")
Back to Journals » Drug Design, Development and Therapy » Volume 15
Gypenosides Inhibit Inflammatory Response and Apoptosis of Endothelial and Epithelial Cells in LPS-Induced ALI: A Study Based on Bioinformatic Analysis and in vivo/vitro Experiments
Authors Tu Q, Zhu Y, Yuan Y, Guo L, Liu L, Yao L, Zou Y, Li J, Chen F
Received 15 October 2020
Accepted for publication 18 December 2020
Published 25 January 2021 Volume 2021:15 Pages 289—303
DOI https://doi.org/10.2147/DDDT.S286297
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Jianbo Sun
Qing Tu,1,* Yabing Zhu,1,* Yuan Yuan,1,* Long Guo,1 Lu Liu,2 Liangfang Yao,1 Yun Zou,1 Jinbao Li,1 Feng Chen1
1Department of Anesthesiology, Shanghai General Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai 200080, People’s Republic of China; 2School of Anesthesiology, Weifang Medical University, Weifang 261053, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Jinbao Li; Feng Chen
Department of Anesthesiology, Shanghai General Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, People’s Republic of China
Email [email protected]; [email protected]
Introduction: Severe inflammatory response leads to poor prognosis of acute lung injury (ALI), the role of gypenosides (GPs) on ALI is not fully clear. The study aimed at investigating the effects of GPs on ALI.
Methods: We firstly established LPS-induced ALI mice model. Then, we tested whether GPs contributed to alleviate inflammatory response and lung injury of ALI in vivo. In order to identify specific mechanisms of the phenomenon, we conducted a bioinformatic analysis of LPS-induced ALI mice based on GEO database to identify hub differentially expressed genes (DEGs). PPI network of the DEGs was used to find hub-genes. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis were conducted based on the DAVID database to identify which pathways the genes enriched. Then, we tested whether GPs inhibited lung injury and inflammatory response via the enriched pathways. We also tested whether GPs inhibited the apoptosis of endothelial and epithelial cells secondary to severe inflammation.
Results: We found GPs significantly alleviated lung injury and improved the survival rate of LPS-induced ALI mice in vivo. Bioinformatic analysis identified 20 hub-genes from DEGs, they were mainly enriched in NF-κB and TNF-α pathways. GPs could reduce the lung injury and inflammatory response via inhibiting NF-κB and TNF-α pathways in vivo. Our results indicated that GPs also inhibited inflammatory response of epithelial and endothelial cells via NF-κB and TNF-α pathways in vitro. Severe inflammatory response could also lead to apoptosis of endothelial and epithelial cells. Our results indicated that GPs effectively inhibited the apoptosis of endothelial and epithelial cells.
Conclusion: Our study suggested GPs contributed to alleviated lung injury in vivo and inhibited inflammation and apoptosis of endothelial and epithelial cells in vitro, providing novel strategies for the prevention and therapy for ALI.
Keywords: gypenosides, lipopolysaccharide, acute lung injury, inflammatory response, apoptosis
Introduction
Acute lung injury (ALI) is a devastating respiratory disorder and is associated with a high mortalityrate, up to 40%–60%.1 Numerous causes may result in ALI, such as sepsis,2 multibacterial pneumonia, severe trauma with shock and multiple transfusions, etc.1 Two major separate barriers protect the lungs from injury during the development of ALI, the alveolar-capillary barrier, the microvascular endothelium and alveolar epithelium. The critical importance of endothelial and epithelial injury during ALI has been well recognized.3 Once the barriers are seriously impaired, the injury becomes out of control, eventually leading to pulmonary edema or even death. Thus, it is important to seek out novel molecular mechanisms involving prevention and treatment of ALI. In animal experiments, directly intratracheal administrated lipopolysaccharide (LPS, an endotoxin produced by gram-negative bacteria) to induce ALI model is widely used.4,5 LPS activates multiple inflammatory pathways, regulating the release of inflammatory mediators,6 leading to a series of inflammatory damage to cells and tissues. LPS challenge is also closely associated with apoptosis of pulmonary endothelial and epithelial cells, while inhibited the apoptosis of endothelial and epithelial cell significantly decreased the lung injury and mortality of ALI.7,8
Gypenosides (GPs) are major ingredients of Gynostemma pentaphylla. GPs exert many pharmacological properties. Recently, researchers raise increasing interests in studying the pharmacological effects of GPs. Researches suggested that GPs’ attenuated neuro-inflammatory level induced by microglial activation via M1/M2 phenotypic alteration.9 A research also indicated GPs had protective effects on autoimmune optic neuritis.10 But they have not provided specific mechanisms. A network pharmacology analysis revealed the anti-inflammatory effects of GPs were mainly enriched in the Janus kinase/signal transducers and activators of transcription (JAK-STAT) pathway and provided potential therapeutic targets of GPs on inflammatory response.11 The progress of ALI is accompanied by various inflammatory pathways activated and constantly magnified, eventually leading to poor prognosis. Some studies have revealed GPs inhibited the apoptosis induced by oxidative stress.12,13 Whether GPs improve lung injury during ALI and the mechanism remains unclear. The study was conducted to investigate potential therapeutic mechanisms of GPs on ALI and provide novel treatments for ALI.
Materials and Methods
Materials and Reagents
GPs were purchased from Jiatian Biotech Co., Ltd (Xi’an, China). The Dulbecco’s Modified Eagle’s Medium (DMEM) or RPMI 1640, fetal bovine serum (#10099-141, Gibco) and penicillin and streptomycin (#15070-063) were purchased from Thermo Fisher Scientific (Walthm, MA, USA). The mouse TNF-α ELISA kit (#1217202) and mouse IL-6 ELISA kit (#1210602) were purchased from Dakewe Biotech Co., Ltd. (Shenzhen, China). The TRizol reagent and cDNA synthesis kit were purchased from Takara (Kusatsu, Japan). The BCA Assay kit and the BeyoClick™ EdU-488 Cell Proliferation Kit (#C0071S) were purchased from Beyotime Biotech Co., Ltd (Shanghai, China). Antibodies of p-p65 (# 3033, 1:1000), p65 (# 8242, 1:1000), cleaved caspase-3 (#9661S, 1:1000), and caspase-3 Antibody (#9662S, 1:1000) were purchased from Cell Signaling Technology (Boston, MA, USA). Bax Antibody (#50599-2-Ig) and Bcl-2 Antibody (#12789-1-AP) were purchased from Proteintech (Chicago, IL, USA). Anti-GAPDH antibody (ab181602, 1:3000) was obtained from Abcam (Cambridge, UK). Ammonium pyrrolidinedithiocarbamate (PDTC, #P8765-1G) was obtained from Sigma-Aldrich (St. Louis, MO, USA). The CCK8 assay kit (# C0038) was obtained from Dojindo Laboratories (Japan). The Annexin V-FITC Apoptosis Detection Kit (#556547) was purchased from BD Pharmingen™ (Franklin Lakes, NJ, US).
Animals
Specific pathogen-free adult male C57BL/6 mice weighing 20–25 g (SPF grade) were obtained from Cyagen Biosciences (Suzhou, China). The mice were housed under the controlled temperature and humidity with a standard day-night cycle, free access to food and water. All procedures were conducted in accordance with the Guide for the Care and Use of Laboratory Animals. Animal experiments were approved by the the Animal Use Committee of Shanghai General Hospital (No. 2019AW009).
Cell Culture
The human pulmonary microvascular endothelial cells (HPMEC) and the MLE-12 (murine lung epithelial cells) were kindly provided by Stem Cell Bank, Chinese Academy of Sciences (Shanghai, China). The cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) or RPMI 1640 supplemented with 10% fetal bovine serum and 1% penicillin and streptomycin at 37°C, 95% humidity, and 5% CO2 incubator.
Histopathological Analysis
LPS-induced ALI mice model was established by intratracheal administration of LPS (5 mg/kg, dilute into 50ul PBS) under inhalation anesthesia with sevoflurane, as a previous study described.14 Mice were sacrificed and lung tissues were collected 8h after LPS challenge. The lower lobes of right lung were collected and fixed in (v/v) 4% paraformaldehyde for 24 h, and then embedded in paraffin, sectioned at 4-μm thickness, stained with H&E solution (Sigma-Aldrich) and then examined under a microscope. The semi-quantitative scoring system was applied to evaluate the intensity of lung injury.15 Two researchers scored the lung injury accordingly, they were blinded to the grouping information.
Measurement of Wet-to-Dry (W/D) Ratio of the Lungs
The wet/dry (W/D) weight ratio of the upper lobe of right lung was also calculated to evaluate the severity of ALI. Upper lobe of the right lung was removed and weighed as wet weight. Next, the lungs were incubated at 60°C for 3 days to remove all moisture and weighed for dry weight, the W/D ratio was finally calculated.
Enzyme-Linked Immunosorbent Assay (ELISA)
The blood samples were collected and immediately placed into sterile ethylenediaminetetraacetic acid test tubes, centrifuged at 3000g for 15 min. The serum was separated and stored in a freezer at −80°C for further assaying. The serum levels of TNF-α and IL-6 were measured by ELISA method according to the manufacturer’s protocols.
Identification of Potential Targets of ALI Based on Bioinformatics Analysis
In order to identify the potential mechanisms of GPs on ALI, we identified differentially expressed genes (DEGs) in LPS-induced ALI mice model based on GEO database. The Linear Model for Microarray Analysis R package (limma; version 3.30.11) was used to identify DEGs. Genes were identified as DEGs with an adjusted P-value < 0.01 and absolute value of fold-change |logFC| >1.5.16 Overlapped genes between the datasets were obtained using a Venn diagram web-tool (https://bioinfogp.cnb.csic.es/tools/venny/index.html). The DEGs were mapped to the STRING database (http://string-db.org/). The protein-protein interaction (PPI) pairs were extracted with median confidence more than 0.4.11 The PPI network was visualized by Cytoscape software (version 3.4.0, http://www.cytoscape.org/). Nodes with higher degree of connectivity tend to be more essential in maintaining the stability of the entire network. The plugin-CytoHubba was used to calculate the degree of each protein node, which indicates the intensity of the protein. Top hub genes were identified by Venn diagram based on the ranking methods of Degree, MCC and Betweenness.17,18 Gene Ontology (GO) annotation analysis and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis of the hub-genes in the study were conducted based on the DAVID database (https://david.ncifcrf.gov/). P < 0.01 and gene counts ≥10 were considered statistically significant.
Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
Total RNA was isolated from upper lobe of left lung tissues, the HPMEC and MLE-12 cells using TRizol reagent (Takara, Kusatsu, Japan), followed by manufacturer’s instructions. Purified RNA concentrations were determined via NanoDrop LITE (Thermo Scientific), then used to reverse transcript into cDNA using commercial cDNA synthesis kit (Takara). Gene expression were measured by real-time PCR with ConnectTM Real-Time PCR System (Bio-Rad, Hercules, CA, USA). The relative change in gene expression was determined according to 2−ΔΔCT formula. Primer sequences are list in Supplementary Table 1.
Western-Blot Analysis
Lower lobe of left lung, the HPMEC and MLE-12 cells were lysed and homogenized in ice-cold RIPA buffer (NCM Biotech, Suzhou, China) and centrifuged at 12,000 g for 15 min at 4°C. Protein concentrations were determined using BCA Assay kit according to manufacturer’s instructions, and equal amounts of protein samples were separated in SDS-PAGE and transferred onto polyvinylidene difluoride (PVDF) membranes (Solarbio Life Sciences, Beijing, China). The PVDF membranes were incubated with respective primary antibodies at 4 °C overnight, after blocked with 5% non-fat milk for 2 h. Immunoreactive bands were visualized by a BIO-RAD ChemiDoc XRS system and densitometric analysis was determined by Image J software (National Institutes of Health, Bethesda, MD, USA).
Cell Viability
The HPMEC were seeded at a density of 10,000 cells per well in 96-well plates and culture in 5% CO2, at 37°C for 24h. Confluent cells were treated with different concentrations of LPS (0.01, 0.05, 0.1, 0.5, 1 μg/mL) and GPs (1, 2.5, 5, 10, 50 μg/mL) for 8h. The cell viability was evaluated using CCK8 assay followed by the manufacturer’s instructions. The absorbance value was measured at 570 nm in a microplate spectrophotometer Epoch reader (Landon, Biotek, UK). The percentage of viable cells was determined using the following formula: % of viable cells = [(absorbance of untreated cells-absorbance of treated cells)/absorbance of untreated cells] × 100. All the experiments were repeated 3 times under the same conditions.
Edu Assay
Cell proliferation was measured by 5-ethynyl-29-deoxyuridine (Edu) assay using BeyoClick™ EdU-488 Cell Proliferation Kit according to manufacturer’s instructions. HPMEC cells seeded into plates with a density of 10 × 105 cells/well. Cells were incubated with 50 μM EdU buffer for 2 h at 37°C, then fixed with 4% formaldehyde for 0.5 h and permeabilized with 0.1% Triton X-100 for 20 min at room temperature. DNA contents were stained with Hoechst 33342 for 10 min at room temperature. Then the results were visualized by a fluorescence microscope.
Flow Cytometry
The HPMEC cells were digested with 0.25% trypsin and centrifuged at 2,000 rpm for 10 min at room temperature. Cell apoptosis was tested using Annexin V-FITC Apoptosis Detection Kit according to the manufacturer’s instructions. Cells were washed with ice-cold PBS and resuspended with Binding buffer (300 μL), then added 5 μL annexin V-FITC and incubated for 15 min at room temperature, adding propidium iodide (PI) staining 5 min before detection.
Statistical Analysis
All data were presented as mean ± SD. Statistical analysis was performed by GraphPad Prism (version 8.2.0, San Diego, CA, USA). For comparisons between two groups, the Student’s t-test (unpaired and paired) was applied. Multiple comparisons were performed by one- or two-way ANOVA. Survival was analyzed by Log-rank (Mantel-Cox) test. A P value < 0.05 was considered statistically significant.
Results
Established LPS-Induced ALI Model
Mice were treated with intratracheal administration of LPS under sevoflurane inhalational anesthesia to establish ALI model as previously described14 (Figure 1A). The mice challenged with LPS exhibited more severity of lung injury then those treated with PBS (Figure 1B and C), indicating that the ALI model was successfully established.
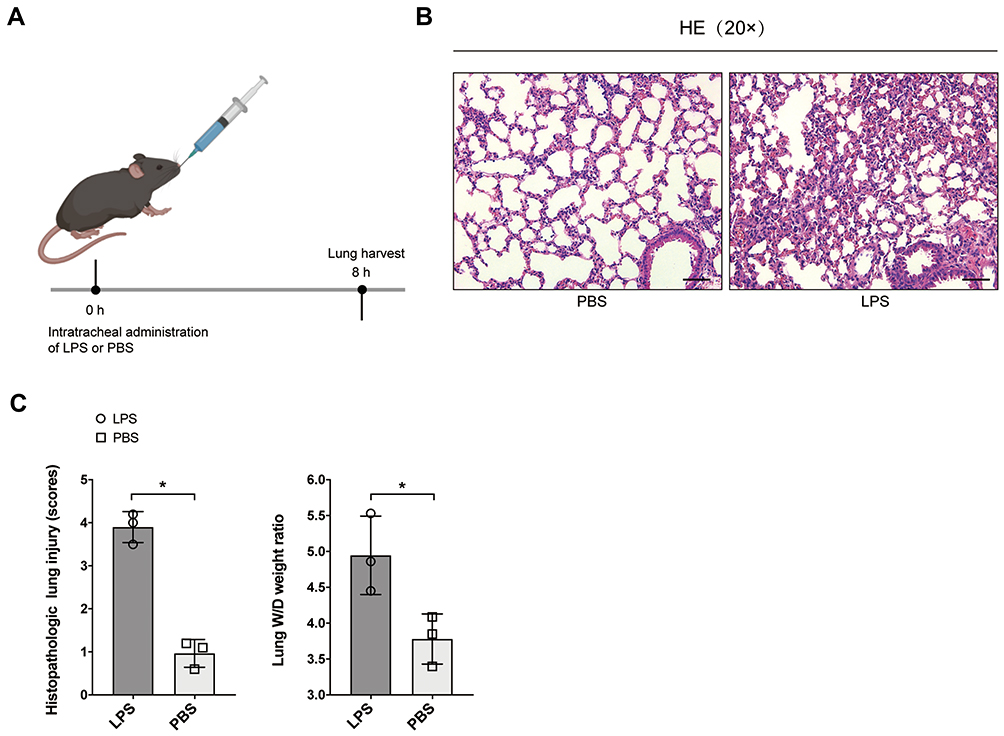 |
Figure 1 Establishment of ALI mice model. (A) Brief description of the experimental process. Mice were treated with intratracheal administration of LPS (5 mg/kg) for ALI model establishment. (B) Representative lung H&E staining of mice (n = 3) treated with PBS or LPS. The histopathologic lung injury scores of mice treated with LPS were significantly higher than that of PBS. (C) The lung W/D weight ratios of mice treated with LPS were also higher than that of PBS. *P < 0.05. |
GPs Alleviated Lung Injury and Improved the Survival Ratio of ALI Mice
Studies suggested that GPs exerted an anti-inflammatory response in various inflammatory disease.19,20 But whether GPs improve lung injury during ALI and its mechanisms remain unclear. Mice were randomly divided into PBS, LPS, GPs and GPs + LPS groups (n = 3). Mice received intratracheal administration of PBS (50ul), intratracheal administration of LPS (5 mg/kg, in 50ul PBS), intraperitoneal injection of GPs (100 mg/kg, in 300ul PBS),21 or intraperitoneal injection of GPs + intratracheal administration of LPS, respectively. A brief description of the experimental process is described in Figure 2A. The H&E staining suggested that GPs alleviated severity of alveolar epithelial and capillary endothelial cells injury, thickening of alveolar membrane, the W/D ratio in LPS + GPs group was also lower than that in LPS group (Figure 2B and C). The serum levels of TNF-α and IL-6 were also lower than that in LPS group, indicating GPs inhibited the inflammatory cytokines release during ALI (Figure 2D). In addition, we also recorded the survival rate of groups (n = 14), the survival rate in LPS group was similar to the previous study.14 However, GPs helped to reduce the mortality in ALI mice (Figure 2E).
 |
Figure 2 GPs alleviated lung injury and improved the survival ratio of ALI mice. (A) Brief description of the experimental process. Mice received intraperitoneal injection of GPs (100 mg/kg) + intratracheal administration of LPS (5 mg/kg) for 8h. (B and C) H&E staining of lung tissue (n = 3) and comparison of histopathologic lung injury scores and W/D ratio. The H&E staining, histopathologic lung injury scores and W/D ratio suggested that GPs alleviated severity of alveolar epithelial and capillary endothelial cells injury, thickening of alveolar membrane. (D) GPs decreased the serum concentrations of TNF-α and IL-1β in vivo. (E) Survival curve between four groups (n = 14). GPs improved the survival rate of ALI mice. Survival rates between groups were analyzed by Log-rank (Mantel-Cox) test, Chi square = 4.218, P = 0.0400. Data are presented as means ± SD. **P < 0.01; *P < 0.05. |
Identification of DEGs and Potential Pathways Involving LPS-Induced ALI Mice
Multiple inflammatory pathways may be activated during ALI. In order to identify specific mechanisms of GPs in improving lung injury during ALI, we conducted a bioinformatics analysis based on GEO database and three gene expression profiles (GSE2411, GSE17355, and GSE18344) were selected (Table 1). Overlapping of 106 DEGs across the datasets were identified by Venn diagram (Supplementary Figure S1). In order to identified hub-genes in the DEGs, we established PPI network of the DEGs by Cytoscape software 3.6.1. The plugin CytoHubba was used to calculate the degree and score of each protein node, which indicates the intensity of protein (Figure 3A). Top hub genes were identified by Venn diagram based on the ranking methods of Degree, MCC and Betweenness (Figure 3B).17,18 Finally, we identified 20 hub-genes, Figure 3C shows the expression levels of the hub-genes in the datasets. The hub-genes were mainly enriched in NF-κB and TNF-α signaling pathway (Figure 3D).
 |
Table 1 Accession Information for Datasets Downloaded from the GEO Database |
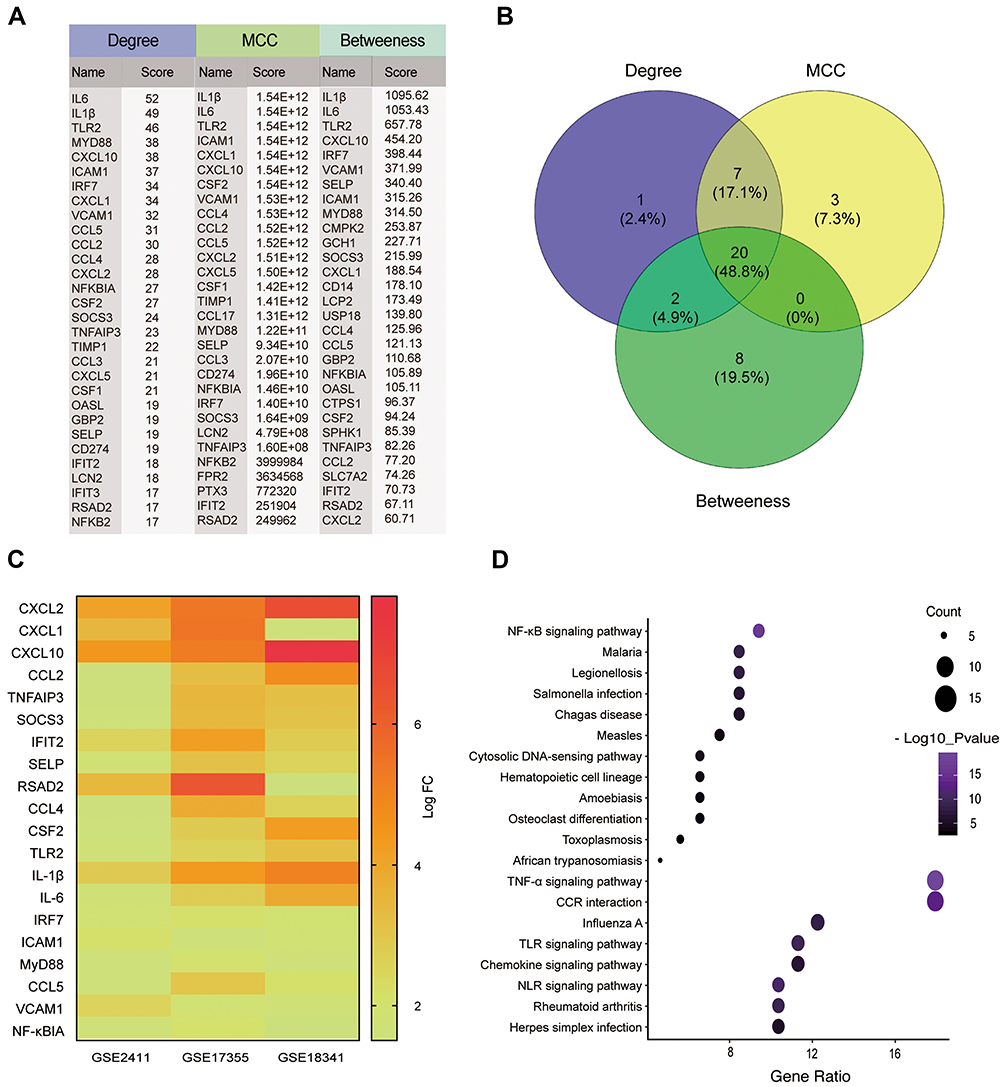 |
Figure 3 Bioinformatics analysis of DEGs. (A) The top 30 ranked DEGs identified by the ranking methods of Degree, MCC and Betweenness by a plugin of CytoHubba software. (B) Venn diagram of hub genes, including CXCL2, CXCL1, CXCL10, CCL2, TNFAIP3, SOCS3, IFIT2, SELP, RSAD2, CCL4, CSF2, TLR2, IL-1β, IL-6, IRF7, ICAM1, Myd88, CCL5, VCAM1, NF-κB. (C) Heat-map of the 20 hub-genes in the selected datasets. (D) KEGG pathway enrichment of the 20 hub-genes, they were mainly enriched in TNF-α and NF-κB signaling pathway. |
Validation of Hub-Genes
Relative mRNA expression of the 20 hub genes above was analyzed by qRT-PCR. Of the 20 hub-genes, 19 were up-regulated (Figure 4), which was in line with their expression levels in the GEO datasets.
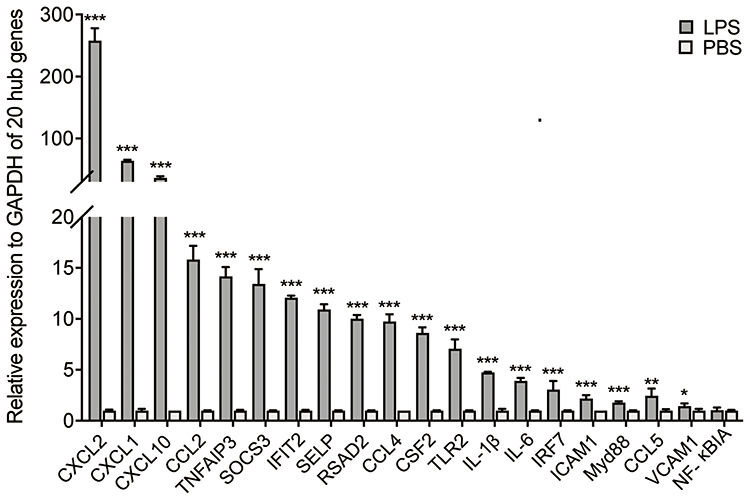 |
Figure 4 Relative mRNA expression of 20 hub genes. In LPS induced ALI mice, 19 of the hub genes were up-regulated, compared to PBS group. Data are presented as means ± SD. ***P < 0.001; **P < 0.01; *P < 0.05. |
GPs Alleviated Inflammatory Response via NF-κB and TNF-α Pathways in vivo
Since the hub-genes were mainly enriched in the NF-κB and TNF-α signaling pathways, we next tested whether GPs alleviated inflammatory response via NF-κB and TNF-α pathways. The Western-Blot results suggested that GPs inhibited the phosphorylation of p65 (Figure 5A). The mRNA levels of TNF-α, IL-1β, IL-6 were also significantly decreased in GPs + LPS group (Figure 5B), suggesting that the GPs exerted anti-inflammatory effects via inhibiting the NF-κB and TNF-α pathway in ALI mice.
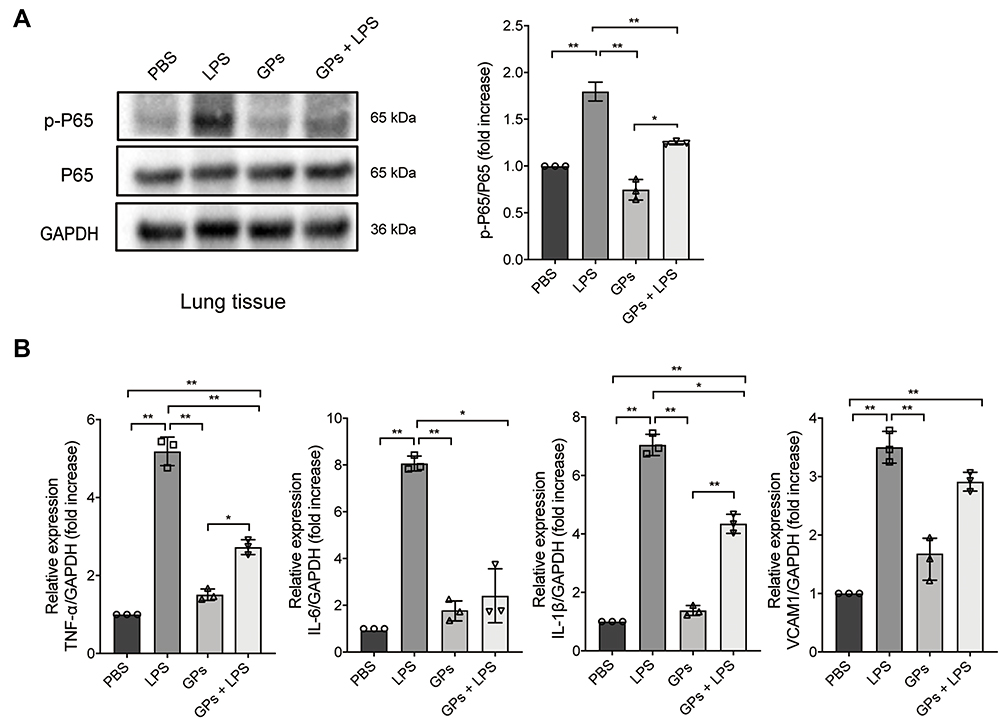 |
Figure 5 GPs inhibited the inflammatory response in vivo. (A) LPS stimulation significantly increased the expression of p-p65, while GPs inhibited the effects in vivo. (B) GPs also inhibited the mRNA expression of cytokines of TNF-α, IL-1β, IL-6, indicating that GPs alleviated the lung injury and inflammatory response in vivo. Data are presented as means ± SD. **P < 0.01; *P < 0.05. |
GPs Alleviated Inflammatory Response in Endothelial via NF-κB and TNF-α Pathways
During the development of ALI, pulmonary microvascular endothelial cells experienced significant injury, causing pulmonary congestion and edema.22 In order to mimic the ALI model in vivo, the HPMEC cells were treated with different concentrations of LPS (0.01 −1 μg/mL) for 8h to determine an optimal dose of LPS. After LPS stimulation, the levels of p-p65 was increased dose dependently (Figure 6A). In line with the results of GPs in vivo, LPS co-treated with GPs could inhibit the expression of p-p65, just as ammonium pyrrolidinedithiocarbamate (PTDC), a widely used NF-κB pathway inhibitor23,24 (Figure 6B and C) and the cytokines of TNF-α, IL-1β, IL-6, VCAM1 in HPMEC (Figure 6D), indicating GPs reduced the cytotoxicity of LPS to HPMEC.
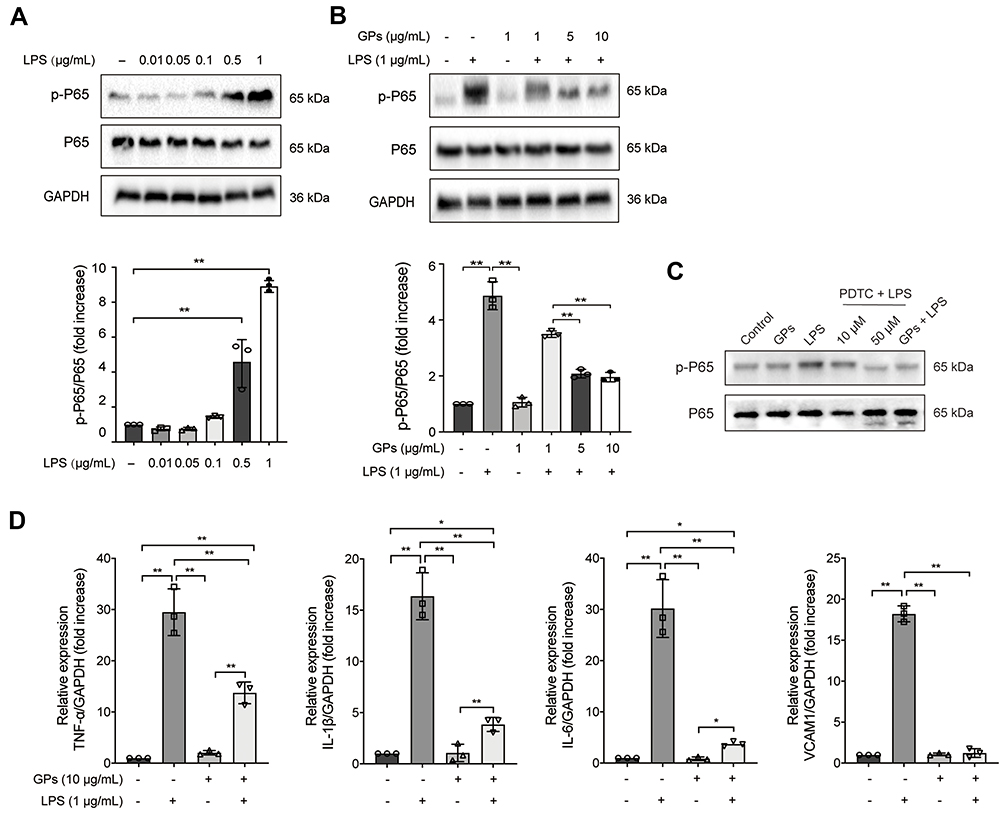 |
Figure 6 GPs alleviated inflammatory response in endothelial cells. (A) HPMEC cells were treated with different concentrations of LPS (0.01, 0.05, 0.1, 0.5 and 1 μg/mL) for 8 h. The results suggested that LPS increased the expression of p-p65 in a dose independent way. (B) LPS (1 μg/mL, 8 h) stimulation significantly increased the expression of p-p65, while LPS co-treated with GPs (1, 5 and 10 μg/mL, 8 h) reduced the level of p-p65. (C). The expression of p-p65 was inhibited by PTDC (a widely used NF-κB pathway inhibitor) and GPs induced by LPS challenge. (D) LPS (1 μg/mL, 8 h) co-treated with GPs (10 μg/mL, 8 h) inhibited the expression of cytokines induced by LPS. Indicating that GPs could alleviate the inflammatory response of endothelial cells. Data are presented as means ± SD. **P < 0.01; *P < 0.05. |
GPs Alleviated Inflammatory Response in Epithelial Cells via NF-κB and TNF-α Pathways
Alveolar epithelial cells could also be significantly damaged during the progress of ALI.25 Based on the above results, we then hypothesized that GPs may alleviate the inflammatory response level of alveolar epithelial cells. Interestingly, GPs also alleviated the expression levels of p-p65 (Figure 7A) and TNF-α, IL-1β, IL-6, VCAM1 (Figure 7B) in the MLE-12, which were similar to the results of the HPMEC.
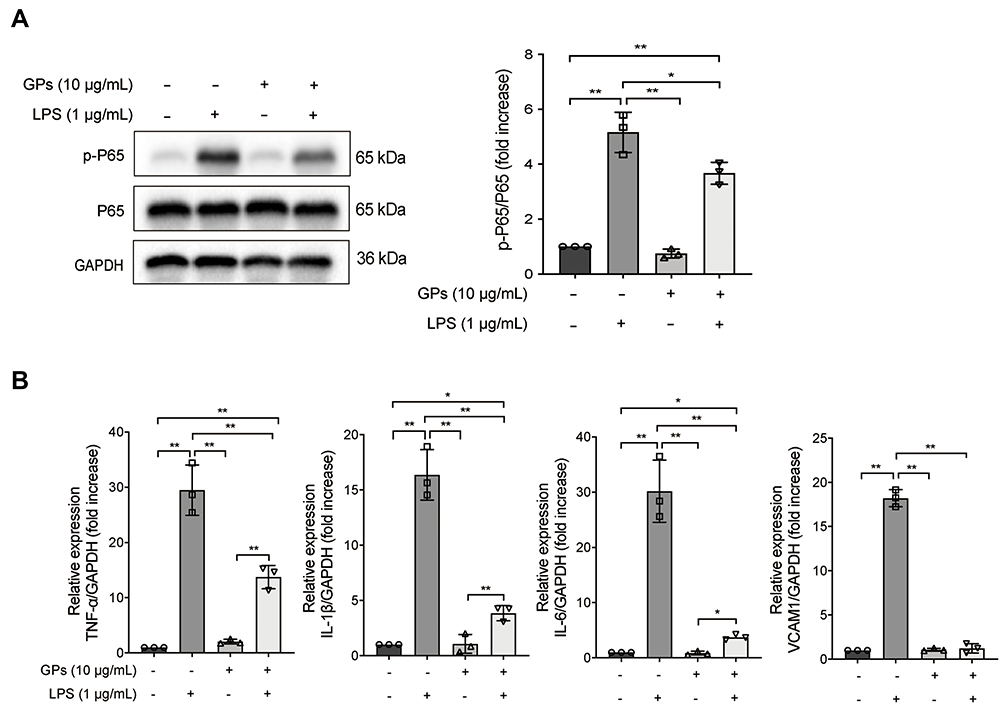 |
Figure 7 GPs alleviated inflammatory response in epithelial cells. (A) GPs (10 μg/mL, 8 h) contributed to inhibit the p-p65 and (B) the cytokines in the MLE-12 induced by LPS (1 μg/mL, 8 h), indicating that GPs could alleviate the inflammatory response in epithelial cells. Data are presented as means ± SD. **P < 0.01; *P < 0.05. |
GPs Improved the Cell Viability and Proliferation of Endothelial Cells Impaired by LPS
Lung endothelium comprised about 50% of all lung cells.26 The integrity of pulmonary endothelial cells played an indispensable role during the repair of ALI. The HPMEC were treated with different concentrations of LPS for 8 h, we found that the cell viability was decreased at 0.1 μg/mL, and reached the worst at 1 μg/mL (Figure 8A). While GPs inhibited the cell viability only at a higher concentration of 50 μg/mL (Figure 8B). Therefore, we treated HPMEC with 1 μg/mL LPS and 10 μg/mL GPs for 8 h. Then, we tested the proliferation levels of HPMEC by Edu assay. The results suggested GPs alleviated the inhibitory effect of LPS on the proliferation of HPMEC (Figure 8C).
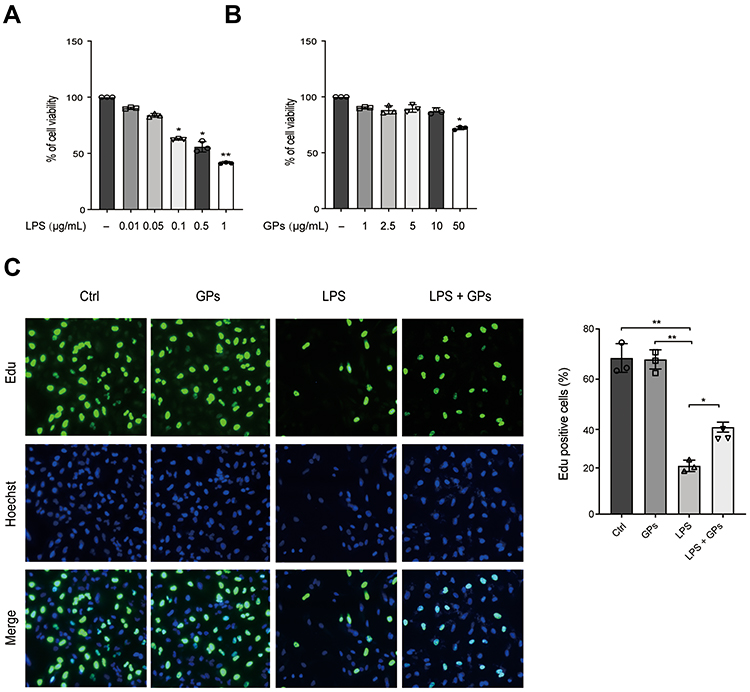 |
Figure 8 GPs improved the cell viability and proliferation of endothelial cells impaired by LPS. (A) HPMEC cells treated with LPS at different concentrations for 8 h showed decreased cell viability at higher concentrations when compared to untreated control cells. (B) HPMEC cells exposed to different concentrations of GPs for 8 h showed decreased cell viability only at higher concentrations (50 μg/mL) when compared to untreated control cells. (C) Edu assay indicated that LPS (1 μg/mL, 8 h) significantly inhibited the proliferation of HPMEC, while GPs (10 μg/mL, 8 h) alleviated the inhibitory effect. Data were represented as mean ± SD (n = 3). **P < 0.01, *P < 0.05. |
GPs Inhibited the Apoptosis of Endothelial and Epithelial Cells Induced by LPS Challenge
Severe inflammatory response leads to the apoptosis of endothelial and epithelial cells during ALI.7,8 Since GPs attenuated the LPS-induced decreased viability of endothelial cells, we then tested whether GPs inhibited the apoptosis of endothelial and epithelial cells induced by LPS challenge. LPS increased the expression of Bax and cleaved caspase-3, decreased the expression of Bcl-2 (Figure 9A), indicating LPS induced apoptosis of HPMEC. We then tested whether GPs inhibited the apoptosis of of HPMEC. The results indicated that GPs could effectively inhibit the apoptosis of HPMEC secondary to LPS challenge (Figure 9B and C).
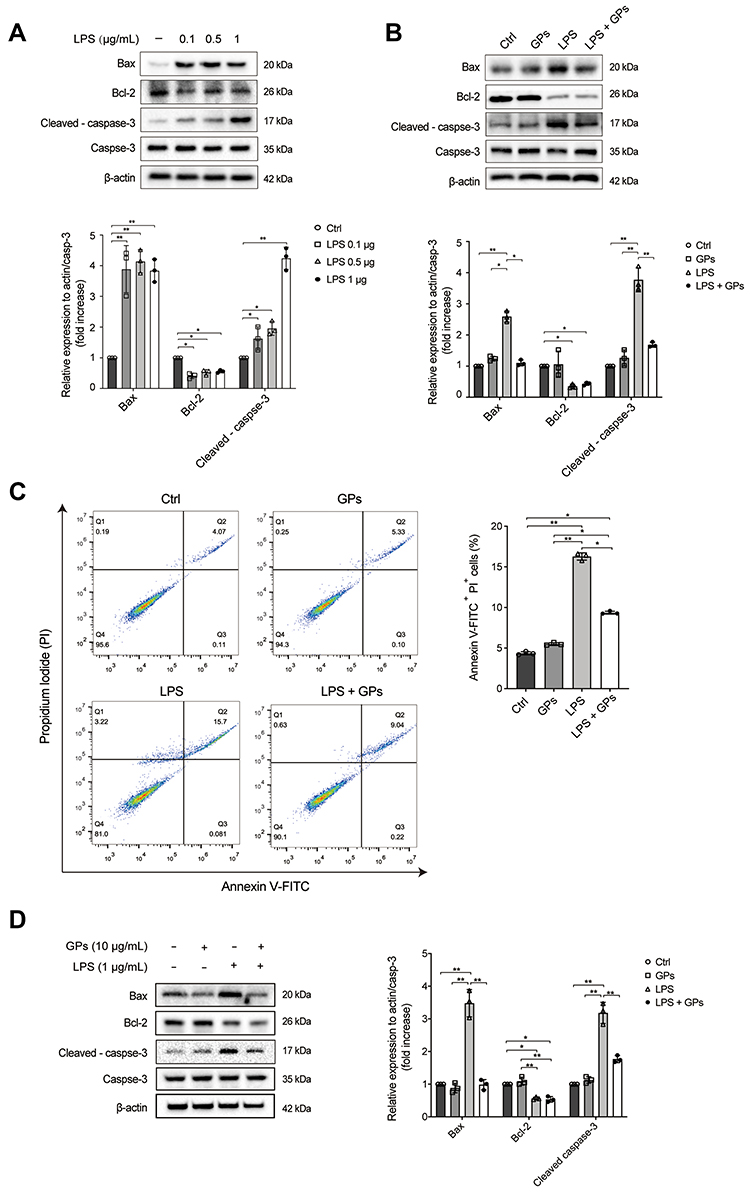 |
Figure 9 GPs inhibited the apoptosis of endothelial and epithelial cells induced by LPS challenge. (A) HPMEC cells were treated with LPS (0.1, 0.5 and 1 μg/mL) for 8 h, LPS increased the expression of Bax and cleaved caspase-3, while decreasing the expression of Bcl-2 in HPMEC. (B) LPS (1 μg/mL, 8 h) co-treated with GPs (10 μg/mL, 8 h) decreased expression of Bax and cleaved caspase-3 induced by LPS challenge in HPMEC cells. (C) GPs (10 μg/mL, 8 h) inhibited the apoptosis of HPMEC induced by LPS (1 μg/mL, 8 h) challenge. (D) GPs (10 μg/mL, 8 h) inhibited the expression of Bax and cleaved caspase-3 in the MLE-12 induced (1 μg/mL, 8 h). Data were represented as mean ± SD (n = 3). **P < 0.01, *P < 0.05. |
Since we found that GPs alleviated the apoptosis of HPMEC, we hypothesized GPS may also inhibit the apoptosis of epithelial cells. The expression of Bax and cleaved caspase-3 increased when treated with LPS in MLE-12, which was in line with the previous study.8 The result also suggested that GPs could decrease the expression of Bax and cleaved caspase-3, which indicated GPs also inhibited the apoptosis of the MLE-12 induced by LPS (Figure 9D), which provided a novel prevention and treatment of alveolar epithelial apoptosis during ALI.
Discussion
ALI is pathologically characterized by widespread lung inflammation response and loss of epithelial and endothelial integrity.27 Macrophages also plays an important role during the development of ALI. In fact, the essence of ALI underlies a series of infectious or non-infectious factors leading to excessive and uncontrolled inflammatory response of alveolar epithelial cells and capillary endothelial cells, leading to alveolar capillary and epithelial damage, eventually resulting in pulmonary edema and acute respiratory distress syndrome.28 So, in the study, we mainly focus on the endothelial and epithelial cells. The alveolar epithelium is regarded as the primary host defense, comes to form robust barrier during serious inflammatory response caused by pathogens in ALI.29,30 Studies suggested that lung injury and inflammatory response may result in apoptosis of endothelial and epithelial cells.7,8 While inhibition of cell apoptosis significantly decreases the lung injury and mortality of ALI.31 Despite the improvements in treating the disease, the mortality of ALI is still in high level. Thus, novel approaches for ALI therapy are necessitated. GPs exert pharmacological effects, such as anti-inflammatory, anti-oxidative stress, etc.11,20 Actually, inflammatory response plays a significant role in endotoxin induced ALI, and severe inflammatory response also leading to apoptosis of endothelial and epithelial cell during ALI, impairing the integrity of alveolar-capillary barrier and the microvascular endothelium and alveolar epithelium barrier. In the study, LPS induced severe injury of lung tissue, and serious expression of inflammatory cytokines in vivo, while GPs effectively alleviated lung injury and improved the survival ratio of ALI mice. Thus, GPs exerted therapeutic effects for ALI. Further, we conducted a bioinformatics analysis of LPS-induced ALI mice to identify hub-genes that could be the potential targets in treating ALI. In the study, we identified the 20 potential targets of LPS-induced ALI, they were mainly enriched in NF-κB and TNF-α signal pathways. In view of this, we hypothesized that GPs may have potential therapeutic effects on ALI via inhibiting NF-κB and TNF-α pathways. Next, we found that GPs inhibited the expression of p-P65 cytokines (TNF-α, IL-1β, IL-6, VCAM1) induced by LPS in vivo, indicating that GPs exerted anti-inflammatory effects via inhibiting NF-κB and TNF-α pathways. Pulmonary vascular endothelial injury is also a hallmark event during ALI, which leads to pulmonary congestion, pulmonary edema and respiratory distress eventually.32 Therefore, we tested the inflammatory response level in HPMEC (human pulmonary microvascular endothelial cells line). LPS dose dependently increased the expression of p-p65 and inflammatory cytokines. However, our results demonstrated that GPs could also alleviate the inflammatory response in HPMEC via inhibiting NF-κB and TNF-α pathways. Inflammation related injury and repair process of alveolar epithelial cells are integral to the pathogenesis of ALI.33 A series of inflammatory response may damage epithelial cells.14,34 We next hypothesized that it may also inhibit the inflammatory response in alveolar epithelial cells. The results suggested GPs effectively inhibited the inflammatory response in MLE-12. Thus, our study suggested that GPs could alleviate inflammatory response induced by LPS in endothelial and epithelial cells via inhibiting NF-κB and TNF-α pathways.
Studies also suggested that GPs induced apoptosis of cancer cells in a high dosage.35 In our study, we found GPs inhibited the cell viability of HPMEC at a dosage of 50 μg/mL. But we did not know the specific mechanism. Research indicated that higher dosage of GPs impaired integrity of cell membrane and membrane potential, finally leading to apoptosis.33 However, lower dosage of GPs exerted an anti-apoptosis effect.12,13 The present study revealed that GPs at 10 μg/mL effectively restrained the apoptosis of the HPMEC and MLE-12 induced by LPS challenge. LPS challenge up-regulated the expression of Bax (a pro-apoptotic protein) and cleaved caspase-3 (an apoptotic effector protein), down-regulated Bcl-2 (an anti-apoptotic protein) in HPMEC, indicating LPS-induced apoptosis in HPMEC. However, the results of Western blots and flow cytometry suggested GPs effectively inhibited the apoptosis of the HPMEC and MLE-12, indicating GPs exerted an anti-apoptosis effects in endothelial and epithelial cells during ALI. But, we are not sure that the protective effects of GPs against apoptosis were directly related to the regulation on NF-κB pathway. However, inhibiting inflammation may reduced the apoptosis of endothelial and epithelial cells during ALI.36,37 Figure 10 provides a brief schematic drawing to illustrate the inhibitory effect of GPs on NF-κB and TNF-α signal pathways and inhibitory of apoptosis of endothelial and epithelial cells.
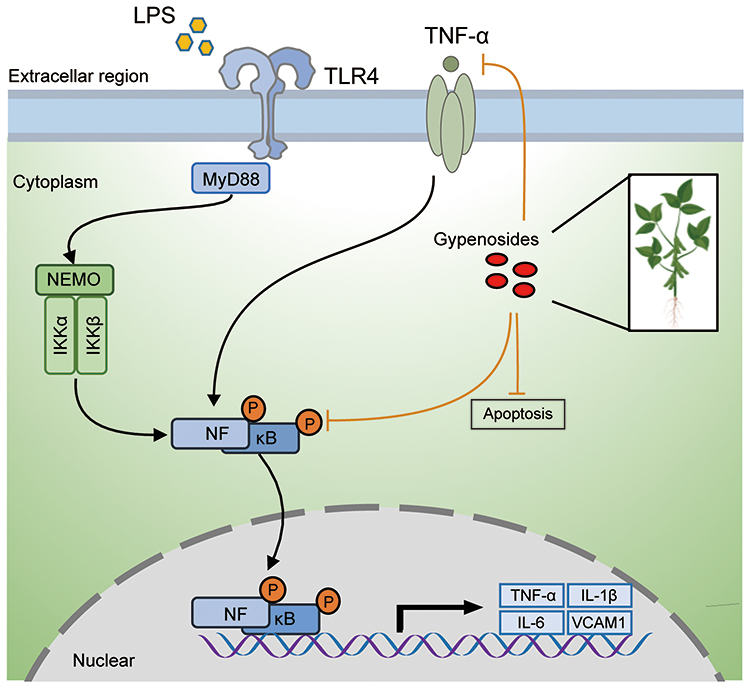 |
Figure 10 Schematic drawing to illustrate the inhibitory effect of GPs on NF-κB and TNF-α signal pathway and inhibitory of apoptosis of endothelial and epithelial cells. GPs could inhibit the phosphorylation of NF-κB, strain the nuclear translocation of NF-κB, thus inhibiting the release of cytokines, such as TNF-α, IL-1β, IL-6 and VCAM1. GPs also help to inhibit the apoptosis of endothelial and epithelial cells induced by LPS challenge, which may play a significant role in the repairment of ALI. |
In spite of positive results of potential therapeutic effects of GPs in ALI in the study, there also existed some limitations. The dosage of intraperitoneal GPs of 100 mg/kg in vivo was applied as previously described,21 maybe the dosage was not optimal. In order to be consistent with in vivo experiments, the HPMEC and MLE-12 were stimulated for 8h, while some experiments detected p-p65 expression in LPS-stimulated cells only after 30 min. However, we detected the high level of p-p65 expression after 8h LPS stimulation in the MLE-12 and HPMECs, which suggested that the high level of inflammatory response. According to the pharmacological components, GPs had similar skeleton and components as ginsenosides, while ginsenosides could enter the cytoplasm by way of pinocytosis.38 For this phenomenon, we speculated that GPs could also enter the cytoplasm by way of pinocytosis, other than the way of second messenger, but the potential mechanism was still unknown. During the development of ALI, many inflammatory pathways activated, including NF-kb and TNF-α pathways. However, there were no existing studies of the effects of GPs on ALI. Our study firstly found that GPs reduced the inflammatory response and intensity of lung injury. In order to figure out the specific effects of GPs on ALI, we conducted a bioinformatic analysis of LPS-induced ALI, NF-kb and TNF-α pathways were the top pathways involved LPS-induced ALI, so we only tested the effects of GPs on these two pathways. Actually, many inflammatory pathways couldbe activated, whether GPs also inhibited other pathways still needs more study to justify. We identified 20 hub-genes, they were mainly enriched in NF-κB and TNF-α pathways, then we tested whether GPs may inhibit these pathways during ALI. Actually, GPs may have therapeutic effect on ALI through a certain specific gene (maybe one of the 20 hub-genes identified in this study), more studies are needed to prove it. A recent study suggested GPs could inhibitcarbon tetrachloride induced liver fibrosis in rats.39 Actually, pulmonary fibrosis is a common phenomenon during the progress of ALI. Therefore, studies should be conducted to provide more evidence of GPs on pulmonary fibrosis.
Conclusion
The present study revealed that GPs inhibited the inflammatory response and apoptosis of endothelial and epithelial cells during ALI. Therefore, the results of our study provided novel approaches for the prevention and therapy of ALI. But the further and specific mechanisms of GPs on ALI are still worth studying.
Acknowledgment
None.
Funding
The study was financially supported by grants from the National Natural Science Foundation of China (No. 81701943 and No. 81971813).
Disclosure
The authors declared that they have no conflicts of interest for this work and no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
1. Ware LB, Matthay MA. The acute respiratory distress syndrome. N Eng J Med. 2000;342(18):1334–1349. doi:10.1056/NEJM200005043421806
2. Chen X, Wang Y, Xie X, et al. Heme oxygenase-1 reduces sepsis-induced endoplasmic reticulum stress and acute lung injury. Mediators Inflamm. 2018:9413876. doi:10.1155/2018/9413876
3. Zhao Y, Ridge K, Zhao J. Acute lung injury, repair, and remodeling: pulmonary endothelial and epithelial biology. Mediators Inflamm. 2017:9081521. doi:10.1155/2017/9081521
4. Kitamura Y, Hashimoto S, Mizuta N, et al. Fas/FasL-dependent apoptosis of alveolar cells after lipopolysaccharide-induced lung injury in mice. Am J Respir Crit Care Med. 2001;163(3 Pt 1):762–769. doi:10.1164/ajrccm.163.3.2003065
5. Gibbons M, Duffin R, Dorward DA, et al. Monocytes control second-phase neutrophil emigration in established lipopolysaccharide-induced murine lung injury. Am J Respir Crit Care Med. 2012;186(6):514–524. doi:10.1164/rccm.201112-2132OC
6. Shen W, Gan J, Xu S, Jiang G, Wu H. Penehyclidine hydrochloride attenuates LPS-induced acute lung injury involvement of NF-kappa B pathway. Pharmacol Res. 2009;60(4):296–302. doi:10.1016/j.phrs.2009.04.007
7. Fang Y, Gao F, Hao J, et al. Molecular mechanisms in lipopolysaccharide-induced pulmonary endothelial barrier dysfunction. Int Immunopharmacol. 2015;29(2):937–946. doi:10.1016/j.intimp.2015.10.010
8. Ji Q, Sun Z, Yang Z, et al. Protective effect of ginsenoside Rg1 on LPS-induced apoptosis of lung epithelial cells. Mol Immunol. 2018;22(18):30935. doi:10.1016/j.molimm.2018.11.003
9. Cai H, Liang Q, Ge G. Gypenoside attenuates β amyloid-induced inflammation in N9 microglial cells via SOCS1 signaling. Neural Plast. 2016;6362707. doi:10.1155/2016/6362707
10. Zhang HK, Ye Y, Zhao ZN, et al. Neuroprotective effects of gypenosides in experimental autoimmune optic neuritis. Int J Ophthalmol. 2017;10(4):541–549. doi:10.18240/ijo.2017.04.07
11. Li K, Li H, Xu W, et al. Research on the potential mechanism of gypenosides on treating thyroid-associated ophthalmopathy based on network pharmacology. Med Sci Monit. 2019;25:4923–4932. doi:10.12659/MSM.917299
12. Zhang H-K, Ye Y, Li K-J, Zhao Z-N, He J-F. Gypenosides prevent H2O2-induced retinal ganglion cell apoptosis by concurrently suppressing the neuronal oxidative stress and inflammatory response. J Mol Neurosci. 2020;70(4):618–630. doi:10.1007/s12031-019-01468-9
13. Chang L, Shi R, Wang X, Bao Y. Gypenoside A protects ischemia/reperfusion injuries by suppressing miR-143-3p level via the activation of AMPK/Foxo1 pathway. Biofactors. 2020;46(3):432–440. doi:10.1002/biof.1601
14. Cao C, Yin C, Shou S, et al. Ulinastatin protects against LPS-induced acute lung injury by attenuating TLR4/NF-κB pathway activation and reducing inflammatory mediators. Shock. 2018;50(5):595–605. doi:10.1097/SHK.0000000000001104
15. Wolthuis EK, Vlaar AP, Choi G, et al. Mechanical ventilation using non-injurious ventilation settings causes lung injury in the absence of pre-existing lung injury in healthy mice. Crit Care. 2009;13(1):R1. doi:10.1186/cc7688
16. Dong P, Yu B, Pan L, et al. Identification of key genes and pathways in triple-negative breast cancer by integrated bioinformatics analysis. Biomed Res Int. 2018;2018:2760918. doi:10.1155/2018/2760918
17. Reimand J, Isserlin R, Voisin V, et al. Pathway enrichment analysis and visualization of omics data using g:profiler, GSEA, cytoscape and enrichmentmap. Nat Protoc. 2019;14(2):482–517. doi:10.1038/s41596-018-0103-9
18. Cline MS, Smoot M, Cerami E, et al. Integration of biological networks and gene expression data using Cytoscape. Nat Protoc. 2007;2(10):
19. Wang F, Dang Y, Wang J, Zhou T, Zhu Y. Gypenosides attenuate lipopolysaccharide-induced optic neuritis in rats. Acta Histochem. 2018;120(4):340–346. doi:10.1016/j.acthis.2018.03.003
20. Wang XS, Yang L, Ying FP, et al. Gypenoside IX suppresses p38 MAPK/Akt/NF-kappa B signaling pathway activation and inflammatory responses in astrocytes stimulated by proinflammatory mediators. J Sep Sci. 2007;40(6):2137–2150. doi:10.1007/s10753-017-0654-x
21. Dong SQ, Zhang QP, Zhu JX, et al. Gypenosides reverses depressive behavior via inhibiting hippocampal neuroinflammation. Biomed Pharmacother. 2018;106:1153–1160. doi:10.1016/j.biopha.2018.07.040
22. Ji Y, Zhang G, Zhu H, et al. Indinavir plus methylprednisolone ameliorates experimental acute lung injury in vitro and in vivo. Shock. 2018;49(2):196–204. doi:10.1097/SHK.0000000000000911
23. Shen YH, Wang LY, Zhang BB, et al. Ethyl rosmarinate protects high glucose-induced injury in human endothelial cells. Molecules. 2018;23(12):3372. doi:10.3390/molecules23123372
24. Hou Y, Liang D, Liu Y, Chen H, Lou X. Up-regulation of DcR3 in microbial toxins-stimulated HUVECs involves NF-κB signaling. BMC Biochem. 2018;19(1):13. doi:10.1186/s12858-018-0102-z
25. Peng XP, Li XH, Li Y, Huang XT, Luo ZQ. The protective effect of oleanolic acid on NMDA-induced MLE-12 cells apoptosis and lung injury in mice by activating SIRT1 and reducing NF-κB acetylation. Int Immunopharmacol. 2019;70:520–529. doi:10.1016/j.intimp.2019.03.018
26. Cheng KT, Xiong S, Ye Z, et al. Caspase-11-mediated endothelial pyroptosis underlies endotoxemia-induced lung injury. J Clin Invest. 2017;127(11):4124–4135. doi:10.1172/JCI94495
27. Johnson ER, Matthay MA. Acute lung injury: epidemiology, pathogenesis, and treatment. J Aerosol Med Pulm Drug Deliv. 2010;23:243–252. doi:10.1089/jamp.2009.0775
28. Matthay MA, Ware LB, Zimmerman GA. The acute respiratory distress syndrome. J Clin Invest. 2012;122(8):2731–2740. doi:10.1172/JCI60331
29. Crosby LM, Waters CM. Epithelial repair mechanisms in the lung. Am J Physiol Lung Cell Mol Physiol. 2010;298:L715–L731. doi:10.1152/ajplung.00361.2009
30. Gropper MA, Wiener-Kronish J. The epithelium in acute lung injury acute respiratory. Curr Opin Crit Care. 2008;14:11–15. doi:10.1097/MCC.0b013e3282f417a0
31. Kawasaki M, Kuwano K, Hagimoto N, et al. Protection from lethal apoptosis in lipopolysaccharide- induced acute lung injury in mice by a caspase inhibitor. Am J Pathol. 2000;157:597–603. doi:10.1016/S0002-9440(10)64570-1
32. Dong W, He B, Qian H, et al. RAB26-dependent autophagy protects adherens junctional integrity in acute lung injury. Autophagy. 2018;14(10):1677–1692. doi:10.1080/15548627.2018.1476811
33. Miyoshi K, Yanagi S, Kawahara K, et al. Epithelial Pten controls acute lung injury and fibrosis by regulating alveolar epithelial cell integrity. Am J Respir Crit Care Med. 2013;187(3):262–275. doi:10.1164/rccm.201205-0851OC
34. Nie Y, Wang Z, Chai G, et al. Dehydrocostus lactone suppresses LPS-induced acute lung injury and macrophage activation through NF-κB signaling pathway mediated by p38 MAPK and Akt. Molecules. 2019;24(8):1510. doi:10.3390/molecules24081510
35. Yan H, Wang X, Niu J, Wang Y, Wang P, Liu Q. Anti-cancer effect and the underlying mechanisms of gypenosides on human colorectal cancer SW-480 cells. PLoS One. 2014;9(4):e95609. doi:10.1371/journal.pone.0095609
36. Yao Y, Jia H, Wang G, Ma Y, Sun W, Li P. miR-297 protects human umbilical vein endothelial cells against LPS-induced inflammatory response and apoptosis. Cell Physiol Biochem. 2019;52(4):696–707. doi:10.33594/000000049
37. Yulis M, Quiros M, Hilgarth R, Parkos CA, Nusrat A. Intracellular Desmoglein-2 cleavage sensitizes epithelial cells to apoptosis in response to pro-inflammatory cytokines. Cell Death Dis. 2018;9(3):389. doi:10.1038/s41419-018-0380-9
38. Wong AS, Che CM, Leung KW. Recent advances in ginseng as cancer therapeutics: a functional and mechanistic overview. Nat Prod Rep. 2015;32(2):256–272. doi:10.1039/c4np00080c
39. Song YN, Dong S, Wei B, Liu P, Zhang YY, Su SB. Metabolomic mechanisms of gypenoside against liver fibrosis in rats: an integrative analysis of proteomics and metabolomics data. PLoS One. 2017;12(3):e0173598. doi:10.1371/journal.pone.0173598
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.