")
Back to Journals » Journal of Asthma and Allergy » Volume 15
Gut Microbiome and Metabolomics Profiles of Allergic and Non-Allergic Childhood Asthma
Authors Zheng P , Zhang K, Lv X, Liu C, Wang Q, Bai X
Received 19 December 2021
Accepted for publication 2 March 2022
Published 6 April 2022 Volume 2022:15 Pages 419—435
DOI https://doi.org/10.2147/JAA.S354870
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Amrita Dosanjh
Ping Zheng,1 Kexing Zhang,2 Xifang Lv,1 Chuanhe Liu,3 Qiang Wang,1 Xuetao Bai1
1China CDC Key Laboratory of Environment and Population Health, National Institute of Environmental Health, Chinese Center for Disease Control and Prevention, Beijing, People’s Republic of China; 2Department of Immunization Program, Xinwu District Center for Disease Control and Prevention, Wuxi, People’s Republic of China; 3Children’s Hospital, Capital Institute of Pediatrics, Beijing, People’s Republic of China
Correspondence: Qiang Wang; Xuetao Bai, China CDC Key Laboratory of Environment and Population Health, National Institute of Environmental Health, Chinese Center for Disease Control and Prevention, 29 Nanwei Road, Xicheng District, Beijing, 100050, People’s Republic of China, Tel +86 10 50930251, Email [email protected]; [email protected]
Purpose: This study aimed to investigate the characteristics of gut bacteria and the derived metabolites among allergic asthmatic children, non-allergic asthmatic children and healthy children without asthma.
Methods: Fecal samples were collected from 57 participants, including 20 healthy children, 27 allergic asthmatic children, and 10 non-allergic asthmatic children. 16S rRNA gene sequencing was conducted for analyzing gut bacterial compositions and untargeted metabolomics was used to analyze the alterations of gut microbe-derived metabolites. The associations between gut bacterial compositions and metabolites were analyzed by the method of Spearman correlation.
Results: The results showed that the compositions and metabolites of gut microbiome were altered both in allergic and non-allergic asthmatics compared with healthy controls. Chao1 (p = 0.025) index reflected a higher bacterial richness and Simpson (p = 0.024) index showed a lower diversity in asthma group. PERMANOVA analysis showed significant differences among the three groups based on unweighted UniFrac distance (p = 0.001). Both allergic and non-allergic asthmatics showed a higher relative abundance of Proteobacteria and a lower relative abundance of genera from Clostridia. More bacteria were altered in non-allergic asthmatics compared with allergic asthmatics. Metabolomics analysis identified that 42 metabolites were significantly associated with allergic asthma, and 58 metabolites were significantly associated with non-allergic asthma (multiple linear regression, p < 0.05). Histamine was 4 folds up-regulated only in the non-allergic asthma group. The relative abundance of Candidatus Accumulib was significantly correlated with the upregulation of histamine. The relative abundance of genera from Clostridia was significantly correlated with the downregulation of lipid and tryptophan metabolism.
Conclusion: The altered gut microbes was associated with the mechanism of asthma attack through metabolites in allergic and non-allergic asthma group, respectively. The result suggested that gut microbiome had an impact on the development of both allergic and non-allergic asthma. The distinct gut microbiome and microbiome-derived metabolites in non-allergic asthma children suggested that gut microbiome might play a critical role in modulation of asthma phenotype.
Keywords: gut microbiome, untargeted metabolomics, allergic childhood asthma, non-allergic childhood asthma, 16S rRNA gene sequencing
Introduction
Childhood asthma is one of the most prevalent chronic pediatric diseases. It affected children’s health all over the world.1 Asthma is a heterogeneous disease with complex pathogenesis, which presents many phenotypes.2 Allergic and non-allergic asthma are the most common phenotypes of asthma. Approximately 80% of children with asthma were reported to be allergic.3 Allergic asthma is mainly induced by inappropriate immune responses of type 2 T helper (Th2) cells, which might be triggered by an increased serum level of total immunoglobulin E (IgE) due to allergen exposures. While non-allergic asthma occurs independently of allergen sensitization.4 Mechanisms of non-allergic asthma development have not been fully elucidated yet.5,6
Apart from allergen triggers, recent studies showed that development of asthma might be modulated by gut microbiome. Hygiene hypothesis firstly suggested a link between microbes and allergy.7,8 Riedler’s study showed that exposures to stables in early lives might have a protective effect not only on allergic but also on non-allergic asthma, which suggested that gut microbiome may be also related with non-allergic asthma based on the hygiene hypothesis.9 Recent studies showed the association between early life gut microorganism colonization and risk of asthma later in life.10–13 As the improvements in multi-omic technologies, gut microbiome has been demonstrated to affect the immune responses in the lung by microbes or microbes-derived metabolites. Surviving gut microbes, fragments of dead bacteria or immune cells (activated by gut microbes or their metabolites) may travel to the lung through lymphatic circulation and trigger the host immune responses.14–16 In addition, metabolites derived from gut microbiome, for instance, short chain fatty acids (SCFAs) could migrate through the bloodstream to modulate lung immune responses.16 Animal experiments illustrated that some gut microbes could regulate differentiation of regulatory T cells (Tregs), and modulate the balance of Th1 and Th2 cells.17–21 While, some gut microbes can also induce Th17 responses.22,23 These findings suggested that a continued cross-talk may exist between gut microbes and immune responses, dysbiosis of gut microbiome may also play a critical role in the recurrent attack of asthma. Furthermore, Zou’s study showed that gut microbiome between allergic and non-allergic asthma patients were different.24 Thus, we hypothesized that gut microbiome has an impact on the attack of both allergic and non-allergic childhood asthma, whereas it plays a different role in allergic and non-allergic asthma.
To confirm our hypothesis, we conducted this study by combining the methods of 16S rRNA gene sequencing and fecal metabolomics to analyze gut microbial compositions and functions in allergic and non-allergic childhood asthma. Fecal metabolomics is the functional readout of gut microbiome.25
Materials and Methods
Study Population
Participants in this study were recruited from a project of National Natural Science Foundation of China, Epigenetic Modification Mechanisms of Black Carbon Impacts on Non-allergic Asthma, performed from 2016 to 2019. A total of 57 children met the criteria of sample collection, and agreed to join in this study. All participants aged from 5 to 14 years old. They all lived in urban areas of Beijing. All asthmatic participants were mild asthma. Asthmatic children were diagnosed according to the Global Initiative for Asthma (GINA, http://ginasthma.org) by pediatricians from Children Hospital of Capital Institute of Pediatrics in Beijing. Briefly, asthma was confirmed as repeated attacks of respiratory symptom (wheezing, shortness of breath, chest tightness, and cough), together with variable expiratory airflow limitation. Lung function and fractional exhaled nitric oxide (FeNO) of asthmatic children was measured by pediatricians. Allergic and non-allergic status of asthmatics was identified by skin prick testing. Children without any history of asthma, allergic disease, and chronic inflammation of the respiratory system were recruited as healthy controls. Height and weight were measured by trained professionals. In addition, all participants were asked to offer information about basic characteristics, and records of disease with a questionnaire based on the questionnaire of the International Study of Asthma and Allergies in Childhood. Food consumption of all participants were investigated by a questionnaire based on the food frequency questionnaire of National Health and Nutrition Examination Survey, which mainly including vegetables, mushrooms and eggs. Carbohydrate intake was defaulted to no difference, as all children shared similar carbohydrate consumption in their diet based on the local diet habits. This study was conducted in accordance with the Declaration of Helsinki and approved by the ethical review committee of the Institute for Environmental Health and Related Product Safety, Chinese Center for Disease Control and Prevention (201,501). All participants and their legal guardians signed the written informed consent.
Fecal Sample Collection
A total of 57 fecal samples were collected between December 2017 to March 2018, including 27 samples from children with allergic asthma, 10 samples from children with non-allergic asthma and 20 samples from healthy children. All participants, who offered feces, were asked to meet the following criteria, including no oral drug intake or no vaccination history in the past one month, no injury or diarrhea in the past two weeks, girls were not in the period of the menstrual cycle when offering feces. Fecal samples of all participants were collected into sterilized tubes and stored at a temperature of −80°C until detected. After removing low quality samples, gut microbiome was detected in 47 samples and metabolomics analysis was detected in 50 samples.
Gut Microbiome Detection and Analysis
Total genome DNA from fecal samples was extracted using the CTAB (Cetyltrimethylammonium Bromide) method. The V4 region of 16S rRNA genes was amplified by using the specific primers of 515F and 806R. Paired end sequencing (2 × 250 bp) was performed on the Illumina HiSeq 2500 (Illumina Inc., USA) by Novogene Bioinformatics Technology Co.
Paired-end reads were assigned to samples based on the unique barcode and truncated by cutting off the barcode and primer sequence. Then they were merged using Fast Length Adjustment of Short reads (FLASH, v1.2.7) to obtain the raw tags.26 The demultiplexed data were conducted using QIIME2.27 Errors were corrected by DADA2.28 Then a tree of phylogenetic diversity analyses was generated. Taxonomy was classified according to the database of SILVA.29 Chao1 and Simpson indices were calculated for evaluating the alpha diversity of gut microbiome. Weighted and unweighted unique fraction metric (UniFrac) distances were calculated to estimate beta diversity.30 Principal coordinates analysis (PCoA) was used to show the clustering of all samples. Linear Discriminant Analysis (LDA) of effect size (LEfSe) was used to discriminate the taxa with statistical and biological difference.31
Metabolomics Detection and Analysis
An untargeted metabolomics analysis on fecal samples was carried out at METABOLON’s Discovery HD4™ Metabolomics Platform (Calibra (DIAN) - Metabolon Joint Laboratory, Hangzhou, China). Samples were prepared and detected according to the operation process of the platform.32 Raw data was extracted, peak-identified and QC processed using Metabolon’s proprietary software. Compounds were identified by comparison to library entries of purified standards. Peaks were quantified using area-under-the-curve. Missing values in samples were filled in with the minimum observed value for each compound, and raw data were normalized to the median of each compound in all samples. Functional pathways were annotated using Kyoto Encyclopedia of Genes and Genomes (KEGG).
Statistical Analysis
Statistical analysis was carried out using R software (v4.0.2). Characteristics of study populations were analyzed by the method of Chi square test, t-test and ANOVA. Differences in gut bacteria diversity and relative abundance among three groups were analyzed by the nonparametric Kruskal–Wallis test. Differences between two groups were analyzed by the Wilcoxon test. Difference of community structure of gut microbiome among groups was analyzed using the method of permutational multivariate ANOVA (PERMANOVA). Metabolomic data were log transformed before statistical analysis. Multiple linear regression was used to identify metabolites that were associated with asthma, by controlling for age, gender and BMI. Spearman correlation was used to analyze the correlation between gut microbes and metabolites. All reported p values were two-tailed and p <0.05 was considered significant.
Results
Characteristics of Study Populations
A total of 57 participants were enrolled in this study, including 27 children with allergic asthma, 10 children with non-allergic asthma and 20 healthy children for control. Of these participants, 54.4% were boys, and 45.6% were girls. The mean age of all participants was 8.0 ± 2.0 years old. All children were Han nationality. Table 1 showed the baseline characteristics of each group. Table 2 showed the details of clinical information of all asthmatic children, including lung function, FeNO and allergens. There was no significant difference in lung function between allergic and non-allergic asthmatic children (t-test, p >0.05). Allergic asthmatic children showed a significantly higher value of FeNO (t-test, p =0.021). Approximately 59.2% allergic asthma children had two or more allergens.
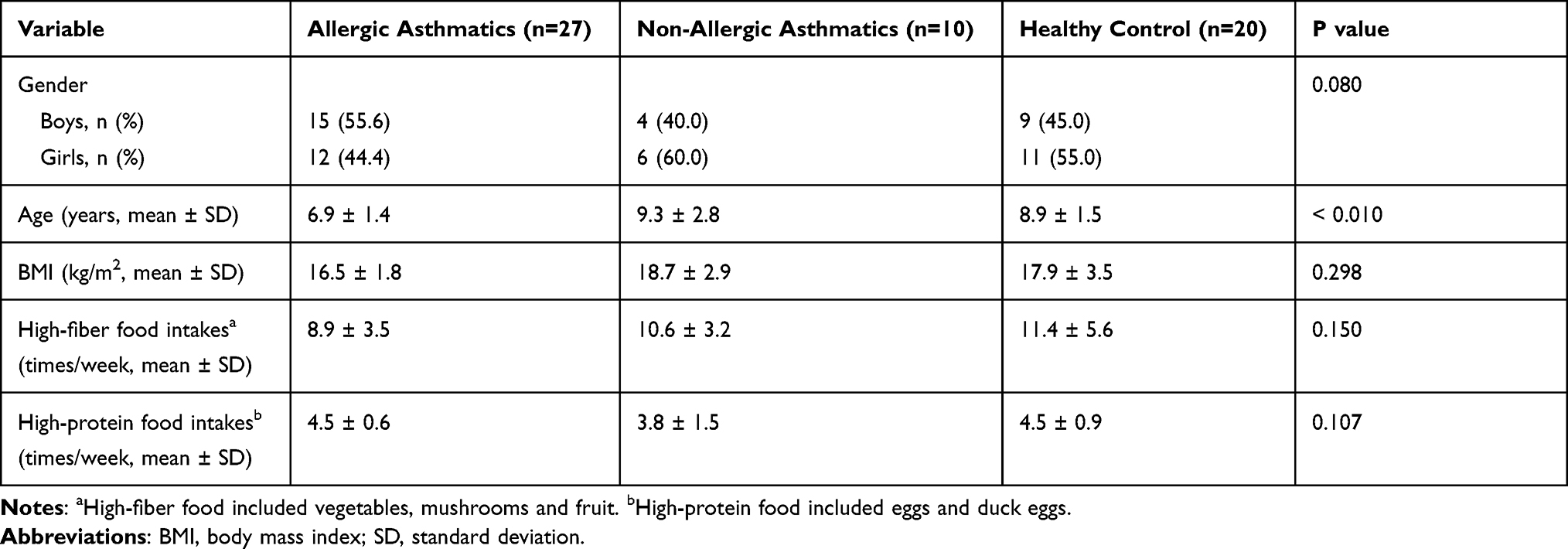 |
Table 1 Baseline Characteristics of All Study Populations |
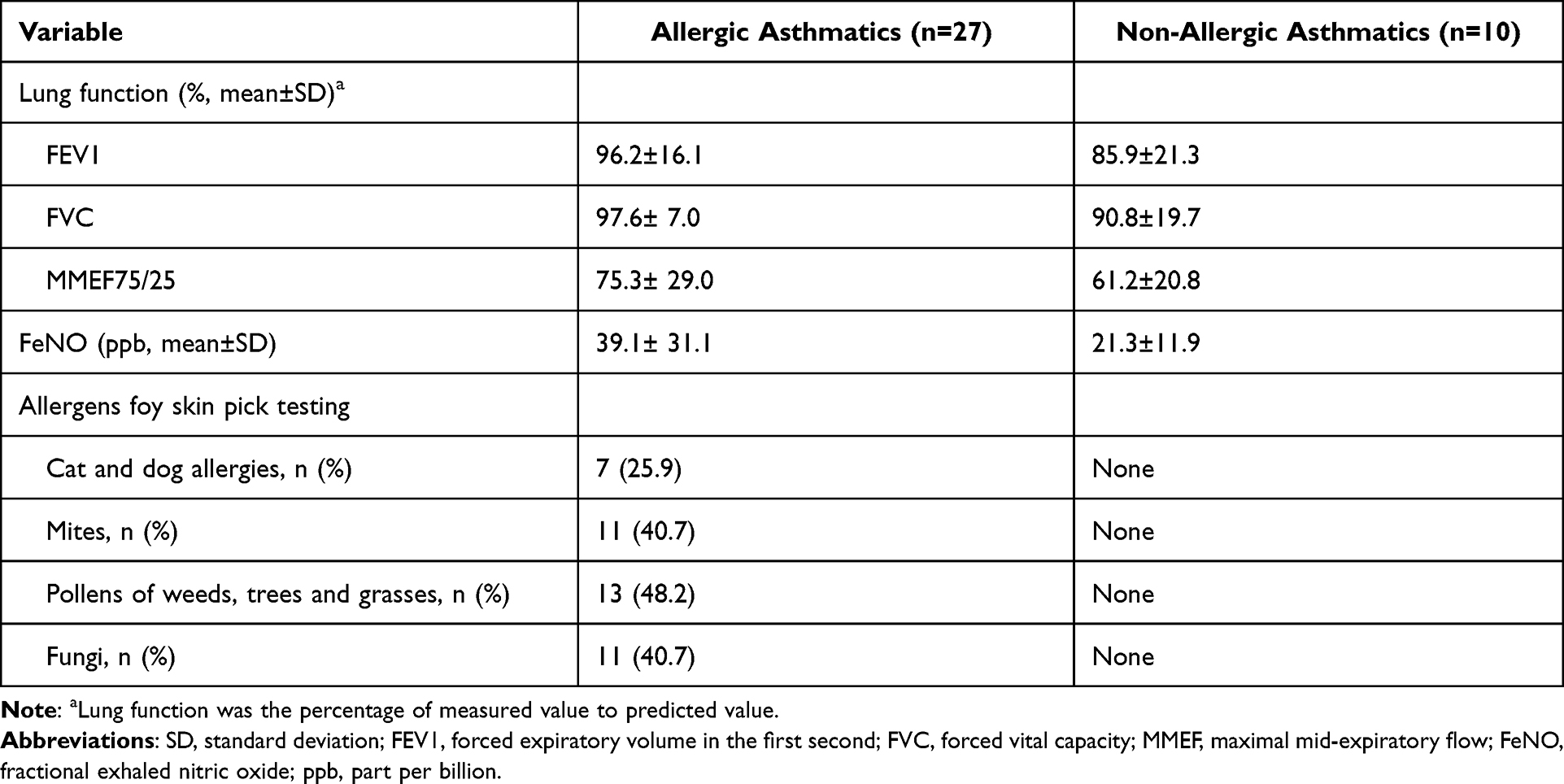 |
Table 2 Clinical Information of Asthmatic Children |
Characteristics of Gut Microbiome
Diversity of Gut Microbiome
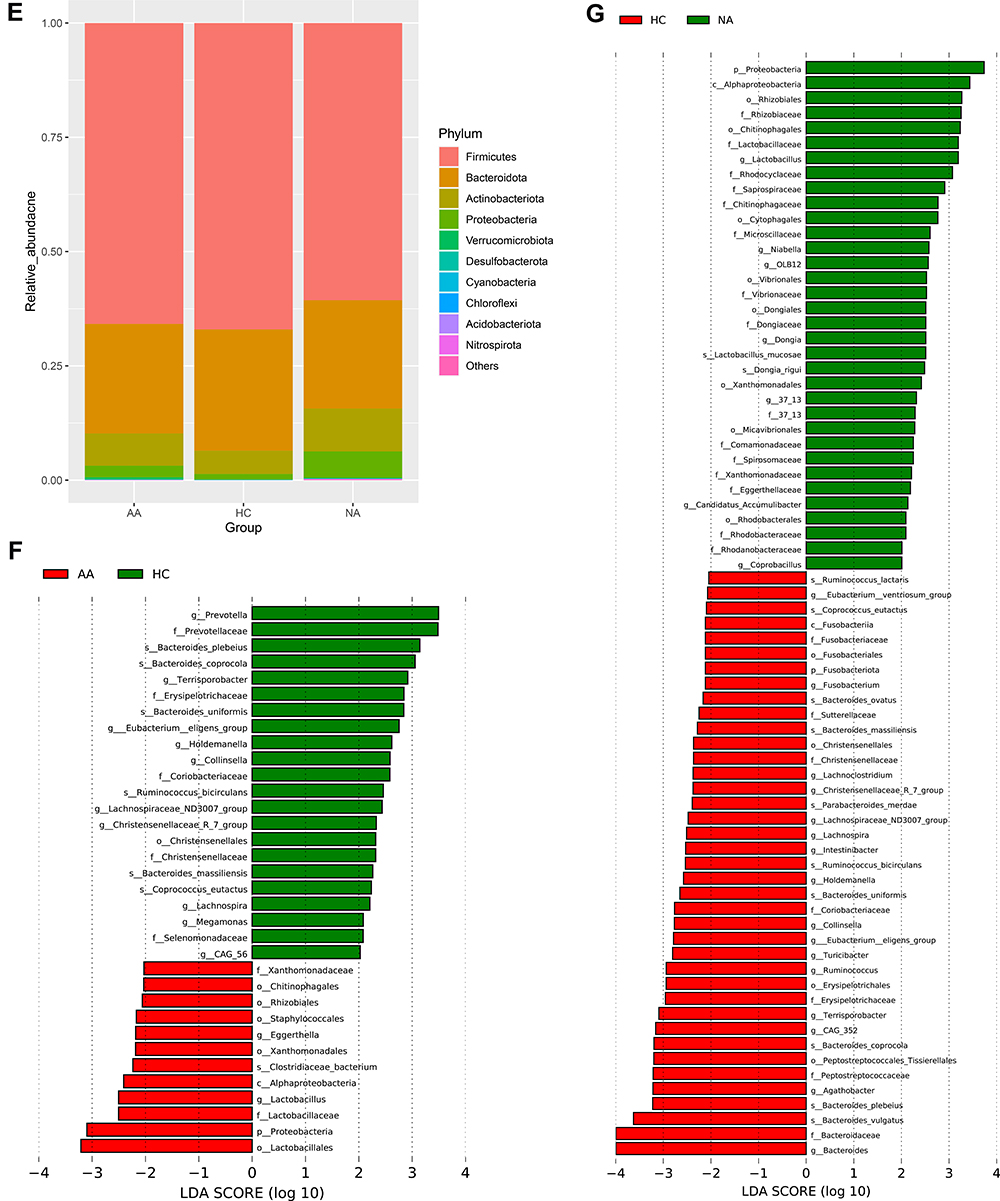
We performed a 16S rRNA genes sequencing to investigate characteristics of gut bacteria in allergic and non-allergic asthmatics. Chao1 (p =0.025) and Simpson (p =0.024) indices showed significant differences among the three groups (Kruskal–Wallis test). Figure 1A and B showed the parameters of alpha diversity by groups. These results reflected a higher bacterial richness and a lower diversity in asthma group. We assessed beta diversity of gut microbiome based on weighted and unweighted UniFrac distance. Figure 1C and D showed beta diversity with the PCoA plot among the three groups. PERMANOVA analysis showed a significant difference among the three groups based on unweighted UniFrac distance (p = 0.001). Figure 1 Continued. Figure 1 Characters of gut microbiome. (A and B) Alpha diversity (Chao1 and Simpson parameter) in allergic, non-allergic asthma and healthy control groups. (C) PCoA plot based on weighted UniFrac distance, (D) PCoA plot based on unweighted UniFrac distance. (E) Relative abundance of top 10 phyla by groups. The height of bars represents relative abundance. (F) LEfSe analysis between allergic asthma and healthy control groups. (G) LEfSe analysis between non-allergic asthma and healthy control groups. The graph of LEfSe distinguished the microbial communities of each group with LDA > 2 and p < 0.05. Abbreviations: AA, allergic asthma group; NA, non-allergic asthma group; HC, healthy control group.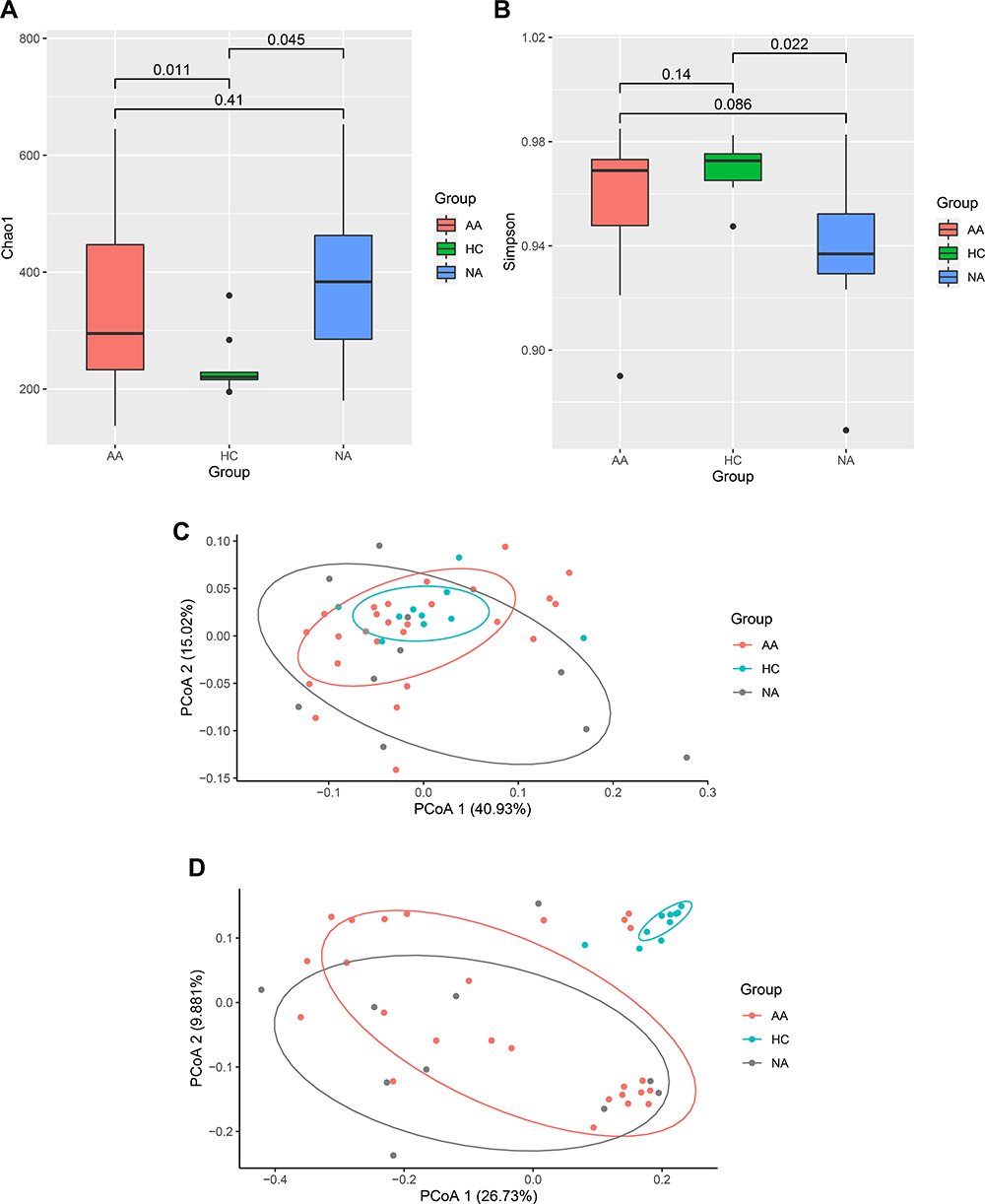
Differential Gut Microbes Among Groups
All of the observed sequences were assigned to 35 phyla. Figure 1E showed the relative abundance of top 10 phyla among the three groups. Firmicutes, Bacteroidetes, Actinobacteria, and Proteobacteria were the dominant phyla of all samples. Proteobacteria showed significant difference among the three groups (Kruskal–Wallis test, p = 0.008). Both allergic (p =0.007) and non-allergic (p =0.019) asthma groups showed significant increases compared with healthy group (Wilcoxon test).
We used LEfSe analysis to discriminate taxa with statistical and biological differences among the three groups, stratified by allergy and non-allergy status. Figure 1F showed the taxa with significant discriminations between allergic asthma and healthy control groups. Figure 1G showed the taxa with significant discriminations between non-allergic asthma and healthy control groups. All discriminated taxa were from phyla of Firmicutes, Bacteroidetes, Actinobacteria and Proteobacteria, except for Fusobacteriota. Difference of Fusobacteriota was induced only by Fusobacterium at genus level, which decreased significantly in non-allergic asthma group. The richest taxon in allergic asthma was Lactobacillales. The next one was phylum of Proteobacteria, including Alphaproteobacteria, Rhizobiales, and Xanthomonadales (Xanthomonadaceae). While in non-allergic asthma group, more taxa (allergic/non-allergic:12/34) ranging from phylum to species were significantly overrepresented, especially more taxa (allergic/non-allergic:5/19) from Proteobacteria showed significant differences. In healthy control group, most of the discriminated taxa were from Bacteroidetes and Firmicutes at phylum level. For phylum of Bacteroidetes, Prevotella and Bacteroides showed significant richness. For phylum of Firmicutes, more bacteria from class of Clostridia showed significant richness, such as Terrisporobacter, Eubacterium eligens group, Lachnospira, Lachnospiraceae ND3007 group, and Christensenellaceae R-7 group.
Then we assessed the alterations in relative abundance at genus level. We found 9 genera of the top 50 genera showed significant differences between allergic asthmatics and healthy controls (8 genera decreased, 1 genus increased), and 14 genera of the top 50 genera showed significant differences between non-allergic asthmatics and healthy controls (13 genera decreased, 1 genus increased) (Wilcoxon test, p < 0.05). Table 3 presented details of the genera with significant differences. We noted that 10 genera among the significantly decreased genera of all asthmatics belonged to Clostridia.
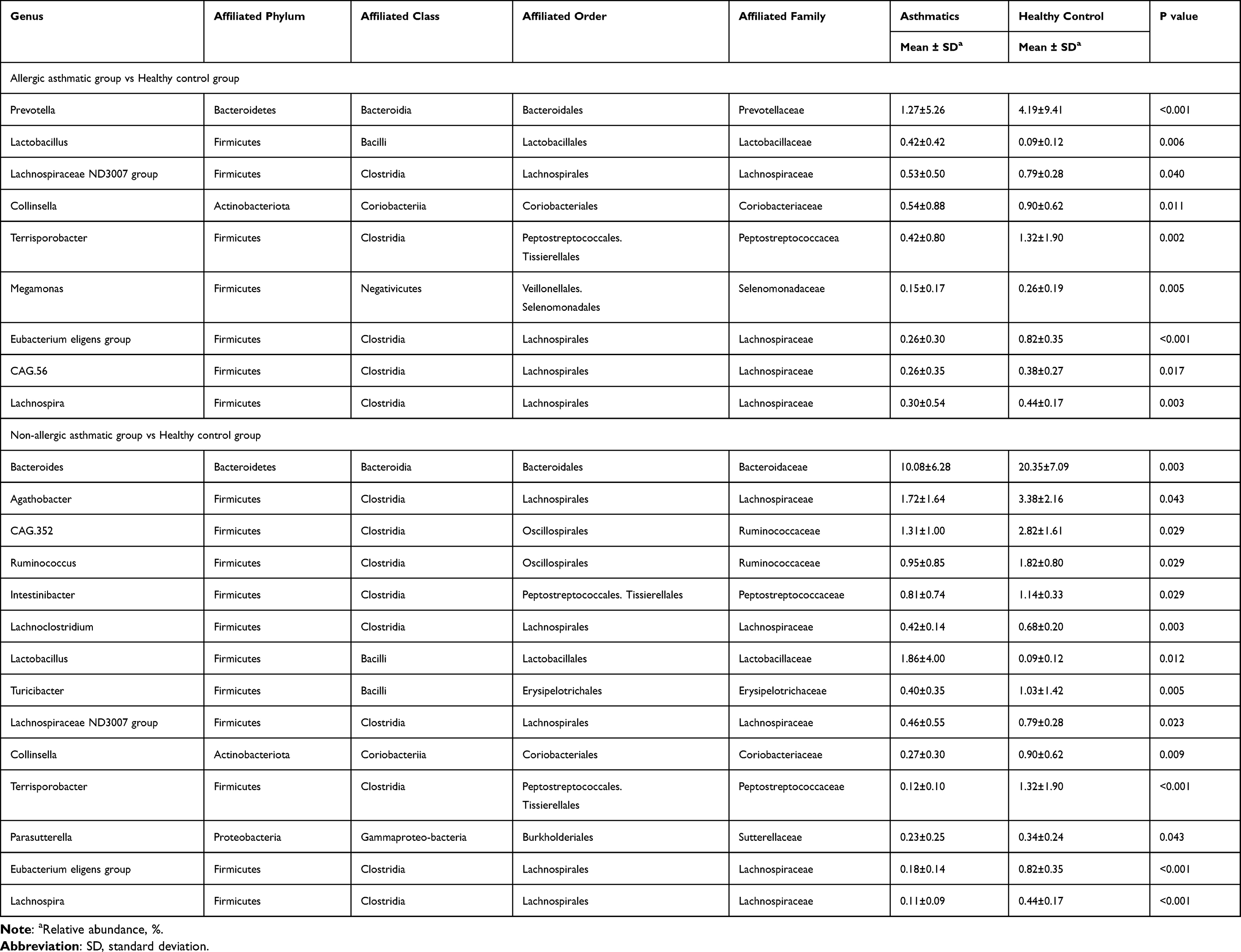 |
Table 3 Genera with Significant Differences in Relative Abundance Between Asthmatic Group and Healthy Control Group (Wilcoxon Test) |
Based on the altered genera obtained from LEfSe and Wilcoxon test analysis, we compared the altered gut bacteria in allergic and non-allergic asthma. Among the significantly increased genera in asthmatics, allergic and non-allergic asthma shared 1 genus (Lactobacillus), while Eggerthella increased only in allergic asthma, Coprobacillus, Niabella, Chitinophagales|37-13, Microscillaceae OLB12, Candidatus Accumulib, and Dogia increased only in non-allergic asthma. Among the significantly decreased genera in asthmatics, allergic and non-allergic asthma group shared 7 genera, including Collinsella, Christensenellaceae R 7 group, Eubacterium eligens group, Lachnospira, Lachnospiraceae ND3007 group, Terrisporobacter, and Holdemanella. Megamonas, Lachnospiraceae CAG-56, and Prevotella only decreased in allergic asthmatics. Intestinibacter, Lachnoclostridium, Ruminococcus, Agathobacter, Eubacterium ventriosum group, Ruminococcaceae CAG-352, Turicibacter, Parasutterella, Bacteroides, and Fusobacterium only decreased in non-allergic asthmatics.
Characteristics of Gut Metabolomics
To further study the functions of gut bacteria, we conducted an untargeted metabolomics analysis. A total of 811 metabolites were found in all samples. We analyzed the associations of asthma status with the metabolites by the method of multiple linear regression, controlling for age, gender and BMI. Among all metabolites, 42 metabolites were significantly associated with allergic asthma (14 of them elevated and 28 of them down regulated), and 58 metabolites were significantly associated with non-allergic asthma (9 of them elevated and 49 of them down regulated) (p < 0.05). Only 6 metabolites showed significantly associations with both allergic and non-allergic asthma. Table 4 showed the pathway of the significantly altered metabolites involved in allergic and non-allergic asthma groups. Supplementary Table 1 showed details of the significantly altered metabolites in allergic and non-allergic asthma groups.
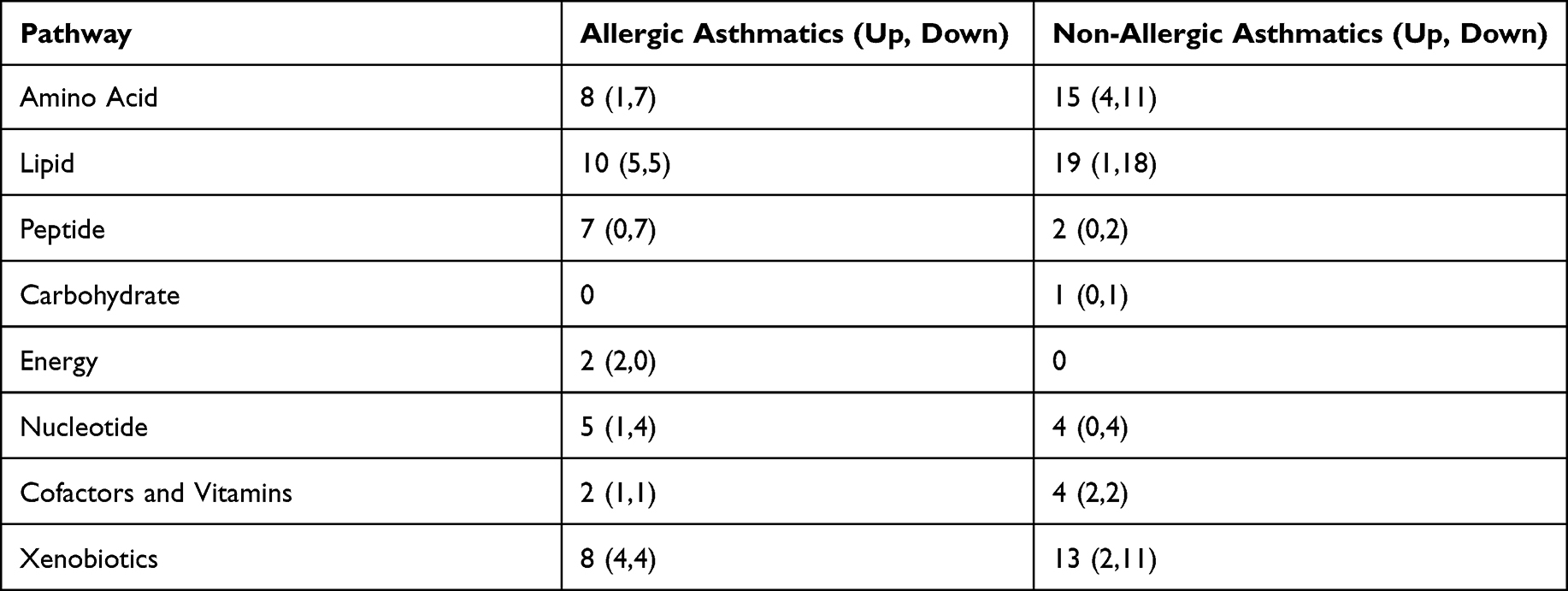 |
Table 4 The Involved Metabolic Pathways of the Altered Metabolites in Allergic and Non-Allergic Asthma Groups |
The altered metabolites mainly involved in amino acid and lipid metabolism. More metabolites of amino acid and lipid metabolism showed significant alterations in non-allergic asthmatic group. For amino acid metabolism, 8 metabolites were significantly altered in allergic asthmatics, while 15 metabolites were significantly altered in non-allergic asthmatics. Only tryptophan and lysine metabolism showed significant alterations in both allergic and non-allergic asthmatic groups. Especially, the metabolites of histidine metabolism, including histamine, N-acetylhistamine, and 4-imidazoleacetate, showed significant increases only in non-allergic asthma group. For fatty acid metabolism, 17 metabolites were significantly decreased in asthmatic groups (p <0.05). Among them, only 4 metabolites were significantly changed in allergic asthmatic group, while 15 ones were significantly changed in non-allergic asthmatic group. Fatty acids metabolism in non-allergic asthmatic group presented more alterations. A marginally significant decrease of SCFAs (p =0.089), and significant decreases in medium chain fatty acids and long chain fatty acids (p <0.05) were observed in non-allergic asthma compared with healthy control group.
Correlation Between Gut Microbiome and Metabolites
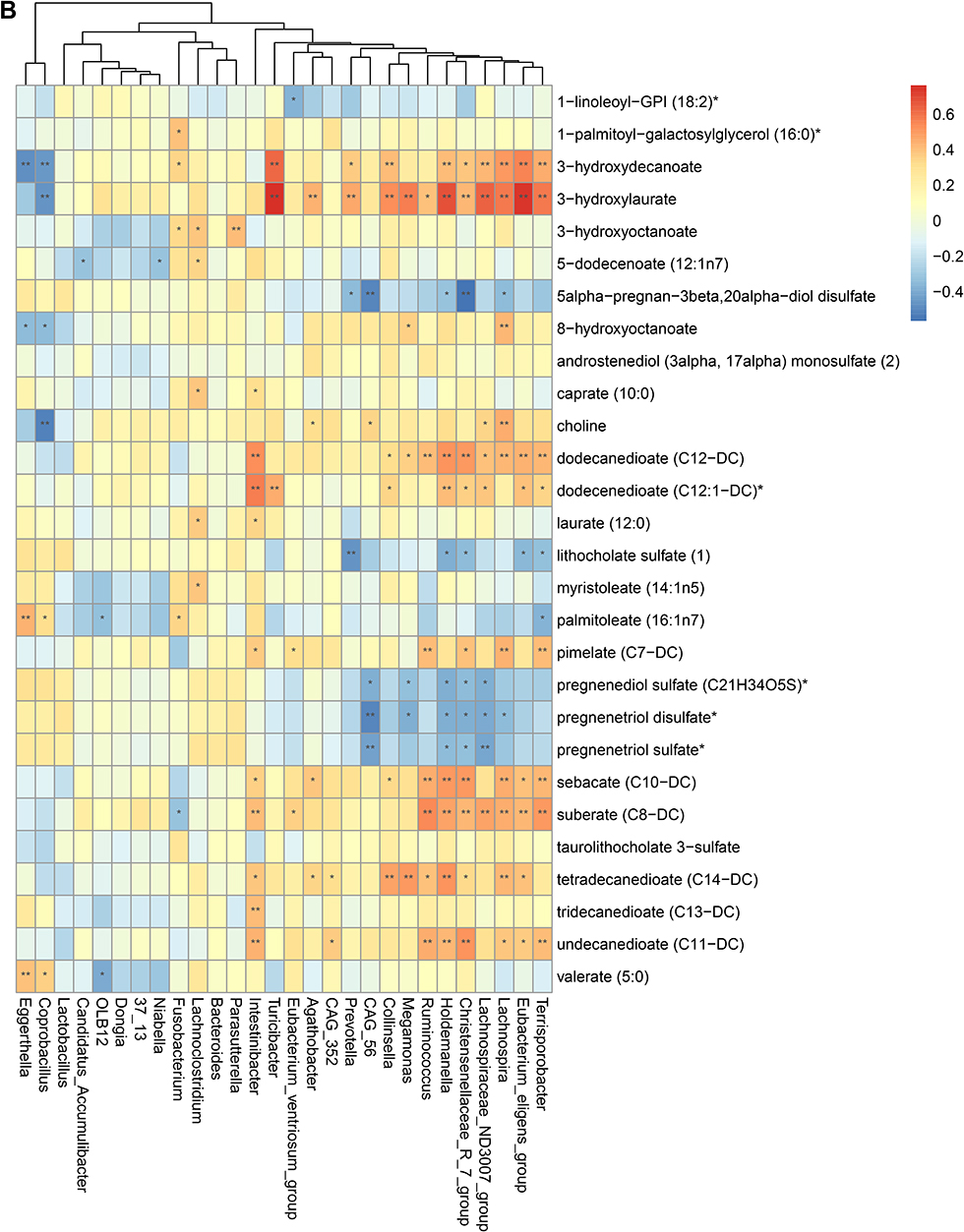
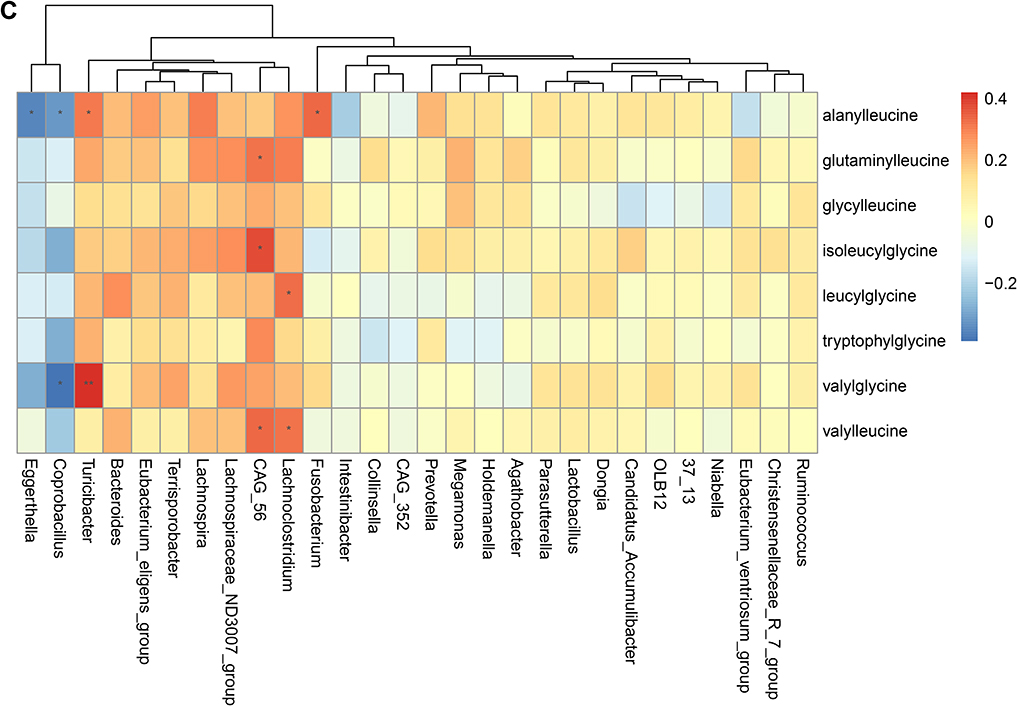
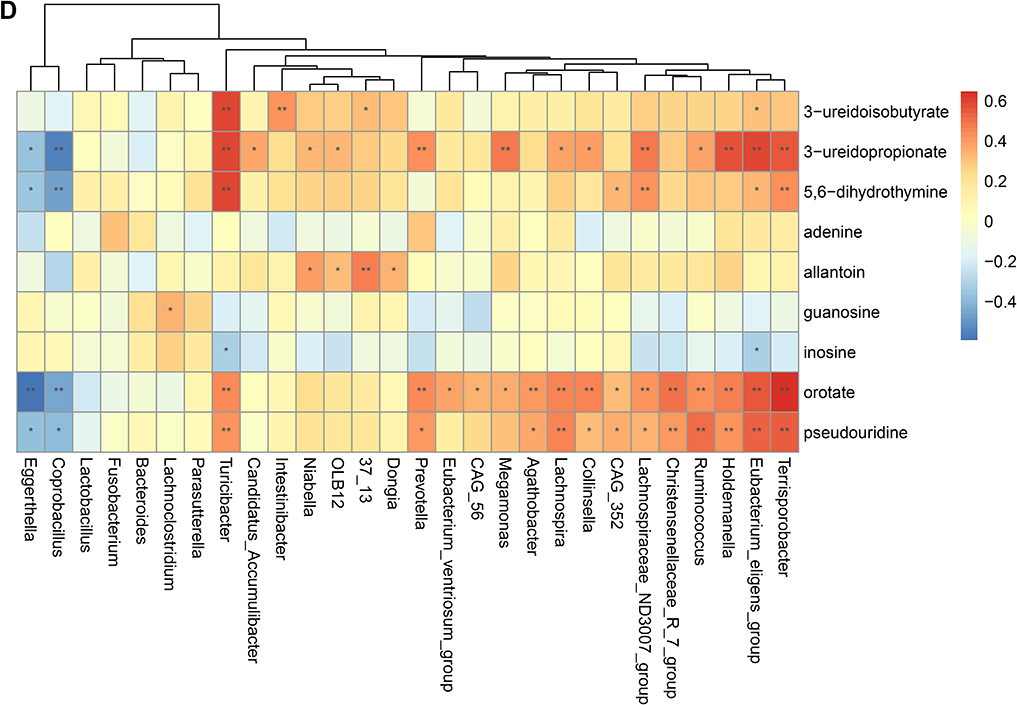
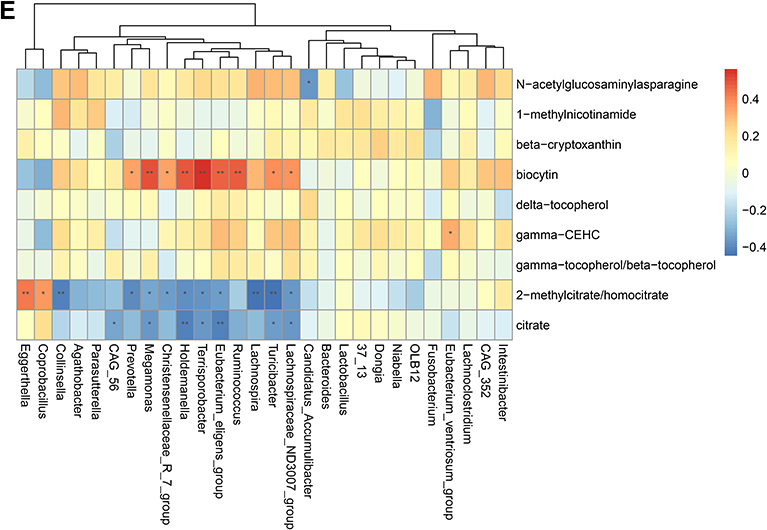
To validate the association between the altered gut microbiome and the altered metabolites in the pathogenesis, we performed a Spearman correlation analysis with the samples tested for both gut microbiome and metabolomics. We analyzed the correlations between the altered genera and the altered metabolites, except xenobiotics metabolites. Figure 2A–E presented the correlations between the altered gut microbes and metabolites. We found that the metabolites of amino acid and lipid metabolism showed more correlations with gut microbes. Figure 2A showed that 28 genera were significantly associated with 20 metabolites of amino acid. Decreased bacteria of Clostridia mainly showed significantly positive correlations with metabolism of tryptophan, tyrosine, lysine, methionine and cysteine. For histidine metabolism, decreased Lachnospira showed a significantly positive correlation with histidine, increased Candidatus Accumulibacter showed a significantly positive correlation with histamine, decreased Christensenellaceae R 7 group, Eubacterium ventriosum group, and Intestinibacter all showed significantly negative correlations with N-acetylhistamine. Christensenellaceae R 7 group, Holdemanella, Turicibacter, and Prevotella showed significantly negative correlations with 4-imidazoleacetate. Figure 2B presented that 24 altered genera showed significant correlations with 26 metabolites of lipid metabolism. Among them, 12 genera from Clostridia showed significant correlations with fatty acid metabolism. Microscillaceae OLB12 was negatively associated with valerate, Eggerthella and Coprobacillus were positively associated with valerate. Candidatus Accumulibacter and Niabella were negatively associated with medium chain fatty acid, Lachnoclostridium was positively associated with medium chain fatty acid. Eggerthella, Coprobacillus, Fusobacterium and Lachnoclostridium showed positive correlations with Long Chain Fatty Acid, Microscillaceae OLB12 and Terrisporobacter showed negative correlations with long chain fatty acid. Figure 2C showed that the altered genera were less associated with peptide metabolism. Figure 2D showed that 22 altered genera were significantly positively associated with 7 nucleotide metabolites, only Eggerthella and Coprobacillus were significantly negatively associated with 4 nucleotide metabolites. Figure 2E presented 11 genera were significantly negatively associated with TCA cycle, 9 genera were significantly positively associated with biocytin. Figure 2 Continued. Figure 2 Continued. Figure 2 Continued. Figure 2 Continued. Figure 2 Spearman correlation heatmap for significantly altered metabolites and genera in asthmatics. *p <0.05, **p <0.01. (A) Correlation heatmap for amino acid. (B) Correlation heatmap for Lipid. (C) Correlation heatmap for peptide. (D) Correlation heatmap for nucleotide. (E) Correlation heatmap for others, including carbohydrate, energy, cofactors and vitamins.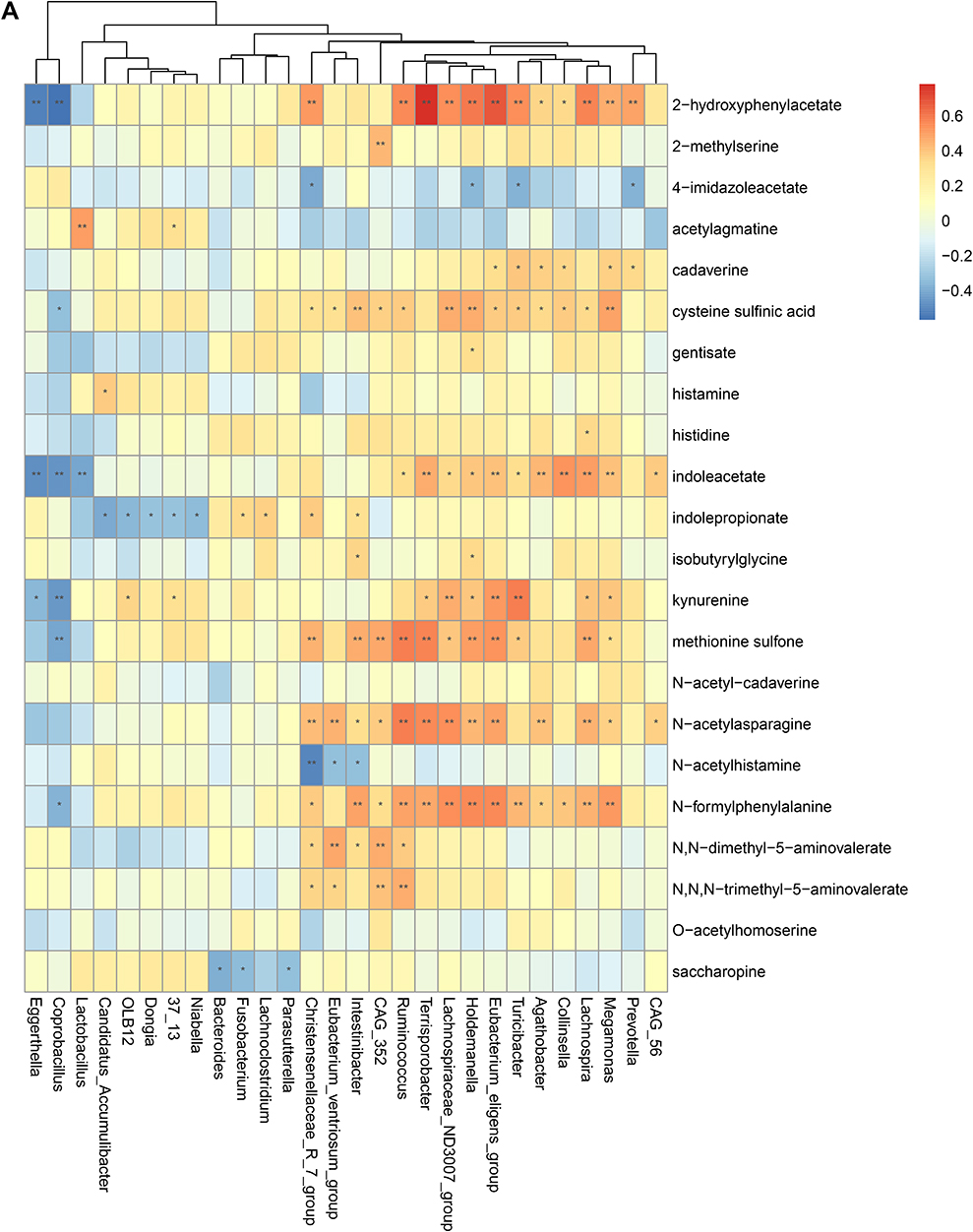
Discussion
In this study, we combined 16S amplicon sequencing and untargeted metabolomics analysis to explore the gut microbiome association with childhood asthma by allergic status. We found that allergic asthmatic children and non-allergic asthmatic children shared some alterations in gut microbes and microbiome-derived metabolites. In addition, we also found that non-allergic asthma had some distinct commensal gut microbes and microbiome-derived metabolites relative to that of allergic asthma.
Firstly, as previous findings we found asthma children had lower gut microbiome diversity than healthy controls.24,33,34 Variation of gut bacterial diversity has been found closely associated with allergic disease including food allergies, diabetes, asthma, even cancer.34–38 Apart from the diversity difference between asthmatic children and healthy controls, we found distinguished Simpson diversity (marginally significant) between allergic and non-allergic asthma children. The heterogeneity of gut microbiome might be related with phenotypic features.39 Lifestyle and diet are important impacting factors of gut microbiome. However, all participating children in this study came from Beijing. They were all students and they were similar in self-reported diets. Therefore, our findings of diversity difference in asthma children suggested that gut microbiome might play a role in asthma phenotype development.
The pathological mechanism of non-allergic asthma has not been fully elucidated yet. Recent studies showed that the human gut microbiome would be an etiological factor of various diseases.39 In this study, gut microbiome was especially altered in asthma children including Clostridia and Proteobacteria. While the altered genera and counts of Clostridia and Proteobacteria were quite different between allergic and non-allergic children. Existing studies have proved that Clostridia and Proteobacteria were related to the pathogenesis of asthma. Clostridia was reported to produce SCFAs which may stimulate the differentiation of Tregs.34–36 Tregs might mediate immunomodulation during asthma development.40 Impaired Tregs would predispose to severe viral bronchiolitis in infancy which was thought to be a major risk of asthma.41 Gut Proteobacteria has been reported to significantly increase in asthmatics.42 Several studies showed that Proteobacteria was higher in the airway of asthmatic subjects.43–45 Brevundimonas vesicularis of Proteobacteria was found enriched in non-allergic asthma.24 More serious dysbiosis in Clostridia and Proteobacteria in non-allergic asthma suggested that they might also play a role in asthma phenotype development. However, few studies explored the host-microbe association by allergic status of asthma. Some certain gut microbes, including Coprobacillus, Niabella, Chitinophagales|37-13, and Microscillaceae OLB12, were enriched only in non-allergic children. The potential roles of these distinct microbes in the development of asthma need to be further studied.
We then further analyzed the metabolome of the gut microbiome for a better view of the microbe-host association. The most interesting finding was that histamine significantly increased only in non-allergic asthma children. Histamine is an important immunomodulator involved in allergic reactions and inflammatory responses. Its functions are determined by the histamine receptor subtypes. It can activate H1R to enhance Th1 and Th2 type immune responses, activate H4R in human immune cells to release pro-inflammatory cytokine and chemokines to modulate immune responses, such as IL-6, TNF-α, IL-8, and also can induce smooth muscle contraction via activating H1R.46–48 Our finding suggested gut bacteria-derived histamine may be related with non-allergic asthma development. Gut histamine was derived from histidine decarboxylation under the action of histidine decarboxylase enzyme (HDC). Some bacteria can secrete HDC, such as Oenococcus oeni and Lactobacillus hilgardii.49,50 Bacterial HDC gene level has been reported significantly elevated in asthmatics, Morganella morganii levels were correspondingly elevated in the gut microbiome.51 We compared relative abundances of the altered microbes with microbe-derived metabolites. We did not find Morganella morganii’s correlation with histamine, however, we only found Candidatus Accumulibacter was positively correlated with histamine. It may be influenced by the sample size. To our knowledge, there was no study reported the relation of Candidatus Accumulibacter and histamine. Candidatus Accumulibacter has been widely used in enhanced biological phosphorus removal (EBPR). At the anaerobic phase of EBPR, SCFAs were absorbed. The role of Candidatus Accumulibacter in non-allergic asthma needs to be further studied.
Apart from the distinct increase of histamine in non-allergic asthmatic children, we found some genera of Clostridia were positively correlated with lipid and tryptophan metabolism. Lipid metabolism were altered in asthmatics in this study, which was consistent with previous finding.52 Lipids have been reported to play an important role in the development of asthma through inflammatory responses.53 Previous study showed that abnormal lipid metabolism was correlated with the severity and IgE levels in asthmatic patients.54 Some microbes of Clostridia have been reported to correlated with asthma by producing SCFAs.19 More lipid metabolites were changed in non-allergic asthma in our study. Combining analyzed with Clostridia dysbiosis in non-allergic asthma, it suggested that Clostridia may also play a role on asthma phenotype development by this pathway. Then we analyzed the metabolites in tryptophan metabolism, kynurenine down regulated in both allergic and non-allergic asthma children. Kynurenine plays an important role in immune responses and has been shown to modulate the systemic cytokine network in asthma patients.55 It is a potent immunomodulatory molecule that may control T-cell immune responses.56–59 Kynurenine can modulate Tregs generation to influence the immune responses.60 Kynurenine pathway metabolism decreased in germ-free mice that lack of gut microbiota.61 Our finding supported that Clostridia might also play a role in the development of asthma by kynurenine pathway.
To our knowledge, few studies explored host-microbe association of childhood asthma by allergic status and fewer studies explored the association with multi-omics research. Our findings in this study added evidence on that host-microbe association might be the etiological factor for the allergic phenotype development of asthma. There were still limitations in this study. Firstly, it was only an association analysis between gut microbiome and metabolomic. It is lacked of direct evidence, such as animal experiment or in vitro study. In addition, the sample size was not big, which was also a limitation of this study.
Conclusion
In this study we observed composition and microbial metabolites of gut microbiome of asthma children by allergic status. Dysbiosis of gut microbiome in allergic and non-allergic asthma suggested that gut microbiome play a critical role in both types of asthma. The distinct altered gut microbes and the microbial derived metabolites among non-allergic asthma children suggested that gut microbiome might play a role in the modulation of asthma phenotype.
Funding
This work was financially supported by the National Natural Science Foundation of China (No. 81573113).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Asher I, Pearce N. Global burden of asthma among children. Int J Tuberc Lung Dis. 2014;18(11):1269–1278. doi:10.5588/ijtld.14.0170
2. Kiley J, Smith R, Noel P. Asthma phenotypes. Curr Opin Pulm Med. 2007;13(1):19–23. doi:10.1097/MCP.0b013e328011b84b
3. Johansson SGO, Bieber T, Dahl R, et al. Revised nomenclature for allergy for global use: report of the Nomenclature Review Committee of the World Allergy Organization, October 2003. J Allergy Clin Immun. 2004;113(5):832–836. doi:10.1016/j.jaci.2003.12.591
4. Romanet Manent S, Charpin D, Magnan A, et al. Allergic vs nonallergic asthma: what makes the difference? Allergy. 2002;57(7):607–613. doi:10.1034/j.1398-9995.2002.23504.x
5. Takejima P, Agondi RC, Rodrigues H, et al. Allergic and nonallergic asthma have distinct phenotypic and genotypic features. Int Arch Allergy Imm. 2017;172(3):150–160. doi:10.1159/000458151
6. Raedler D, Ballenberger N, Klucker E, et al. Identification of novel immune phenotypes for allergic and nonallergic childhood asthma. J Allergy Clin Immun. 2015;135(1):81–91. doi:10.1016/j.jaci.2014.07.046
7. Strachan DP, Taylor EM, Carpenter RG. Family structure, neonatal infection, and hay fever in adolescence. Arch Dis Child. 1996;74(5):422–426. doi:10.1136/adc.74.5.422
8. Stiemsma LT, Reynolds LA, Turvey SE, et al. The hygiene hypothesis: current perspectives and future therapies. Immunotargets Ther. 2015;4:143–157. doi:10.2147/ITT.S61528
9. Riedler J, Braun-Fahrländer C, Eder W, et al. Exposure to farming in early life and development of asthma and allergy: a cross-sectional survey. Lancet. 2001;358(9288):1129–1133. doi:10.1016/S0140-6736(01)06252-3
10. Stokholm J, Blaser MJ, Thorsen J, et al. Maturation of the gut microbiome and risk of asthma in childhood. Nat Commun. 2018;9(1):141. doi:10.1038/s41467-017-02573-2
11. Olszak T, An D, Zeissig S, et al. Microbial exposure during early life has persistent effects on natural killer T cell function. Science. 2012;336(6080):489–493. doi:10.1126/science.1219328
12. Stiemsma LT, Turvey SE. Asthma and the microbiome: defining the critical window in early life. Allergy Asthma Clin Immunol. 2017;13:3. doi:10.1186/s13223-016-0173-6
13. Sokolowska M, Frei R, Lunjani N, et al. Microbiome and asthma. Asthma Res Pract. 2018;4:1. doi:10.1186/s40733-017-0037-y
14. Marsland BJ, Trompette A, Gollwitzer ES. The gut-lung axis in respiratory disease. Ann Am Thorac Soc. 2015;12(Suppl 2):S150–S156. doi:10.1513/AnnalsATS.201503-133AW
15. Anand S, Mande SS. Diet, Microbiota and gut-lung connection. Front Microbiol. 2018;9. doi:10.3389/fmicb.2018.02147
16. Bingula R, Filaire M, Radosevic-Robin N, et al. Desired turbulence? Gut-lung axis, immunity, and lung cancer. J Oncol. 2017;2017:1–15. doi:10.1155/2017/5035371
17. Atarashi K, Tanoue T, Oshima K, et al. Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature. 2013;500(7461):232–236. doi:10.1038/nature12331
18. Karimi K, Inman MD, Bienenstock J, et al. Lactobacillus reuteri-induced regulatory T cells protect against an allergic airway response in mice. Am J Respir Crit Care Med. 2009;179(3):186–193. doi:10.1164/rccm.200806-951OC
19. Furusawa Y, Obata Y, Fukuda S, et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature. 2013;504(7480):446–450. doi:10.1038/nature12721
20. Mazmanian SK, Liu CH, Tzianabos AO, et al. An immunomodulatory molecule of symbiotic bacteria directs maturation of the host immune system. Cell. 2005;122(1):107–118. doi:10.1016/j.cell.2005.05.007
21. Belkaid Y, Hand TW. Role of the microbiota in immunity and inflammation. Cell. 2014;157(1):121–141. doi:10.1016/j.cell.2014.03.011
22. Salzman NH. The role of the microbiome in immune cell development. Ann Allergy Asthma Immunol. 2014;113(6):593–598. doi:10.1016/j.anai.2014.08.020
23. Schiavi E, Gleinser M, Molloy E, et al. The surface-associated exopolysaccharide of bifidobacterium longum 35624 plays an essential role in dampening host proinflammatory responses and repressing local TH17 responses. Appl Environ Microb. 2016;82(24):7185–7196. doi:10.1128/AEM.02238-16
24. Zou XL, Wu JJ, Ye HX, et al. Associations between gut microbiota and asthma endotypes: a cross-sectional study in South China based on patients with newly diagnosed asthma. J Asthma Allergy. 2021;14:981–992. doi:10.2147/JAA.S320088
25. Zierer J, Jackson MA, Kastenmüller G, et al. The fecal metabolome as a functional readout of the gut microbiome. Nat Genet. 2018;50(6):790–795. doi:10.1038/s41588-018-0135-7
26. Magoc T, Salzberg SL. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics. 2011;27(21):2957–2963. doi:10.1093/bioinformatics/btr507
27. Bolyen E, Rideout JR, Dillon MR, et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat Biotechnol. 2019;37:852–857. doi:10.1038/s41587-019-0209-9
28. Callahan BJ, McMurdie PJ, Rosen MJ, et al. DADA2: high-resolution sample inference from Illumina amplicon data. Nat Methods. 2016;13(7):581–583. doi:10.1038/nmeth.3869
29. Quast C, Pruesse E, Yilmaz P, et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 2012;41:D590–D596. doi:10.1093/nar/gks1219
30. Lozupone CA, Hamady M, Kelley ST, et al. Quantitative and qualitative β diversity measures lead to different insights into factors that structure microbial communities. Appl Environ Microb. 2007;73(5):1576–1585. doi:10.1128/AEM.01996-06
31. Segata N, Izard J, Waldron L, et al. Metagenomic biomarker discovery and explanation. Genome Biol. 2011;12(6):R60. doi:10.1186/gb-2011-12-6-r60
32. Kamarajan P, Rajendiran TM, Kinchen J, et al. Head and neck squamous cell carcinoma metabolism draws on glutaminolysis, and stemness is specifically regulated by glutaminolysis via aldehyde dehydrogenase. J Proteome Res. 2017;16(3):1315–1326. doi:10.1021/acs.jproteome.6b00936
33. Wang Q, Li F, Liang B, et al. A metagenome-wide association study of gut microbiota in asthma in UK adults. Bmc Microbiol. 2018;18(1):114. doi:10.1186/s12866-018-1257-x
34. Begley L, Madapoosi S, Opron K, et al. Gut microbiota relationships to lung function and adult asthma phenotype: a pilot study. BMJ Open Respir Res. 2018;5(1):e324. doi:10.1136/bmjresp-2018-000324
35. Chen CC, Chen KJ, Kong MS, et al. Alterations in the gut microbiotas of children with food sensitization in early life. Pediatr Allergy Immunol. 2016;27(3):254–262. doi:10.1111/pai.12522
36. Zimmermann P, Messina N, Mohn WW, et al. Association between the intestinal microbiota and allergic sensitization, eczema, and asthma: a systematic review. J Allergy Clin Immun. 2019;143(2):467–485. doi:10.1016/j.jaci.2018.09.025
37. Mokhtari P, Metos J, Anandh Babu PV. Impact of type 1 diabetes on the composition and functional potential of gut microbiome in children and adolescents: possible mechanisms, current knowledge, and challenges. Gut Microbes. 2021;13(1):1–18. doi:10.1080/19490976.2021.1926841
38. Zheng Y, Fang Z, Xue Y, et al. Specific gut microbiome signature predicts the early-stage lung cancer. Gut Microbes. 2020;11(4):1030–1042. doi:10.1080/19490976.2020.1737487
39. Manor O, Dai CL, Kornilov SA, et al. Health and disease markers correlate with gut microbiome composition across thousands of people. Nat Commun. 2020;11(1):5206. doi:10.1038/s41467-020-18871-1
40. Martin-Orozco E, Norte-Munoz M, Martinez-Garcia J. Regulatory T cells in allergy and asthma. Front Pediatr. 2017;5:117. doi:10.3389/fped.2017.00117
41. Lynch JP, Werder RB, Curren BF, et al. Long-lived regulatory T cells generated during severe bronchiolitis in infancy influence later progression to asthma. Mucosal Immunol. 2020;13(4):652–664. doi:10.1038/s41385-020-0268-8
42. McDonnell L, Gilkes A, Ashworth M, et al. Association between antibiotics and gut microbiome dysbiosis in children: systematic review and meta-analysis. Gut Microbes. 2021;13(1):1–18. doi:10.1080/19490976.2020.1870402
43. Hilty M, Burke C, Pedro H, et al. Disordered microbial communities in asthmatic airways. PLoS One. 2010;5(1):e8578. doi:10.1371/journal.pone.0008578
44. Huang YJ, Nelson CE, Brodie EL, et al. Airway microbiota and bronchial hyperresponsiveness in patients with suboptimally controlled asthma. J Allergy Clin Immun. 2011;127(2):372–381. doi:10.1016/j.jaci.2010.10.048
45. Hufnagl K, Pali-Schöll I, Roth-Walter F, et al. Dysbiosis of the gut and lung microbiome has a role in asthma. Semin Immunopathol. 2020;42(1):75–93. doi:10.1007/s00281-019-00775-y
46. Shi Z, Fultz RS, Engevik MA, et al. Distinct roles of histamine H1- and H2-receptor signaling pathways in inflammation-associated colonic tumorigenesis. Am J Physiol-Gastr L. 2019;316(1):G205–G216.
47. Marek Jutel TWSK, Watanabe T, Klunker S. Histamine regulates T-cell and antibody responses by differential expression of H1 and H2 receptors. Nature. 2001;413(6854):420–424. doi:10.1038/35096564
48. Thangam EB, Jemima EA, Singh H, et al. The role of histamine and histamine receptors in mast cell-mediated allergy and inflammation: the hunt for new therapeutic targets. Front Immunol. 2018;9:1873. doi:10.3389/fimmu.2018.01873
49. Coton M, Romano A, Spano G, et al. Occurrence of biogenic amine-forming lactic acid bacteria in wine and cider. Food Microbiol. 2010;27(8):1078–1085. doi:10.1016/j.fm.2010.07.012
50. Thomas CM, Hong T, van Pijkeren JP, et al. Histamine derived from probiotic Lactobacillus reuteri suppresses TNF via modulation of PKA and ERK signaling. PLoS One. 2012;7(2):e31951. doi:10.1371/journal.pone.0031951
51. Barcik W, Pugin B, Westermann P, et al. Histamine-secreting microbes are increased in the gut of adult asthma patients. J Allergy Clin Immun. 2016;138(5):1491–1494. doi:10.1016/j.jaci.2016.05.049
52. Crestani E, Harb H, Charbonnier L, et al. Untargeted metabolomic profiling identifies disease-specific signatures in food allergy and asthma. J Allergy Clin Immun. 2020;145(3):897–906. doi:10.1016/j.jaci.2019.10.014
53. Reinke SN, Gallart-Ayala H, Gómez C, et al. Metabolomics analysis identifies different metabotypes of asthma severity. Eur Respir J. 2017;49(3):1601740. doi:10.1183/13993003.01740-2016
54. Jiang T, Dai L, Li P, et al. Lipid metabolism and identification of biomarkers in asthma by lipidomic analysis. Biochim Biophys Acta Mol Cell Biol Lipids. 2021;1866(2):158853. doi:10.1016/j.bbalip.2020.158853
55. Luukkainen A, Karjalainen J, Hurme M, et al. Relationships of indoleamine 2,3-dioxygenase activity and cofactors with asthma and nasal polyps. Am J Rhinol Allergy. 2014;28(1):e5–e10. doi:10.2500/ajra.2014.28.4013
56. Sinclair LV, Neyens D, Ramsay G, et al. Single cell analysis of kynurenine and system L amino acid transport in T cells. Nat Commun. 2018;9(1):1981. doi:10.1038/s41467-018-04366-7
57. Frumento G, Rotondo R, Tonetti M, et al. Tryptophan-derived catabolites are responsible for inhibition of T and natural killer cell proliferation induced by indoleamine 2,3-dioxygenase. J Exp Med. 2002;196(4):459–468. doi:10.1084/jem.20020121
58. Opitz CA, Litzenburger UM, Sahm F, et al. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature. 2011;478(7368):197–203. doi:10.1038/nature10491
59. Lanz TV, Becker S, Mohapatra SR, et al. Suppression of Th1 differentiation by tryptophan supplementation in vivo. Amino Acids. 2017;49(7):1169–1175. doi:10.1007/s00726-017-2415-4
60. Chen W, Liang X, Peterson AJ, et al. The indoleamine 2,3-dioxygenase pathway is essential for human plasmacytoid dendritic cell-induced adaptive T regulatory cell generation. J Immunol. 2008;181(8):5396–5404. doi:10.4049/jimmunol.181.8.5396
61. Gao J, Xu K, Liu H, et al. Impact of the gut microbiota on intestinal immunity mediated by tryptophan metabolism. Front Cell Infect Microbiol. 2018;8:13. doi:10.3389/fcimb.2018.00013
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.