")
Back to Journals » Journal of Hepatocellular Carcinoma » Volume 7
Gut Microbiota, Peroxisome Proliferator-Activated Receptors, and Hepatocellular Carcinoma
Authors Yu Q, Wu L , Ji J, Feng J , Dai W, Li J, Wu J, Guo C
Received 26 August 2020
Accepted for publication 10 October 2020
Published 29 October 2020 Volume 2020:7 Pages 271—288
DOI https://doi.org/10.2147/JHC.S277870
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Ahmed Kaseb
Qiang Yu,1,2 Liwei Wu,2 Jie Ji,2 Jiao Feng,2 Weiqi Dai,1– 3 Jingjing Li,1,2 Jianye Wu,1 Chuanyong Guo1,2
1Department of Gastroenterology, Putuo People’s Hospital, Tongji University School of Medicine, Shanghai 200060, People’s Republic of China; 2Department of Gastroenterology, Shanghai Tenth People’s Hospital, Tongji University School of Medicine, Shanghai 200072, People’s Republic of China; 3Shanghai Tongren Hospital, Shanghai Jiaotong University School of Medicine, Shanghai 200336, People’s Republic of China
Correspondence: Jianye Wu; Chuanyong Guo
Department of Gastroenterology, Putuo People’s Hospital, Tongji University School of Medicine, Shanghai 200060, People’s Republic of China
Email [email protected]; [email protected]
Abstract: Hepatocellular carcinoma (HCC) is one of the most common malignant tumors in the world. HCC incidence rate is sixth and mortality is fourth worldwide. However, HCC pathogenesis and molecular mechanisms remain unclear. The incidence of HCC is associated with genetic, environmental, and metabolic factors. The role of gut microbiota in the pathogenesis of HCC has attracted researchers’ attention because of anatomical and functional interactions between liver and intestine. Studies have demonstrated the involvement of gut microbiota in the development of HCC and chronic liver diseases, such as alcoholic liver disease (ALD), nonalcoholic fatty liver disease (NAFLD), and liver cirrhosis. Peroxisome proliferator-activated receptors (PPARs) are a group of receptors with diverse biological functions. Natural and synthetic PPAR agonists show potential for treatment of NAFLD, liver fibrosis, and HCC. Recent studies have demonstrated that PPARs take part in gut microbiota inhabitation and adaptation. This manuscript reviews the role of gut microbiota in the development of HCC and precancerous diseases, the role of PPARs in modulation of gut microbiota and HCC, and potential of gut microbiota for HCC diagnosis and treatment.
Keywords: gut microbiota, hepatocellular carcinoma, PPARs, carcinogenesis
Introduction
More than 1×1014 microorganisms colonize the human gastrointestinal tract, including bacteria (about 1×104 bacterial species), archaea, fungi, and viruses.1 Sequencing results for 1267 human intestinal microbial samples from three continents have shown that human gastrointestinal tract contains more than 9 million genes, which is 150 times the number of all human genes. Among these genes, more than 99% are bacterial, thus intestinal microbiota are also called intestinal microflora.2 Most of the bacteria in the intestine belong to five phyla: Bacteroidetes, Firmicutes, Actinobacteria, Proteobacteria, and Verrucomicrobia. Bacteroidetes and Firmicutes account for 90–95% of all gut microorganisms in healthy people.1 Gut microbiota play a key role in human health through modulation of metabolism and immunity.3 Thus, gut microbiota are considered to be the “forgotten organ”4 Increasing evidence has demonstrated the involvement of gut microbiota in human diseases, such as inflammatory bowel disease (IBD),5 type 2 diabetes mellitus (T2DM),6 obesity,7 Alzheimer’s disease,8 and heart failure.9 The underlying mechanisms might be related to microbiota dysbiosis, alternation of bacterial metabolite production, and host immune disorder.10 Gut microbiota is a dynamic system, which can be influenced by a serious of factors, including age, immune system formation, geographical location, and short- and long-term dietary structure.11
Liver cancer is one of the most common malignant tumors of the digestive system and is characterized by a high mortality rate.12–14 In 2018, there were 841,080 new liver cancer cases in the world, including 596,574 in males and 244,506 in females. A total of 781,631 patients died of liver cancer in 2018, of which 548,375 were males and 233,256 were females. Liver cancer became the sixth most common cancer in the world and the fourth leading cause of cancer death worldwide.15 Hepatocellular carcinoma (HCC) makes up 75–85% of primary liver cancer cases. The pathogenesis of HCC is complex and involves a series of factors.16,17 In China, HCC is mainly attributed to hepatitis B virus (HBV) infection.18–20 In USA, the predominant HCC etiology is NAFLD.21,22 Commonly used diagnostic methods for HCC include abdominal ultrasonography, computed tomography (CT), magnetic resonance imaging (MRI), selective hepatic angiography, and detection of serum alpha-fetoprotein and α-L-fucosidase.23 Liver biopsy is the gold standard for the diagnosis of HCC. However, HCC diagnosis is usually performed with non-invasive techniques, such as dynamic-MRI and/or dynamic-CT. During dynamic-MRI or dynamic-CT detection, comparing with surrounding liver, HCC nodule shows hypersignal intensity in the arterial phase (wash-in), and hypodensity or hyposignal intensity in the venous phase (wash-out). The detection of nodules with wash-in and wash-out in liver cirrhosis patients has an approximately 95% positive predictive value (PPV) for HCC diagnosis.24 If the patient is non-fibrotic or has no typical HCC imaging manifestations (wash-in and wash-out), then liver biopsy is recommended.25 However, liver biopsy is not suitable for patients with coagulopathy and hypertension and its sensitivity is not high enough for the diagnosis of early HCC. In addition, there are no early biomarkers and specific symptoms in early stages, resulting in diagnosis at advanced stages for the majority of HCC patients. Hence, a noninvasive diagnostic method for HCC at an early stage is urgently needed. Increasing evidence has indicated the potential of gut microbiota as a novel diagnostic tool for HCC and other precancerous diseases.26,27
In the past decade, the findings in experimental and clinical studies demonstrated the role of gut microbiota in the different stages of liver diseases and the development of liver cirrhosis and HCC.28 The majority of HCC develop in patients with liver cirrhosis.28 Pathological changes in liver cirrhosis, such as portal hypertension and decreased gastric acid secretion, directly destroy intestinal barrier and indirectly affect the composition of gut microbiota, promoting the pathological bacteria translocation and the progression of liver diseases.29 The mechanisms by which gut microbiota promotes the hepatocarcinogenesis involves the leaky gut, gut microbiota dysbiosis, activated lipopolysaccharide (LPS)- Toll-like receptor 4 (TLR4) signaling and the alternation of bacterial metabolites.30 Modulation of gut microbiome via administration of probiotics or antibiotics might suppress the occurrence and progression of HCC in animal models.31–33 T-cell checkpoint inhibition is considered the breakthrough in cancer immunotherapy. The anti-programmed cell death-1 (PD1) agent nivolumab has been approved for advanced HCC treatment after sorafenib failure.34 A recent review indicated that gut microbiota could modulate the efficiency of PD-1 inhibition in melanoma and might influence the efficiency of immune check point therapy in HCC.28 The present manuscript focuses on the mechanisms by which gut microbiota promotes the development of HCC and the potential of gut microbiome as a novel diagnostic biomarker and therapeutic target for HCC.
Intestinal Barrier and Gut-Liver Axis
Intestinal barrier prevents harmful substances and pathogens from entering the human body and maintains its stability. A normal intestinal barrier consists of mechanical, biological, immune, and chemical barriers. The mechanical barrier includes the intestinal mucus layer, peristalsis, and epithelium. Normal peristalsis of the small intestine can prevent bacteria from remaining near the intestinal mucosa for too long and reduces the chance of bacteria passing through the mucosa to reach the epithelium. Intestinal flora competes with pathogenic microorganisms for nutrition and forms a biological barrier on the surface of intestinal mucosa, which can prevent the invasion and colonization of pathogenic microorganisms. Some gut bacteria secrete bacteriostatic substances, bacteriocins, and organic acids, which can kill pathogenic bacteria and neutralize toxins.35 Gut microbiota have been reported to play a crucial role in the maturation of human immune system by regulating maturation and differentiation of T, B, and dendritic cells and maintaining gut homeostasis. In turn, intestinal cells can regulate intestinal flora through antimicrobial peptides secreted by Paneth cells.36,37
The immune barrier consists of intestinal mucosa lymphoid tissue and secretory antibodies of intestinal plasma cells (sIgA).38 S-IgA produced by the gut-associated lymphoid tissue (GALT) can selectively coat Gram-negative bacteria in the gut, form an antigen-antibody complex, block the combination of bacterial and epithelial cell receptors, stimulate secretion of intestinal mucus, and accelerate the flow of the mucus layer, which can effectively prevent bacterial adhesion to intestinal mucosa.39 The chemical barrier consists of mucus secreted by the intestinal epithelium, digestive fluid, and bacteriostatic substances produced by resident bacteria.1,40
The “gut-liver” axis theory based on a strong anatomical and functional interaction between the liver and the gut was first proposed in 1998.41 The liver has two independent blood supply sources: the hepatic artery and portal vein. The portal vein makes up approximately 70–75% of liver’s blood supply. The portal vein brings the blood from the spleen and intestines to the liver and contains nutrients absorbed by the digestive tract as well as metabolites and antigens of gut microbiota, such as lipopolysaccharides (LPSs) and bacterial DNA. Bile secreted by the hepatocytes is essential for the digestion and absorption of lipids and fat soluble vitamins. In normal conditions, after specific receptors like nucleotide-binding oligomerization domain-like receptors (NLRs) and Toll-like receptors (TLRs) recognize bacterial metabolites, the liver clears intestinal bacteria and their products, such as LPSs, which are mediators of inflammation and antigens, in order to maintain a stable internal environment.42 Hence, normal liver function is a part of the intestinal barrier. Of note, constant exposure to low-level bacterial metabolites suppresses the activation of immune cells by TLRs, which is called “endotoxin tolerance,” and activates immune suppression via cytokines, such as transforming growth factor beta (TGFβ), interleukin (IL)-10, and hepatocyte growth factor (HGF).43 In physiological conditions, gut microbiota can regulate hepatic lipogenesis, bile acid metabolism, oxidation, and levels of inflammation mediators in the liver. In turn, liver regulates gut microbiota through secretion of bile.44
In addition, the liver and gut microbiota affect each other in pathological conditions. The clinical studies indicated that patients with poorer liver function have higher gut permeability and mucosal impairment.45,46 High levels of portal vein LPSs were observed in patients with liver cirrhosis46 due to overgrowth of intestinal bacteria and dysbiosis, which are attributed to reduced gastric acid and bile acid secretion and low intestinal motility disturbance caused by liver cirrhosis.47 The LPSs activate Kupffer cells (KCs) and hepatic stellate cells (HSCs) in the liver, leading to overexpression of inflammatory factors, including tumor necrosis factor (TNF-α) and interleukin (IL)-6, and inflammatory response and oxidative stress of the liver, finally causing hepatocyte DNA damage and the occurrence and accumulation of mutations.30 In turn, over release of inflammation mediators by KCs and HSCs aggravates intestinal mucosal injury, while portal hypertension results in the edema of intestinal mucosa, which increases its permeability.48,49 The next section will investigate the effects of gut microbiota on the occurrence and progression of HCC and its underlying mechanisms.
Mechanism for Intestinal Microorganism Promotion in HCC and Precancerous Diseases
Recent studies have revealed that alteration of the intestinal barrier and composition of gut microbiota, such as LPSs and deoxycholic acid (DCA), promote the development of CLD and HCC by inducing chronic liver inflammation and injury. The peroxisome proliferator-activated receptor (PPAR) pathway has been reported to modulate microbial inhabitation and adaptation, which might influence the onset and progression of HCC.
Leaky Gut
In pathological conditions, the structure of the gut microbial community is disturbed, leading to the reduction of beneficial microbial organisms, overgrowth of pathobionts or potentially harmful microorganisms, and loss of microbial organisms.50 This process is called dysbiosis. Gut microbiota dysbiosis, reduced bile acid secretion, overexpression of inflammatory factors in the intestine, and other factors might destroy the intestinal barrier and increase permeability of the intestinal mucosa, leading to translocation of gut bacteria and high levels of bacterial toxins and metabolites in the portal vein, which activates the immune response in the liver. This phenomenon is known as a leaky gut.51,52 HCC is a consequence of a vicious cycle of chronic liver injury, inflammation, and regeneration and is the terminal stage of CLD, including chronic viral hepatitis, alcoholic liver disease (ALD), non-alcoholic fatty liver disease (NAFLD), and non-alcoholic steatohepatitis (NASH).53 Moreover, the majority of HCCs occur in patients with liver fibrosis and cirrhosis. Hence, CLD, liver fibrosis, and liver cirrhosis are regarded as precancerous HCC diseases.54 A recent study indicated that the leaky gut and gut microbiota dysbiosis are observed in patients with CLD, and liver cirrhosis and might contribute to the occurrence and progression of HCC in mice.30 It should be noted that in hepatocarcinogenesis induced by diethylnitrosamine (DEN)+ carbon tetrachloride (CCl4), gut sterilization had a strong effect on the inhibition of HCC formation in the late stages and a mild effect in the early stages, demonstrating that tumor-promoting signals induced by the leaky gut mainly occur during late stages of hepatocarcinogenesis.33
LPS is a cell wall component of Gram-negative bacteria and is known as an endotoxin. It is released only when the bacteria die. LPS binds to its Toll-like receptor 4 (TLR4) expressed by hepatocytes, KCs, and hepatic stellate cells, promoting hepatic inflammation and subsequent liver fibrogenesis and hepatocarcinogenesis.33 Dapito et al33 have found that administration of low dose LPSs through subcutaneous osmotic pumps for 3 months promoted hepatocarcinogenesis induced by DEN plus CCl4 in mice. The LPS treatment increased the release of inflammatory factors, tumor number, and size. Tumor growth was suppressed after the LPS levels were reduced by the treatment of cocktail antibiotics.33 When the intestinal barrier is destroyed by a series of factors, the permeability of intestinal mucosa increases and LPSs and other gut microbiota metabolites translocate from the intestine to the portal vein, leading to subsequent liver injury. LPSs can also enter circulating blood, which is called intestinal endotoxemia.55 Hence, LPS levels in the portal vein and circulating blood to some extent reflect the permeability of intestinal mucosa. High levels of LPSs have been observed in patients with CLD and HCC and in animal models. It has been reported that patients infected with HBV and hepatitis C virus (HCV) have higher LPS serum levels than the uninfected individuals.56 Ethanol and its metabolite acetaldehyde can destroy the intestinal barrier and increase the LPS levels in the portal vein and serum in rodent models administered with acute and chronic ethanol treatment.57 Accordingly, plasma LPS concentrations are elevated in patients with ALD.58 Insulin resistance plays a key role in the pathogenesis of NAFLD. In a high fat diet-induced mouse obesity model, LPS serum levels increased two- to three-fold, inducing insulin resistance by upregulating inflammatory pathways.59,60 Accordingly, an increase in intestinal permeability was observed in patients with NAFLD due to alternation of gut epithelial tight junction.61 More importantly, a study conducted by Lin et al46 have demonstrated that LPS levels in the portal vein were positively correlated with the severity of liver cirrhosis (Child-Turcotte-Pugh scores) and highest levels were observed in patients with Child-Turcotte-Pugh cirrhosis stage C. Moreover, serum LPS levels were elevated in patients with HCC and HCC animal models.62,63 In addition, Bellot et al64 have found that plasma levels of bacterial DNA, which activate Toll-like receptor (TLR)-9, were elevated in patients with CLD. These findings demonstrate that the livers of CLD and HCC patients were exposed to high LPS levels and other bacterial products due to a leaky gut, which might promote the onset and progression of HCC.
Dysbiosis
The qualitative and quantitative alternations have been observed in gut microbiota of CLD and HCC patients, including a change in bacterial abundance, loss of beneficial bacteria, and increase in pathogens.65 This process is known as dysbiosis. Dysbiosis can affect the progression of CLD and HCC by altering microbiota metabolites, such as LPSs, short-chain fatty acids (SCFAs), and deoxycholic acid (DCA). By performing 16S rRNA gene sequencing, researchers have attempted to figure out the difference in gut microbiota between CLD or HCC patients and healthy individuals. The relative findings and references have been listed in Table 1.66–74
The above findings indicate that gut microbiota dysbiosis in CLD patients is disease-specific. However, the majority of CLD patients experience a stage of liver cirrhosis in the process of developing HCC. Research on gut microbiota dysbiosis in liver cirrhosis patients has included patients with diverse underlying CLD, demonstrating that at least some of the microbiota dysbiosis features in liver cirrhosis are common to different aetiologies, and the features are driven by end-stage liver disease features, including reduced bile secretion, portal hypertension, and changes in intestinal immune barrier.27,30 Grat et al have reported an abundance of Escherichia coli in patients with liver cirrhosis and HCC.75 Increased levels of Streptococcus, Veillonella, Clostridium, Prevotella, and Enterobacteriaceae, Streptococcaceae, Veillonellaceae, Pasteurellaceae, and Fusobacteriaceae families were observed in patients with liver cirrhosis, as well as decreased levels of Bacteroides, Eubacterium, and Alistipes and Lachnospiraceae and Bacteroidaceae families27,76 (Table 1). In addition, dysbiosis in patients with decompensated liver cirrhosis was more obvious than that in patients with compensated liver cirrhosis, demonstrating that it is the cirrhosis stage and not the underlying CLD that drives gut microbiota dysbiosis in liver cirrhosis.77
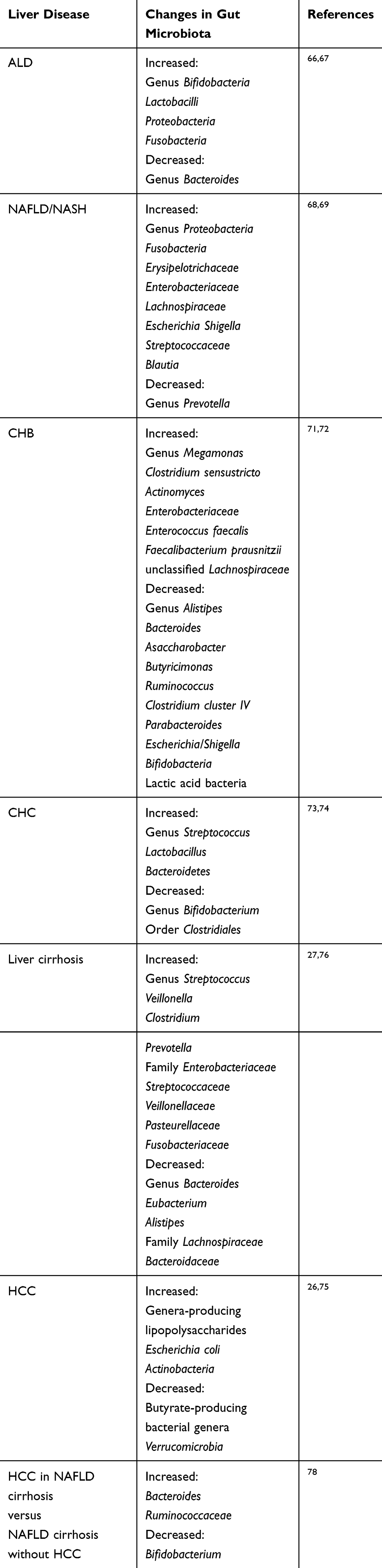 |
Table 1 Changes in Gut Microbiota for Different Liver Diseases |
Many HCC patients have been diagnosed at advanced stages due to the lack of effective strategy for early diagnosis. However, recent research on gut microbiota dysbiosis in HCC patients makes early diagnosis possible. A study conducted by Ren et al26 collected fecal samples from healthy individuals and liver cirrhosis and HCC patients in East, Central, and Northwest China. Then, fecal microbial diversity and composition were identified. Gut microbiota diversity in cirrhosis patients was lower than that in healthy individuals. However, diversity then increased from cirrhosis to early HCC with cirrhosis. Compared to healthy individuals, the levels of butyrate-producing genera were decreased, while genera-producing LPSs was increased in patients with early HCC (Table 1). Thirty microbial markers were identified using five-fold cross-validation in a random forest model. Most importantly, these markers were further verified in patients from Central and Northwest China, achieving a cross-regional validation. However, this study has several limitations, influencing the interpretation of results to some extent. First, in the discovery cohort, the patients with early HCC had impaired liver function and higher portal hypertension compared with patients with liver cirrhosis. It might be the severity of liver dysfunction rather than existence of HCC that caused the difference in gut microbiota between the two groups. Second, in the validation cohort, the authors only enrolled healthy individuals and patients with HCC. The diagnostic efficacy of these microbial markers should be further investigated by studies conducted on patients with HCC and patients with CLD without HCC.
In a study conducted on NAFLD patients, fecal microbial diversity was found to decrease from healthy individuals to patients with NAFLD. However, no difference in fecal microbial diversity was found between patients with NAFLD-related cirrhosis and HCC and patients with NAFLD-related cirrhosis without HCC.78 Compared with patients with NAFLD-related cirrhosis without HCC, patients with NAFLD-related cirrhosis and HCC have higher levels of Bacteroides and Ruminococcaceae and lower levels of Bifidobacterium.78 However, in another study conducted on NASH patients, no difference in fecal microbial composition was found between patients with and without HCC.79 The different findings in these studies might be due to the differences in techniques to analyze the samples, enrollment of cohorts, ethnics, geographical position and underlying liver diseases.80 It should be noted that even in studies on mice, the location of facility might influence the composition of gut microbiota. Moreover, a recent study indicated that gut microbiota in laboratory mice was significantly different from that in wild mice.81
Hence, in order to compare the findings and generalize conclusions in different studies, sample collection, bacterial lysis, DNA purification sequencing, bioinformatics and statistical analysis should be standardized in the future studies on gut microbiota in HCC patients.82 The efficacy and stability of gut microbiota as a diagnostic tool for HCC needs to be further validated in populations from different continents. The combination of microbial markers and current diagnostic strategies for HCC might promote the early diagnosis of HCC.26
LPS-TLR4 Pathway
It has been reported that leaky gut leads to high portal and plasma LPS levels in patients and animal models with CLD and HCC. Liver is the first target of gut microbe-associated molecular patterns (MAMPs). Pattern recognition receptors (PRRs), such as TLR4 and NLRs in the liver recognize MAMPs, especially LPSs. TLR4 is expressed in hepatocytes, HSCs, KCs, and endothelial cells. Activation of the LPS-TLR4 pathway leads to an inflammatory response in the liver.83 A functional study conducted in germ-free, gut-sterilized, or TLR-deficient rodents demonstrated that the LPS-TLR4 pathway plays a significant role in hepatocarcinogenesis. Gabele et al84 have found that dextran sulfate treatment can lead to high plasma LPS levels due to the leaky gut, promoting liver fibrosis and subsequent hepatocarcinogenesis in mice with NASH. Chronic administration of low dose LPSs can increase the size and number of tumors in DEN+CCl4-induced HCC. Moreover, cancer size and tumor number in germ-free or antibiotics-administered mice were reduced in the same model. In addition, inhibition of TLR4 reduced the tumor size and number but had no effect on tumor incidence.33
Cancer-promoting effect of the LPS/TLR4 pathway is attributed to multiple mechanisms. TLR4 in HSCs is activated after recognizing LPSs, resulting in nuclear factor kappa-B (NF-κB)-mediated overexpression of hepatomitogen epiregulin, which promotes mitosis. Furthermore, HSCs activated by LPSs secrete extracellular matrix, especially collagen.85 HSC activation and excessive collagen deposition play essential roles in pathogenesis of liver fibrosis and subsequent liver cirrhosis.86 In addition, epiregulin-deficiency suppresses hepatocarcinogenesis in mice administered with DEN and CCl4.33 In addition, activated HSCs secrete vascular endothelial growth factor (VEGF). VEGF promotes angiogenesis, which plays a key role in hepatocarcinogenesis.85,87
Activated LPS/TLR4 can suppress hepatocyte apoptosis via the NF-κB-mediated mechanism. Cleaved caspase-3 is a biomarker that promotes cell apoptosis. Cleaved caspase-3 levels were negatively correlated with tumor size in TLR4-deficient and gut-sterilized mice.33 In addition, activation of the LPS/TLR4 pathway in KCs resulted in overexpression of inflammatory factors, including IL-6 and TNF-α, leading to TNF-α- and IL-6-dependent compensatory hepatocyte proliferation and reduction in hepatocyte apoptosis.88
Recent studies have demonstrated that the LPS-TLR4 pathway is related to metastasis and poor prognosis in liver cancer patients.89 Jing et al have found that activation of the LPS-TLR4 pathway in HCC cells can enhance tumor cell invasive potential and induce NF-κB-mediated epithelial–mesenchymal transition, which is essential for tumor metastasis. In addition, the LPS/TLR4 and AKT+MAPK pathways collaborate to regulate cell proliferation, nitric oxide synthase (NOS) expression, and chemoresistance of HepG2 cells.90
Microbiota Metabolites
In addition to LPSs, it has been reported that other bacterial metabolites, such as DCA and SCFA, regulate HCC progression. Bile acids (BA) are synthesized by hepatocytes and discharged into the intestine through the common bile duct. The intestinal microorganisms metabolize conjugated bile acids into unconjugated primary bile acids and further metabolize them into secondary bile acids in the colon. On the one hand, bile acids exert direct antimicrobial effects by damaging the member of bacterium. On the other hand, bile acids exert indirect antimicrobial effects by increasing farnesoid X-activated receptor (FXR)-induced intestinal anti-microbial peptide synthesis.91,92 Bile acids regulate the growth and adhesion of gut microbiota, playing a key role in gut microbiota homeostasis.93 About 95% of the bile acids are absorbed by the intestinal epithelium and then enter the liver through the portal vein. They are then metabolized by the hepatocytes and secreted into the bile to complete the enterohepatic circulation of bile acids. The enterohepatic circulation of bile acids connects liver, intestine, and microbiota together.
Nuclear receptor FXR is the major BA-sensing receptor. BA modulates the proliferation of intestinal epithelial cells, maintaining the integrity of epithelial barrier in a FXR-dependent manner.94 FXR in hepatocytes has anti-inflammatory and liver generation-promoting effects.95,96 Meanwhile, FXR is the master regulator of bile acids. In physiological conditions, FXR modulates the BA synthesis and transport (discussed elsewhere).97 Cholesterol 7α-hydroxylase (CYP7A1) is the rate-limiting enzyme of bile acid synthesis. FXR is the transcriptional repressor of CYP7A1.91 After sensing bile acids, intestinal epithelial FXR is activated. The activated FXR increases the expression of fibroblast growth factor 15/19 (FGF 15/19), leading to the activation of hepatic fibroblast factor receptor4 (FGFR4). The activated FGFR4 suppresses the expression of CYP7A1, inhibiting the bile acids synthesis in the liver.98 In physiological conditions, FXR-FGF15/19-FGFR4 pathway plays a key role in BA homeostasis. Whole-body FXR-deficiency in mice promotes spontaneous hepatocarcinogenesis.99 In FXR-null mice, intestinal selective reactivation of FXR/FGF15 pathway restored BA homeostasis and inhibited spontaneous HCC development.100 During gut microbiota dysbiosis and inflammation, intestinal FXR is suppressed, leading to the inhibition of FGF19-FGFR4 signaling.101 The enterohepatic circulation of BA is disrupted, high-level BA in the enterocyte might aggravate the intestinal inflammation.98 Meanwhile, the suppressed FGF19-FGFR4 pathway increase hepatic BA synthesis by modulating c-Jun N-terminal kinase (JNK)–extracellular-signal-regulated kinase (ERK)–CYP7A1 pathway.102 Moreover, during the hepatic inflammation, hepatic nuclear factor kappa-B (NF-κB) signaling is activated, suppressing the expression of FXR in the liver. Inactivated FXR increased the hepatic BA synthesis by regulating downstream small heterodimer partner (SHP)-CYP7A1 pathway.103 Meanwhile, the hepatic BA transporters controlled by FXR are suppressed when FXR is inactivated.103 As a consequence, hepatic cholestasis and inflammation are aggravated, which might promote hepatocarcinogenesis.91
It has been reported that high-level bile acids may produce cytotoxicity by inducing cell necrosis.104 DCA is a secondary bile acid, which is produced after 7α-dehydroxylation of primary bile acids by the gut microbiota. In recent years, the role of DCA in the progression of CLD and HCC has been demonstrated. Increased levels of plasma DCA were found in NASH-induced HCC mouse model, which is induced by the administration of high-fat diet and dimethylbenz(a)anthracene (DMBA).105 Accordingly, increased abundance of Gram-positive bacterial strains, particularly the Clostridium genus, which is capable of producing DCA, was observed in mice fed with a high-fat diet. Conversely, no HCC was observed in mice treated with DMBA and a normal diet. In addition, HCC formation was suppressed after endogenous production of DCA was inhibited by administration of vancomycin, while HCC formation was promoted by the treatment with diets containing DCA.105 Lipoteichoic acid (LPA) is a component of Gram-positive bacterial cell wall and an agonist of Toll-like receptor 2 (TLR2). LPA collaborates with DCA to activate TLR2 in HSCs, leading to upregulation of senescence-associated secretory phenotype (SASP) and cyclooxygenase-2 (Cox-2). Cox-2-mediated prostaglandin-2 can inhibit antitumor immunity through prostaglandin EP4 receptor, thus promoting HCC progression.106
In addition, bile acid metabolism can regulate HCC growth by recruiting C-X-C chemokine receptor type 6+ natural killer T (CXCR6+ NKT) cells. Chemokine C-X-C motif ligand 16 (CXCL16) expressed in the sinusoidal endothelial cells is the solo ligand of CXCR6. CXCL16 can be activated by the primary bile acid and is suppressed by the secondary bile acid. Activated NKT cells secrete interferon-γ, kill tumor cells in a CD1d-dependent manner, and suppress tumor growth in the liver. Inhibition of conversion from primary to secondary bile acid via administration of vancomycin can induce NKT cell recruitment and suppress tumor growth, which can be reversed by oral supplementation with secondary bile acid.107
SCFA is a substance that can be absorbed by the intestinal tract and is produced by intestinal flora fermentation of a variety of human indigestible polysaccharides such as cellulose, including propionic and butyric acids. Propionic acid is mainly produced by Bacteroidetes and butyric acid is generally produced by Firmicutes. SCFA is the direct energy source for intestinal epithelial cells that can reduce apoptosis and maintain the integrity of the mechanical barrier. SCFA can also reduce pH in the intestine, inhibit growth and colonization of pathogenic bacteria, and suppress inflammatory reactions.108 Moreover, SCFA has been reported to regulate hepatocyte proliferation and differentiation and modulate T-reg cell differentiation via an epigenetic mechanism, reducing the inflammatory response in the liver.109 In addition, Ortega et al110 have found that a butyric acid prodrug tributyrin induces apoptosis in HCC cells by upregulating p53 in the nucleus. These properties suggest that SCFA metabolic disorder can be related to the development of HCC.
PPARs and Gut Microbiota
Peroxisome proliferator-activated receptors (PPARs) belong to a superfamily of nuclear receptors that can be activated by their specific ligands. PPAR ligands include endogenous ligands, such as 15-deoxy-Δ12,14-prostaglandin J2 (15d-PGJ2),111,112 SCFAs,113 and free fatty acids,114 as well as exogenous ligands, such as thiazolidinediones (TZDs),115 resveratrol,116 and honokiol.117 PPARs have been reported to play key roles in modulation of a variety of biological activities, including lipid and carbohydrate metabolism, bile acid synthesis, inflammation, and cell cycle.118–120 Three subtypes of PPARs have been identified in mammals: PPARα, PPARβ/δ, and PPARγ. PPARα is mainly expressed in the liver, heart, kidney, gastrointestinal tract, and adipose tissue.121 PPARβ/δ is expressed in the muscle, intestine, heart, and adipose tissue. PPARγ is expressed in the adipose tissue, colon, and immune cells.122
A recent study demonstrated that in order to colonize and survive in the gastrointestinal tract, gut microbiota modulate the host immune response by regulating the PPAR pathway in the intestinal epithelial and immune modulatory cells (Figure 1).123 Enterococcus faecalis is transferred from mother to child after birth. Are et al have found that co-culture with Enterococcus faecalis isolated from newborn babies can regulate PPARγ1 phosphorylation, enhancing DNA binding and transcriptional activation of downstream IL-10 gene in colonic cell lines and mouse primary colonic epithelial cells.124 IL-10 is an anti-inflammatory cytokine that plays a key role in gut homeostasis. In addition to PPARγ, PPARα activation also promotes IL-10 release in the intestinal epithelium.125 After binding with IL-10, gut macrophages convert to anti-inflammatory phenotype C–X–3–C motif chemokine receptor macrophages, modulating the immune response to maintain host intestinal barrier and gut microbial homeostasis. Loss of IL-10 receptors induces spontaneous colitis.126 In addition, intestinal pathogens also modulate intestinal inflammatory response via the PPAR pathway to colonize the gut. Kundu et al127 have found that Salmonella enterica serovar Typhimurium (S. Typhimurium) decreased the expression of PPARγ in a TLR4-independent manner in mouse intestinal epithelium, leading to overexpression of inflammatory transcription factors NF-κB and AP-1 and downstream TNF-α and IL-6. The intestinal inflammatory response helps S. Typhimurium to colonize the host gut and induce colitis.
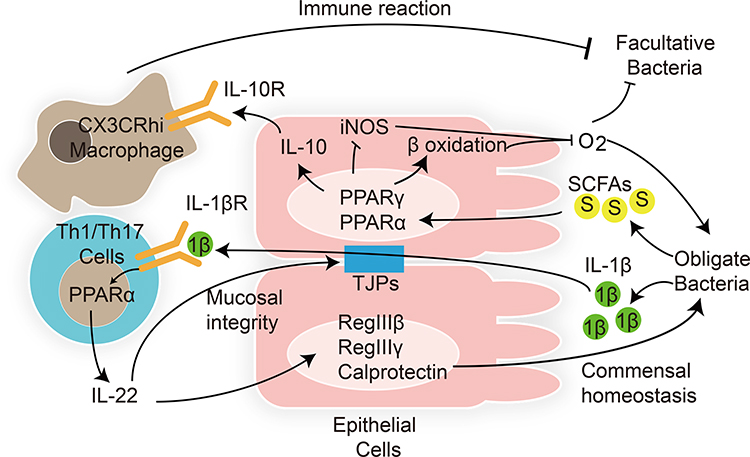 |
Figure 1 Interactions between gut microbiota and host PPARs in microbial inhabitation and adaptation. Lines ending in bars represent inhibition and lines ending in arrowheads represent activation.Notes: Adapted from Hasan A U, Rahman A, Kobori H. Interactions between Host PPARs and Gut Microbiota in Health and Disease. Int J Mol Sci, 2019. 20(2).123 |
As PPAR ligands, SCFAs can regulate gut homeostasis via the PPAR pathway. The overgrowth of facultative anaerobic Enterobacteriaceae is considered a marker of dysbiosis. Activation of the PPARγ pathway by butyrate activates β-oxidation of colonocytes and inhibits the expression of inducible nitric oxide synthase (NOS) in the colon. Moreover, T-reg cell expansion induced by SCFAs can cooperate with the activated PPARγ pathway to limit the luminal bioavailability of oxygen and suppress the growth of facultative anaerobic bacteria Escherichia and Salmonella.128
Clostridia-related segmented filamentous bacteria release the inflammatory mediator interleukin (IL)-1β. IL-1β activates intestinal T helper 1 and 17 (Th1 and Th17) cells via the PPARα pathway.125 Activated Th1 and Th17 cells produce the inflammatory cytokine interleukin (IL)-22. On one hand, IL-22 is essential for maintenance of gut barrier integrity and intestinal epithelial regeneration. On the other hand, IL-22 activates anti-microbial peptide RegIIIβ, RegIIIγ, and calprotectin expression in epithelial cells, which is essential for intestinal mucosal immunity.129 Decreased levels of IL-22, RegIIIβ, RegIIIγ, and calprotectin increased intestinal inflammation susceptibility and gut dysbiosis in PPARα-deficient mouse, which could be reversed by PPARα agonist GW7647.
Yu et al130 have reported increased incidence of HCC in PPARγ-deficient (PPARγ±) mice compared to wild-type (PPARγ+/+) mice in a DEN-induced HCC model. A rosiglitazone (a type of TZD) treatment suppressed the incidence of HCC in PPARγ+/+ mice, but not in PPARγ± mice. Moreover, overexpression of PPARγ induced by adenoviral infection in Hep3B cells inhibited cell proliferation, induced G2/M cell cycle arrest, and triggered extrinsic and intrinsic apoptosis.130 These findings demonstrate that PPARγ acts as an antioncogene in hepatocarcinogenesis. However, the direct effects of PPARγ on gut microbiota in hepatocarcinogenesis have not been investigated.115
Although FXR is the master regulator of bile acids, PPAR can also modulate their metabolism. In hepatocyte, PPARα is a target gene of FXR.91 Cytochrome P450 enzymes (CYPs), sulfotransferases (SULTs) and UDP-glucuronosyltransferases (UGTs) are responsible for BA detoxification and can be activated by PPARα131 During liver inflammation, hepatic nuclear factor kappa-B (NF-κB) pathway is activated, leading to the inhibition of hepatic FXR.91 The inhibited FXR suppresses BA detoxification by decreasing the expression of PPARα and downstream CYPs, SULTs and UGTs.131 Moreover, suppressed PPARα pathway inhibited the expression of multidrug resistance protein 2 (MDR2), MDR3, multidrug resistance-associated protein 3 (MRP3) and MRP4. MRP3 and MRP4 regulate BA efflux to general circulation.132 MDR2 and MDR3 modulate the canicular biliary secretion of phosphatidylcholine.133 Combined with the cholestasis induced by the inhibition of FXR during liver inflammation as we discussed in Microbiota Metabolites, liver injury caused by high-level BA are aggravated, which might promote the progression of HCC.91
Although there have been no studies focusing on the direct effects of PPARs on the onset and progression of HCC by modulating gut microbiota, recent research has demonstrated the protective effects of natural and synthetic PPAR agonists against CLD by reversing leaky gut and gut dysbiosis.
Targeting Gut Microbiota for HCC Prevention
The above findings suggest that gut microbiota have an essential role in progression of CLD and hepatocarcinogenesis in animal models and patients. Gut microbiota seems to be a promising target for the treatment of precancerous disease and HCC prevention. Studies conducted in animal models have indicated that administration of antibiotics and probiotics can prevent hepatocarcinogenesis induced by NAFLD and chemical toxins. Moreover, it has been reported that gut microbiota can modulate the curative effect of targeted therapy.134,135
Antibiotics
Antibiotics including norfloxacin and rifaximin have been commonly used in the clinic to prevent encephalopathy and treat enterogenous infections, such as spontaneous peritonitis in patients with advanced liver cirrhosis or HCC. The suppressive effects of antibiotics on hepatocarcinogenesis are attributed to: 1) amelioration of the leaky gut by reducing the number of intestinal bacteria, including pathogens and potential pathogens and suppressing liver inflammation; 2) production reduction of bacterial metabolites by some antibiotics that promote hepatocarcinogenesis.30 For instance, vancomycin can inhibit DCA production by eliminating Gram-positive bacteria.105 Oral supplementation with a cocktail of antibiotics containing ampicillin, neomycin, metronidazole, and vancomycin reduced the tumor size and number in HCC mice induced by diethylnitrosamine (DEN)+ carbon tetrachloride (CCl4) or dimethylolbutanoic acid (DMBA) + high-fat diet (HFD).33,105 It should be noted that suppressive effects of antibiotics on carcinogenesis are more effective at advanced stages than the early stages, suggesting the feasibility of antibiotics administration for HCC patients with advanced liver cirrhosis and those at high risk of HCC. However, long-term treatment by broad-spectrum antibiotics might lead to a decrease in probiotics levels and an increase in drug-resistant bacteria in the intestine, promoting gut microbiota dysbiosis. In addition, the side effects of antibiotics, such as vancomycin nephrotoxicity, restrict their long-term administration.
Hence, an antibiotic with mild side effects, high long-term safety, or even life-long administration for HCC prevention is needed. Rifaximin is a potent broad-spectrum antibiotic that cannot be absorbed by the human body and has extremely high intestinal concentrations and mild side effects.136 Rifaximin is commonly used in the clinic to treat enterocolitis or traveler’s diarrhea and prevent encephalopathy in patients with advanced liver disease and HCC. More importantly, no resistance to rifaximin has been observed in patients receiving long-term treatment. In a DEN/CCl4-induced HCC mouse model, the rifaximin treatment effectively reduced tumor size and number. In addition, rifaximin effectively ameliorated portal hypertension and reduced the incidence of spontaneous peritonitis in patients with liver cirrhosis, thus prolonging their survival.137,138 However, the effects of rifaximin on the development of HCC in patients with advanced liver disease remain unclear and more experimental and clinical studies are needed.
Probiotics
Probiotics are active microorganisms that are beneficial to the host as they colonize the human body and change the composition of certain types of host flora. They can promote absorption of nutrients and maintain intestinal health by regulating the immune function and intestinal flora balance. Recent studies have confirmed that probiotics can ameliorate CLD in patients and animal models and suppress hepatocarcinogenesis in animal models. Administration of a VSL#3 mixture containing Streptococcus, Bifidobacterium, and Lactobacillus in patients or rodent models can improve insulin resistance, reduce LPS serum levels, depress the total hepatic fatty acid content and liver inflammation, and attenuate liver injury.139,140 In a clinical study conducted on patients with HBV-induced liver cirrhosis without overt hepatic encephalopathy, 3-month oral administration of probiotics (containing Clostridium butyricum and B. infantis) improved patient cognition. There was an increased abundance of beneficial Clostridium butyricum and B. infantis and a decreased abundance of opportunistic pathogens Enterococcus and Enterobacteriaceae. The intestinal barrier was improved after the probiotics treatment, resulting in a decreased level of venous ammonia and increased cognitive ability in patients with liver cirrhosis.141 Accordingly, Dhiman et al142 have found that the VSL#3 treatment in patients with liver cirrhosis reduces the risk of hospitalization for hepatic encephalopathy and decreases the severity of cirrhosis.
The VSL#3 pretreatment ameliorated gut microbiota dysbiosis, suppressed intestinal inflammation, reduced serum LPS levels, and inhibited HCC growth and multiplicity in HCC rats induced by DEN.143 Degirolamo et al have found that VSL#3 treatment increased fecal BA excretion and hepatic BA synthesis by inhibiting gut-liver FXR/FGF15 pathway in mice.144 The effect of VSL#3 on FXR/FGF15 pathway in hepatocarcinogenesis should be further investigated. In a subcutaneous mouse tumor model, the administration of a probiotics mixture Prohep containing Lactobacillus rhamnosus GG, viable Escherichia coli Nissle 1917, and heat-inactivated VSL#3 suppressed tumor growth and reduced tumor size and weight. An increased amount of beneficial Prevotella and Oscillibacter was observed in the treatment group, which produced anti-inflammatory cytokine IL-10. Prohep administration suppressed tumor angiogenesis by reducing the Th17 polarization and secretion of IL-17.145 Aflatoxins are carcinogenic fungal metabolites that can induce HCC. Gratz et al have found that treatment with a probiotic Lactobacillus rhamnosus strain GG, which binds aflatoxins, reduced the hepatotoxicity of aflatoxins by increasing their excretion via the fecal route in rats.32 The above findings demonstrate that the suppressive effects of probiotics on hepatocarcinogenesis are attributed to amelioration of gut microbiota dysbiosis, improvement of intestinal barrier, inhibition of liver inflammation, modulation of host immune system, and reduction of carcinogen toxicity. Probiotics demonstrate good safety for CLD treatment, as all patients with decompensated cirrhosis tolerated the probiotics treatment.146 However, there are limited studies on the effects of probiotics on HCC patients. In addition, the majority of probiotics cannot colonize the host digestive tract. The probiotics are also used in different combinations in different studies, making it difficult to compare their effectiveness.
Fecal Microbiota Transplantation (FMT)
Fecal microbiota transplantation (FMT) refers to the process of transplanting functional bacteria from the feces of healthy people into the intestinal tract of patients, thereby reconstructing intestinal microflora with normal structure and function.147 In third-century China, Ge Hong used stool from healthy people to treat food poisoning and diarrhea.148 The use of FMT as a treatment for Clostridium difficile infection (CDI) has been approved by the US Food and Drug Administration (FDA) in 2013. FMT cure rate for treating recurrent and refractory CDI is nearly 90%, which is 2–3 times that of the standard antibiotics therapy.149 In recent years, FMT has shown potential for treatment of inflammatory bowel disease (IBD), irritable bowel syndrome (IBS), obesity, and idiopathic thrombocytopenic purpura.147,150 It has been reported that transplantation of intestinal microbiota from lean donors can increase insulin sensitivity in patients with metabolic syndrome. Increased gut microbiota diversity and abundance of butyrate-producing intestinal microbiota have been observed after the treatment.151 The effects of FMT on NASH and liver cirrhosis are currently being evaluated in clinical trials.152 FMT might suppress hepatocarcinogenesis by ameliorating gut microbiota dysbiosis, reducing the release of LPSs and other cytotoxic products, and suppressing liver inflammation.148 This hypothesis needs to be verified with more animal experiments and clinical trials. Moreover, it has not been determined whether gut microbiota restoration by FMT is permanent or transient. Most importantly, the safety of FMT has not been demonstrated. The majority of patients with advanced liver disease have a suppressed immune system. Therefore, they might be infected with pathogens, viruses, and fungi through FMT.
Prokinetics
Portal hypertension is observed in the majority of patients with advanced liver cirrhosis and HCC, leading to hyperemia and edema of the intestinal mucosa, which influences periodic peristalsis of the small intestine.153 In addition, liver injury can cause gastrointestinal dysfunction in patients with liver cancer, further aggravating the impact on gastric emptying and small intestine motility. Liver dysfunction can also induce sympathetic nerve excitation, inhibit the parasympathetic nerve, and have adverse effects on gastrointestinal motility, absorption, secretion, and other activities. Gut dysmotility caused by the above factors leads to bacterial overgrowth in the intestine and subsequent LPS translocation. The administration of cisapride-a prokinetic can effectively reduce intestinal permeability, improve intestinal transit, and suppress bacterial overgrowth and LPS translocation in animal models and patients with liver cirrhosis.154,155 Similar protective effects have been observed in nonselective β-adrenergic blockers (such as propranolol), which reduce sympathetic activity.156,157 Furthermore, cohort study results158,159 have demonstrated that long-term administration of propranolol reduces the risk of developing HCC in patients with liver cirrhosis. In addition, propranolol has been reported to suppress proliferation and induce apoptosis of HepG2 and HepG2.2.15 liver cancer cells in vitro.160
PPAR Agonists
Sun et al161 have reported that a water-insoluble polysaccharide (WIP) isolated from the sclerotium of Poria cocos improves hyperglycemia, insulin resistance, hyperlipidemia, and liver steatosis in mice with NAFLD by activating the PPARγ pathway. The WIP treatment increased the SCFA-producing Lachnospiracea, Alloprevotella, Parabacteroides, Clostridium IV, Ruminococcus, and Bacteroides and decreased pro-inflammatory Megamonas and Proteus. It also maintained intestinal integrity, demonstrated by the decreased LPS plasma levels.
In a study conducted on rats with ethanol-induced liver disease, a selective PPARδ agonist MBX-8025 ameliorated liver injury. The MBX-8025 treatment restored bile acid homeostasis and increased hydrogen‐producing Rikenellaceae. Hydrogen protected cells from oxidative stress. MBX-8025 decreased pathogenic Enterococcaceae and Coriobacteriaceae. Enterococcaceae is related to liver failure and increased LPS serum levels. Coriobacteriaceae can impair cholesterol homeostasis. The MBX-8025 treatment also reversed ethanol-induced gut barrier dysfunction.162
Synthetic PPARγ agonists TZDs have been reported to have anti-hepatoma effects in vivo and in vitro. Rosiglitazone suppresses BEL-7402 and Huh7 cell proliferation by upregulating phosphatase and tensin homolog (PTEN) and downregulating Cox-2 via activation of the PPARγ pathway.163 Rosiglitazone inhibits the migration of BEL-7404 cells via PPARγ-mediated upregulation of PTEN and downregulation of phosphorylated Akt and focal adhesion kinase.164 Troglitazone has been reported to induce HepG2 cell apoptosis in vivo.165 Moreover, in an orthotopic metastasis mouse model, rosiglitazone was found to suppress lung metastasis in MHCC97L cells.166 Endogenous PPARγ ligand 15d-PGJ2 inhibited proliferation and induced apoptosis of LM3, SMMC-7721, and Huh-7 cells via ROS-mediated JNK activation and Akt downregulation.167 Natural PPARγ agonist avicularin has been reported to induce apoptosis and inhibit migration and invasion of SMMC7721 and Bel7402 via PPARγ activation induced by ERK and AMPK. Furthermore, avicularin can inhibit lung metastasis in Bel7402 cells.168
A PPARα agonist fenofibrate has been reported to suppress expression of CYP7A1 by inhibiting hepatocyte nuclear factor 4 (HNF-4) in vivo.169 Fenofibrate was found to increase the biliary phosphatidylcholine secretion in rat hepatocytes by activating MDR3.170 More importantly, fenofibrate treatment improved cholestasis in patients with primary biliary cirrhosis and primary biliary cholangitis.171,172 The effect of fenofibrate on bile acids metabolism in hepatocarcinogenesis should be further investigated.
Based on the findings discussed above, it can be concluded that PPAR agonists have anti-hepatoma effects and can potentially be used for treating CLD by modulating gut microbiota. However, the effects of PPAR agonists on the endogenous hepatocarcinogenesis driven by the leaky gut and dysbiosis remain unclear and need further investigation.
Modulation of Targeted Therapy
Sorafenib, approved by the FDA in 2007 for the targeted therapy of advanced liver cancer, is a small molecule oral multi kinase inhibitor. Sorafenib has dual antitumor effects. On one hand, it acts on serine/threonine kinase and receptor tyrosine kinase in tumor cells and blood vessels and directly inhibits tumor growth by inhibiting the Raf/MEK/ERK signal transduction pathway.173,174 On the other hand, it can block the formation of tumor neovascularization by inhibiting VEGF and platelet-derived growth factor (PDGF) receptors and indirectly inhibit tumor cell growth.175–177 A recent study demonstrated that gut microbiota regulate the VEGF-C secreted by villus macrophages, which is essential for lacteal integrity in mice.178 More importantly, in a mouse obesity-driven choroidal neovascularization model, gut microbiota dysbiosis induced by high-fat diet destroyed the intestinal barrier and increased the production of VEGF-A, leading to pathological angiogenesis.179 These findings indicate that gut microbiota might influence the effectiveness of sorafenib by regulating the expression of VEGF, which still needs to be verified via animal experiments and clinical trials.
Conclusion
A large number of studies have demonstrated the contribution of gut microbiota to the progression of CLD and hepatocarcinogenesis via multiple mechanisms. However, the direct effect of gut microbiota on hepatocarcinogenesis has not been elucidated. The impaired gut barrier and alteration of gut microbiota and their metabolites, such as LPS and DCA, result in chronic liver inflammation and injury, promoting the development of HCC. Studies on dysbiosis in HCC patients suggest the potential of gut microbiota as a noninvasive tool for early diagnosis of HCC. The administration of antibiotics, probiotics, FMT, and prokinetics, which target gut microbiota, might be safe therapeutic options for HCC prevention and treatment. The PPARs can modulate microbial inhabitation and adaptation. PPAR agonists show potential for treating CLD by reversing leaky gut and dysbiosis, indicating the possibility of their use in HCC prevention and treatment (Figure 2). Gut microbiota might modulate efficiency of HCC-targeted therapy. However, animal model findings cannot be directly translated to human patients since CLD and HCC development cannot be perfectly modeled in animals. The clinical trials on patients need to be well designed and administration of antibiotics, probiotics, and proton pump inhibitors before interference should be taken into consideration. Further efforts to determine the roles of gut microbiota during the onset and progression of HCC will assist in finding novel effective and safe strategies for HCC diagnosis, prevention, and treatment.
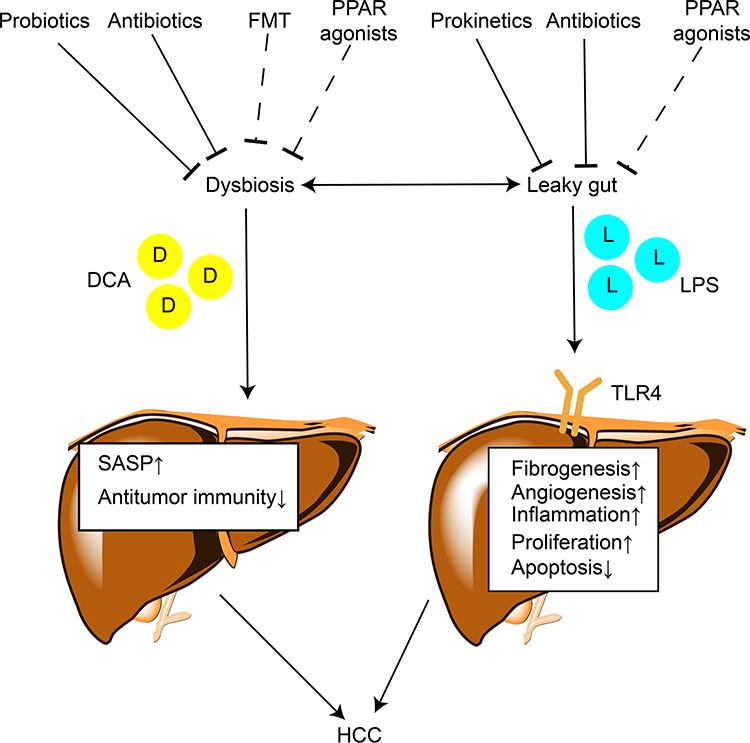 |
Figure 2 The mechanisms by which gut microbiota contributes to hepatocarcinogenesis and therapeutic targets. Lines ending in bars represent inhibition and lines ending in arrowheads represent activation. Dotted lines ending in bars mean that the antitumor effect of this strategy needs further validation. |
Funding
This study was supported by: (1) National Natural Science Foundation of China (No. 81670472); (2) the Yangfan Project of Shanghai Science and Technology Commission (No. 20YF1443300); (3) the Natural Science Foundation of Shanghai (No. 19ZR1447700); (4) the Health System Innovation Project of Shanghai Putuo Science and Technology Commission (No. PTKWWS201801, No. PTKWWS201903); (5) WBN Hepatology Research Fund of China Hepatitis Prevention and Treatment Foundation (No. CFHPC2019031).
Disclosure
The authors declare no conflicts of interest.
References
1. Sebastian Domingo JJ, Sanchez Sanchez C. From the intestinal flora to the microbiome. Rev Esp Enferm Dig. 2018;110(1):51–56.
2. Li J, Jia H, Cai X, et al. An integrated catalog of reference genes in the human gut microbiome. Nat Biotechnol. 2014;32(8):834–841.
3. Clemente JC, Ursell L, Parfrey L, et al. The impact of the gut microbiota on human health: an integrative view. Cell. 2012;148(6):1258–1270. doi:10.1016/j.cell.2012.01.035
4. Dietert RR, Dietert JM. The microbiome and sustainable healthcare. Healthcare (Basel). 2015;3(1):100–129. doi:10.3390/healthcare3010100
5. Khan I, Ullah N, Zha L, et al. Alteration of gut microbiota in Inflammatory Bowel Disease (IBD): cause or Consequence? IBD treatment targeting the gut microbiome. Pathogens. 2019;8:3.
6. Wang X, Xu X, Xia Y. Further analysis reveals new gut microbiome markers of type 2 diabetes mellitus. Antonie Van Leeuwenhoek. 2017;110(3):445–453. doi:10.1007/s10482-016-0805-3
7. Chen X, Devaraj S. Gut microbiome in obesity, metabolic syndrome, and diabetes. Curr Diab Rep. 2018;18(12):129. doi:10.1007/s11892-018-1104-3
8. Sun M, Ma K, Wen J, et al. A review of the brain-gut-microbiome axis and the potential role of microbiota in Alzheimer’s disease. J Alzheimers Dis. 2020;73(3):849–865. doi:10.3233/JAD-190872
9. Branchereau M, Burcelin R, Heymes C. The gut microbiome and heart failure: a better gut for a better heart. Rev Endocr Metab Disord. 2019;20(4):407–414. doi:10.1007/s11154-019-09519-7
10. Young VB. The role of the microbiome in human health and disease: an introduction for clinicians. BMJ. 2017;356:j831. doi:10.1136/bmj.j831
11. Org E, Parks BW, Joo JWJ, et al. Genetic and environmental control of host-gut microbiota interactions. Genome Res. 2015;25(10):1558–1569. doi:10.1101/gr.194118.115
12. Wu L, Li J, Liu T, et al. Quercetin shows anti-tumor effect in hepatocellular carcinoma LM3 cells by abrogating JAK2/STAT3 signaling pathway. Cancer Med. 2019;8(10):4806–4820. doi:10.1002/cam4.2388
13. Dai W, Xu L, Yu X, et al. OGDHL silencing promotes hepatocellular carcinoma by reprogramming glutamine metabolism. J Hepatol. 2020;72(5):909–923. doi:10.1016/j.jhep.2019.12.015
14. Liu T, Li S, Wu L, et al. Experimental study of hepatocellular carcinoma treatment by shikonin through regulating PKM2. J Hepatocell Carcinoma. 2020;7:19–31. doi:10.2147/JHC.S237614
15. Bray F, Ferlay J, Soerjomataram I, et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. doi:10.3322/caac.21492
16. Feng J, Li J, Wu L, et al. Emerging roles and the regulation of aerobic glycolysis in hepatocellular carcinoma. J Exp Clin Cancer Res. 2020;39(1):126.
17. Kew MC. Hepatocellular carcinoma: epidemiology and risk factors. J Hepatocell Carcinoma. 2014;1:115–125. doi:10.2147/JHC.S44381
18. Sherman M. Hepatocellular carcinoma: epidemiology, surveillance, and diagnosis. Semin Liver Dis. 2010;30(1):3–16. doi:10.1055/s-0030-1247128
19. Feng J, Wu L, Ji J, et al. PKM2 is the target of proanthocyanidin B2 during the inhibition of hepatocellular carcinoma. J Exp Clin Cancer Res. 2019;38(1):204.
20. Li S, Li J, Dai W, et al. Genistein suppresses aerobic glycolysis and induces hepatocellular carcinoma cell death. Br J Cancer. 2017;117(10):1518–1528. doi:10.1038/bjc.2017.323
21. Bertot LC, Adams LA. Trends in hepatocellular carcinoma due to non-alcoholic fatty liver disease. Expert Rev Gastroenterol Hepatol. 2019;13(2):179–187. doi:10.1080/17474124.2019.1549989
22. Welzel TM, Graubard BI, Quraishi S, et al. Population-attributable fractions of risk factors for hepatocellular carcinoma in the United States. Am J Gastroenterol. 2013;108(8):1314–1321. doi:10.1038/ajg.2013.160
23. Bertino G, et al. Hepatocellular carcinoma serum markers. Semin Oncol. 2012;39(4):410–433.
24. Ayuso C, Rimola J, Vilana R, et al. Diagnosis and staging of hepatocellular carcinoma (HCC): current guidelines. Eur J Radiol. 2018;101:72–81. doi:10.1016/j.ejrad.2018.01.025
25. Russo FP, Imondi A, Lynch EN, et al. When and how should we perform a biopsy for HCC in patients with liver cirrhosis in 2018? A review. Dig Liver Dis. 2018;50(7):640–646. doi:10.1016/j.dld.2018.03.014
26. Ren Z, Li A, Jiang J, et al. Gut microbiome analysis as a tool towards targeted non-invasive biomarkers for early hepatocellular carcinoma. Gut. 2019;68(6):1014–1023.
27. Qin N, Yang F, Li A, et al. Alterations of the human gut microbiome in liver cirrhosis. Nature. 2014;513(7516):59–64. doi:10.1038/nature13568
28. Schwabe RF, Greten TF. Gut microbiome in HCC - Mechanisms, diagnosis and therapy. J Hepatol. 2020;72(2):230–238. doi:10.1016/j.jhep.2019.08.016
29. Sanduzzi Zamparelli M, Rocco A, Compare D, et al. The gut microbiota: a new potential driving force in liver cirrhosis and hepatocellular carcinoma. United European Gastroenterol J. 2017;5(7):944–953. doi:10.1177/2050640617705576
30. Yu LX, Schwabe RF. The gut microbiome and liver cancer: mechanisms and clinical translation. Nat Rev Gastroenterol Hepatol. 2017;14(9):527–539.
31. de Moreno de LeBlanc A, Matar C, Perdigon G. The application of probiotics in cancer. Br J Nutr. 2007;98(Suppl 1):S105–10. doi:10.1017/S0007114507839602
32. Gratz S, Täubel M, Juvonen RO, et al. Lactobacillus rhamnosus strain GG modulates intestinal absorption, fecal excretion, and toxicity of aflatoxin B(1) in rats. Appl Environ Microbiol. 2006;72(11):7398–7400. doi:10.1128/AEM.01348-06
33. Dapito DH, Mencin A, Gwak G-Y, et al. Promotion of hepatocellular carcinoma by the intestinal microbiota and TLR4. Cancer Cell. 2012;21(4):504–516. doi:10.1016/j.ccr.2012.02.007
34. Finkelmeier F, Waidmann O, Trojan J. Nivolumab for the treatment of hepatocellular carcinoma. Expert Rev Anticancer Ther. 2018;18(12):1169–1175. doi:10.1080/14737140.2018.1535315
35. Trivedi PJ, Adams DH. Gut-liver immunity. J Hepatol. 2016;64(5):1187–1189. doi:10.1016/j.jhep.2015.12.002
36. Chung H, Pamp SJ, Hill JA, et al. Gut immune maturation depends on colonization with a host-specific microbiota. Cell. 2012;149(7):1578–1593.
37. Zhao Q, Elson CO. Adaptive immune education by gut microbiota antigens. Immunology. 2018;154(1):28–37. doi:10.1111/imm.12896
38. Shi T, Wei J, Liu G, Han M, Liu T. [Researches on the change of intestinal barrier function in patients with ulcerative colitis]. Zhonghua Yi Xue Za Zhi. 2015;95(24):1941–1943. Chinese.
39. Ren Z, et al. Progress in mycotoxins affecting intestinal mucosal barrier function. Int J Mol Sci. 2019;20:11.
40. Chelakkot C, Ghim J, Ryu SH. Mechanisms regulating intestinal barrier integrity and its pathological implications. Exp Mol Med. 2018;50(8):103. doi:10.1038/s12276-018-0126-x
41. Marshall JC. The gut as a potential trigger of exercise-induced inflammatory responses. Can J Physiol Pharmacol. 1998;76(5):479–484. doi:10.1139/y98-049
42. Thaiss CA, Zmora N, Levy M, Elinav E. The microbiome and innate immunity. Nature. 2016;535(7610):65–74.
43. Heymann F, Tacke F. Immunology in the liver–from homeostasis to disease. Nat Rev Gastroenterol Hepatol. 2016;13(2):88–110.
44. Jiang J-W, Chen X-H, Ren Z, et al. Gut microbial dysbiosis associates hepatocellular carcinoma via the gut-liver axis. Hepatobiliary Pancreat Dis Int. 2019;18(1):19–27. doi:10.1016/j.hbpd.2018.11.002
45. Rainer F, Horvath A, Sandahl TD, et al. Soluble CD163 and soluble mannose receptor predict survival and decompensation in patients with liver cirrhosis, and correlate with gut permeability and bacterial translocation. Aliment Pharmacol Ther. 2018;47(5):657–664. doi:10.1111/apt.14474
46. Lin RS, Lee F-Y, Lee S-D, et al. Endotoxemia in patients with chronic liver diseases: relationship to severity of liver diseases, presence of esophageal varices, and hyperdynamic circulation. J Hepatol. 1995;22(2):165–172. doi:10.1016/0168-8278(95)80424-2
47. Miettinen TA. Lipid absorption, bile acids, and cholesterol metabolism in patients with chronic liver disease. Gut. 1972;13(9):682–689. doi:10.1136/gut.13.9.682
48. Yajima S, Morisaki H, Serita R, et al. Tumor necrosis factor-alpha mediates hyperglycemia-augmented gut barrier dysfunction in endotoxemia. Crit Care Med. 2009;37(3):1024–1030.
49. Wiest R, Garcia-Tsao G. Bacterial translocation (BT) in cirrhosis. Hepatology. 2005;41(3):422–433. doi:10.1002/hep.20632
50. Petersen C, Round JL. Defining dysbiosis and its influence on host immunity and disease. Cell Microbiol. 2014;16(7):1024–1033. doi:10.1111/cmi.12308
51. Obrenovich MEM. Leaky gut leaky brain?. Microorganisms. 2018;6:4.
52. Compare D, Coccoli P, Rocco A, et al. Gut–liver axis: the impact of gut microbiota on non alcoholic fatty liver disease. Nutr Metab Cardiovasc Dis. 2012;22(6):471–476. doi:10.1016/j.numecd.2012.02.007
53. Niu Z-S, Niu X-J, Wang W-H, et al. Latest developments in precancerous lesions of hepatocellular carcinoma. World J Gastroenterol. 2016;22(12):3305–3314. doi:10.3748/wjg.v22.i12.3305
54. Di Tommaso L, Sangiovanni A, Borzio M, et al. Advanced precancerous lesions in the liver. Best Pract Res Clin Gastroenterol. 2013;27(2):269–284. doi:10.1016/j.bpg.2013.03.015
55. Posteraro B, Paroni Sterbini F, Petito V, et al. Liver injury, endotoxemia, and their relationship to intestinal microbiota composition in alcohol-preferring rats. Alcohol Clin Exp Res. 2018;42(12):2313–2325. doi:10.1111/acer.13900
56. Sandler NG, Koh C, Roque A, et al. Host response to translocated microbial products predicts outcomes of patients with HBV or HCV infection. Gastroenterology. 2011;141(4):
57. Ghosh Dastidar S, et al. Rodent models of alcoholic liver disease: role of binge ethanol administration. Biomolecules. 2018;8:1.
58. Fukui H, Brauner B, Bode JC, et al. Plasma endotoxin concentrations in patients with alcoholic and non-alcoholic liver disease: reevaluation with an improved chromogenic assay. J Hepatol. 1991;12(2):162–169. doi:10.1016/0168-8278(91)90933-3
59. Cani PD, Amar J, Iglesias MA, et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes. 2007;56(7):1761–1772. doi:10.2337/db06-1491
60. Caricilli AM, Saad MJ. The role of gut microbiota on insulin resistance. Nutrients. 2013;5(3):829–851. doi:10.3390/nu5030829
61. Miele L, Valenza V, La Torre G, et al. Increased intestinal permeability and tight junction alterations in nonalcoholic fatty liver disease. Hepatology. 2009;49(6):1877–1887. doi:10.1002/hep.22848
62. Joshi K, Kohli A, Manch R, Gish R. Alcoholic liver disease: high risk or low risk for developing hepatocellular carcinoma? Clin Liver Dis. 2016;20(3):563–580.
63. Nolan JP. The role of intestinal endotoxin in liver injury: a long and evolving history. Hepatology. 2010;52(5):1829–1835. doi:10.1002/hep.23917
64. Bellot P, García-Pagán JC, Francés R, et al. Bacterial DNA translocation is associated with systemic circulatory abnormalities and intrahepatic endothelial dysfunction in patients with cirrhosis. Hepatology. 2010;52(6):2044–2052. doi:10.1002/hep.23918
65. Wei X, Jiang S, Chen Y, et al. Cirrhosis related functionality characteristic of the fecal microbiota as revealed by a metaproteomic approach. BMC Gastroenterol. 2016;16(1):121. doi:10.1186/s12876-016-0534-0
66. Dubinkina VB, et al. Links of gut microbiota composition with alcohol dependence syndrome and alcoholic liver disease. Microbiome. 2017;5(1):141.
67. Puri P, Liangpunsakul S, Christensen JE, et al. The circulating microbiome signature and inferred functional metagenomics in alcoholic hepatitis. Hepatology. 2018;67(4):1284–1302. doi:10.1002/hep.29623
68. Shen F, Zheng R-D, Sun X-Q, et al. Gut microbiota dysbiosis in patients with non-alcoholic fatty liver disease. Hepatobiliary Pancreat Dis Int. 2017;16(4):375–381. doi:10.1016/S1499-3872(17)60019-5
69. Boursier J, Mueller O, Barret M, et al. The severity of nonalcoholic fatty liver disease is associated with gut dysbiosis and shift in the metabolic function of the gut microbiota. Hepatology. 2016;63(3):764–775. doi:10.1002/hep.28356
70. Zhu L, Baker SS, Gill C, et al. Characterization of gut microbiomes in nonalcoholic steatohepatitis (NASH) patients: a connection between endogenous alcohol and NASH. Hepatology. 2013;57(2):601–609. doi:10.1002/hep.26093
71. Wang J, Wang Y, Zhang X, et al. Gut microbial dysbiosis is associated with altered hepatic functions and serum metabolites in chronic Hepatitis B patients. Front Microbiol. 2017;8:2222.
72. Lu H, Wu Z, Xu W, et al. Intestinal microbiota was assessed in cirrhotic patients with hepatitis B virus infection. Intestinal microbiota of HBV cirrhotic patients. Microb Ecol. 2011;61(3):693–703. doi:10.1007/s00248-010-9801-8
73. Inoue T, Nakayama J, Moriya K, et al. Gut dysbiosis associated with Hepatitis C virus infection. Clin Infect Dis. 2018;67(6):869–877. doi:10.1093/cid/ciy205
74. Aly AM, Adel A, El-Gendy AO, et al. Gut microbiome alterations in patients with stage 4 hepatitis C. Gut Pathog. 2016;8(1):42. doi:10.1186/s13099-016-0124-2
75. Grat M, Wronka KM, Krasnodębski M, et al. Profile of gut microbiota associated with the presence of hepatocellular cancer in patients with liver cirrhosis. Transplant Proc. 2016;48(5):1687–1691. doi:10.1016/j.transproceed.2016.01.077
76. Chen Y, et al. Characterization of fecal microbial communities in patients with liver cirrhosis. Hepatology. 2011;54(2):562–572.
77. Bajaj JS, Betrapally NS, Gillevet PM. Decompensated cirrhosis and microbiome interpretation. Nature. 2015;525(7569):E1–2. doi:10.1038/nature14851
78. Ponziani FR, Bhoori S, Castelli C, et al. Hepatocellular carcinoma is associated with gut microbiota profile and inflammation in nonalcoholic fatty liver disease. Hepatology. 2019;69(1):107–120. doi:10.1002/hep.30036
79. Sydor S, et al. Altered microbiota diversity and bile acid signaling in cirrhotic and noncirrhotic NASH-HCC. Clin Transl Gastroenterol. 2016;48(5):e00131. doi:10.14309/ctg.0000000000000131
80. Gopalakrishnan V, Helmink BA, Spencer CN, et al. The influence of the gut microbiome on cancer, immunity, and cancer immunotherapy. Cancer Cell. 2018;33(4):570–580. doi:10.1016/j.ccell.2018.03.015
81. Rosshart SP, Vassallo BG, Angeletti D, et al. Wild mouse gut microbiota promotes host fitness and improves disease resistance. Cell. 2017;171(5):1015–1028 e13. doi:10.1016/j.cell.2017.09.016
82. Elinav E, Garrett WS, Trinchieri G, et al. The cancer microbiome. Nat Rev Cancer. 2019;19(7):371–376. doi:10.1038/s41568-019-0155-3
83. Yang J, Li M, Zheng QC. Emerging role of Toll-like receptor 4 in hepatocellular carcinoma. J Hepatocell Carcinoma. 2015;2:11–17.
84. Achiwa K, Ishigami M, Ishizu Y, et al. DSS colitis promotes tumorigenesis and fibrogenesis in a choline-deficient high-fat diet-induced NASH mouse model. Biochem Biophys Res Commun. 2016;470(1):15–21. doi:10.1016/j.bbrc.2015.12.012
85. Weber SN, Bohner A, Dapito DH, et al. TLR4 deficiency protects against hepatic fibrosis and diethylnitrosamine-induced pre-carcinogenic liver injury in fibrotic liver. PLoS One. 2016;11(7):e0158819. doi:10.1371/journal.pone.0158819
86. Feng J, Chen K, Xia Y, et al. Salidroside ameliorates autophagy and activation of hepatic stellate cells in mice via NF-κB and TGF-β1/Smad3 pathways. Drug Des Devel Ther. 2018;12:1837–1853. doi:10.2147/DDDT.S162950
87. Moawad AW, Szklaruk J, Lall C, et al. Angiogenesis in hepatocellular carcinoma; pathophysiology, targeted therapy, and role of imaging. J Hepatocell Carcinoma. 2020;7:77–89. doi:10.2147/JHC.S224471
88. Yu LX, Yan HX, Liu Q, et al. Endotoxin accumulation prevents carcinogen-induced apoptosis and promotes liver tumorigenesis in rodents. Hepatology. 2010;52(4):1322–1333.
89. Liu W-T, Jing -Y-Y, Yu G-F, et al. Toll like receptor 4 facilitates invasion and migration as a cancer stem cell marker in hepatocellular carcinoma. Cancer Lett. 2015;358(2):136–143. doi:10.1016/j.canlet.2014.12.019
90. Hsiao -C-C, Chen P-H, Cheng C-I, et al. Toll-like receptor-4 is a target for suppression of proliferation and chemoresistance in HepG2 hepatoblastoma cells. Cancer Lett. 2015;368(1):144–152. doi:10.1016/j.canlet.2015.08.004
91. Jia W, Xie G, Jia W. Bile acid-microbiota crosstalk in gastrointestinal inflammation and carcinogenesis. Nat Rev Gastroenterol Hepatol. 2018;15(2):111–128.
92. Ridlon JM, Kang D-J, Hylemon PB, et al. Gut microbiota, cirrhosis, and alcohol regulate bile acid metabolism in the gut. Dig Dis. 2015;33(3):338–345. doi:10.1159/000371678
93. Gadaleta RM, van Erpecum KJ, Oldenburg B, et al. Farnesoid X receptor activation inhibits inflammation and preserves the intestinal barrier in inflammatory bowel disease. Gut. 2011;60(4):463–472. doi:10.1136/gut.2010.212159
94. Inagaki T, Moschetta A, Lee Y-K, et al. Regulation of antibacterial defense in the small intestine by the nuclear bile acid receptor. Proc Natl Acad Sci U S A. 2006;103(10):3920–3925. doi:10.1073/pnas.0509592103
95. Drafahl KA, McAndrew CW, Meyer AN, et al. The receptor tyrosine kinase FGFR4 negatively regulates NF-kappaB signaling. PLoS One. 2010;5(12):e14412. doi:10.1371/journal.pone.0014412
96. Schaap FG, Trauner M, Jansen PL. Bile acid receptors as targets for drug development. Nat Rev Gastroenterol Hepatol. 2014;11(1):55–67. doi:10.1038/nrgastro.2013.151
97. Stanimirov B, Stankov K, Mikov M. Bile acid signaling through farnesoid X and TGR5 receptors in hepatobiliary and intestinal diseases. Hepatobiliary Pancreat Dis Int. 2015;14(1):18–33. doi:10.1016/S1499-3872(14)60307-6
98. Gadaleta RM, Cariello M, Sabbà C, Moschetta A. Tissue-specific actions of FXR in metabolism and cancer. Biochim Biophys Acta. 2015;1851(1):30–39. doi:10.1016/j.bbalip.2014.08.005
99. Yang F, Huang X, Yi T, Yen Y, Moore DD, Huang W. Spontaneous development of liver tumors in the absence of the bile acid receptor farnesoid X receptor. Cancer Res. 2007;67(3):863–867. doi:10.1158/0008-5472.CAN-06-1078
100. Degirolamo C, Modica S, Vacca M, et al. Prevention of spontaneous hepatocarcinogenesis in farnesoid X receptor-null mice by intestinal-specific farnesoid X receptor reactivation. Hepatology. 2015;61(1):161–170. doi:10.1002/hep.27274
101. Nijmeijer RM, Gadaleta RM, van Mil SWC, et al. Farnesoid X receptor (FXR) activation and FXR genetic variation in inflammatory bowel disease. PLoS One. 2011;6(8):e23745. doi:10.1371/journal.pone.0023745
102. Holt JA, et al. Definition of a novel growth factor-dependent signal cascade for the suppression of bile acid biosynthesis. Genes Dev. 2003;17(13):1581–1591. doi:10.1101/gad.1083503
103. Halilbasic E, Claudel T, Trauner M. Bile acid transporters and regulatory nuclear receptors in the liver and beyond. J Hepatol. 2013;58(1):155–168. doi:10.1016/j.jhep.2012.08.002
104. Allen K, Jaeschke H, Copple BL. Bile acids induce inflammatory genes in hepatocytes: a novel mechanism of inflammation during obstructive cholestasis. Am J Pathol. 2011;178(1):175–186. doi:10.1016/j.ajpath.2010.11.026
105. Yoshimoto S, Loo TM, Atarashi K, et al. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature. 2013;499(7456):97–101. doi:10.1038/nature12347
106. Loo TM, Kamachi F, Watanabe Y, et al. Gut microbiota promotes obesity-associated liver cancer through PGE 2 -mediated suppression of antitumor immunity. Cancer Discov. 2017;7(5):522–538. doi:10.1158/2159-8290.CD-16-0932
107. Ma C, Han M, Heinrich B, et al. Gut microbiome-mediated bile acid metabolism regulates liver cancer via NKT cells. Science. 2018;360:6391.
108. Chen T, Kim CY, Kaur A, et al. Dietary fibre-based SCFA mixtures promote both protection and repair of intestinal epithelial barrier function in a Caco-2 cell model. Food Funct. 2017;8(3):1166–1173. doi:10.1039/C6FO01532H
109. Furusawa Y, Obata Y, Fukuda S, et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature. 2013;504(7480):446–450. doi:10.1038/nature12721
110. Ortega JF, de Conti A, Tryndyak V, et al. Suppressing activity of tributyrin on hepatocarcinogenesis is associated with inhibiting the p53-CRM1 interaction and changing the cellular compartmentalization of p53 protein. Oncotarget. 2016;7(17):24339–24347. doi:10.18632/oncotarget.8248
111. Wu L, Tang Q, Yin X, et al. The therapeutic potential of adipose tissue-derived mesenchymal stem cells to enhance radiotherapy effects on hepatocellular carcinoma. Front Cell Dev Biol. 2019;7:267. doi:10.3389/fcell.2019.00267
112. Li J, Guo C, Wu J. 15-Deoxy–(12,14)-Prostaglandin J2 (15d-PGJ2), an endogenous ligand of PPAR-gamma: function and mechanism. PPAR Res. 2019;2019:7242030. doi:10.1155/2019/7242030
113. Lukovac S, et al. Differential modulation by Akkermansia muciniphila and Faecalibacterium prausnitzii of host peripheral lipid metabolism and histone acetylation in mouse gut organoids. mBio. 2014;5:4.
114. Wang N, Kong R, Luo H, et al. Peroxisome proliferator-activated receptors associated with nonalcoholic fatty liver disease. PPAR Res. 2017;2017:6561701. doi:10.1155/2017/6561701
115. Hsu HT, Chi CW. Emerging role of the peroxisome proliferator-activated receptor-gamma in hepatocellular carcinoma. J Hepatocell Carcinoma. 2014;1:127–135.
116. Yeh C-B, Hsieh M-J, Lin C-W, et al. The antimetastatic effects of resveratrol on hepatocellular carcinoma through the downregulation of a metastasis-associated protease by SP-1 modulation. PLoS One. 2013;8(2):e56661. doi:10.1371/journal.pone.0056661
117. Rajendran P, Li F, Shanmugam MK, et al. Honokiol inhibits signal transducer and activator of transcription-3 signaling, proliferation, and survival of hepatocellular carcinoma cells via the protein tyrosine phosphatase SHP-1. J Cell Physiol. 2012;227(5):2184–2195. doi:10.1002/jcp.22954
118. Issemann I, Green S. Activation of a member of the steroid hormone receptor superfamily by peroxisome proliferators. Nature. 1990;347(6294):645–650.
119. Peters JM, Barnes R, Bennett L, et al. Localization of the gene for familial partial lipodystrophy (Dunnigan variety) to chromosome 1q21-22. Nat Genet. 1998;18(3):292–295. doi:10.1038/ng0398-292
120. Xiang S, Chen K, Xu L, et al. Bergenin exerts hepatoprotective effects by inhibiting the release of inflammatory factors, apoptosis and autophagy via the PPAR-gamma pathway. Drug Des Devel Ther. 2020;14:129–143. doi:10.2147/DDDT.S229063
121. Tyagi S, Sharma S, Gupta P, et al. The peroxisome proliferator-activated receptor: A family of nuclear receptors role in various diseases. J Adv Pharm Technol Res. 2011;2(4):236–240. doi:10.4103/2231-4040.90879
122. Tailleux A, Wouters K, Staels B. Roles of PPARs in NAFLD: potential therapeutic targets. Biochim Biophys Acta. 2012;1821(5):809–818. doi:10.1016/j.bbalip.2011.10.016
123. Hasan AU, Rahman A, Kobori H. Interactions between Host PPARs and gut microbiota in health and disease. Int J Mol Sci. 2019;20(2).
124. Are A, Aronsson L, Wang S, et al. Enterococcus faecalis from newborn babies regulate endogenous PPARgamma activity and IL-10 levels in colonic epithelial cells. Proc Natl Acad Sci U S A. 2008;105(6):1943–1948. doi:10.1073/pnas.0711734105
125. Manoharan I, Suryawanshi A, Hong Y, et al. Homeostatic PPARalpha signaling limits inflammatory responses to commensal microbiota in the intestine. J Immunol. 2016;196(11):4739–4749. doi:10.4049/jimmunol.1501489
126. Zigmond E, Bernshtein B, Friedlander G, et al. Macrophage-restricted interleukin-10 receptor deficiency, but not IL-10 deficiency, causes severe spontaneous colitis. Immunity. 2014;40(5):720–733. doi:10.1016/j.immuni.2014.03.012
127. Kundu P, et al. Absence of intestinal PPARgamma aggravates acute infectious colitis in mice through a lipocalin-2-dependent pathway. PLoS Pathog. 2014;10(1):e1003887.
128. Byndloss MX, Olsan EE, Rivera-Chávez F, et al. Microbiota-activated PPAR-gamma signaling inhibits dysbiotic Enterobacteriaceae expansion. Science. 2017;357(6351):570–575. doi:10.1126/science.aam9949
129. Zenewicz LA, Yin X, Wang G, et al. IL-22 deficiency alters colonic microbiota to be transmissible and colitogenic. J Immunol. 2013;190(10):5306–5312. doi:10.4049/jimmunol.1300016
130. Yu J, Shen B, Chu ESH, et al. Inhibitory role of peroxisome proliferator-activated receptor gamma in hepatocarcinogenesis in mice and in vitro. Hepatology. 2010;51(6):2008–2019. doi:10.1002/hep.23550
131. Li T, Chiang JY. Bile acid signaling in metabolic disease and drug therapy. Pharmacol Rev. 2014;66(4):948–983.
132. Zhang Y, Zhang Y, Klaassen CD, et al. Alteration of bile acid and cholesterol biosynthesis and transport by Perfluorononanoic Acid (PFNA) in mice. Toxicol Sci. 2018;162(1):225–233. doi:10.1093/toxsci/kfx237
133. Wang R, Sheps JA, Liu L, et al. Hydrophilic bile acids prevent liver damage caused by lack of biliary phospholipid in Mdr2−/−mice. J Lipid Res. 2019;60(1):85–97. doi:10.1194/jlr.M088070
134. Alexander JL, Wilson ID, Teare J, et al. Gut microbiota modulation of chemotherapy efficacy and toxicity. Nat Rev Gastroenterol Hepatol. 2017;14(6):356–365. doi:10.1038/nrgastro.2017.20
135. Zheng Y, Wang T, Tu X, et al. Gut microbiome affects the response to anti-PD-1 immunotherapy in patients with hepatocellular carcinoma. J Immunother Cancer. 2019;7(1):193. doi:10.1186/s40425-019-0650-9
136. Koo HL, DuPont HL. Rifaximin: a unique gastrointestinal-selective antibiotic for enteric diseases. Curr Opin Gastroenterol. 2010;26(1):17–25. doi:10.1097/MOG.0b013e328333dc8d
137. Menshawy A, Mattar O, Barssoum K, et al. Safety and efficacy of rifaximin in prophylaxis of spontaneous bacterial peritonitis: a systematic review and meta-analysis. Curr Drug Targets. 2019;20(4):380–387. doi:10.2174/1389450119666180924145156
138. Vlachogiannakos J, Viazis N, Vasianopoulou P, et al. Long-term administration of rifaximin improves the prognosis of patients with decompensated alcoholic cirrhosis. J Gastroenterol Hepatol. 2013;28(3):450–455.
139. Li Z, et al. Probiotics and antibodies to TNF inhibit inflammatory activity and improve nonalcoholic fatty liver disease. Hepatology. 2003;37(2):343–350. doi:10.1053/jhep.2003.50048
140. Loguercio C, Federico A, Tuccillo C, et al. Beneficial effects of a probiotic VSL#3 on parameters of liver dysfunction in chronic liver diseases. J Clin Gastroenterol. 2005;39(6):540–543. doi:10.1097/01.mcg.0000165671.25272.0f
141. Xia X, Chen J, Xia J, et al. Role of probiotics in the treatment of minimal hepatic encephalopathy in patients with HBV-induced liver cirrhosis. J Int Med Res. 2018;46(9):3596–3604.
142. Dhiman RK, Rana B, Agrawal S, et al. Probiotic VSL#3 reduces liver disease severity and hospitalization in patients with cirrhosis: a randomized, controlled trial. Gastroenterology. 2014;147(6):1327–37 e3. doi:10.1053/j.gastro.2014.08.031
143. Zhang H-L, Yu L-X, Yang W, et al. Profound impact of gut homeostasis on chemically-induced pro-tumorigenic inflammation and hepatocarcinogenesis in rats. J Hepatol. 2012;57(4):803–812. doi:10.1016/j.jhep.2012.06.011
144. Degirolamo C, Rainaldi S, Bovenga F, et al. Microbiota modification with probiotics induces hepatic bile acid synthesis via downregulation of the Fxr-Fgf15 axis in mice. Cell Rep. 2014;7(1):12–18. doi:10.1016/j.celrep.2014.02.032
145. Li J, Sung CYJ, Lee N, et al. Probiotics modulated gut microbiota suppresses hepatocellular carcinoma growth in mice. Proc Natl Acad Sci U S A. 2016;113(9):E1306–E1315. doi:10.1073/pnas.1518189113
146. Marlicz W, et al. The effect of short term treatment with probiotic VSL#3 on various clinical and biochemical parameters in patients with liver cirrhosis. J Physiol Pharmacol. 2016;67(6):867–877.
147. Smits LP, Bouter KEC, de Vos WM, et al. Therapeutic potential of fecal microbiota transplantation. Gastroenterology. 2013;145(5):946–953. doi:10.1053/j.gastro.2013.08.058
148. Bibbo S, Ianiro G, Gasbarrini A, Cammarota G. Fecal microbiota transplantation: past, present and future perspectives. Minerva Gastroenterol Dietol. 2017;63(4):420–430.
149. Hamilton MJ, Weingarden AR, Sadowsky MJ, et al. Standardized frozen preparation for transplantation of fecal microbiota for recurrent Clostridium difficile infection. Am J Gastroenterol. 2012;107(5):761–767. doi:10.1038/ajg.2011.482
150. Paramsothy S, Kamm MA, Kaakoush NO, et al. Multidonor intensive faecal microbiota transplantation for active ulcerative colitis: a randomised placebo-controlled trial. Lancet. 2017;389(10075):1218–1228. doi:10.1016/S0140-6736(17)30182-4
151. Vrieze A, Van Nood E, Holleman F, et al. Transfer of intestinal microbiota from lean donors increases insulin sensitivity in individuals with metabolic syndrome. Gastroenterology. 2012;143(4):913–6 e7. doi:10.1053/j.gastro.2012.06.031
152. Wang J-W, Kuo C-H, Kuo F-C, et al. Fecal microbiota transplantation: review and update. J Formos Med Assoc. 2019;118(Suppl 1):S23–S31. doi:10.1016/j.jfma.2018.08.011
153. Bhat M, Arendt BM, Bhat V, et al. Implication of the intestinal microbiome in complications of cirrhosis. World J Hepatol. 2016;8(27):1128–1136. doi:10.4254/wjh.v8.i27.1128
154. Pardo A, et al. Effect of cisapride on intestinal bacterial overgrowth and bacterial translocation in cirrhosis. Hepatology. 2000;31(4):858–863.
155. Madrid AM, Hurtado C, Venegas M, et al. Long-Term treatment with cisapride and antibiotics in liver cirrhosis: effect on small intestinal motility, bacterial overgrowth, and liver function. Am J Gastroenterol. 2001;96(4):1251–1255. doi:10.1111/j.1572-0241.2001.03636.x
156. Perez-Paramo M, Muñoz J, Albillos A, et al. Effect of propranolol on the factors promoting bacterial translocation in cirrhotic rats with ascites. Hepatology. 2000;31(1):43–48. doi:10.1002/hep.510310109
157. Senzolo M, Fries W, Buda A, et al. Oral propranolol decreases intestinal permeability in patients with cirrhosis: another protective mechanism against bleeding? Am J Gastroenterol. 2009;104(12):3115–3116. doi:10.1038/ajg.2009.457
158. Nkontchou G, Aout M, Mahmoudi A, et al. Effect of long-term propranolol treatment on hepatocellular carcinoma incidence in patients with HCV-associated cirrhosis. Cancer Prev Res (Phila). 2012;5(8):1007–1014. doi:10.1158/1940-6207.CAPR-11-0450
159. Herrera I, Pascual S, Zapater P, et al. The use of beta-blockers is associated with a lower risk of developing hepatocellular carcinoma in patients with cirrhosis. Eur J Gastroenterol Hepatol. 2016;28(10):1194–1197. doi:10.1097/MEG.0000000000000677
160. Wang F, Liu H, Wang F, et al. Propranolol suppresses the proliferation and induces the apoptosis of liver cancer cells. Mol Med Rep. 2018;17(4):5213–5221.
161. Sun SS, et al. An insoluble polysaccharide from the sclerotium of Poria cocos improves hyperglycemia, hyperlipidemia and hepatic steatosis in ob/ob mice via modulation of gut microbiota. Chin J Nat Med. 2019;17(1):3–14.
162. Chu H, Jiang L, Gao B, et al. The selective PPAR-delta agonist seladelpar reduces ethanol-induced liver disease by restoring gut barrier function and bile acid homeostasis in mice. Transl Res. 2020. doi:10.1016/j.trsl.2020.06.006
163. Cao LQ, Wang X-L, Wang Q, et al. Rosiglitazone sensitizes hepatocellular carcinoma cell lines to 5-fluorouracil antitumor activity through activation of the PPARgamma signaling pathway. Acta Pharmacol Sin. 2009;30(9):1316–1322. doi:10.1038/aps.2009.119
164. Zhang W, Wu N, et al. PPARgamma activator rosiglitazone inhibits cell migration via upregulation of PTEN in human hepatocarcinoma cell line BEL-7404. Cancer Biol Ther. 2006;5(8):1008–1014. doi:10.4161/cbt.5.8.2887
165. Zhou YM, Wen YH, Kang XY, et al. Troglitazone, a peroxisome proliferator-activated receptor gamma ligand, induces growth inhibition and apoptosis of HepG2 human liver cancer cells. World J Gastroenterol. 2008;14(14):2168–2173.
166. Shen B, Chu ESH, Zhao G, et al. PPARgamma inhibits hepatocellular carcinoma metastases in vitro and in mice. Br J Cancer. 2012;106(9):1486–1494. doi:10.1038/bjc.2012.130
167. Chen K, Dai W, Wang F, et al. Inhibitive effects of 15-deoxy-Delta(12),(14)-prostaglandin J2 on hepatoma-cell proliferation through reactive oxygen species-mediated apoptosis. Onco Targets Ther. 2015;8:3585–3593.
168. Han M, Gao H, Ju P, et al. Hispidulin inhibits hepatocellular carcinoma growth and metastasis through AMPK and ERK signaling mediated activation of PPARgamma. Biomed Pharmacother. 2018;103:272–283. doi:10.1016/j.biopha.2018.04.014
169. Marrapodi M, Chiang JY. Peroxisome proliferator-activated receptor alpha (PPARalpha) and agonist inhibit cholesterol 7alpha-hydroxylase gene (CYP7A1) transcription. J Lipid Res. 2000;41(4):514–520.
170. Ghonem NS, Ananthanarayanan M, Soroka CJ, et al. Peroxisome proliferator-activated receptor α activates human multidrug resistance transporter 3/ATP-binding cassette protein subfamily B4 transcription and increases rat biliary phosphatidylcholine secretion. Hepatology. 2014;59(3):1030–1042. doi:10.1002/hep.26894
171. Levy C, Peter JA, Nelson DR, et al. Pilot study: fenofibrate for patients with primary biliary cirrhosis and an incomplete response to ursodeoxycholic acid. Aliment Pharmacol Ther. 2011;33(2):235–242. doi:10.1111/j.1365-2036.2010.04512.x
172. Cheung AC, Lapointe-Shaw L, Kowgier M, et al. Combined ursodeoxycholic acid (UDCA) and fenofibrate in primary biliary cholangitis patients with incomplete UDCA response may improve outcomes. Aliment Pharmacol Ther. 2016;43(2):283–293. doi:10.1111/apt.13465
173. Ray EM, Sanoff HK. Optimal therapy for patients with hepatocellular carcinoma and resistance or intolerance to sorafenib: challenges and solutions. J Hepatocell Carcinoma. 2017;4:131–138. doi:10.2147/JHC.S124366
174. Hsu CH, et al. Sorafenib in advanced hepatocellular carcinoma: current status and future perspectives. J Hepatocell Carcinoma. 2014;1:85–99.
175. Wilhelm S, Carter C, Lynch M, et al. Discovery and development of sorafenib: a multikinase inhibitor for treating cancer. Nat Rev Drug Discov. 2006;5(10):835–844.
176. Feng J, Dai W, Mao Y, et al. Simvastatin re-sensitizes hepatocellular carcinoma cells to sorafenib by inhibiting HIF-1alpha/PPAR-gamma/PKM2-mediated glycolysis. J Exp Clin Cancer Res. 2020;39(1):24. doi:10.1186/s13046-020-1528-x
177. Li S, Dai W, Mo W, et al. By inhibiting PFKFB3, aspirin overcomes sorafenib resistance in hepatocellular carcinoma. Int J Cancer. 2017;141(12):2571–2584. doi:10.1002/ijc.31022
178. Suh SH, Choe K, Hong SP, et al. Gut microbiota regulates lacteal integrity by inducing VEGF-C in intestinal villus macrophages. EMBO Rep. 2019;20:4.
179. Andriessen EM, Wilson AM, Mawambo G, et al. Gut microbiota influences pathological angiogenesis in obesity-driven choroidal neovascularization. EMBO Mol Med. 2016;8(12):1366–1379. doi:10.15252/emmm.201606531
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.