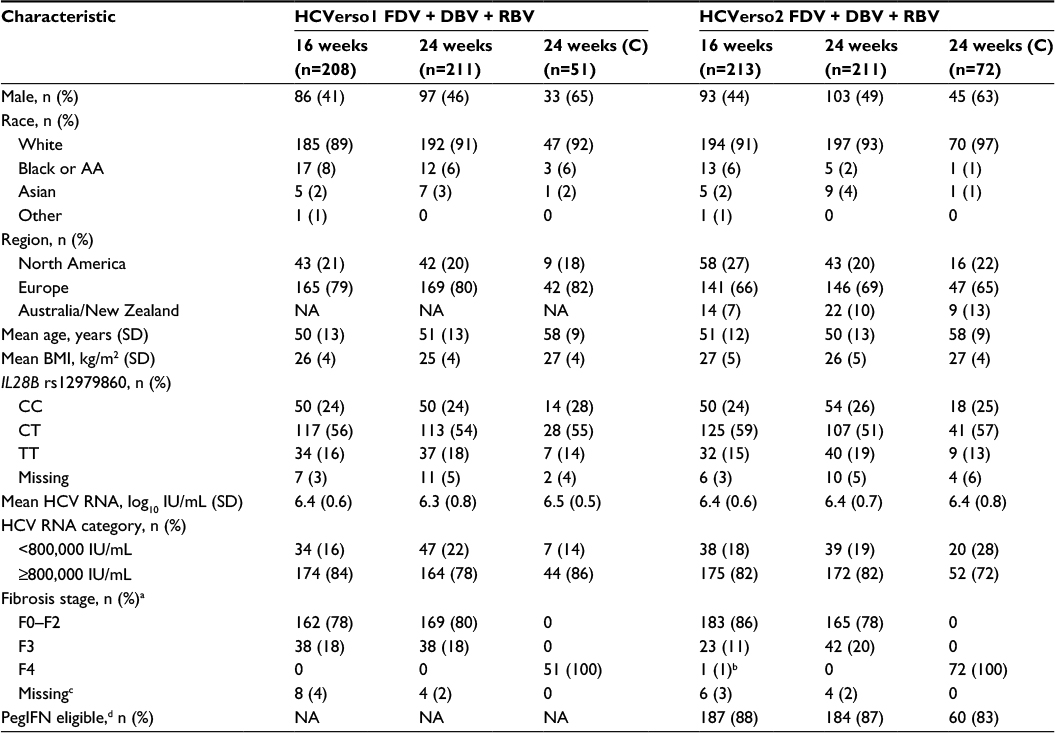 | Table 1 Baseline demographics and clinical characteristics Notes: aFibroScan results were used to determine stage of fibrosis for patients without a liver biopsy (<9.5 kPa = F0–F2; ≥9.5–<13 kPa = F3; ≥13 kPa = F4). bAssignment to Arm 3, cirrhosis, was based on investigator’s assessment for the presence of cirrhosis. This patient had no clinical evidence of cirrhosis per investigator; however, screening FibroScan was ≥13kPa. cIn patients with missing fibrosis data, cirrhosis was excluded, but further details on fibrosis staging were not available. dPegIFN eligibility was based on the investigators’ clinical judgment and determined at the randomization visit. Abbreviations: AA, African-American; BMI, body mass index; C, compensated cirrhosis; DBV, deleobuvir; FDV, faldaprevir; HCV, hepatitis C virus; NA, not applicable; PegIFN, pegylated interferon-α; RBV, ribavirin; SD, standard deviation. |
Virologic response
Results for the primary endpoint of SVR12 are shown in Figure 2. Compared with the historical control, the adjusted SVR12 rates after treatment with faldaprevir + deleobuvir + ribavirin were superior for the 24-week arm of HCVerso1 and for both the 16- and 24-week arms of HCVerso2 (Figure 2A and C). In HCVerso1, 24 weeks of treatment resulted in a higher SVR12 rate than 16 weeks of treatment among patients without cirrhosis (Figure 2B). In HCVerso2, there was no significant difference in SVR12 rates between the 16- and 24-week treatment arms (Figure 2D). Among patients with compensated cirrhosis, the SVR12 rate was 72.5% in HCVerso1 and 73.6% in HCVerso2. Further virologic endpoints are summarized in Table 2. In both studies, the Week 4 responses were similar in the 16- and 24-week non-cirrhotic treatment groups. In the 16-week groups, SVR12 was higher among patients with HCV RNA undetected at Week 4 than among those with HCV RNA <25 IU/mL detected at Week 4. Reasons for not achieving SVR12 are summarized in Table 3. The frequency of on-treatment virologic failure (mostly occurring before 16 weeks) was similar in the 16- and 24-week noncirrhotic treatment groups (7%–9%) and slightly higher among patients with cirrhosis (13%–14%). Premature discontinuation for reasons other than lack of efficacy (mostly for AEs or “withdrawal”) (Figure 1) accounted for a lack of SVR12 in between 5% and 10% of patients across treatment groups. Relapse (among patients who completed planned treatment and had undetectable HCV RNA at the EOT) was the most common reason for lack of SVR12 among patients in the 16-week treatment groups. The rate of relapse was numerically lower in the 24-week (noncirrhotic) treatment groups than in the 16-week treatment groups.
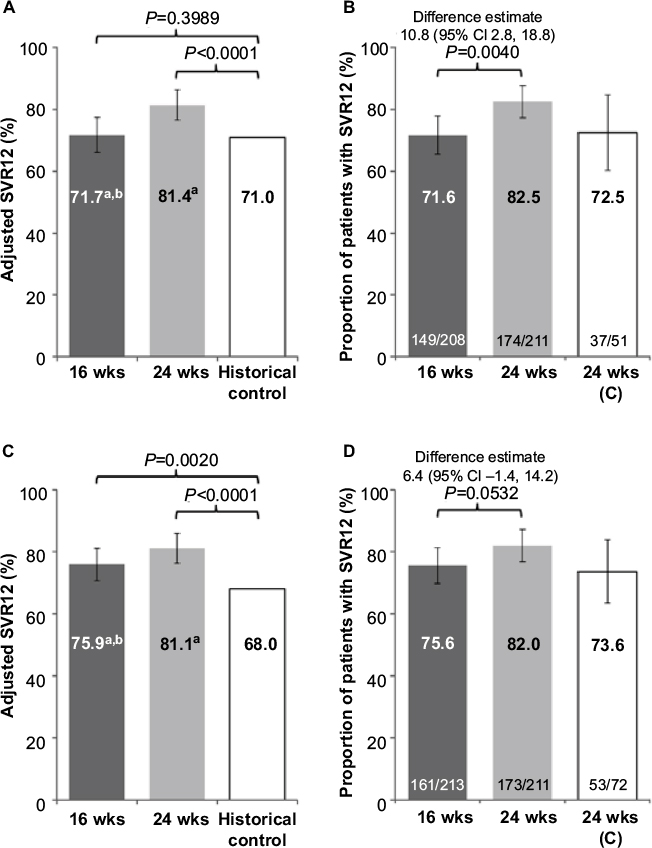 | Figure 2 Adjusted SVR12 versus historical control for HCVerso1 (A) and HCVerso2 (C), and SVR12 by treatment group for HCVerso1 (B) and HCVerso2 (D) Notes: aAdjusted for inclusion of patients with cirrhosis (11% in historical trials of approved DAAs and PegIFN). bCombination of the rates for 16 weeks in patients without cirrhosis and 24 weeks in patients with cirrhosis. Error bars indicate 95% CI values. Abbreviations: C, compensated cirrhosis; CI, confidence interval; SVR12, sustained virologic response at 12-week posttreatment; wks, weeks. |
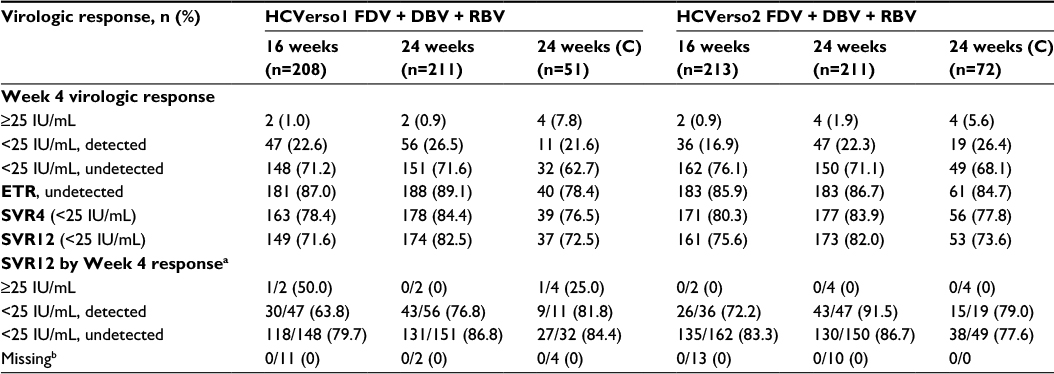 | Table 2 Virologic response rates (intention-to-treat population) Notes: an/N (%), where N = number with indicated Week 4 virologic response; n = number with SVR12. bNone had virologic breakthrough. Abbreviations: C, compensated cirrhosis; DBV, deleobuvir; ETR, end of treatment response (HCV RNA <25 IU/mL, target not detected at the end of planned treatment); FDV, faldaprevir; HCV, hepatitis C virus; RBV, ribavirin; SVR4/12, virologic response at 4 or 12 weeks after the end of treatment. |
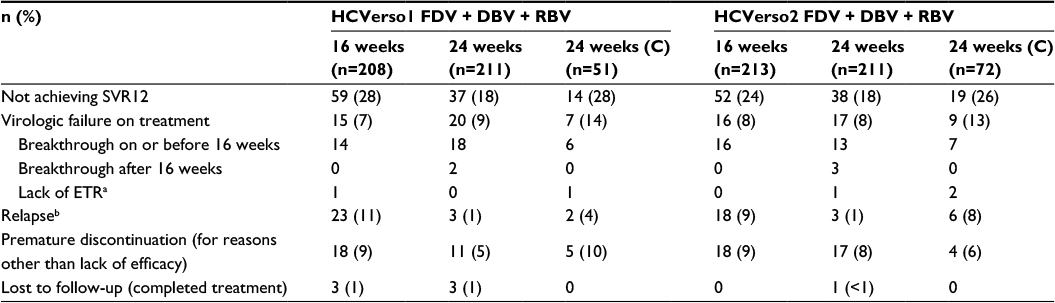 | Table 3 Reasons for not achieving SVR12 Notes: aPatients without virologic breakthrough who had HCV RNA detectable at the EOT. bRelapse rates, based on the number of patients who completed planned treatment and had HCV RNA undetectable at EOT, were: HCVerso1 13% (23/175), 2% (3/167), and 5% (2/37) in the 16-, 24-, and 24-week cirrhotic groups, respectively; HCVerso2 10% (18/174), 2% (3/169), and 11% (6/56) in the 16-, 24-, and 24-week cirrhotic groups, respectively. Abbreviations: C, compensated cirrhosis; DBV, deleobuvir; EOT, end of treatment; ETR, end of treatment response (HCV RNA <25 IU/mL, target not detected at the end of planned treatment); FDV, faldaprevir; HCV, hepatitis C virus; RBV, ribavirin; SVR12, sustained virologic response at 12-week posttreatment. |
In HCVerso1, among patients without cirrhosis, the rates of SVR12 were higher for 24 weeks than 16 weeks of treatment across several baseline subgroups, including patients with nonCC IL28B genotype, patients with baseline viral load ≥800,000 IU/mL, and patients 40–65 years of age (Figure 3). Differences were less pronounced in HCVerso2, where only patients with baseline viral load ≥800,000 IU/mL showed a clear difference in SVR12 rates between 16- and 24-week treatment arms. Multivariate analysis identified the associations between SVR12 and different baseline factors in the two studies (Table S3). In HCVerso1, there was a significant association between SVR12 and duration of treatment (P=0.0226, odds ratio [OR] 24-week noncirrhotic vs 16-week noncirrhotic 2.079, 95% CI 1.230–3.513; OR 24-week cirrhotic vs 16-week noncirrhotic 1.195, 95% CI 0.552–2.586), IL28B rs12979860 (CC vs nonCC genotype, P=0.0051, OR 2.538, 95% CI 1.322–4.873), sex (male vs female, P=0.0089, OR 0.517, 95% CI 0.315–0.847), and region (North America vs Europe, P=0.0029, OR 0.435, 95% CI 0.252–0.753). In HCVerso2, factors found to be significantly associated with SVR12 were IL28B rs12979860 (CC vs nonCC genotype P=0.0108, OR 2.239, 95% CI 1.205–4.163), baseline HCV RNA (≥800,000 vs <800,000 IU/mL, P=0.0195, OR 0.436, 95% CI 0.217–0.875), and sex (male vs female, P=0.0071, OR 0.514, 95% CI 0.316–0.835).
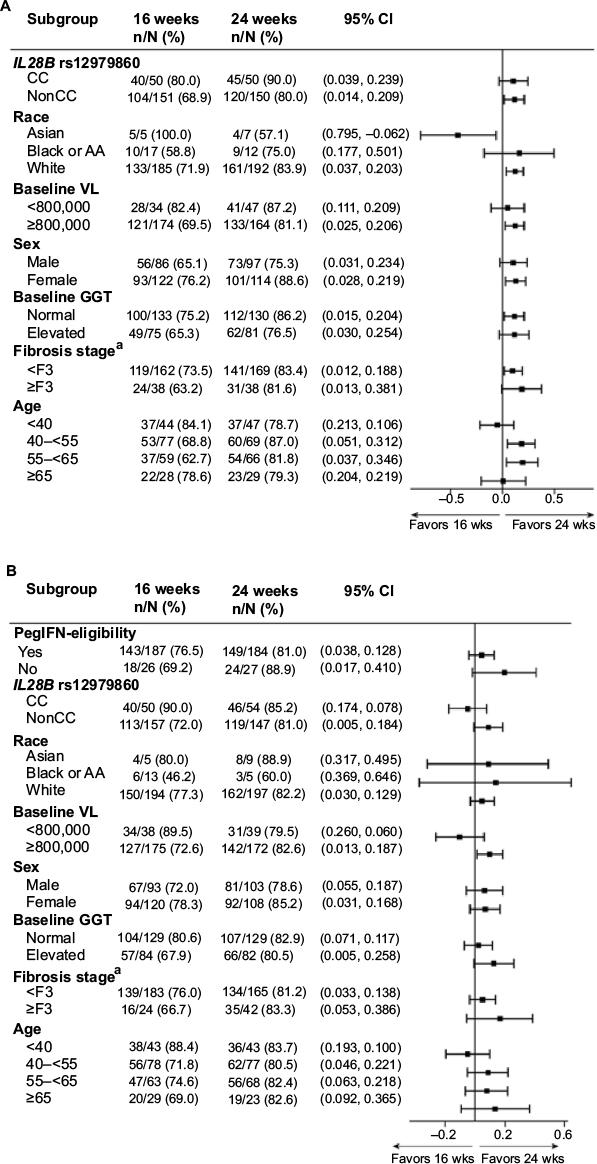 | Figure 3 SVR12 for 24-week versus 16-week regimen by subgroups in HCVerso1 (A) and HCVerso2 (B) Notes: aNo patient in the 16-week or 24-week non-cirrhotic arms had fibrosis stage F4. Abbreviations: AA, African-American; CI, confidence interval; GGT, gamma-glutamyl transferase; PegIFN, peginterferon-α; VL, viral load; wks, weeks. |
Resistance
Baseline genotypic analysis of HCV GT-1b NS3/4A and NS5B population-based sequences was available for samples from 460 and 462 patients, respectively, in HCVerso1, and for 490 patients each in HCVerso2. Resistance-associated variants (RAVs) were detected in baseline samples from a minority of these patients. In the NS3 domain, no RAVs were detected at R155 or A156 in either study; D168E was detected in seven (1.5%) samples in HCVerso1 and in three (0.6%) samples in HCVerso2. In the NS5B domain, the most frequently detected baseline RAVs in HCVerso1 and HCVerso2 were A421V (43 [9%] and 40 [8%], respectively), V499A (69 [15%] and 77 [16%], respectively), and V499T/I/L (28 [6%] and 33 [7%], respectively), whereas no baseline RAVs were detected at NS5B P495. SVR12 rates for patients with NS5B A421V or V499A polymorphisms are summarized in Table 4.
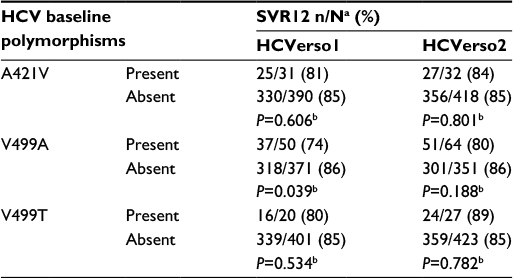 | Table 4 SVR12 rates among patients with HCV NS5B resistance-associated variants at baseline Notes: aN = patients evaluable for SVR12, excluding those who discontinued treatment prematurely for reasons other than virologic failure. bTwo-sided Fisher’s exact test comparing percentages of patients who achieved a virologic response with and without a specific baseline amino acid variant (“present” does not include the variants detected as a mixture with other amino acids). Abbreviations: HCV, hepatitis C virus; SVR12, sustained virologic response at 12-week posttreatment. |
HCV genotypic analysis of samples from patients who received active treatment and failed to achieve SVR12 was available for 86 patients in HCVerso1 and 93 patients in HCVerso2. Overall, the most frequent (>10%) treatment-emergent RAVs detected were: in the NS3 domain, D168V (27 [31%] and 32 [34%] in HCVerso1 and HCVerso2, respectively) and D168T (11 [13%] in HCVerso1); and in the NS5B domain, P495L (34 [40%] and 37 [39%] in HCVerso1 and HCVerso2, respectively) and P495Q (9 [10.5%] in HCVerso1). All on-treatment failures had RAVs in NS3 and/or NS5B and most frequently in both (92.5% [37/40] in HCVerso1 and 97.5% [39/40] in HCVerso2) (Figure S2). Among patients with relapse, RAVs at both loci were detected in most samples (68% [17/25] and 54% [14/26] in HCVerso1 and HCVerso2, respectively), with RAVs at a single locus (either NS3 or NS5B) detected in 24% (6/25) in HCVerso1, 27% (7/26) in HCVerso2, and no RAVs detected in the remaining relapse samples. Among patients who discontinued active treatment prematurely, most had no RAVs detected among their post baseline samples (81% [17/21] in HCVerso1 and 56% [15/27] in HCVerso2).
Safety and tolerability
The incidence of AEs, serious AEs, and AEs leading to discontinuation of study treatment was similar across treatment groups for both studies (Table 5). Most AEs were considered related to treatment. The most common AEs were nausea, vomiting, and diarrhea. AEs were mostly mild or moderate with onset within the first 8 weeks of treatment. AEs led to discontinuation of all medication in 6%–8% of patients. The most frequent AEs leading to discontinuation were nausea (7 [1.5%] patients in HCVerso1 and 3 [0.6%] in HCVerso2) and vomiting (7 [1.5%] patients in HCVerso1 and 6 [1.2%] in HCVerso2). Rash and photosensitivity were mostly mild: for photosensitivity, 89/100 (89%) cases in HCVerso1 and 84/100 (84%) cases in HCVerso2 were mild; for rash, 109/111 (98%) cases in HCVerso1 and 149/157 (95%) cases in HCVerso2 were mild. The most frequently reported serious AE was anemia (three patients in HCVerso1 and four in HCVerso2). There was one death, in HCVerso1, which was due to an accidental fall and not considered related to the study treatment.
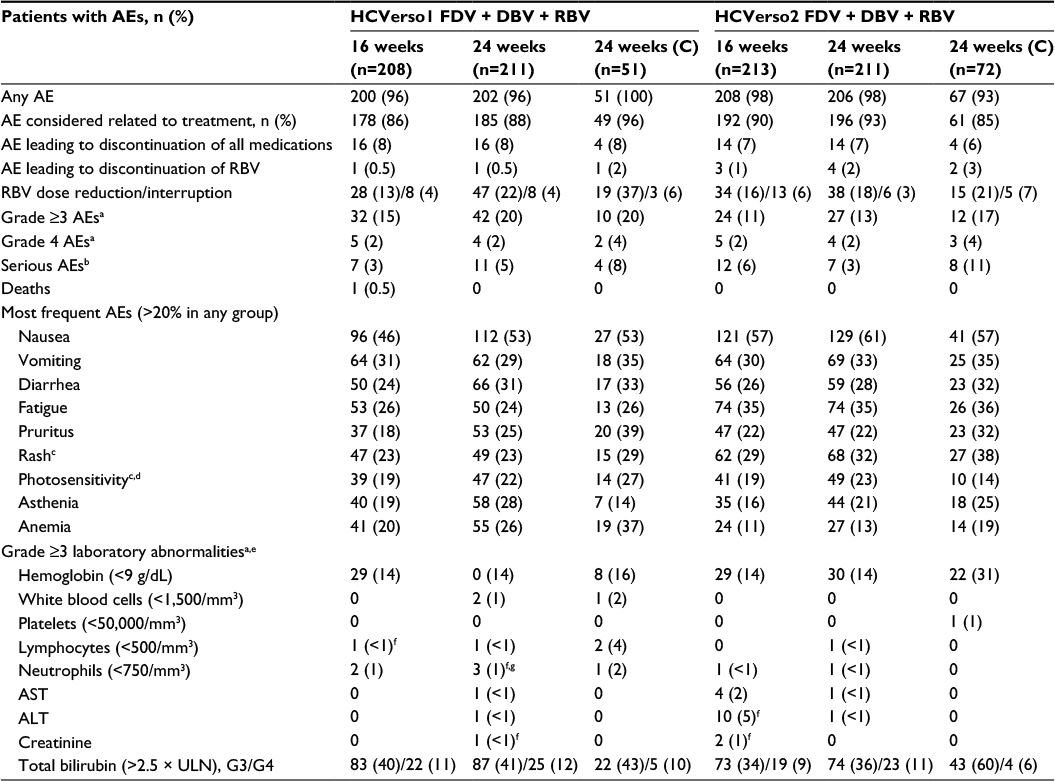 | Table 5 Overview of AEs Notes: aSeverity based on Division of AIDS Table for Grading the Severity of Adult and Pediatric Adverse Events: Grade 3 = severe, Grade 4 = potentially life-threatening. bHCVerso 1 serious AEs included the following: in the 16-week group, anemia, decreased appetite, dizziness, and fatigue (1), agranulocytosis (1), non-Hodgkin’s lymphoma (1), pancreatic carcinoma (1), loss of consciousness (1), fall and multiple injuries (1), and pericarditis (1); in the 24-week group, agranulocytosis and febrile neutropenia (1), duodenal ulcer, gastroesophageal reflux disease, bacterial gastritis, and gingivitis (1), acute renal failure (1), anemia (1), depression (1), psychotic disorder (1), syncope, fall, and hematoma (1), Escherichia urinary tract infection, anemia, and pyrexia (1), postgastric surgery syndrome (1), thrombosis (1), and arthritis (1); in the 24-week cirrhotic group, diarrhea (1), gastrointestinal hemorrhage, hyperbilirubinemia, and prerenal failure (1), cholelithiasis (1), and urethral stenosis (1). HCVerso2 serious AEs included the following: in the 16-week group, anemia (1), anemia, diarrhea, fatigue, and acute prerenal failure (1), vomiting and diarrhea (1), vomiting and hematemesis (1), pancreatitis (1), tonsillitis (1), hepatocellular carcinoma (1), prostate cancer (1), depression and suicide attempt (1), bipolar I disorder (1), depression and insomnia (1), and acute renal failure (1); in the 24-week group, anemia and thrombocytosis (1), pneumonia (1), basal cell carcinoma (1), depression (1), drug eruption (1), drug hypersensitivity (1), and fall, jaw fracture, loss of consciousness, and malaise (1); in the 24-week cirrhotic group, anemia and pneumonia (1), cellulitis, febrile neutropenia, hyperbilirubinemia, and hepatocellular carcinoma (1), gastric ulcer (1), gastrointestinal hemorrhage and portal hypertensive gastropathy (1), pericarditis (1), amnesia (1), photosensitivity reaction (1), and hypertensive crisis (1). cComposite term Boehringer Ingelheim customized MedDRA query. dPatients were instructed to avoid direct sun exposure as much as possible and to apply UVA and UVB sun-blocking cream to any uncovered skin daily during treatment. eAll Grade 3 unless otherwise indicated. fOne Grade 4. gOccurred at or after Week 12. Abbreviations: AE, adverse event; ALT, alanine aminotransferase; AST, aspartate aminotransferase; C, compensated cirrhosis; DBV, deleobuvir; FDV, faldaprevir; RBV, ribavirin; ULN, upper limit of normal; UVA, ultraviolet A; UVB, ultraviolet B. |
There was one case of Grade 4 neutropenia that occurred after 12 weeks of treatment in HCVerso1 and was considered by the investigator to be probably related to the study drugs (Table 5). Grade 3 or 4 bilirubin elevations were observed in around half of all patients (Table 5). Bilirubin elevations were characterized mainly by unconjugated bilirubin, and bilirubin levels returned to baseline shortly after the end of active treatment (Figure S3). In HCVerso1, no patient experienced both ALT ≥3 × upper limit of normal (ULN) or baseline and total bilirubin >2 × ULN with a direct:indirect bilirubin ratio >1. In HCVerso2, one patient with baseline cirrhosis experienced a cholestatic injury pattern after 12 weeks of trial treatment with ALT 5 × ULN, total bilirubin >2 × ULN with a direct:indirect bilirubin ratio >1, and alkaline phosphatase elevations following administration of amoxicillin/clavulanate potassium (Augmentin®); the internal hepatic advisory committee determined that the case was not study drug-induced liver injury, but was more likely related to amoxicillin/clavulanate potassium use. All laboratory abnormalities in this patient resolved with discontinuation of medications.
Discussion
In these two large Phase III clinical studies, 24 weeks of the IFN-free combination of faldaprevir, deleobuvir, and ribavirin in treatment-naïve, HCV GT-1b-infected patients resulted in higher adjusted rates of SVR12 than historical control rates based on a first-generation protease inhibitor plus PegIFN and ribavirin. Results for the 16-week treatment arms differed between the two studies. In HCVerso2, SVR12 following 16 weeks of treatment remained superior to the historical control and was not significantly lower than the SVR12 after 24 weeks of treatment. However, in HCVerso1, 16 weeks of treatment resulted in an SVR12 rate that was significantly lower than that achieved with 24 weeks of treatment (72% vs 82%, respectively, P=0.0040) and was not superior to the historical control. Notably, the rate of relapse in the 16-week treatment arms was higher than in the 24-week treatment arms in both studies. Assessment of SVR12 rates according to virologic response at Week 4 showed higher response rates among patients who had HCV RNA undetected at Week 4 than among those with HCV RNA <25 IU/mL detected at Week 4 in the 16-week treatment arm. The impact of Week 4 responses on SVR12 was less pronounced in the 24-week treatment arms, including among those with cirrhosis. The studies included a total of 123 patients with compensated cirrhosis; these patients achieved SVR12 rates of 73% in HCVerso1 and 74% in HCVerso2 after 24 weeks of treatment. Data from studies using other all-oral DAA regimen suggest that patients with cirrhosis may require longer treatment durations to achieve SVR than patients without cirrhosis.2 However, failure to achieve SVR12 in patients with cirrhosis in the HCVerso studies was mostly due to virologic breakthrough or premature discontinuation due to AEs, with a low rate of posttreatment relapse, suggesting that an extended treatment duration would not improve the response rate.
RAVs present prior to the initiation of treatment have the potential to compromise the response to antiviral treatment. Consistent with previous observations,26 NS3 R155 and D168 RAVs associated with resistance to faldaprevir were infrequently detected at baseline in the HCVerso studies. The NS5B RAVs A421V and V499A were more common at baseline (83/922 [9%] and 146/922 [16%], respectively). The baseline RAVs NS5B A421V and V499A have been shown to confer low-level resistance to deleobuvir in vitro (<sixfold decrease in potency).17 Baseline A421V had no impact on SVR12. A reduction in SVR12 may be associated with baseline V499A among a small subset of patients, as was noted in HCVerso1 (74% [37/50] SVR12 with V499A vs 86% [318/371] SVR12 without V499A, P=0.039). Virologic failure resulted in the selection of multiple RAVs in most cases: samples from the majority of patients with virologic failure had RAVs in both the NS3 and NS5B domains, predominantly encoding NS3 D168V and NS5B P495L. Faldaprevir Phase 1b/2 studies have indicated that NS3 D168 RAVs have shorter posttreatment persistence and are less fit compared with NS3 R155 RAVs,27,28 such as the GT-1a R155K RAV, which has been reported to have a median time to loss of 9.8 months.29 From earlier studies of deleobuvir,17,30 P495 RAVs selected during breakthrough were rapidly outgrown by wild-type virus in the absence of drug selective pressure; for example, P495 RAVs persisted up to 8 weeks after the EOT and were no longer detected by the end of follow-up (68 weeks after EOT).30
Overall, the tolerability profile for the faldaprevir, deleobuvir, ribavirin combination was less favorable than that recently reported for other all-DAA regimens.31 Reported AEs in both the studies were predominantly mild or moderate in severity (with onset within the first 8 weeks of treatment), were similar in the 16- and 24-week treatment arms, and were consistent with the safety profiles previously reported for deleobuvir and faldaprevir.19–21 Photosensitivity and rash both occurred in around 20% of patients and were mostly mild. The most frequently reported AEs were gastrointestinal. Nausea and vomiting were the most common AEs leading to premature discontinuation of all treatments. Bilirubin elevations were common, but were mainly the result of an increase in unconjugated bilirubin, were not associated with ALT elevations, and returned to baseline levels immediately after the EOT. There were no cases of drug-induced liver injury. One patient experienced Grade 4 neutropenia. Across clinical studies with deleobuvir, Grade 4 neutropenia has been reported for four out of approximately 1,400 patients. All events occurred after at least 12 weeks of treatment, which is not typical of drug-induced neutropenia. Three patients fully recovered after treatment discontinuation. The remaining patient recovered after a 5-day treatment discontinuation, following which treatment was reinitiated with no recurrence of neutropenia.
Limitations
The HCVerso studies have some limitations. The use of historical controls is subject to potential bias, particularly when, as in these studies, adjustments are needed to account for differences between control and study patient populations. In addition, the field of HCV treatment has moved rapidly since the initiation of these studies, such that the historical controls used are already not representative of the current standard of care. Historical controls were based on treatment regimen comprising a first-generation protease inhibitor plus PegIFN/ribavirin. New all-oral DAA combinations are now available for patients with HCV GT-1b infection and have demonstrated the potential for SVR12 rates of >90% with treatment durations as short as 12 weeks.11,13 Therefore, based on the results of the HCVerso studies, Boehringer Ingelheim has decided not to move forward in this therapeutic area.
Conclusions
In conclusion, in treatment-naïve, noncirrhotic patients with HCV GT-1b infection, faldaprevir + deleobuvir + ribavirin for 16 or 24 weeks resulted in SVR12 rates of between 71.6% and 82.5%. Although the adjusted SVR12 rates for 24 weeks of treatment were significantly higher than historical controls in both studies, they were lower than expected. No justification could be made for the shorter and more desirable 16-week treatment duration because it resulted in a higher rate of relapse than the 24-week arms in both studies.
Acknowledgments
We thank the patients, the investigators, and all our colleagues at Boehringer Ingelheim who worked to provide the data reported here, and DDL Diagnostics Laboratory (Rijswijk, The Netherlands) for HCV population-based sequencing. Medical writing assistance, supported financially by Boehringer Ingelheim Pharma GmbH & Co. KG, was provided by Esther Race of Choice Healthcare Solutions during the preparation of this manuscript.
Disclosure
CS has served on the advisory boards for Abbott, AbbVie, Achillion, BI, BMS, Janssen, Gilead, Merck/MSD, Novartis, Roche, and Vertex; the speaker bureaus for Abbott, AbbVie, BI, BMS, Falk, Gilead, Janssen, Merck/MSD, Novartis, Roche, and Siemens; and has received research support from Abbott, Gilead, Janssen, Merck/MSD, Roche, and Siemens.
FCastelli has received research support from Abbott, Astellas, BMS, Boehringer Ingelheim, ViiV Healthcare, Schering, Roche, Janssen, Novartis, and Pfizer.
PA has served on the advisory boards for AbbVie, Boehringer Ingelheim, Gilead, Janssen, Merck, and Roche; has acted as a consultant for BMS and Merck; and has received research support from Gilead Sciences, Merck, and Roche.
MB has acted as a consultant for Boehringer Ingelheim and as a speaker for BMS, Gilead, Janssen, Merck, and Novartis.
MC has received research support from BMS and Gilead Science; has served on the advisory boards for AbbVie, Achillion, Alfa Wasserman, Bayer, BMS, GenSpera, Gilead Science, GSK, Janssen, Jennerex, Lundbeck, Merck, Novartis, Roche, Tibotec, and Vertex; and has acted as a speaker for Bayer, BMS, Gilead Science, Janssen, Merck, Novartis, Roche, Sanofi, Tibotec, and Vertex.
SP has acted as a consultant and speaker for Abbott, BMS, Boehringer Ingelheim, Gilead Sciences, GSK, Merck, Novartis, Roche, Sanofi, Tibotec, and Vertex; and has received research support from BMS, Gilead, Merck, and Roche.
FCalinas has served on the advisory boards for AbbVie, BMS, Gilead Sciences, Janssen, Merck, and Roche; the speaker bureaus for BMS, Gilead Sciences, Janssen, Merck, and Roche; and has acted as a consultant for Boehringer Ingelheim and Intercept.
MP has served on the advisory boards, speaker bureaus, or as a consultant for Boehringer Ingelheim, Gilead Sciences, GSK, Janssen, Merck, Vertex, and ViiV Healthcare; received medicines, equipment, or administrative support from Merck; and has received research support from BMS, Gilead Sciences, Novartis, and Roche.
AO and MS report no conflicts of interest.
JOS, MR, SA, QD, and RV are employees of Boehringer Ingelheim. GK was employed by Boehringer Ingelheim and is currently employed by Gilead Sciences. FJM was employed by Boehringer Ingelheim and is currently employed by AbbVie.
DN has received research support from AbbVie, BI, BMS, Genentech, Gilead, Janssen, Kadmon, Merck, and Vertex.
References
Mohd Hanafiah K, Groeger J, Flaxman AD, Wiersma ST. Global epidemiology of hepatitis C virus infection: new estimates of age-specific antibody to HCV seroprevalence. Hepatology. 2013;57(4):1333–1342. | ||
EASL. EASL recommendation on treatment of hepatitis C; 2014. Available from: http://files.easl.eu/easl-recommendations-on-treatment-of-hepatitis-C.pdf. Accessed November 12, 2014. | ||
El-Kamary SS, Jhaveri R, Shardell MD. All-cause, liver-related, and non-liver-related mortality among HCV-infected individuals in the general US population. Clin Infect Dis. 2011;53(2):150–157. | ||
Negro F, Alberti A. The global health burden of hepatitis C virus infection. Liver Int. 2011;31(Suppl 2):1–3. | ||
Gower E, Estes CC, Hindman S, Razavi-Shearer K, Razavi H. Global epidemiology and genotype distribution of the hepatitis C virus infection. J Hepatol. 2014;61(1 Suppl):S45–S57. | ||
INCIVEK™ (telaprevir) [prescribing information]. MA: Vertex Pharmaceuticals, Inc; 2013. Available from: http://www.accessdata.fda.gov/drugsatfda_docs/label/2013/201917s012lbl.pdf. Accessed November 2014. | ||
VICTRELIS™ (Boceprevir) [prescribing information]. NJ: Merck & Co., Inc.; 2012. Available from: http://www.merck.com/product/usa/pi_circulars/v/victrelis/victrelis_pi.pdf. Accessed November 12, 2014. | ||
Pegasys™ (pegylated interferon alfa-2a) [prescribing information]. CA: Genentech USA, Inc. Available from: http://www.accessdata.fda.gov/drugsatfda_docs/label/2014/103964s5261lbl.pdf. Accessed November 12, 2014. | ||
OLYSIO™ (simeprevir) [prescribing information]. USA: Janssen Products; 2014. Available from: http://www.accessdata.fda.gov/drugsatfda_docs/label/2014/205123s002lbledt.pdf. Accessed November 2014. | ||
SOVALDI™ (sofosbuvir) [prescribing information]. CA: Gilead Sciences Inc.; 2013. Available from: http://www.accessdata.fda.gov/drugsatfda_docs/label/2013/204671s000lbl.pdf. Accessed November 2014. | ||
HARVONI (ledispavir and sofosbuvir) [prescribing information]. CA: Gilead Sciences Inc.; 2014. Available from: http://www.accessdata.fda.gov/drugsatfda_docs/label/2014/205834s000lbl.pdf. Accessed November 2014. | ||
DAKLINZA (daclatasvir) [prescribing information]. Princeton, NJ: Bristol-Myers Squibb; 2015. Available from: http://www.accessdata.fda.gov/drugsatfda_docs/label/2015/206843Orig1s000lbl.pdf. Acccessed August 2015. | ||
VIEKIRA PAK (ombitasvir, paritaprevir, and ritonavir tablets; dasabuvir tablets) [prescribing information]. IL: AbbVie Inc.; 2014. Available from: http://www.accessdata.fda.gov/drugsatfda_docs/label/2014/206619lbl.pdf. Accessed August 2015. | ||
Pawlotsky JM. New hepatitis C therapies: the toolbox, strategies, and challenges. Gastroenterology. 2014;146(5):1176–1192. | ||
White PW, Llinas-Brunet M, Amad M, et al. Preclinical characterization of BI 201335, a C-terminal carboxylic acid inhibitor of the hepatitis C virus NS3-NS4A protease. Antimicrob Agents Chemother. 2010;54(11):4611–4618. | ||
Manns MP, Bourliere M, Benhamou Y, et al. Potency, safety, and pharmacokinetics of the NS3/4A protease inhibitor BI201335 in patients with chronic HCV genotype-1 infection. J Hepatol. 2011;54(6):1114–1122. | ||
Larrey D, Lohse AW, Trepo C, et al. Antiviral effect, safety, and pharmacokinetics of five-day oral administration of deleobuvir (BI 207127), an investigational hepatitis C virus RNA polymerase inhibitor, in patients with chronic hepatitis C. Antimicrob Agents Chemother. 2013;57(10):4727–4735. | ||
LaPlante SR, Bos M, Brochu C, et al. Conformation-based restrictions and scaffold replacements in the design of hepatitis C virus polymerase inhibitors: discovery of deleobuvir (BI 207127). J Med Chem. 2014;57(5):1845–1854. | ||
Zeuzem S, Asselah T, Angus P, et al. Faldaprevir (BI 201335), deleobuvir (BI 207127) and ribavirin oral therapy for treatment-naive HCV genotype 1: SOUND-C1 final results. Antivir Ther. 2013;18(8):1015–1019. | ||
Zeuzem S, Soriano V, Asselah T, et al. Faldaprevir and deleobuvir for HCV genotype 1 infection. N Engl J Med. 2013;369(7):630–639. | ||
Zeuzem S, Dufour JF, Buti M, et al. Interferon-free treatment of chronic hepatitis C with faldaprevir, deleobuvir and ribavirin: SOUND-C3, a Phase 2b study. Liver Int. 2015;35(2):417–421. | ||
Administration UFaD. Division of AIDS Table for Grading the Severity of Adult and Pediatric Adverse Events. version 1.0, December 2004; clarification August 2009. Available from: http://www.accessdata.fda.gov/drugsatfda_docs/label/2014/206619lbl.pdf. Accessed November 2014. | ||
Sherman KE, Flamm SL, Afdhal NH, et al. Response-guided telaprevir combination treatment for hepatitis C virus infection. N Engl J Med. 2011;365(11):1014–1024. | ||
Poordad F, McCone Jr J, Bacon BR, et al. Boceprevir for untreated chronic HCV genotype 1 infection. N Engl J Med. 2011;364(13):1195–1206. | ||
Research CfDEa. Virology Review (Addendum) NDA: 202258. Available from: http://www.accessdata.fda.gov/drugsatfda_docs/nda/2011/202258Orig1s000MicroR.pdf. Accessed January, 2015. | ||
Berger KL, Triki I, Cartier M, et al. Baseline hepatitis C virus (HCV) NS3 polymorphisms and their impact on treatment response in clinical studies of the HCV NS3 protease inhibitor faldaprevir. Antimicrob Agents Chemother. 2014;58(2):698–705. | ||
Berger KL, Lagace L, Triki I, et al. Viral resistance in hepatitis C virus genotype 1-infected patients receiving the NS3 protease inhibitor faldaprevir (BI 201335) in a phase 1b multiple-rising-dose study. Antimicrob Agents Chemother. 2013;57(10):4928–4936. | ||
Berger KL BR, Cartier M, Marquis, et al. Analysis of baseline polymorphisms and persistence of emergent variants from Phase Ib and II trials evaluating the HCV NS3 protease inhibitor faldaprevir (BI 201335). Hepatology (Baltimore, Md). 2012;56(S1):574A. | ||
Sullivan JC, De Meyer S, Bartels DJ, et al. Evolution of treatment-emergent resistant variants in telaprevir phase 3 clinical trials. Clin Infect Dis. 2013;57(2):221–229. | ||
Larrey D, Lohse AW, de Ledinghen V, et al. Rapid and strong antiviral activity of the non-nucleosidic NS5B polymerase inhibitor BI 207127 in combination with peginterferon alfa 2a and ribavirin. J Hepatol. 2012;57(1):39–46. | ||
Afdhal N, Zeuzem S, Kwo P, et al. Ledipasvir and sofosbuvir for untreated HCV genotype 1 infection. N Engl J Med. 2014;370(20):1889–1898. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.