")
Back to Journals » Therapeutics and Clinical Risk Management » Volume 12
Growth hormone therapy for Prader–Willi syndrome: challenges and solutions
Authors Grugni G , Sartorio A, Crinò A
Received 1 February 2016
Accepted for publication 31 March 2016
Published 2 June 2016 Volume 2016:12 Pages 873—881
DOI https://doi.org/10.2147/TCRM.S70068
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Garry Walsh
Graziano Grugni,1,2 Alessandro Sartorio,1,2 Antonino Crinò3
1Division of Auxology, 2Experimental Laboratory for Auxo-endocrinological Research, San Giuseppe Hospital, Istituto Auxologico Italiano, Verbania, 3Autoimmune Endocrine Diseases Unit, Bambino Gesù Children’s Hospital, Rome, Italy
Abstract: Prader–Willi syndrome (PWS) is characterized by a dysregulation of growth hormone (GH)/insulin-like growth factor I axis, as the consequence of a complex hypothalamic involvement. PWS’ clinical picture seems to resemble the classic non-PWS GH deficiency (GHD), including short stature, excessive body fat, decreased muscle mass, and impaired quality of life. GH therapy is able to ameliorate the phenotypic appearance of the syndrome, as well as to improve body composition, physical strength, and cognitive level. In this regard, however, some pathophysiologic and clinical questions still remain, representing a challenge to give the most appropriate care to PWS patients. Data about the prevalence of GHD in PWS children are not unequivocal, ranging from 40% to 100%. In this context, to establish whether the presence (or not) of GHD may have a different effect on clinical course during GH therapy may be helpful. In addition, the comparison of GH effects in PWS children diagnosed as small for gestational age with those obtained in subjects born appropriate for gestational age is of potential interest for future trials. Emerging information seems to demonstrate the maintenance of beneficial effects of GH therapy in PWS subjects after adolescent years. Thus, GH retesting after achievement of final height should be taken into consideration for all PWS patients. However, it is noteworthy that GH administration exerts positive effects both in PWS adults with and without GHD. Another critical issue is to clarify whether the genotype–phenotype correlations may be relevant to specific outcome measures related to GH therapy. Moreover, progress of our understanding of the role of GH replacement and concomitant therapies on bone characteristics of PWS is required. Finally, a long-term surveillance of benefits and risks of GH therapy is strongly recommended for PWS population, since most of the current studies are uncontrolled and of short duration.
Keywords: PWS, GH deficiency, GH therapy, transition phase
Introduction
Prader–Willi syndrome (PWS) is considered the most common syndromic cause of life-threatening obesity, occurring in approximately one in 10,000–30,000 live births.1 PWS is associated with the loss of expression of paternal alleles in the PWS region of chromosome 15, without sex differences in prevalence. Three main genetic mechanisms are responsible for PWS: deletion of the paternal chromosome 15 (del15q11-q13), maternal uniparental disomy of chromosome 15 (UPD15), and imprinting defects.2 Individuals with del15q11-q13 can be further grouped in two classes, the larger type I deletion and the smaller type II deletion.1,2
The syndrome affects multiple body systems, and the major clinical manifestations are hyperphagia and early onset of severe obesity with its comorbidities (if uncontrolled), musculoskeletal issues, dysmorphic features, multiple hormonal abnormalities, and cognitive, behavioral, and psychiatric disturbances.3 Endocrine issues include growth hormone (GH)/insulin-like growth factor I (IGF-I) axis dysfunction, hypogonadism, premature adrenarche, central hypothyroidism, and central adrenal insufficiency (rare).4 It is noteworthy that the clinical picture of PWS varies during the entire lifespan, requiring a multidisciplinary approach to ensure the more appropriate management of these patients.1,4 The early infancy period is dominated by muscular hypotonia with feeding difficulties and failure to thrive, followed in later infancy or early childhood by excessive appetite leading to obesity, in the absence of intervention. Other features of the childhood period include developmental delay, temperature instability, decreased pain sensitivity, underdevelopment of sex organs, strabismus, scoliosis, hypopigmentation, respiratory abnormalities (central/obstructive apnea, excessive daytime sleepiness), acromicria, characteristic facial features, and personality problems. Adolescence is characterized by compulsive eating and increased prevalence of obesity, incomplete and delayed puberty, progression of scoliosis, sleep disorders, skin picking, worsening of learning difficulties, and maladaptive behavior, including psychiatric illness. Obesity becomes more prominent during adulthood, frequently accompanied by its comorbidities: type 2 diabetes mellitus, metabolic syndrome, gallstones, hepatic steatosis, and cardiovascular and respiratory problems. Hypogonadism and osteoporosis are present in the great majority of PWS adults, together with a high propensity to develop psychosis and self-injurious behavior. Overall, the PWS phenotype seems to be related to a complex hypothalamic dysfunction, confirmed by the presence of lack of satiety, impaired thermoregulation, endocrine abnormalities, high pain threshold, and central sleep apnea.4,5
Short stature is a characteristic feature of subjects with PWS. Approximately 50% of subjects are born small for gestational age (SGA).6 A typical pattern of growth is found, with reduced linear growth velocity in childhood, lack of the pubertal growth acceleration, and compromised final adult height.7 Considered individually, final height is almost always lower than the midparental height.
Altogether, PWS patients are viewed as affected by GH insufficiency, although the exact mechanisms of GH deficiency (GHD) are not fully understood. In addition, a variable percentage of subjects show stimulated GH values within the normal range. In spite of these observations, the bulk of evidence demonstrates the beneficial effects of GH replacement therapy, which includes amelioration of the phenotypic appearance of the syndrome (height, face, head circumference, acromicria), and improvement in body composition, physical strength, and cognitive level.8,9 In this context, however, some pathophysiologic and clinical questions still remain open, representing a challenge to ensure the most appropriate care to patients with PWS.10
Starting from an updated overview of GH secretory aspects and GH therapeutic effects in PWS, the present review is aimed at highlighting those areas not fully investigated in these subjects, with special reference to age, genotype, and bone health. The computerized databases PubMed, MEDLINE, and Google Scholar were searched to identify articles examining the pathophysiology of GH secretion and the effects of GH treatment in PWS subjects. We used the keywords growth hormone (GH), somatotropin, somatropin, somatotrophin, somatrophin, Prader–Willi syndrome, Prader–Willi, bone, bone mineral density, osteoporosis, osteopenia, genotype, and the Boolean functions AND and OR during searches. In addition, we also reviewed the references of all published original articles. Concerning GH therapy, studies of short duration were excluded (<6 months).
GH and childhood
Impaired GH responsiveness to a variety of GH secretagogues, as well as decreased 24-hour spontaneous GH secretion and reduced IGF-I levels, has been documented in a large number of PWS children.11,12 In the international KIGS (Pfizer International Growth) database, GHD was detected in 74% of 424 children with PWS, while IGF-I values were reduced in nearly all subjects.13 More recently, it has been found that 84% of 113 children and 81.4% of 102 prepubertal patients of a French PWS cohort were GH deficient after stimulation tests.14 However, the prevalence of GHD in PWS children remains still controversial, ranging from 40% to 100%.11,14 This discrepancy may be attributed to several factors, such as the limitation of GH testing, the different stimulation tests used, and/or the different methods of GH assay. In addition, GH levels are age-dependent, and it may be that GHD is a developing feature in PWS. In this respect, the youngest PWS children seem to have impaired hypothalamic GH-releasing hormone (GHRH) secretion with a normal GH pituitary reserve, while only some of them may be defined as true pituitary GH deficient and a small percentage shows a normal GH secretion.15 These findings seem to support the hypothesis that GH pituitary reserve, which is still preserved in young patients, gradually declines with age, suggesting that GHD in PWS may be due to an evolving process.16 Another critical point is related to the concomitant presence of obesity, which is characterized per se by reduced GH secretion. A higher prevalence of GHD is detected in obese PWS children compared with nonobese subjects.16 Nevertheless, nonobese children and adolescents with PWS showed a lower GH-stimulated response in comparison to age- and body mass index (BMI)-matched healthy subjects, both in terms of immunoreactive and bioactive levels.17 According to the altered GH secretory pattern, subnormal IGF-I levels were found in most of PWS children, without significant correlation with BMI, different from what was observed in simple obesity.9,11,18 Altogether, the combination of reduced GH and IGF-I levels indicates that a true GH/IGF-I axis dysfunction is present in the great majority of children with PWS. Consequently, GH testing is not required before starting GH treatment, and the positive effects of its administration are detectable in all children with PWS, independent from the GH secretory status. In this context, it remains to be explained why there is such a good GH response in people with PWS overall. In addition, it seems to be of interest to establish whether the presence (or not) of GHD may have a different impact on clinical course during GH therapy.10
The benefits of GH treatment in children with PWS are well established, including positive action on linear growth, body composition, motor function, and psychomotor development.19,20 The greater effects are observed in the first years of life, thus recommending the early institution of GH therapy (between 4 and 6 months of age) for PWS children.9,10 During childhood, long-term GH therapy allows the complete recovery from the growth defect, normalizing the final adult height, and is associated with an increase of lean body mass, adiponectin levels, and head circumference.21–23 GH administration is effective in increasing muscle strength and motor skills, as well as improving mental speed, flexibility, health-related quality of life, and cognition and behavioral issues.24–26 It is noteworthy, however, that body composition cannot be fully normalized by GH therapy, although the positive effects on lean body mass are maintained over the years.22,27 In addition, percent fat standard deviation (SD) score is reported to improve significantly during the first year of GH treatment, returning however to baseline level after 8 years of therapy.22 In this context, replacement GH dosage seems to exert a crucial role, considering that GH amount commonly used in non-PWS GHD is usually ineffective on body composition.27,28 This finding may be due, at least in part, to the significant percentage of PWS children born SGA, a condition requiring a higher dose of GH. Taking into account this observation, the comparison of GH effects in PWS children diagnosed as SGA with those obtained in subjects born appropriate for gestational age may represent a challenge for future trials.
The dosage of GH actually recommended in children with PWS is 1 mg/m2/daily, to be reached within the first weeks and months, after a first period of treatment at a lower dose. The goal of GH treatment is to obtain a significant increase of serum IGF-I concentrations within the upper limits of the normal range (maximum + 2 SDS), in order to avoid potential side effects due to exaggerated GH therapy.9,10 However, recent studies have demonstrated that immunoreactive IGF-I levels are not strictly correlated with IGF bioactivity during GH treatment, thus making immunoreactive IGF-I concentration an unreliable index for GH dosage titration in PWS children.29
GH replacement treatment is generally well tolerated, the effects of this therapy being well documented across the last 15 years. Nevertheless, fatal events in young patients with PWS have been reported during the first months of GH administration, with respiratory infections being the most common cause of death.30 In this respect, a causal role of GH therapy in reported deaths seems to be excluded, but the question still remains open and additional surveillance studies about this issue are required.9,10 Another challenge for the future is to confirm the safety of starting GH therapy early in the life. For this purpose, a complete clinical and instrumental assessment should be performed before starting GH treatment, including polysomnography, ear, nose, and throat evaluation, and thyroid function. Although long-term GH administration is not reported to impair respiratory function during sleep, a close polysomnography and ear, nose, and throat monitoring for the entire duration of GH therapy is recommended.31 In case of lymphoid tissue hyperplasia, adenoidectomy/tonsillectomy should be taken into consideration either before or during GH treatment. Starting from the age of 2.1 to 4.5 years, weight increase is observed, and PWS children will become obese if caloric intake is not restricted.32 Especially in subjects with rapid weight gain, a strict surveillance of glucose and insulin homeostasis during GH treatment is necessary, in conjunction with appropriate environmental, dietary, and lifestyle changes.33 In order to minimize the potential risks of GH therapy, however, it is recommended to exclude from this treatment children with severe obesity and those with uncontrolled diabetes. Finally, although scoliosis is not considered a contraindication for GH therapy, a careful follow-up is required in case of its worsening.
GH and transition phase
The transition from adolescence to adulthood is a developmental stage characterized by a wide set of physical and psychosocial changes. It begins in late puberty and finishes with full adult maturation, thus lasting from 3 to 10 years.34
The effects of GH during the transition period are mainly related to the maintenance of body proportions, cardiovascular function, and favorable metabolic indices, as well as to optimize quality of life.35 In the general population, individuals with untreated GHD during the transition period show impaired somatic and metabolic maturation, including altered body composition, low bone mineral density (BMD), unfavorable lipid profile, and decreased energy/quality of life. In the light of these data, reevaluation of pituitary function during transition phase is generally performed in patients not affected by PWS.34,35 As previously mentioned, the clinical picture of PWS subjects is similar to those observed in individuals with nonsyndromic GHD.1,2,4 In this context, however, few data about GH secretion throughout the transitional period in PWS are available. With GHRH plus arginine as provocative agents, 33% of a group of 24 PWS patients evaluated using BMI-specific cutoff values during the transition phase met diagnostic criteria for severe GHD.36 In another study, two of seven PWS subjects (28%) had GH peak levels after GHRH plus arginine test <9 μg/L.37 More recently, GHD was demonstrated in five of twelve PWS subjects aged 18 to 25 years.38
Although controlled studies evaluating the effects of continuous GH treatment during the transition phase are not available in PWS to date, a prolonged positive effect can be assumed by inference from GHD of nonsyndromic cause and observational studies in PWS.9 In this regard, it has been demonstrated that GH replacement therapy is able to reduce fat mass and to increase fat-free mass in PWS adults, including patients in the transition phase.8 An increase of 19% in exercise capacity, evaluated using treadmill exercise testing, was reported in twelve PWS adults (including five transition patients) after 1 year of GH therapy.39 Long-term GH treatment in 15 PWS adults (seven in the transition phase) ameliorated body composition, muscle size, and quality, and increased muscle strength and exercise tolerance.40 Improvement in mental speed and flexibility and motor performance tests during GH administration was detected in 19 PWS individuals with a median age of 25 years, with a rapid deterioration in physical and social status as well as overall functioning after GH therapy withdrawal.41 Moreover, it has been reported that cessation of GH therapy in young PWS patients worsened BMI, with a tendency toward increases in visceral adipose tissue.42
Altogether, these nongrowth-stimulating effects of GH should be taken into consideration during the transition phase. The challenge is to demonstrate that GH therapy after completion of linear growth will lead to an improvement in morbidity and mortality in PWS individuals. Thus, a reevaluation of the GH secretory pattern may be beneficial in all PWS patients after achievement of final height (Table 1). To avoid the potential negative effects of GH treatment, however, PWS individuals with severe obesity, poorly controlled diabetes mellitus, untreated severe obstructive apnea, and active psychosis should be excluded.
 | Table 1 Current indications for use of GH therapy in PWS |
GH and adulthood
Evidence of GHD in PWS adults is beginning to emerge, although study findings differ largely on its prevalence in these patients. Depending on the different study groups and the provocative tests used for the evaluation of GH secretion, 8%–55% of adults with PWS met criteria for severe GHD, according to BMI-specific cutoffs (Table 1).36,37,43,44 Impaired GH secretion in PWS adults, however, is revealed not only by deficits in secretion amplitude parameters but also by deconvolution analysis, as demonstrated by abnormalities in the shape of the secretory response which is more delayed when compared to obese controls.45 The hypothesis that an imbalance of GH/IGF-I axis occurred in PWS adults is reinforced by the presence of reduced IGF-I concentrations in the majority of subjects, as observed in pediatric age.9,11,18,36,37,44
Similar to subjects with nonsyndromic GHD, PWS adults have reduced muscle mass, muscle strength, and BMD, decreased heart geometry, increased cardiovascular risk profile, and altered psychological status.10,40,46,47 In this context, available data demonstrate a positive effect of GH treatment in these patients on body composition, skeletal muscle characteristics and motor performance, peak expiratory flow, nocturnal respiratory abnormalities, metabolic markers, and psychological well-being.8,39,40,48–55 It is noteworthy that GH administration leads to a significant improvement of IGF-I levels, lean body mass, cardiovascular outcomes, strength development, and exercise capacity both in PWS adults with and without GHD.39,40,54 This observation could be of interest in the strategic perspective of the use of GH therapy in PWS adults, since the beneficial effects deriving from GH administration seem to be independent from a concomitant diagnosis of GHD. Future research may shed additional light on this question, particularly about the need, or not, to determine the presence of GHD after attainment of adult height.
Another critical point is to determine how GH therapy should be prolonged in adult life. Until now, the majority of research on PWS has mainly focused on clinical picture in pediatric age and early adulthood. Conversely, data about older PWS people are scanty. In this regard, there is concern that, in the absence of hormonal replacement, the risk of premature aging or dementia in individuals older than 40 years may be increased,56,57 mainly in the subgroup of female PWS due to UPD15 with a history of psychosis.58 Interestingly, previous studies suggest that GH and IGF-I availability may have favorable effects on cognitive function in PWS adults as well as in non-PWS individuals at increased risk of cognitive decline and dementia.41,59 However, it has not yet been established whether GH/IGF-I axis dysfunction may really act as a determining factor in the pathogenesis of cognitive deterioration.60 In this light, further additional studies are requested to determine the role of GH therapy in promoting brain health in pathological aging adults, including PWS subjects. Moreover, the potential adverse effects of long-term GH administration need to be carefully considered. For the purpose of avoiding the possible negative effects of GH treatment, the Consensus Guidelines for GH therapy in PWS recommended that GH administration should be continued for as long as demonstrated benefits outweigh the risks, excluding adults with severe obesity and active psychosis.9 Few side effects, however, are generally reported in PWS adults during GH therapy, but the data available so far are insufficient. The great majority of the current literature involves uncontrolled and short-term studies, and only a small number of PWS subjects has been evaluated.8,9 The most frequent adverse effects include lower extremity edema and altered glucose and insulin homeostasis.61 In this context, only a slight increase in fasting glucose and trends toward hyperinsulinemia and higher insulin resistance are generally reported.8 Nevertheless, GH treatment must be suspended in PWS adults with uncontrolled diabetes mellitus.9 Data regarding the effects of GH substitutive therapy on respiratory system and heart function are still inconclusive. Information about the relationship between GH treatment and sleep-disordered breathing in PWS adults is limited to a single study which reported that only one out of ten subjects had worsened apnea/hypopnea index after 6 weeks of therapy.62 In this regard, patients with severe apnea must be adequately treated with noninvasive ventilation before starting GH therapy. To date, few studies have analyzed the cardiovascular characteristics of PWS adults in detail. Obese PWS adults have been found to harbor hypokinetic cardiac features resembling those seen in GH-deficient subjects (ie, lower systolic function, decreased cardiac mass, and lower chronotropic response to an adrenergic stimulus).46 Impaired microcirculatory function and occasional sinus node dysfunction have also been described.63 Favorable effects on cardiovascular features have been observed in both short- and long-term GH treatment.49,54 In both cases, however, there is evidence of a trend toward a decline of diastolic and systolic indices of the left ventricle. Because of the paucity of data in older PWS individuals, it is unclear if these subtle longitudinal modifications of functional cardiac parameters are related to the natural history of the syndrome or may be a consequence of GH administration. For this purpose, a close cardiac monitoring is mandatory during long-term GH replacement for PWS adults.
An association of GH therapy with a higher incidence of cancer has been reported in the general population.64 The safety record of GH, however, remains good, supported by a wide range of sources from around the world.65 The relevance of risk of cancer in PWS subjects is currently unknown, and malignancies are not commonly reported as cause of death. Nevertheless, this circumstance may be simply related to the early mortality of PWS, since neoplastic disease is generally observed after middle age. In this context, it is widely recognized that mortality rate of adults with PWS is higher than in the general population but is generally associated with obesity and its complications, including cardiovascular diseases and respiratory failure.58,66–68 In spite of this, strict alertness on the possible association between GH treatment and cancer is warranted in PWS adults, and further surveillance protocols are needed in the future. It is of particular importance to avoid high IGF-I levels during GH therapy, as epidemiological studies in healthy adults without GHD have suggested associations between serum IGF-I levels in the upper part of the normal range or above and some forms of cancer.65
GH and genotype
A number of studies have mentioned differences in some of the clinical features of PWS depending on the underlying genetic abnormality, particularly between del15q11-q13 and UPD15.1,2,10 With regard to the GH secretory pattern, PWS adults with del15q11-q13 achieve higher GHRH + arginine-induced GH peak compared to patients with UPD15.36,43,69 Similar data have been found in pediatric age after clonidine stimulation, arginine test, and insulin tolerance test.15,70,71 Interestingly, transition patients with UPD15 genotype had a higher incidence of GHD than those with del15q11-q13 (80% versus 25%).69 In this context, the deconvolution-based assessment of pituitary GH secretion shows the most delayed GH response in UPD15 subjects, both in children and adults.45,72 On the other hand, stimulated GH levels and integrated GH secretion are similar in PWS individuals carrying type I and type II deletion.69
Because data on genotype–phenotype relationship may be relevant to specific outcome measures related to GH therapy, for example, metabolic syndrome,73 studies specifically designed to examine this issue represent a challenge for the future research in PWS.9 The aim is to establish if the different GH secretory patterns among PWS individuals carrying the major genetic subtypes may have a true clinical significance.
GH and bone
Prepubertal PWS children not treated with GH have a normal BMD, when adjusted for their short stature,74,75 while adolescent and adult subjects show decreased BMD and bone mineral content, resulting in a high risk of osteoporosis.11,76 Reduced BMD in PWS is correlated with an increased frequency of fractures both in the long bones and in the bones of hands and feet.66,77 At the metacarpal level, PWS patients have an altered bone geometry leading to a reduced strength.78 The pathological bone characteristics of PWS individuals are believed to be secondary to a complex interaction of different factors, including deficiency of sex steroids, GH/IGF-I axis insufficiency, low muscular activity, and deficient intakes of both vitamin D and calcium as a result of calorie-restricting diets and obesity.79,80 However, other hormonal agents are involved in the regulation of BMD, such as adiponectin, leptin, and ghrelin.81 Total adiponectin and high-molecular weight adiponectin are negatively associated to BMD, whereas both leptin and ghrelin seem to be positively correlated to BMD, albeit data on ghrelin are still contradictory. In this context, PWS is known to be associated with high levels of adiponectin, while hyperleptinemia and hyperghrelinemia may be the expression of a state of leptin- and ghrelin resistance.82,83 Finally, data from animal model suggest a critical role of the q11-q13 region on paternal chromosome 15, which contains the Snord116 cluster, in the regulation of skeletal phenotype of PWS.84
Few data about the effects of GH administration on BMD and bone geometry in PWS are currently available in the literature. de Lind van Wijngaarden et al have previously shown that 24 months of GH therapy had no effect on BMD in 46 PWS prepubertal children, apart from a temporary decrease of total body BMD in the first 6 months of treatment.75 More recently, another study has reported that BMD of total body and lumbar spine and bone mineral apparent density of the lumbar spine remain stable during long-term GH therapy in prepubertal children with PWS but decrease during adolescence.85 As far as adulthood is concerned, it has been observed that GH treatment for 2 years is unable to ameliorate low BMD in individuals with PWS.86 Nevertheless, data about GH substitutive therapy and BMD in PWS subjects are not unequivocal. Other authors have observed that GH administration exerts a beneficial effect on bone mineralization of PWS patients.87,88 To the same extent, PWS adults who received GH have larger and consequently stronger metacarpal bones, as a result of stimulation of subperiosteal bone growth induced by GH and IGF-I.78 The discrepancies among results on this matter may be related to the different clinical characteristics of the study groups, including degree of obesity, lifestyle factors (eg, muscular activity, calcium intake, and vitamin D levels), duration of GH treatment, and lack of adequate gonadal hormone levels during pubertal period, due to delayed timing of sex steroid therapy. In this respect, the challenge is to ensure an appropriate management of the endocrine issues in PWS patients, including prolonged GH therapy during transition phase (described in the “GH and transition phase” section) and concomitant sex hormone replacement at appropriate age, as well as to provide proper dietary and motor activity plans at the appropriate time.6,10
Conclusion
Current evidence supports the view that GH replacement for PWS exerts a beneficial effect on physical aspects, cognition, and behavioral phenotype in both pediatric and adult populations. However, there is little literature on several topics related to the role of GH therapy in lifelong care of these patients. This manuscript has taken into consideration a number of questions, which remain still open (Table 2). In this light, further research is required to improve our understanding of the physiopathology of GH/IGF-I axis during the entire lifespan of PWS subjects. A more detailed knowledge of the therapeutic targets of GH administration is also needed, with the aim to optimize the clinical management of these individuals. At the same time, the role of concomitant therapies (sex steroids, dietary treatment, physical therapy) in the effects of GH administration should be better clarified. Since most of the current studies are uncontrolled and of short duration, a long-term surveillance of benefits and risks of GH therapy is strongly recommended for PWS population, in order to obtain definitive data on the safety of this treatment.
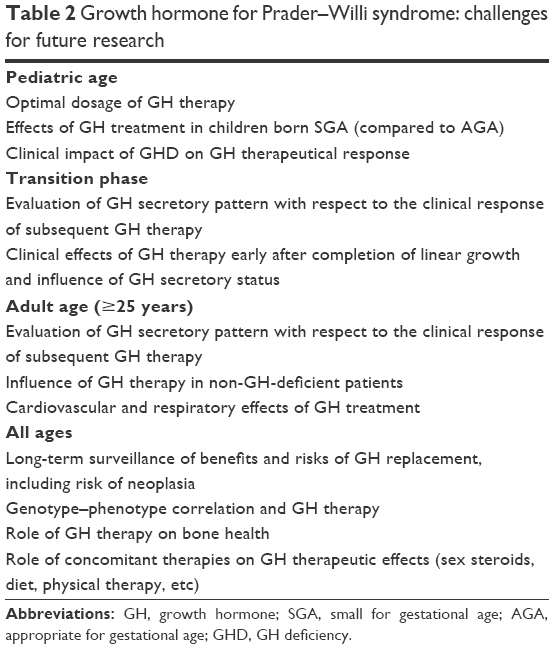 | Table 2 Growth hormone for Prader–Willi syndrome: challenges for future research |
Disclosure
The authors report no conflicts of interest in this work.
References
Cassidy SB, Schwartz S, Miller JL, Driscoll DJ. Prader-Willi syndrome. Genet Med. 2012;14(1):10–26. | ||
Angulo MA, Butler MG, Cataletto ME. Prader-Willi syndrome: a review of clinical, genetic, and endocrine findings. J Endocrinol Invest. 2015;38(12):1249–1263. | ||
Grugni G. Growth hormone in health and disease: Prader-Willi syndrome – GH therapy and bone. Nat Rev Endocrinol. 2013;9(6):320–321. | ||
Cataletto M, Angulo M, Hertz G, Whitman B. Prader-Willi syndrome: a primer for clinicians. Int J Pediatr Endocrinol. 2011;2011(1):12. | ||
Kim JH, Choi JH. Pathophysiology and clinical characteristics of hypothalamic obesity in children and adolescents. Ann Pediatr Endocrinol Metab. 2013;18(4):161–167. | ||
Lionti T, Reid SM, White SM, Rowell MM. A population-based profile of 160 Australians with Prader-Willi syndrome: trends in diagnosis, birth prevalence and birth characteristics. Am J Med Genet A. 2015;167A(2):371–378. | ||
Butler MG, Lee J, Manzardo AM, et al. Growth charts for non-growth hormone treated Prader-Willi syndrome. Pediatrics. 2015;135(1):e126–e135. | ||
Sanchez-Ortiga R, Klibanski A, Tritos NA. Effects of recombinant human growth hormone therapy in adults with Prader-Willi syndrome: a meta-analysis. Clin Endocrinol (Oxf). 2012;77(1):86–93. | ||
Deal CL, Tony M, Höybye C, Allen DB, Tauber M, Christiansen JS; 2011 Growth Hormone in Prader-Willi Syndrome Clinical Care Guidelines Workshop Participants. Growth Hormone Research Society Workshop summary: consensus guidelines for recombinant human growth hormone therapy in Prader-Willi syndrome. J Clin Endocrinol Metab. 2013;98(6):E1072–E1087. | ||
Goldstone AP, Holland AJ, Hauffa BP, Hokken-Koelega AC, Tauber M; speakers contributors at the Second Expert Meeting of the Comprehensive Care of Patients with PWS. Recommendations for the diagnosis and management of Prader-Willi Syndrome. J Clin Endocrinol Metab. 2008;93(11):4183–4197. | ||
Burman P, Ritzen EM, Lindgren AC. Endocrine dysfunction in Prader-Willi syndrome: a review with special reference to GH. Endocr Rev. 2001;22(6):787–799. | ||
Aycan Z, Baş VN. Prader-Willi syndrome and growth hormone deficiency. J Clin Res Pediatr Endocrinol. 2014;6(2):62–67. | ||
Tauber M, Cutfield W. KIGS highlights: growth hormone treatment in Prader-Willi Syndrome. Horm Res. 2007;68(Suppl 5):48–50. | ||
Diene G, Mimoun E, Feigerlova E, et al. French Reference Centre for PWS. Endocrine disorders in children with Prader-Willi syndrome: data from 142 children of the French database. Horm Res Paediatr. 2010;74(2):121–128. | ||
Di Giorgio G, Grugni G, Fintini D, et al. Growth hormone response to standard provocative stimuli and combined tests in very young children with Prader-Willi syndrome. Horm Res Paediatr. 2014;81(3):189–195. | ||
Cohen M, Harrington J, Narang I, Hamilton J. Growth hormone secretion decreases with age in paediatric Prader-Willi syndrome. Clin Endocrinol (Oxf). 2015;83(2):212–215. | ||
Grugni G, Crinò A, Pagani S, et al. GH secretory pattern in non-obese children and adolescents with Prader-Willi syndrome. J Pediatr Endocrinol Metab. 2011;24(7–8):477–481. | ||
Corrias A, Bellone J, Beccaria L, et al. GH/IGF-I axis in Prader-Willi syndrome: evaluation of IGF-I levels and of the somatotroph responsiveness to various provocative stimuli. Genetic Obesity Study Group of Italian Society of Pediatric Endocrinology and Diabetology. J Endocrinol Invest. 2000;23(2):84–89. | ||
Angulo MA, Castro-Magana M, Lamerson M, Arguello R, Accacha S, Khan A. Final adult height in children with Prader-Willi syndrome with and without human growth hormone treatment. Am J Med Genet A. 2007;143A(13):1456–1461. | ||
Wolfgram PM, Carrel AL, Allen DB. Long-term effects of recombinant human growth hormone therapy in children with Prader-Willi syndrome. Curr Opin Pediatr. 2013;25(4):509–514. | ||
Lindgren AC, Lindberg A. Growth hormone treatment completely normalizes adult height and improves body composition in Prader-Willi syndrome: experience from KIGS (Pfizer International Growth Database). Horm Res. 2008;70(3):182–187. | ||
Bakker NE, Kuppens RJ, Siemensma EP, et al. Eight years of growth hormone treatment in children with Prader-Willi syndrome: maintaining the positive effects. J Clin Endocrinol Metab. 2013;98(10):4013–4022. | ||
Bakker NE, Siemensma EP, Koopman C, Hokken-Koelega AC. Dietary energy intake, body composition and resting energy expenditure in prepubertal children with Prader-Willi syndrome before and during growth hormone treatment: a randomized controlled trial. Horm Res Paediatr. 2015;83(5):321–331. | ||
Siemensma EP, Tummers-de Lind van Wijngaarden RF, Festen DA, et al. Beneficial effects of growth hormone treatment on cognition in children with Prader-Willi syndrome: a randomized controlled trial and longitudinal study. J Clin Endocrinol Metab. 2012;97(7):2307–2314. | ||
Reus L, Pelzer BJ, Otten BJ, et al. Growth hormone combined with child-specific motor training improves motor development in infants with Prader-Willi syndrome: a randomized controlled trial. Res Dev Disabil. 2013;34(10):3092–3103. | ||
Böhm B, Ritzén EM, Lindgren AC. Growth hormone treatment improves vitality and behavioural issues in children with Prader-Willi syndrome. Acta Paediatr. 2015;104(1):59–67. | ||
de Lind van Wijngaarden RFA, Siemensma EPC, Festen DAM, et al. Efficacy and safety of long-term continuous growth hormone treatment in children with Prader-Willi syndrome. J Clin Endocrinol Metab. 2009;94(11):4205–4215. | ||
Carrel AL, Myers SE, Whitman BY, Allen DB. Benefits of long-term GH therapy in Prader-Willi syndrome: a 4-year study. J Clin Endocrinol Metab. 2002;87(4):1581–1585. | ||
Bakker NE, van Doorn J, Renes JS, Donker GH, Hokken-Koelega AC. IGF-1 levels, complex formation, and IGF-I bioactivity in growth hormone-treated children with Prader-Willi syndrome. J Clin Endocrinol Metab. 2015;100(8):3041–3049. | ||
Tauber M, Diene G, Molinas C, Hébert M. Review of 64 cases of death in children with Prader-Willi syndrome. Am J Med Genet A. 2008;146A(7):881–887. | ||
Berini J, Spica Russotto V, Castelnuovo P, et al. Growth hormone therapy and respiratory disorders: long-term follow-up in PWS children. J Clin Endocrinol Metab. 2013;98(9):E1516–E1523. | ||
Miller JL, Lynn CH, Driscoll DC, et al. Nutritional phases in Prader-Willi syndrome. Am J Med Genet A. 2011;155A(5):1040–1049. | ||
Crinò A, Di Giorgio G, Manco M, Grugni G, Maggioni A. Effects of growth hormone therapy on glucose metabolism and insulin sensitivity indices in prepubertal children with Prader-Willi syndrome. Horm Res. 2007;68(2):83–90. | ||
Clayton PE, Cuneo RC, Juul A, Monson JP, Shalet SM, Tauber M; European Society of Paediatric Endocrinology. Consensus statement on the management of the GH-treated adolescent in the transition to adult care. Eur J Endocrinol. 2005;152(2):165–170. | ||
Inzaghi E, Cianfarani S. The challenge of growth hormone deficiency diagnosis and treatment during the transition from puberty into adulthood. Front Endocrinol (Lausanne). 2013;4:34. | ||
Grugni G, Marzullo P, Ragusa L, et al. Impairment of GH responsiveness to combined GH-releasing hormone and arginine administration in adult patients with Prader-Willi syndrome. Clin Endocrinol (Oxf). 2006;65(4):492–499. | ||
van Nieuwpoort IC, Sinnema M, Castelijns JA, Twisk JW, Curfs LM, Drent ML. The GH/IGF-I axis and pituitary function and size in adults with Prader-Willi syndrome. Horm Res Paediatr. 2011;75(6):403–411. | ||
Rigamonti AE, Grugni G, Marazzi N, Bini S, Bidlingmaier M, Sartorio A. Unaltered ratio of circulating levels of growth hormone/GH isoforms in adults with Prader–Willi syndrome after GHRH plus arginine administration. Growth Horm IGF Res. 2015;25(4):168–173. | ||
Gondoni LA, Vismara L, Marzullo P, et al. Growth hormone therapy improves exercise capacity in adult patients with Prader-Willi syndrome. J Endocrinol Invest. 2008;31(9):765–772. | ||
Lafortuna CL, Minocci A, Capodaglio P, et al. Skeletal muscle characteristics and motor performance after 2-year growth hormone treatment in adults with Prader-Willi syndrome. J Clin Endocrinol Metab. 2014;99(5):1816–1824. | ||
Hoybye C, Thoren M, Bohm B. Cognitive, emotional, physical and social effects of growth hormone treatment in adults with Prader-Willi syndrome. J Intellect Disabil Res. 2005;49(Pt 4):245–252. | ||
Oto Y, Tanaka Y, Abe Y, et al. Exacerbation of BMI after cessation of growth hormone therapy in patients with Prader-Willi syndrome. Am J Med Genet A. 2014;164A(3):671–675. | ||
Grugni G, Crinò A, Bertocco P, Marzullo P. Body fat excess and stimulated growth hormone levels in adult patients with Prader-Willi syndrome. Am J Med Genet A. 2009;149(4):726–731. | ||
Sode-Carlsen R, Farholt S, Rabben KF, et al. Body composition, endocrine and metabolic profiles in adults with Prader-Willi syndrome. Growth Horm IGF Res. 2010;20(3):179–184. | ||
Grugni G, Marostica E, Crinò A, Marzullo P, De Nicolao G, Sartorio A. Deconvolution-based assessment of pituitary GH secretion stimulated with GHRH+arginine in Prader-Willi adults and obese controls. Clin Endocrinol (Oxf). 2013;79(2):224–231. | ||
Marzullo P, Marcassa C, Campini R, et al. The impact of growth hormone/insulin-like growth factor–I axis and nocturnal breathing disorders on cardiovascular features of adult patients with Prader-Willi syndrome. J Clin Endocrinol Metab. 2005;90(10):5639–5646. | ||
Thomas JDJ, Monson JP. Adult GH deficiency throughout lifetime. Eur J Endocrinol. 2009;161(Suppl 1):S97–S106. | ||
Hoybye C. Five-years growth hormone (GH) treatment in adults with Prader-Willi syndrome. Acta Paediatr. 2007;96(3):410–413. | ||
Marzullo P, Marcassa C, Campini R, et al. Conditional cardiovascular response to growth hormone therapy in adult patients with Prader-Willi syndrome. J Clin Endocrinol Metab. 2007;92(4):1364–1371. | ||
Bertella L, Mori I, Grugni G, et al. Quality of life and psychological well-being in GH-treated, adult PWS patients: a longitudinal study. J Intellect Disabil Res. 2007;51(Pt 4):302–311. | ||
Mogul HR, Lee PD, Whitman BY, et al. Growth hormone treatment of adults with Prader-Willi syndrome and growth hormone deficiency improves lean body mass, fractional body fat, and serum triiodothyronine without glucose impairment: results from the United States multicenter trial. J Clin Endocrinol Metab. 2008;93(4):1238–1245. | ||
Sode-Carlsen R, Farholt S, Rabben KF, et al. Growth hormone treatment in adults with Prader-Willi syndrome: the Scandinavian study. Endocrine. 2012;41(2):191–199. | ||
Butler MG, Smith BK, Lee J, et al. Effects of growth hormone treatment in adults with Prader-Willi syndrome. Growth Horm IGF Res. 2013;23(3):81–87. | ||
Marzullo P, Marcassa C, Minocci A, et al. Long-term echocardiographic and cardioscintigraphic effects of growth hormone treatment in adults with Prader-Willi syndrome. J Clin Endocrinol Metab. 2015;100(5):2106–2114. | ||
Hoybye C. Growth hormone treatment of Prader-Willi syndrome has long-term, positive effects on body composition. Acta Paediatr. 2015;104(4):422–427. | ||
Whittington J, Holland A. Behavior and psychiatric conditions in people with PWS. In: Hoybye C, editor. Prader-Willi Syndrome. New York: Nova Science Publishers; 2013:85–94. | ||
Sinnema M, Schrander-Stumpel CT, Verheij HE, Meeuwsen M, Maaskant MA, Curfs LM. Dementia in a woman with Prader-Willi syndrome. Eur J Med Genet. 2010;53(3):145–148. | ||
Whittington JE, Holland AJ, Webb T. Ageing in people with Prader-Willi syndrome: mortality in the UK population cohort and morbidity in an older sample of adults. Psychol Med. 2015;45(3):615–621. | ||
Baker LD, Barsness SM, Borson S, et al. Effects of growth hormone-releasing hormone on cognitive function in adults with mild cognitive impairment and healthy older adults: results of a controlled trial. Arch Neurol. 2012;69(11):1420–1429. | ||
Gasperi M, Castellano AE. Growth hormone/insulin-like growth factor I axis in neurodegenerative diseases. J Endocrinol Invest. 2010;33(8):587–591. | ||
Vogt KS, Emerick JE. Growth hormone therapy in adults with Prader-Willi syndrome. Diseases. 2015;3:56–67. | ||
Miller J, Silverstein J, Shuster J, Driscoll DJ, Wagner M. Short-term effects of growth hormone on sleep abnormalities in Prader-Willi Syndrome. J Clin Endocrinol Metab. 2006;91(2):413–417. | ||
Patel S, Harmer JA, Loughnan G, Skilton MR, Steinbeck K, Celermajer DS. Characteristics of cardiac and vascular structure and function in Prader–Willi syndrome. Clin Endocrinol (Oxf). 2007;66(6):771–777. | ||
Deodati A, Ferroli BB, Cianfarani S. Association between growth hormone therapy and mortality, cancer and cardiovascular risk: systematic review and meta-analysis. Growth Horm IGF Res. 2014;24(4):105–111. | ||
Allen DB, Backeljauw P, Bidlingmaier M, et al. GH safety workshop position paper: a critical appraisal of recombinant human GH therapy in children and adults. Eur J Endocrinol. 2015;174(2):P1–P9. | ||
Butler JV, Whittington JE, Holland AJ, Boer H, Clarke D, Webb T. Prevalence of, and risk factors for, physical ill-health in people with Prader-Willi syndrome: a population-based study. Dev Med Child Neurol. 2002;44(4):248–255. | ||
Grugni G, Crinò A, Bosio L, et al. The Italian National Survey for Prader-Willi syndrome: an epidemiologic study. Am J Med Genet A. 2008;146A(7):861–872. | ||
Lionti T, Reid SM, Rowell MM. Prader-Willi syndrome in Victoria: mortality and causes of death. J Paediatr Child Health. 2012;48(6):506–511. | ||
Grugni G, Giardino D, Crinò A, et al. Growth hormone secretion among adult patients with Prader-Willi syndrome due to different genetic subtypes. J Endocrinol Invest. 2011;34(7):493–497. | ||
Whitman B, Carrel A, Bekx T, Weber C, Allen D, Myers S. Growth hormone improves body composition and motor development in infants with Prader-Willi syndrome after six months. J Pediatr Endocrinol Metab. 2004;17(4):591–600. | ||
Oto Y, Obata K, Matsubara K, et al. Growth hormone secretion and its effect on height in pediatric patients with different genotypes of Prader-Willi syndrome. Am J Med Genet A. 2012;158A(6):1477–1480. | ||
Marostica E, Grugni G, De Nicolao G, et al. The GHRH+arginine stimulated pituitary GH secretion in children and adults with Prader–Willi syndrome shows age- and BMI-dependent and genotype-related differences. Growth Horm IGF Res. 2013;23(6):261–266. | ||
Grugni G, Crinò A, Bedogni G, et al. Metabolic syndrome in adult patients with Prader-Willi syndrome. Nutr Metab Cardiovasc Dis. 2013;23(11):1134–1140. | ||
Edouard T, Deal C, Van Vliet G, et al. Muscle-bone characteristics in children with Prader-Willi syndrome. J Clin Endocrinol Metab. 2012;97(2):E275–E281. | ||
de Lind van Wijngaarden RF, Festen DA, Otten BJ, et al. Bone mineral density and effects of growth hormone treatment in prepubertal children with Prader-Willi syndrome: a randomized controlled trial. J Clin Endocrinol Metab. 2009:94(10):3763–3771. | ||
Vestergaard P, Kristensen K, Bruun JM, et al. Reduced bone mineral density and increased bone turnover in Prader-Willi syndrome compared with controls matched for sex and body mass index – a cross-sectional study. J Pediatr. 2004;144(5):614–619. | ||
Kroonen LT, Herman M, Pizzutillo PD, Macewen GD. Prader-Willi syndrome: clinical concerns for the orthopaedic surgeon. J Pediatr Orthop. 2006;26(5):673–679. | ||
Longhi S, Grugni G, Gatti D, et al. Adults with Prader–Willi syndrome have weaker bones: effect of treatment with GH and sex steroids. Calcif Tissue Int. 2015;96(2):160–166. | ||
Rubin DA, Nowak J, McLaren E, Patiño M, Castner DM, Dumont-Driscoll MC. Nutritional intakes in children with Prader-Willi syndrome and non-congenital obesity. Food Nutr Res. 2015;59:29427. | ||
Pereira-Santos M, Costa PR, Assis AM, Santos CA, Santos DB. Obesity and vitamin D deficiency: a systematic review and meta-analysis. Obes Rev. 2015;16(4):341–349. | ||
Biver E, Salliot C, Combescure C, et al. Influence of adipokines and ghrelin on bone mineral density and fracture risk: a systematic review and meta-analysis. J Clin Endocrinol Metab. 2011;96(9):2703–2713. | ||
Grugni G, Guzzaloni G, Morabito F. Impairment of GH responsiveness to GH-releasing hexapeptide (GHRP-6) in Prader-Willi syndrome. J Endocrinol Invest. 2001;24(5):340–348. | ||
McAllister CJ, Whittington JE, Holland AJ. Development of the eating behaviour in Prader-Willi syndrome: advances in our understanding. Int J Obes (Lond). 2011;35(2):188–197. | ||
Khor EC, Fanshawe B, Qi Y, et al. Prader-Willi critical region, a non-translated, imprinted central regulator of bone mass: possible role in skeletal abnormalities in Prader-Willi syndrome. PLoS One. 2016;11(1):e0148155. | ||
Bakker NE, Kuppens RJ, Siemensma EP, et al. Bone mineral density in children and adolescents with Prader-Willi syndrome: a longitudinal study during puberty and 9 years of growth hormone treatment. J Clin Endocrinol Metab. 2015;100(4):1609–1618. | ||
Jørgensen AP, Ueland T, Sode-Carlsen R, et al. Two years of growth hormone treatment in adults with Prader-Willi syndrome do not improve the low BMD. J Clin Endocrinol Metab. 2013;98(4):E753–E760. | ||
Khare M, Gold JA, Wencel M, et al. Effect of genetic subtypes and growth hormone treatment on bone mineral density in Prader-Willi syndrome. J Pediatr Endocrinol Metab. 2014;27(5–6):511–518. | ||
Nakamura Y, Murakami N, Iida T, Asano S, Ozeki S, Nagai T. Growth hormone treatment for osteoporosis in patients with scoliosis of Prader-Willi syndrome. J Orthop Sci. 2014;19(6):877–882. | ||
Corneli G, Di Somma C, Prodam F, et al. Cut-off limits of the GH response to GHRH plus arginine test and IGF-I levels for the diagnosis of GH deficiency in late adolescents and young adults. Eur J Endocrinol. 2007;157(6):701–708. | ||
Corneli G, Di Somma C, Baldelli R, et al. The cut-off limits of the GH response to GH-releasing hormone-arginine test related to body mass index. Eur J Endocrinol. 2005;153(2):257–264. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.