")
Back to Journals » Nanotechnology, Science and Applications » Volume 13
Green One-Step Synthesis of Medical Nanoagents for Advanced Radiation Therapy
Authors Salado-Leza D , Porcel E , Yang X , Štefančíková L, Bolsa-Ferruz M, Savina F, Dragoe D , Guerquin-Kern JL, Wu TD, Hirayama R , Remita H, Lacombe S
Received 29 April 2020
Accepted for publication 30 June 2020
Published 7 August 2020 Volume 2020:13 Pages 61—76
DOI https://doi.org/10.2147/NSA.S257392
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Israel (Rudi) Rubinstein
Daniela Salado-Leza,1,2 Erika Porcel,1 Xiaomin Yang,1 Lenka Štefančíková,1 Marta Bolsa-Ferruz,1 Farah Savina,1 Diana Dragoe,3 Jean-Luc Guerquin-Kern,4 Ting-Di Wu,4 Ryoichi Hirayama,5 Hynd Remita,6 Sandrine Lacombe1
1Université Paris Saclay, CNRS UMR 8214, Institut des Sciences Moléculaires d’Orsay, 91405 Orsay, France; 2Cátedra CONACyT, Faculty of Chemical Sciences, Autonomous University of San Luis Potosí, 78210 San Luis Potosí, Mexico; 3Université Paris Saclay, CNRS UMR 8182, Institut de Chimie Moléculaire et des Matériaux d’Orsay, 91405 Orsay, France; 4Paris-Saclay University, Multimodal Imaging Center (UMS 2016/US 43) CNRS, INSERM, Institut Curie, 91405 Orsay, France; 5Department of Charged Particle Therapy Research, National Institute of Radiological Sciences, National Institutes for Quantum and Radiological Science and Technology, 263-8555 Chiba, Japan; 6Université Paris Saclay, CNRS UMR 8000, Institut de Chimie Physique, 91405 Orsay, France
Correspondence: Sandrine Lacombe
Université Paris-Saclay, CNRS UMR 8214, Institut des Sciences Moléculaires d’Orsay, André Riviére Street, Building 520, Orsay Cedex 91405, France
Tel +33 1 6915 8263
Email [email protected]
Purpose: Metal-based nanoparticles (M-NPs) have attracted great attention in nanomedicine due to their capacity to amplify and improve the tumor targeting of medical beams. However, their simple, efficient, high-yield and reproducible production remains a challenge. Currently, M-NPs are mainly synthesized by chemical methods or radiolysis using toxic reactants. The waste of time, loss of material and potential environmental hazards are major limitations.
Materials and Methods: This work proposes a simple, fast and green strategy to synthesize small, non-toxic and stable NPs in water with a 100% production rate. Ionizing radiation is used to simultaneously synthesize and sterilize the containing NPs solutions. The synthesis of platinum nanoparticles (Pt NPs) coated with biocompatible poly(ethylene glycol) ligands (PEG) is presented as proof of concept. The physicochemical properties of NPs were studied by complementary specialized techniques. Their toxicity and radio-enhancing properties were evaluated in a cancerous in vitro model. Using plasmid nanoprobes, we investigated the elementary mechanisms underpinning radio-enhancement.
Results and Discussion: Pt NPs showed nearly spherical-like shapes and an average hydrodynamic diameter of 9 nm. NPs are zero-valent platinum successfully coated with PEG. They were found non-toxic and have the singular property of amplifying cell killing induced by γ-rays (14%) and even more, the effects of carbon ions (44%) used in particle therapy. They induce nanosized-molecular damage, which is a major finding to potentially implement this protocol in treatment planning simulations.
Conclusion: This new eco-friendly, fast and simple proposed method opens a new era of engineering water-soluble biocompatible NPs and boosts the development of NP-aided radiation therapies.
Keywords: platinum nanoparticles, radiolytic method, environmentally-friendly process, nanomedicine, radiotherapy
Introduction
The addition of metallic compounds was proposed two decades ago as a novel strategy to enhance the biological efficiency of medical radiations.1 Biston et al showed the efficiency of combining cisplatin, a commonly used platinum antineoplastic drug, with conventional radiotherapy to improve gliomas treatment.2 To selectively increase the toxic effects of radiation, agents capable of concentrating in the tumor have been introduced such as nanoparticles (NPs). Indeed, a wide variety of nanostructured materials have been proposed in the fight against cancer. Organic, inorganic and hybrid NPs are been studied as adjuvants to enhance the diagnosis and treatment of tumors. In particular, metal-based NPs endowed with intrinsic physicochemical properties (eg luminescence or magnetism) and functional surfaces, could be used as: i) contrast agents for imaging, ii) vectors for targeted drug delivery, iii) compounds for anticancer drug development, iv) radio-enhancers for improved radiotherapy or v) as multi-modal platforms for theranostic. For instance, biogenic NPs containing gold (Au NPs) and silver (Ag NPs) have been largely studied due to their potential use as anticancer nano-therapeutics, showing remarkable results killing, among others; colorectal and pulmonary cancer cells.3–5 However, a suitable radio-enhancer candidate is a non-toxic high-Z nanoparticle (HZ-NPs) that only under irradiation acts as an adjuvant anticancer agent. Hainfeld et al demonstrated that harmless glucose coated Au NPs extend the life of mice treated with 160 kV X-rays.6,7 Moreover, Yan Li et al proved that porous Pt NPs enhance conventional radiation therapy using 250 kV X-rays.8 Currently, clinical trials are in progress to evaluate the efficiency of hafnium oxide-based NPs (NBTXR3, Nanobiotix – France),9 gadolinium (AGuIX, NH TherAguix – France),10,11 and gold (CYT-6091 CytImmune – USA) nanoagents as radio-enhancing or antineoplastic compounds.12
There is theoretical and experimental evidence demonstrating that the radio-enhancing effect of HZ-NPs is due to the induction of nanosize damage triggered by the production of electron bursts and radical clusters in the vicinity of the agent, far from the nucleus.13–15 More interestingly, the use of metallic NPs was found to be effective not only in the case of conventional radiation treatments (using X-rays or γ-rays) but also with ion beams, a modality that is superior to conventional radiotherapy. Shortly, particle therapy (so-called ion beam therapy) is based on the use of charged particles (protons or carbon ions) accelerated to high energies (70 to 400 MeV/a.m.u.) to eradicate the intractable radio-resistant tumours.16,17 Compared to conventional radiotherapy, it presents two main advantages: i) a precise ballistic effect with a finite range and ii) a maximum dose deposition at the end of the ion tracks (Bragg peak), which prevents damage in the surrounding healthy tissue. The relative biological efficiency of carbon ions is two to four times superior to the effect of electromagnetic radiations (X and γ-rays) that prevents any resistance to the treatment.18 In this context, Kaur et al showed that the addition of glucose-capped Au NPs improve the treatment of HeLa cells by 62 MeV carbon ions.19 Kim et al demonstrated that the effect of 40 MeV protons is enhanced in the presence of gold and iron oxide NPs using a CT26 mouse model.20 Even if the physicochemical mechanism proposed in that work was controversial,21,22 it evidenced for the first time, the radio-enhancement of proton treatment using metallic NPs. Recently, antibody-functionalized gold and other HZ-NPs (eg Gd2O3 NCs, Fe2O3 NPs, Pt NDs and Bi NRs) were tested, using colon, lung and breast cancer cell models treated by protons (2150 MeV) and carbon ions (100 MeV/a.m.u, LET= 50 keV/μm).23–25 Our group demonstrated that mono- and bimetallic gold, platinum as well as gadolinium-based nanoagents increase the induction of complex biodamage when photons (60Co γ-rays),26,27 proton (150 MeV)28 or medical carbon beams (276 MeV/a.m.u) are used as ionizing radiations.29 We also demonstrated that gadolinium-based NPs amplify the cell killing induced by ion beams.30
Considering the tremendous potential of metallic NPs to improve the performances of conventional and ion beam radiation therapies, the production of ready-to-use stable colloidal solutions, is critical. Radio-enhancing nanoagents composed of gold, hafnium, gadolinium and also platinum, are commonly produced via chemical procedures.31–34 The use of chemicals, such as organic surfactants in non-polar solvents to control NPs size and shape may exhibit environmental hazards, which limits the safe-by-design of nanoagents and contributes to their toxicity. Hence, multiple steps that include ligand exchange and phase transfer procedures are required to produce hydrophilic, stable, biocompatible and non-toxic agents ready-to-use in clinic. These procedures are time consuming and result in an intrinsic loss of material.
Radiolysis is an alternative method to produce metallic NPs of controlled size and shape in solution or on supports. For instance, the solvated electrons and hydrogen radicals produced in situ by the radiolysis of the solution are strong reducing species. This method presents the following major benefits: limited use of chemical reductants, high production yields of metallic compounds under mild conditions (temperature and pressure) and NPs size monodispersity.35 Different types of NPs have been synthesized following this methodology (Table 1).36–44 However, in most of the cases, potentially toxic chemicals are used to control NPs formation and the final products are not adapted to straightforward medical use. Therefore, the development of efficient protocols able to produce non-toxic metallic nanoagents remains a challenge.
 |
Table 1 Overview of High-Z-Based NPs Synthesized by Radiolysis and Their Applications |
In this work, we developed a simple one-pot radiolysis-driven method to synthesize with a 100% production rate, biocompatible metallic NPs dispersed in a sterile water solution, ready to use in clinic. The production of sub-10 nm Pt NPs coated with poly(ethylene glycol) (PEG), a non-toxic polymeric coating approved by the FDA and the EU for internal consumption is presented. The efficiency of these nanoagents to improve the effects of medical radiation beams was demonstrated and quantified on HeLa cells treated by γ-rays and medical carbon ions. To elucidate the molecular-scale mechanisms induced by nanoparticles, experiments were performed using plasmids as bio-nanoprobes. This new green, simple, reproducible and efficient method to produce stable and ready-to-use colloidal solutions containing metallic nanoagents has been recently registered for being patented (FR1900008).
Materials and Methods
Synthesis of PtPEG NPs
PEGylated Pt NPs were obtained by γ-ray water radiolysis as described in the French Patent Application FR1900008. In brief, an aqueous solution containing tetraammine platinum (II) chloride (Pt(NH3)4Cl2.H2O) (Sigma-Aldrich) and poly(ethylene glycol) (H(OCH2CH2)nOH, MW = 1000 g.mol−1) (Fluka) was de-aerated by bubbling with nitrogen and then exposed to a panoramic 60Co gamma source at the maximum dose rate of 95.5 Gy.min−1. The optimized PtPEG NPs, synthesized with a PEG:Pt molar ratio of ca. 100 and a Pt concentration of 10−2 mol.L−1, were used without further purification in all cellular and molecular scale experiments. The as-synthesized colloidal PtPEG NPs, stored at 4°C, remain stable in pure water for a couple of months.
To guarantee long-term nanoagents storage, NPs followed a lyophilization process. PtPEG NPs were stored at −80°C overnight and freeze-dried at −50°C (40 Bar) for 48 h using a laboratory Alpha 1–2 LDplus lyophilized (CHRIST, Germany). After 2 days, the vials were sealed and removed. The physicochemical properties of re-suspended PtPEG NPs remained constant.
Physicochemical Characterization
The PtPEG NPs core size and morphology were determined using a JEOL 2200FS Transmission Electron Microscope operated at 200 kV. Drops of fresh as-synthesized PtPEG NPs were deposited on carbon-formvar copper grids (200 mesh, Agar Scientific Ltd) and observed after natural drying. The NPs size distribution and roundness were obtained by analyzing more than 2000 particles using the ImageJ 1.46r software. The roundness corresponds to the inverse of the major_axis/minor_axis aspect ratio, which is equal to  . A value of 1.0 indicates its proximity to a circle (spherical shape descriptor).
. A value of 1.0 indicates its proximity to a circle (spherical shape descriptor).
The hydrodynamic diameter and surface charge, in terms of zeta potential, of the PtPEG NPs were determined using a Zetasizer Nano-ZS (Malvern Instrument Ltd) equipped with a He-Ne laser (λ = 633 nm). Samples from three independent NPs batches, diluted at 5×10−4 mol.L−1 concentration, were analyzed at room temperature.
The chemical composition and electronic states of NPs were analyzed by X-ray photoelectron spectroscopy (XPS) and Fourier transform infrared spectroscopy (FT-IR). The samples were prepared by drop casting. The XPS spectrometer used (K Alpha, Thermo Fisher Scientific, base pressure in the low 10−9 mbar range) was equipped with a monochromatic aluminum source (Al Kα, 1486.7 eV). A spot size of 400 µm was used, which corresponds to an irradiated zone of approximately 1 mm2. The hemispherical analyzer was operated at 0° take-off angle in the Constant Analyzer Energy (CAE) mode, with a pass energy of 200 eV and a step of 1 eV for the acquisition of survey scans, and a pass energy of 50 eV and a step of 0.1 eV for the acquisition of narrow windows. Charge compensation was done by means of a “dual beam” flood gun. The recorded spectra were processed with the Avantage software, using a peak-fitting routine with Shirley background and symmetrical 7030% mixed Gaussian-Lorentzian peak shapes, and a Doniach-Sunjic type function was used to fit the Pt metal asymmetric peaks.
The FT-IR analysis was performed using the attenuated total reflection (ATR) module of an Alpha FT-IR Spectrometer (Bruker Optik GmbH). The transmittance FT-IR spectra were measured from 4000 to 400 cm−1 (OPUS software, spectral resolution of ~4 cm−1). Three independent batches of NPs were analyzed. The resulting FT-IR spectra are the average of 60 scans.
Irradiation Facilities
A 60Co panoramic gamma source (Energy = 1.25 MeV and LET = 0.2 keV.μm−1) was used to synthesize the PtPEG NPs (95.5 Gy.min−1, 10 kGy per 10−3 mol.L−1 of PtII) and to irradiate the bio-nanoprobe (DNA plasmids) (481 Gy.h−1, from 0 up to 500 Gy). The dose rates were determined by Fricke dosimetry. All samples were irradiated with a dose variation below 10%. HeLa cells were irradiated with a GSR-D1 137Cs gamma irradiator (Energy = 0.662 MeV and LET = 0.2 keV.μm−1) with a dose rate of 1 Gy.min−1 and doses ranged from 0 up to 5 Gy. The dose difference was <5%. At the Heavy Ion Medical Accelerator Centre in Chiba, Japan, HeLa cells were exposed to C6+ ions at the maximum of the Spread-Out Bragg Peak (5 mm SOBP) (Initial Energy = 290 MeV/u.m.a, average LET = 110 keV.μm−1), with a dose rate of approximately 14 Gy.min−1 and doses ranging from 0 up to 2.5 Gy. The beam delivered a uniform flux within 2% variation in a 10 cm diameter circle.
In vitro experimentation
Cell Culture Conditions
HeLa cells (cervical cancer-derived cells), purchased commercially from the American Type Culture Collection (CCL-2TM, LGC Standards S.a.r.l. - France Office), were grown in monolayer using Dulbecco’s Modified Eagle Medium (DMEM) (Life Technologies) enriched with 4.5 g/L glucose and supplemented with 10% heat-inactivated Fetal Bovine Serum (FBS), 1% antibiotics (100 U/mL penicillin and 100 μg/mL streptomycin) and 1% glutamine (all supplements from PAA Laboratories) and cultured at 37°C in a humidified 95% air-5% CO2 atmosphere.
Cytotoxicity Determination
HeLa cells were incubated for 6 h with an enriched medium containing PtPEG NPs at two different Pt molar concentrations, 5×10−4 and 10−3 mol.L−1(ca. 1.08×1010 and 2.16×1010 NPs per cell). After a standard 14 days culture time,45 the obtained colonies were scored and the plating efficiency was compared to a control sample consisting of HeLa cells incubated with an equal amount of pure water (vehicle). The analysis of variance was performed using the ANOVAOneWay method provided by OriginPro 8.5.
Uptake Quantification
Around 2×106 HeLa cells were incubated for 6 h with PtPEG NPs at a final Pt concentration of 5×10−4 mol.L−1. After incubation the culture medium was removed, cells were rinsed with a phosphate-buffered saline solution (PBS, pH 7.4) and collected by trypsinization. Cells were then counted and centrifuged (7 min at 1200 rpm). The pellets were dissolved in aqua regia and finally diluted in ultrapure water. The internalized Pt content (μg.L−1) in HeLa cells was thus determined by Inductively Coupled Plasma (ICP) using an Optical Emission Spectrophotometer (OES) Varian 720 ES.
Intracellular Localization
Nanoscale Secondary Ion Mass Spectrometry (Nano-SIMS) experiments were performed with a CAMECA NanoSIMS 50™. About 35,000 HeLa cells were plated on a 1.0 cm2 Melinex film support (Agar Scientific Ltd., Stansted, Essex, UK) and left to attach at 37°C in a humidified 95% air-5% CO2 atmosphere for 2 h. The cells were grown overnight and then incubated for 6 hours with PtPEG NPs at a Pt concentration of 5×10−4 mol.L−1. After incubation, non-internalized NPs were removed by rinsing with PBS and new enriched medium was added. The Melinex supports were then cut in small squares (9 mm2) and cells were fixed by slam freezing on a metal mirror pre-cooled to −180°C with liquid nitrogen. The cryofixation was performed in a Leica EM CPC system. The samples were dehydrated following a freeze-drying protocol which started at −130°C. After reaching 0°C, dehydrated samples were embedded in resin and polymerised at 60°C for 8 h. After polymerization several sections of ca. 0.2 μm were deposited on a silicon holder and analyzed. The primary incident ion beam was generated from a cesium source with energy of 16 keV and a probe size of ca. 50 nm in diameter. The magnetic field was set to detect simultaneously sputtered platinum ions (Pt) on one of the detectors and sputtered phosphorus ions (P−) on another one. The mass spectrometer was calibrated with a control sample consisting of a Pt layer deposed on a gold film. The 194Pt− secondary ion was selected as the isotopic Pt marker. It did not present any interference with other cell components.
Clonogenic Assay
Cell survival was assessed by a standard 14 days colony formation assay (CFA). Exponentially grown HeLa cells (2×105) were plated in T25 culture flasks (BioLite 25 cm2 cell culture treated flask, Thermo Scientific) and maintained overnight in a 5% CO2 incubator at 37°C. Cells were then incubated for 6 h with enriched culture medium containing PtPEG NPs at a final Pt concentration of 5×10−4 mol.L−1. An equivalent water volume was added into control samples. Afterwards, the cells were irradiated in fresh medium under atmospheric conditions at room temperature, independently by γ-rays (137Cs) and C6+ ions. Immediately after exposure, the cells were trypsinized and re-seeded into 10 cm Petri dishes (Falcon 3002) at different densities in order to achieve ca. 100 colonies per dish. Formed colonies were fixed and stained with a 0.5% methylene blue 50% (v/v) methanol solution. The colonies containing more than 50 cells were scored and the calculated survival fractions were normalized to the survival fraction of the corresponding control. The HeLa plating efficiency was found close to 65%.
Bio-Nanoprobe Sample Preparation and Analysis
The bio-nanoprobe samples were prepared as described in our previous works with minor modifications.26 Prior to irradiation, 500 ng of the pBR322 DNA plasmid (Sigma-Aldrich) diluted in a TE buffer solution (10 mmol.L−1 Tris-HCl (pH = 7.6) and 1 mmol.L−1 ethylenediaminetetraacetic acid (EDTA)) were incubated for 1 h with PtPEG NPs (Pt 2.42×10−4 mol.L−1), or with the same amount of ultra-pure water (18.2 MΩ·cm at 25°C) in control samples. The contribution of indirect effects was highlighted by adding dimethylsulfoxide (DMSO) at a final concentration of 1 mol.L−1 to control samples and to samples containing PtPEG NPs. Aliquots of 18 μL were then exposed to γ-rays (doses ranged from 0 up to 500 Gy) under aerobic conditions. After irradiation, the three conformations of DNA plasmids were separated by electrophoresis (75 V, 101 mA and 7 W) in a 1% agarose gel, loaded with ethidium bromide (0.003%), for 3 h at room temperature. The lines were then revealed under UV light (λ= 302 nm) and recorded by a CCD camera. ImageQuant 5.0 software was used to quantify the intensity of each DNA line corresponding to each DNA conformation. The yields of single and double-strand breaks were calculated as described elsewhere.26
Results and Discussion
Synthesis and Characterization of PtPEG NPs
The mechanisms underpinning the formation of PtPEG NPs are the following.46–48 The excitation and ionization of water molecules by high-energy radiation (γ-rays, X-rays, electrons or ion beams) leads to the production of radiolytic species (Equation 1). These species are homogeneously distributed in the bulk.47,49
(1)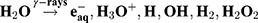
The solvated electrons eaq− and hydrogen atoms H∙ are strong reducing agents (E°(H2O/eaq−) = –2.87 V and E°(H+/H∙) = –2.31 V). They efficiently reduce dissolved metal ions (Mn+) (Equation 2 and 3).
(2) (3)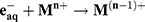
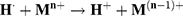
Thus, the PtII precursor was reduced to the zero-valent state following the multi-step reaction: PtII → PtI and PtI → Pt0. The uniform energy deposited in the medium led to a homogeneous distribution of nucleation sites in the solution and the formation of metal clusters during the coalescence step with a remarkable monodispersity (Equation 4).
(4)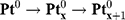
PEG was used to limit the nucleation and growth steps, and increase the NPs stability in suspensions. The surface coating with PEG chains was achieved in situ, a remarkable advantage of our method.
The UV-vis absorption spectra showed that before irradiation the aqueous solution containing tetraammine platinum (II) chloride and PEG displayed an absorption peak at 240 nm and a small shoulder around 290 nm, which correspond to the ligand-to-metal charge transfer (LMCT) band of the precursor (Figure S1).50 Supplementary Figures and Tables are described at the Supporting Information file. After irradiation, the appearance of a stretched absorption spectrum with a single broad peak centered at 270 nm, indicated the formation of small Pt aggregates.47,50 The color of the solution changed from colorless before irradiation to dark brown after irradiation (inset in Figure S1). The spectrum and the color remained unchanged for several weeks indicating the complete reduction of the PtII precursor and the formation of stable PtPEG NPs.
High resolution-transmission electron microscopy (HR-TEM) (Figure 1AC) revealed that PtPEG NPs were homogeneously formed as sub-10 nm semi-spherical nanoparticles (roundness = 0.66 ± 0.17 nm, Figure S2) with an average core diameter of 3.2 ± 0.8 nm (Figure 1D). It corresponds to the assembly of ca. 1000 Pt atoms. We have actually shown that the core’s size and shape, of the PEGylated metal-based NPs synthesized in pure water by our method using γ-rays, strongly depend on the chemicals nature, ie the metallic precursor (oxidation state and coordination) and stabilizer (functionality and chain length), their molar ratio and radiation dose.26,27 In this work, the NPs obtained using an optimized PEG-OH:PtII molar ratio of ca. 100 showed the greatest qualities such as; colloidal stability and size homogeneity.
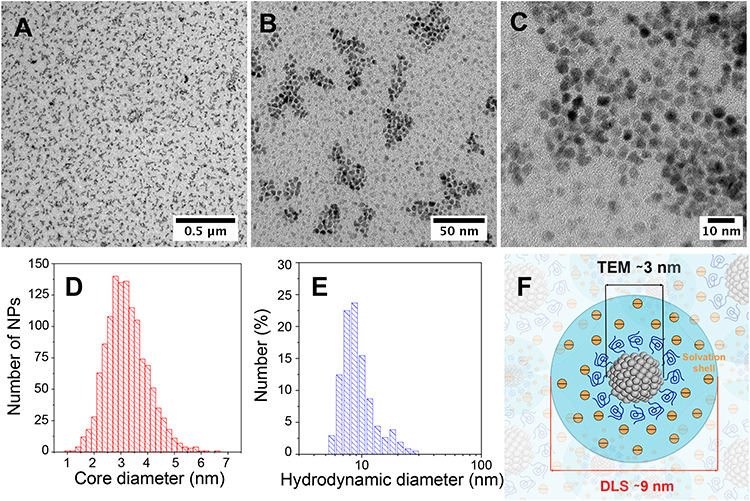 |
Figure 1 HR-TEM images of PtPEG NPs synthesized in water with an initial PtII concentration of 10−2 mol.L−1 and a PEG:Pt molar ratio of ca. 100, measured at different magnifications. Scale bars: (A) 0.5 μm, (B) 50 nm and (C) 10 nm. Size distribution of PtPEG NPs determined by (D) HR-TEM and (E) DLS. (F) Scheme of PtPEG NPs in aqueous solution. |
Hence, Dynamic Light Scattering (DLS) measurements (Figure 1E) of the as-synthesized PtPEG NPs showed an average hydrodynamic diameter of 8.8 ± 3.1 nm and a homogenous distribution (PdI= 0.238 ± 0.004). The zeta potential was found close to −16.6 ± 5.1 mV in pure water (pH 6.2). The PZC or isoelectric point was observed at pH = 3, a result that reflects the good stability of the PtPEG NPs at the pH of the intracellular fluids (between 6.8 and 7.4). The negative zeta potential is attributed to the –OH functional end-group of the PEG coating, which favors electrostatic and steric repulsions between particles preventing their aggregation in polar solvents and biological relevant solutions such as PBS, NaCl and TE. In addition, the observed negative charge suggests that PEG molecules thereby remain grafted at all pH values. This size and grafting, schematically represented in Figure 1F, is compatible with passive accumulation of NPs in tumors by the enhanced permeability and retention (EPR) effect.51 The freshly prepared PEGylated NPs were stable in aqueous solution for a couple of months at 4°C. To optimize the long-term storage, they were successfully lyophilized using a two-step freeze-drying method. The shelf life of lyophilized NPs was found higher than 6 months when stored at 4°C. Their re-suspension in various biocompatible buffers (eg PBS, NaCl and TE) did not modify their characteristics.
XPS was used to probe the result of the in situ PEGylation and verify the reduction of PtII into Pt0. The survey XPS spectrum (Figure S3) revealed the presence of Pt, C, O, N and Cl, which confirmed the purity and elemental composition of the NPs. For a better clarity, in Figure 2, narrow windows on the energy range of each element are presented. Figure 2A displays the two spin-orbit components of Pt, Pt-4f7/2 and Pt-4f5/2.52,53 The peaks were deconvoluted using Shirley background, symmetrical 7030% mixed Gaussian-Lorentzian shapes and a Doniach-Sunjic type function for the fit of metallic Pt peaks. The peaks at 71.2 and 74.4 eV correspond to zero-valent Pt atoms located at the NPs’ bulk. The peaks at 70.4 and 73.7 eV correspond to Pt atoms located at the surface. This measurement confirms the complete reduction of PtII into Pt0. Interestingly, the full width at half maximum (FWHM) of the peaks increased by about 33% (FWHM from 1.2 to 1.6 eV) in comparison to the peaks of solid Pt. It indicates a strong interaction between the metallic surface and the PEG polymer.54 The peak centered at 286.0 eV (Figure 2B) is characteristic of carbon adjacent to oxygen in an ether environment (C-O-C) and the small shoulder at a lower binding energy corresponds to the carbon-carbon bond in PEG.55,56 Moreover, at 286.0 eV, an overlap between the peaks characteristic of aminated carbons (C-N) and those given by C-O may also indicate the presence of the amine group at the NPs surface.57,58 The tail observed at higher binding energy (BE), from ca. 287 to 288 eV in Figure 2B, may indicate the presence, in a very small amount, of C=O bonds.57,59 The peak at 532.3 eV (Figure 2C) corresponds to the C-O-C (ether) and C-O-H (hydroxyl) oxygen.55 The position and FWHM of the peaks remained unchanged, which indicates the formation of a PEG monolayer56,60 perpendicular to the NP surface.61 Interestingly, the N-1s peak at 398.2 eV (Figure 2D) broadened after irradiation (Table S1). This suggests the contribution of several amine species. The chemical shift of the peak to a lower BE (from 400.2 eV to 398.2 eV after synthesis) suggests an interaction of nitrogen-containing functional groups with the NP surface.61
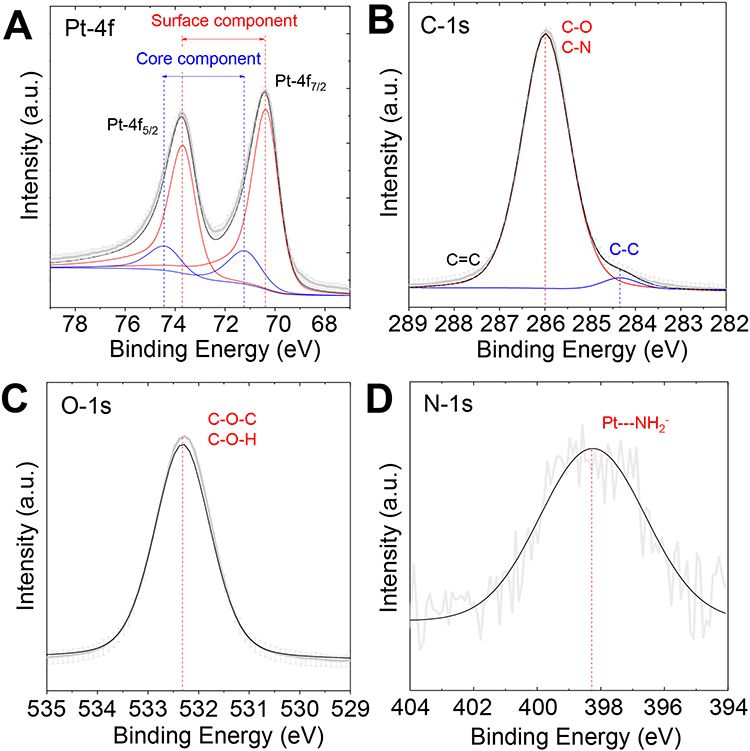 |
Figure 2 XPS spectra of PtPEG NPs in the ranges of (A) Pt-4f, (B) C-1s, (C) O-1s and (D) N-1s core levels. |
The analysis of the surface chemistry was completed using FT-IR spectroscopy (Figure 3). The initial compounds and the mixed solution were characterized before and after irradiation. The band located at 3424 cm−1 corresponds to the hydroxyl stretching mode νO-H of the bonded hydrogen of the native PEG-OH (Figure 3A).62 The peak centered at 2885 cm−1 is characteristic of the alkyl stretching mode (νCH2) and the peaks in the region from 1000 to 1300 cm−1 are characteristic of the stretching vibrational modes of ether (νC-O). The Pt(NH3)4Cl2 precursor (Figure 3B) displayed bands at 3242 and 3135 cm−1, which are attributed to the asymmetric and symmetric vibrational N-H stretching modes (νNH3). From 1564 to 842 cm−1 the bending and rocking vibrational modes (δHNH and ρNH3) were observed. The IR band at 510 cm−1 corresponds to the Pt-N stretching mode.63
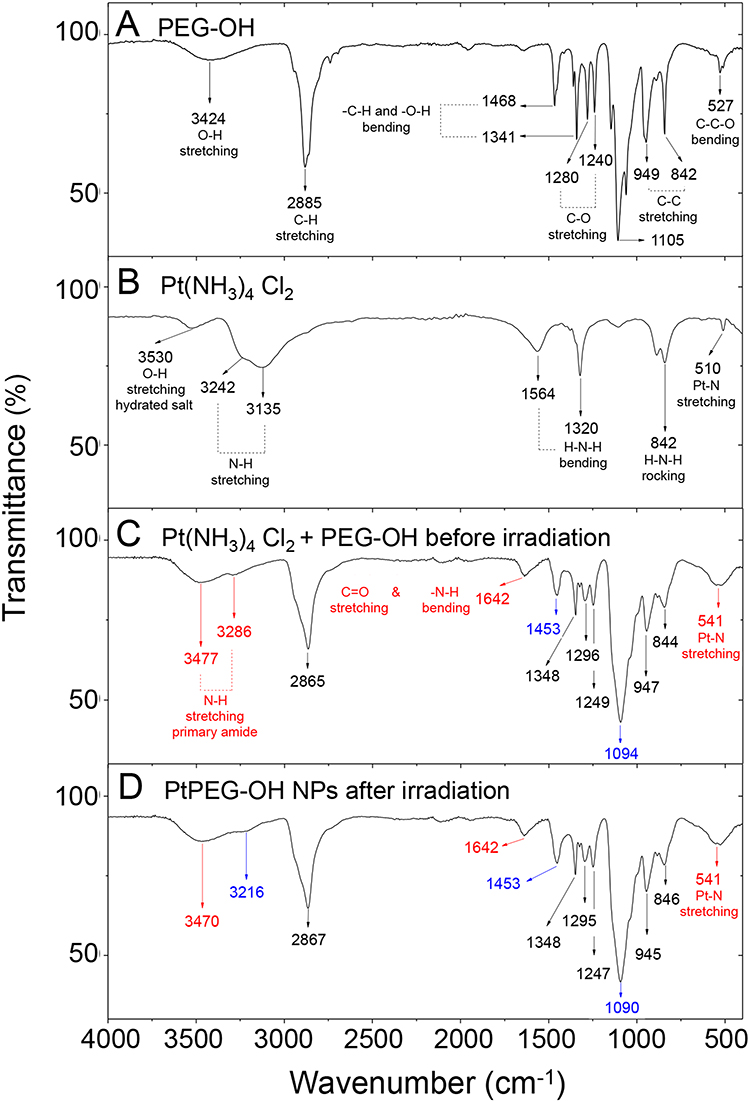 |
Figure 3 FT-IR spectra of (A) pure PEG-OH, (B) pure PtII precursor, (C) PtII precursor mixed with PEG-OH before irradiation and (D) PtPEG NPs obtained after irradiation of the PtII precursor mixed with PEG-OH. |
The interaction of the PtII precursor with PEG-OH before irradiation was confirmed by FT-IR (Figure 3C). The peaks observed at 3477 and 3286 cm−1 are characteristic of the N-H stretching mode (νNH) of a new amide group. In particular, the IR band at 1642 cm−1 is attributed to the C=O stretching vibrational mode of primary amides. This signal overlaps with the 1620 to 1655 cm−1 region, which corresponds to the N-H bending vibrational mode of this functional group. At 1453 cm−1, the C-N stretching mode (νCN) of primary amides was also observed. All these features converge and indicate stable intermediate species are formed before irradiation. The peak at 541 cm−1 is attributed to the Pt-N bond and confirms this statement.64 Its broadening is attributed to the overlap with the C=O bending vibrational mode of amides (535–615 cm−1 region). We suggest that the Pt(NH3)4Cl2 precursor reacts with PEG to form an amide linker due to an initial PEG activation. In the presence of oxygen, this activation may take place via dehydrogenation (oxidation) of the hydroxyl-end group of the PEG. The oxidation of PEG is thermodynamically favourable.65 In the present case, the reaction may also be catalyzed at room temperature by Pt.66–68 PEG may be oxidized into aldehydes (-CHO) or carboxylic acids (–COOH), and thus react preferentially with the ammonium ligand (–NH3+) of the Pt precursor69 to form a stable conjugation shell (Figure S4). After irradiation, minor changes were observed and the bonds visible by IR remained unchanged (eg C-O, C=O, O-H and N-H) (Figure 3D). This result suggests that the PEGylation of small Pt NPs is due to Pt-H-N moieties or electrostatic interactions between amide species and Pt, as proposed in other work.70
Cytotoxicity, Uptake and Localization of PtPEG NPs in Cells
The cytotoxicity of PtPEG NPs in HeLa cells was evaluated by performing CFA. This assay essentially tests every single cell in the population for its ability to proliferate. The CFA is used to determine cell survival after exposure to ionizing radiation and thus, it is the method of choice to evaluate the toxicity of potential radio-nanoenhancers prior to irradiation. The plating efficiencies (PE) of the control culture and of cells incubated for 6 h with PtPEG NPs at a Pt concentration of 5×10−4 mol.L−1, were similar (~65%, p= 0.42). It indicates that PtPEG NPs are harmless for HeLa cells at this concentration. However, significant cytotoxicity (~30%) appeared at a concentration of 10−3 mol.L−1 (PE ~42%, p= 0.02). Therefore, cells were irradiated at the harmless 5×10−4 mol.L−1 Pt concentration.
Differing from other cytotoxic assays, such as those using tetrazolium reagents (eg MTS) to evaluate cells viability as a function of their metabolic activity, a CFA describes the NPs concentration range to safely fight cancer cells through combining nanomedicine and radiation therapy. For instance, it has been shown that HfO2-based NPs do not decrease the viability of adherent normal human primary prostate epithelial cells (HPrEC) in terms of their metabolic activity in a wide concentration range (up to 1000 μg.mL−1) but, determined by CFA, they significantly induced cytotoxicity at 1000 μg.mL−1.71 Indeed, the toxicity determination is a challenging step for NPs clinical translation. Even if green strategies are used to synthesize metal-based NPs, they are susceptible to experiencing, eg metallic ions dissolution that induces genotoxicity and produces toxic reactive oxygen species.72,73 The methodology here presented, assure that PEGylated Pt NPs are appropriate capped with a robust and stable biocompatible shell, which may minimize metallic ions release and non-specific interactions.
The uptake of PtPEG NPs at a Pt concentration of 5×10−4 mol.L−1 in HeLa cells was quantified, after 6 h of incubation. The ICP-OES analysis showed that 0.495 Pt μg was internalised by ca. 2×106 cells, which corresponds to ~5.178×105 NPs per cell.
The intracellular mapping of NPs was investigated by Nano-SIMS. The Nano-SIMS analysis was performed on HeLa cells loaded with PtPEG NPs at the non-toxic conditions mentioned before. This technique allows for detecting different chemical elements simultaneously (Figure 4A). The distribution images of phosphorus (P−) and platinum (Pt−) are presented in Figure 4B–D (square frame of 50 µm × 50 µm). The morphological cell structure was derived from the P− image (Figure 4B). Phosphorus is essentially contained in DNA, RNA and phospholipids. So its detection is used to distinguish individual cells and delineate the nucleus from the cytoplasm. The 194Pt− image (Figure 4C) shows the distribution of Pt compounds. Figure 4D presents the merged image of 194Pt− and P−. The Nano-SIMS analysis clearly demonstrated the presence of Pt inside the cell. PtPEG NPs were seen in the cytoplasm exclusively but not in the nucleus. This is in agreement with other works performed by the group with Pt complexes75 and other studies with metal-containing NPs.76–78 Note that PtPEG NPs preferentially accumulated at cytoplasmic sites poor in phosphorus (region indicated with an arrow). This was observed in other works where 15N-labeled peptide vectors co-localized in HeLa cytoplasmic zones with a low phosphorous concentration.79
 |
Figure 4 (A) Schematic representation of the Nano-SIMS principle. Nano-SIMS images of a HeLa cell loaded with PtPEG NPs. Panels correspond to the intracellular location of (B) P− and (C) 194Pt−. (D) Merged image of 194Pt− and P−. The arrow points out the location of NPs in the cell cytoplasm. |
Effect of PtPEG NPs on Radiation-Induced Cell Killing
The impact of PtPEG NPs on cell killing induced by medical carbon ions (provided by the HIMAC particle therapy centre, Chiba, Japan) of 290 MeV/u.m.a incident energy and an average linear energy transfer LET of 110 keV.μm−1 (5 mm SOBP mode) was evaluated by performing clonogenic assay. For comparison, similar experiments were performed with γ-rays. The survival curves of the controls and cells incubated with PtPEG NPs (Pt 5×10−4 mol.L−1, 6 h) are shown in Figure 5A. The surviving fractions (SF) of cells were determined for doses ranging from 0 up to 5 Gy, and from 0 up to 2.5 Gy for γ-rays and carbon ions, respectively.
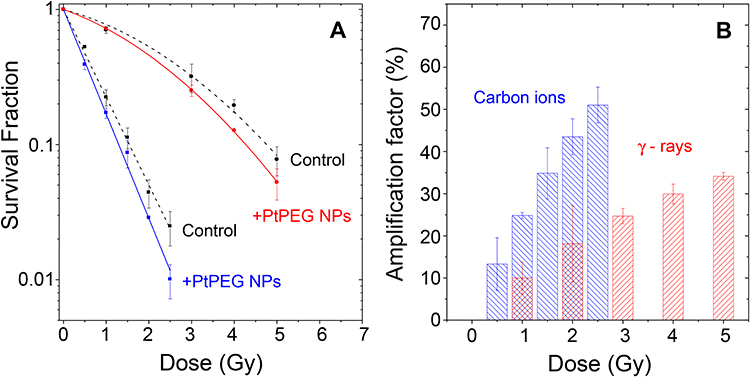 |
Figure 5 (A) Surviving fractions of HeLa cells irradiated by γ-rays (●, ●) and carbon ions (■, ■) in controls and in the presence of PtPEG NPs as a function of the radiation dose. (B) Amplification factors as a function of the radiation doses for γ-rays (red columns) and carbon ions (blue columns). |
The cell response curves were simulated using the linear quadratic (LQ) model:
(5)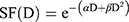
where the parameter α is attributed to the induction of directly lethal damage and β to the additive sub-lethal lesions leading to cell death.80,81 The analysis shows that PtPEG NPs induced an increase of α from 0.15 to 0.26 with γ-rays and from 1.50 to 1.78 with carbon ion radiation. Contrary β remained nearly constant. So, the presence of PtPEG NPs during carbon irradiation enhances cell killing due to the increased induction of directly lethal damages (α). The efficiency of PtPEG NPs to amplify cancer cell killing was quantified using the amplification factor (AF), and the dose-enhancing factor (DEF). AF was used to quantify the efficiency of NPs to enhance cell killing at a defined dose point (D). It is equivalent to the radiation sensitising enhancement ratio (SER) commonly used to describe cell-specific radiosensitization effects.77 AF was calculated as follows:
(6)
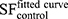 and
and 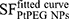 correspond, respectively, to the survival fraction of the control and the survival fraction of the cells loaded with NPs obtained at the same irradiation dose. The values are reported in Figure 5B. Interestingly, at 2 Gy, the AF increased by 44% with incident carbon ions and 14% with γ-rays. This clearly shows that the amplification of cell killing with carbon ions is stronger than with γ-rays at the same dose.
correspond, respectively, to the survival fraction of the control and the survival fraction of the cells loaded with NPs obtained at the same irradiation dose. The values are reported in Figure 5B. Interestingly, at 2 Gy, the AF increased by 44% with incident carbon ions and 14% with γ-rays. This clearly shows that the amplification of cell killing with carbon ions is stronger than with γ-rays at the same dose.
DEF is commonly used to assess the dose enhancement in vitro for a given survival fraction, commonly 10% of survival (SF = 10%).82
(7)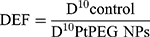
where D10 corresponds to the dose required to reach 10% survival. DEFs of 1.1 and 1.19 were found for γ rays and carbon ion irradiations, respectively. This corresponds to dose enhancements of about 10% and 16%, respectively, confirming that PtPEG NPs are more efficient for improving the lethality of carbon ions than γ-rays. It indicates that lower radiation doses are needed to obtain the same effect, in particular for carbon ions.
We evaluated the relative biological efficiency (RBE) at 10% of survival using γ-rays as a reference beam.83
(8)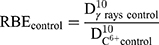
and
(9)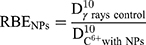
The RBE was found close to 3.09 for free HeLa cells, which is in good agreement with other works.2 In the presence of PtPEG NPs, the RBE value was 3.68 which is a ~19% increase. This result clearly demonstrates that the treatment of HeLa cells with PtPEG NPs increased the RBE of carbon ions.
Nanoscale Mechanisms of PtPEG NPs Activated by Radiation
The interaction of ionizing radiation with biomolecules leads to bond cleavages in nucleic acids, lipids and proteins. It is commonly accepted that complex damage to sizes in the nanometer range is the most lethal for cells. In order to characterize the impact of NPs on the induction of complex lesions, we used DNA plasmids as bio-nanoprobes and quantified the induction of double-strand breaks (DSB). The average number of nanosized damage per plasmid induced in the bio-nanoprobes free of NPs (control) and in the presence of PtPEG NPs at different radiation doses is presented in Figure 6. In some experiments, dimethyl sulfoxide (DMSO), a well-known radical scavenger with redox potential 0.16 V, was added to disentangle the effect of water radicals, in particular the highly reactive hydroxyl radicals (OH∙).84
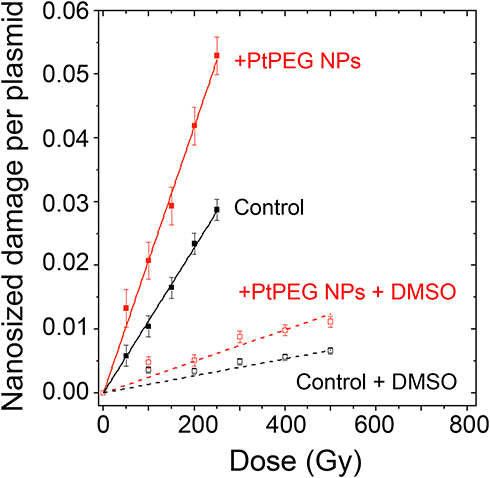 |
Figure 6 Nanosized damage induced by γ-rays in the bio-nanoprobes free from NPs (■) and in the presence of PtPEG NPs (■), as a function of the irradiationdose. Results obtained in the presence of DMSO in samples free from NPs (□) or with PtPEG NPs (□). |
The induction of nanosized damage increased linearly with the irradiation dose for all samples. It indicates that the damage was induced by single ionising events.85 In the presence of PtPEG NPs, the induction of nanosized damage was strongly amplified and it sharply decreased in the presence of DMSO (dotted lines). This is in agreement with previous studies performed with metal-based NPs activated by photons26,84 or by incident ions.27–29
The yields of nanosized damage in NPs free samples (10)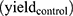 and NPs loaded samples
and NPs loaded samples 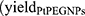 ) correspond to the slopes of the respective dose-response curves. The values are reported in Table 2. The efficiency of PtPEG NPs to amplify the induction of nanosized damage was quantified using the molecular amplification factor (mAF):
) correspond to the slopes of the respective dose-response curves. The values are reported in Table 2. The efficiency of PtPEG NPs to amplify the induction of nanosized damage was quantified using the molecular amplification factor (mAF):

 |
Table 2 Yields of Controls and Plasmids Loaded with PtPEG NPs with an NP:Plasmid Ratio Close 4:1. DMSO Was Added in Some Experiments. The mAF is Also Reported |
In the presence of PtPEG NPs, the induction of nanosized damage was enhanced by 83% (from 11.40×10−5 to 20.90×10−5 damage per plasmid per Gy). On the other hand, the presence of DMSO decreased the induction of nanosized damage (from 20.90×10−5 to 2.48×10−5), which indicates that the amplification of complex damage triggered by PtPEG NPs is mediated in 90% by water radicals (OH∙).
In summary, the amplification of complex damage by Pt NPs is attributed to a sequence of physical and chemical processes.15,29 Briefly, Pt NPs are activated (ionized) by the incident radiation (eg photons or particles) and related secondary electrons produced along the track. The probability of ionization and consecutively the number of electrons emitted locally increases with the atomic number (Z) of the compounds in the NPs. In addition, relaxation processes such as Auger de-excitation, plasmon de-excitation and electron capture take part in the emission of electrons.86–89 The excitation and ionization of water molecules close to the NPs lead to the production of highly reactive radicals nano-clusters. The interaction of these radical clusters with biomolecules will cause nanosized lesions. The activation of PtPEG NPs and the induction of spatially confined perturbations are toxic for cells.
Conclusion
This work presents a simple, fast, scalable and eco-friendly method to produce non-toxic metallic nanoagents dispersed in sterile water solution with a remarkable production rate of 100%, ready-to-use in clinic. As proof-of-concept, the synthesis of small negatively charged PEGylated Pt NPs is presented. The method consists of a one-step radiolytic protocol, using gamma radiation, combined with in situ PEGylation.
As a major characteristic, the PEGylated Pt NPs have the singular property to improve the performance of medical radiations, particularly the effect of particle therapy beams as shown by the 44% amplification of cell killing induced by carbon ions. The enhancement of complex biodamage confirms that the amplification of radiation effects is due to a cascade of early-stage mechanisms that follow the activation of nanoparticles by radiation. This finding is important to improve the composition of future nanoagents and simulate treatment planning.
Gamma radiation is currently used in a large scale by private stakeholders to decontaminate food or sterilise medical instrumentation. Therefore, the production of non-toxic metallic nanoagents using this singular method may be transposable to industrial production in the medium term.
Dense metallic materials are well detected in computed tomography (CT). Moreover, PEGylated Pt NPs may be functionalized with elements active in Positron-emission tomography (PET) or Magnetic resonance imaging (MRI) (eg 89Zr for PET, Gd for MRI). Thus, the remarkable competitiveness of this synthesis reinforces the development of advanced radiation strategies such as “image guided radiation therapy”.
Acknowledgments
The authors acknowledge the Mexican National Council for Science and Technology (CONACyT) for supporting D. Salado-Leza through a PhD CONACyT – French Government scholarship (216823/312283). We thank the China Scholarship Council for granting X. Yang (CSC, N° 201607040068), the European Commission for grants PEOPLE-ITN-ARGENT (no. 608163) and PEOPLE-IEF-NANOHAPY (no. 624370), the Japanese Society for the Promotion of Sciences (JSPS) for a visiting grant offered to M. Bolsa-Ferruz. Part of this research was conducted at NIRS-HIMAC. Our sincere gratitude to Laure Catala (Institut de Chimie Moléculaire et des Matériaux d’Orsay, Univertisé Paris Saclay) for DLS measurements, to Sergio Marco and Sylvain Trepout (Laboratoire d’Imagerie Integrative, Institut Curie INSERM U759) for TEM observations. We thank Stephan Roux and Gloria Jiménez Sánchez (Université de Franche-Comté, Institut UTINAM-UMR CNRS 6213) for ICP-OES determination. The French team acknowledges Evelyne Sage (Institut Curie, Orsay, France) for supporting cells γ-irradiation experiments.
Author Contributions
DS-L synthesized and characterized the nanoparticles, designed the experimental protocols, performed samples irradiation with γ-rays, prepared cells for Nano-SIMS and ICP-OES measurements, determined NPs cytotoxicity, performed data analysis and interpretation, wrote the original draft and actively participated in reviewing and editing the final version of this manuscript. EP was engaged in the study design and experimental data interpretation, as well as, in the in vitro irradiation assays. XY performed NPs lyophilization and evaluated their physicochemical properties and stability post-lyophilization. LS and MB-F performed cells irradiation by carbon ions and contributed to data analysis and interpretation. FS supported UV-vis characterization. DD performed XPS measurements and contributed to data interpretation. J-LG-K and TW supported Nano-SIMS samples preparation and performed the acquisition and images analysis. HR was remarkably engaged in nanoparticles design, synthesis, characterization and validation. SL, as the head of the group, obtained the funding; participated substantially in study design, formal analysis, project administration, validation and manuscript edition. All authors contributed to data analysis, drafting or revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
Daniela Salado-Leza, Erika Porcel, Hynd Remita and Sandrine Lacombe report a patent FR1900008 pending. The authors report no other possible conflicts of interest in this work.
References
1. Kobayashi K, Frohlich H, Usami N, Takakura K, Le Sech C. Enhancement of X-ray-induced breaks in DNA bound to molecules containing platinum: a possible application to hadrontherapy. Radiat Res. 2002;157(1):32–37. doi:10.1667/0033-7587(2002)157[0032:EOXRIB]2.0.CO;2
2. Biston M-C, Joubert A, Adam J-F, et al. Cure of fisher rats bearing radioresistant F98 glioma treated with cis-platinum and irradiated with monochromatic synchrotron X-Rays. Cancer Res. 2004;64(7):2317–2323. doi:10.1158/0008-5472.CAN-03-3600
3. Barabadi H, Vahidi H, Damavandi Kamali K, et al. Emerging theranostic gold nanomaterials to combat lung cancer: a systematic review. J Clust Sci. 2020;31(2):323–330. doi:10.1007/s10876-019-01650-4
4. Barabadi H, Vahidi H, Damavandi Kamali K, et al. Emerging theranostic silver nanomaterials to combat colorectal cancer: a systematic review. J Clust Sci. 2020;31(2):311–321. doi:10.1007/s10876-019-01668-8
5. Barabadi H, Hosseini O, Damavandi Kamali K, et al. Emerging theranostic silver nanomaterials to combat lung cancer: a systematic review. J Clust Sci. 2020;31:1. doi:10.1007/s10876-019-01639-z
6. Hainfeld JF, Slatkin DN, Smilowitz HM. The use of gold nanoparticles to enhance radiotherapy in mice. Phys Med Biol. 2004;49(18):N309N315. doi:10.1088/0031-9155/49/18/N03
7. Hainfeld JF, Dilmanian FA, Slatkin DN, Smilowitz HM. Radiotherapy enhancement with gold nanoparticles. J Pharm Pharmacol. 2008;60(8):977–985. doi:10.1211/jpp.60.8.0005
8. Li Y, Yun KH, Lee H, Goh SH, Suh YG, Choi Y. Porous platinum nanoparticles as a high-Z and oxygen generating nanozyme for enhanced radiotherapy in vivo. Biomaterials. 2019;197:12–19. doi:10.1016/j.biomaterials.2019.01.004
9. Bonvalot S, Le Pechoux C, De Baere T, et al. First-in-human study testing a new radioenhancer using nanoparticles (NBTXR3) activated by radiation therapy in patients with locally advanced soft tissue sarcomas. Clin Cancer Res. 2017;23(4):908–917. doi:10.1158/1078-0432.CCR-16-1297
10. Lux F, Tran VL, Thomas E, et al. AGuIX ® from bench to bedside-transfer of an ultrasmall theranostic gadolinium-based nanoparticle to clinical medicine. Br J Radiol. 2019;92(1093):1–19.
11. Verry C, Sancey L, Dufort S, et al. Treatment of multiple brain metastases using gadolinium nanoparticles and radiotherapy: NANO-RAD, a Phase I study protocol. BMJ Open. 2019;9:2. doi:10.1136/bmjopen-2018-023591
12. Libutti SK, Paciotti GF, Byrnes AA, et al. Phase I and pharmacokinetic studies of CYT-6091, a novel PEGylated colloidal gold-rhTNF nanomedicine. Clin Cancer Res. 2010;16(24):6139–6149. doi:10.1158/1078-0432.CCR-10-0978
13. Porcel E, Kobayashi K, Usami N, Remita H, Le Sech C, Lacombe S. Photosensitization of plasmid-DNA loaded with platinum nano-particles and irradiated by low energy X-rays. J Phys Conf Ser. 2011;261(1):012004. doi:10.1088/1742-6596/261/1/012004
14. Butterworth KT, McMahon SJ, Currell FJ, Prise KM. Physical basis and biological mechanisms of gold nanoparticle radiosensitization. Nanoscale. 2012;4(16):4830. doi:10.1039/c2nr31227a
15. Wang Y, Liu J, Ma X, Liang XJ. Nanomaterial-assisted sensitization of oncotherapy. Nano Res. 2018;11(6):2932–2950. doi:10.1007/s12274-017-1961-0
16. Durante M, Loeffler JS. Charged particles in radiation oncology. Nat Rev Clin Oncol. 2010;7(1):37–43. doi:10.1038/nrclinonc.2009.183
17. Vitolo V, Fiore MR, Barcellini A, et al. Carbon ion radiotherapy in the management of the tumors of the peripheral nervous system. Anticancer Res. 2019;39(2):909–913. doi:10.21873/anticanres.13193
18. IAEA and ICRU. Relative biological effectiveness in ion beam therapy. In: International Atomic Energy Agency, Relative Biological Effectiveness in Ion Beam Therapy, Technical Reports Series. Vienna: IAEA; 2008:461.
19. Kaur H, Pujari G, Semwal MK, Sarma A, Avasthi DK. In vitro studies on radiosensitization effect of glucose capped gold nanoparticles in photon and ion irradiation of HeLa cells. Nucl Instruments Methods Phys Res Sect B Beam Interact with Mater Atoms. 2013;301:7–11. doi:10.1016/j.nimb.2013.02.015
20. Kim J-K, Seo S-J, Kim K-H, et al. Therapeutic application of metallic nanoparticles combined with particle-induced x-ray emission effect. Nanotechnology. 2010;21(42):425102. doi:10.1088/0957-4484/21/42/425102
21. Dollinger G. Comment on ‘Therapeutic application of metallic nanoparticles combined with particle-induced x-ray emission effect.’ Nanotechnology. 2011;22(24):248001. doi:10.1088/0957-4484/22/24/248001
22. Le Sech C, Kobayashi K, Usami N, Furusawa Y, Porcel E, Lacombe S. Comment on “Enhanced relative biological effectiveness of proton radiotherapy in tumor cells with internalized gold nanoparticles”. Appl Phys Lett. 2012;100(2):026101. doi:10.1063/1.3675570
23. Li F, Li Z, Jin X, et al. Radiosensitizing effect of gadolinium oxide nanocrystals in NSCLC cells under carbon ion irradiation. Nanoscale Res Lett. 2019;14:1. doi:10.1186/s11671-019-3152-2
24. Abdul Rashid R, Zainal Abidin S, Khairil Anuar MA, et al. Radiosensitization effects and ROS generation by high Z metallic nanoparticles on human colon carcinoma cell (HCT116) irradiated under 150 MeV proton beam. OpenNano. 2019;4:100027. doi:10.1016/j.onano.2018.100027
25. Li S, Bouchy S, Penninckx S, et al. Antibody-functionalized gold nanoparticles as tumor-targeting radiosensitizers for proton therapy. Nanomedicine. 2019;14(3):317–333. doi:10.2217/nnm-2018-0161
26. Salado-Leza D, Traore A, Porcel E, et al. Radio-enhancing properties of bimetallic Au: Pt nanoparticles: experimental and theoretical evidence. Int J Mol Sci. 2019;20(22):5648. doi:10.3390/ijms20225648
27. Yang X, Salado‐Leza D, Porcel E, et al. A facile one‐pot synthesis of versatile PEGylated platinum nanoflowers and their application in radiation therapy. Int J Mol Sci. 2020;21(5):1–20.
28. Schlatholter T, Eustache P, Porcel E, et al. Improving proton therapy by metal-containing nanoparticles: nanoscale insights. Int J Nanomedicine. 2016;11:1549. doi:10.2147/IJN.S99410
29. Porcel E, Liehn S, Remita H, et al. Platinum nanoparticles: a promising material for future cancer therapy? Nanotechnology. 2010;21(8):85103. doi:10.1088/0957-4484/21/8/085103
30. Porcel E, Tillement O, Lux F, et al. Gadolinium-based nanoparticles to improve the hadrontherapy performances. Nanomedicine. 2014;10(8):1601–1608. doi:10.1016/j.nano.2014.05.005
31. Alric C, Miladi I, Kryza D, et al. The biodistribution of gold nanoparticles designed for renal clearance. Nanoscale. 2013;5(13):5930–5939. doi:10.1039/c3nr00012e
32. Maggiorella L, Barouch G, Devaux C, et al. Nanoscale radiotherapy with hafnium oxide nanoparticles. Futur Oncol. 2012;8(9):1167–1181. doi:10.2217/fon.12.96
33. Mignot A, Truillet C, Lux F, et al. A top-down synthesis route to ultrasmall multifunctional gd-based silica nanoparticles for theranostic applications. Chem A Eur J. 2013;19(19):6122–6136. doi:10.1002/chem.201203003
34. Pedone D, Moglianetti M, De Luca E, Bardi G, Pompa PP. Platinum nanoparticles in nanobiomedicine. Chem Soc Rev. 2017;46(16):4951–4975.
35. Abedini A, Daud AR, Abdul Hamid MA, Kamil Othman N, Saion E. A review on radiation-induced nucleation and growth of colloidal metallic nanoparticles. Nanoscale Res Lett. 2013;8(1):474. doi:10.1186/1556-276X-8-474
36. Akar B, Pushpavanam K, Narayanan E, Rege K, Heys JJ. Mechanistic investigation of radiolysis-induced gold nanoparticle formation for radiation dose prediction. Biomed Phys Eng Express. 2018;4(6):549–559. doi:10.1088/2057-1976/aac280
37. Abdelghany AM, Abdelrazek EM, Badr SI, Abdel-Aziz MS, Morsi MA. Effect of gamma-irradiation on biosynthesized gold nanoparticles using chenopodium murale leaf extract. J Saudi Chem Soc. 2017;21(5):528–537. doi:10.1016/j.jscs.2015.10.002
38. Biswal J, Ramnani SP, Shirolikar S, Sabharwal S. Seedless synthesis of gold nanorods employing isopropyl radicals generated using gamma radiolysis technique. Int J Nanotechnol. 2010;7(912):907–918. doi:10.1504/IJNT.2010.034697
39. Nguyen TKL, Nguyen ND, Dang VP, et al. Synthesis of platinum nanoparticles by gamma co-60 ray irradiation method using chitosan as stabilizer. Adv Mater Sci Eng. 2019;2019:1–6. doi:10.1155/2019/9624374
40. Cele T, Maaza M, Gibaud A. Synthesis of platinum nanoparticles by gamma radiolysis. MRS Adv. 2018;3(4243):2537–2557. doi:10.1557/adv.2018.233
41. Keita B, Nadjo L, De Cointet C, Amblard J, Belloni J. STM investigation of platinum oligomer clusters prepared by radiolysis. Chem Phys Lett. 1996;249(56):297–303. doi:10.1016/0009-2614(95)01425-X
42. Gharibshahi E, Saion E, Ashraf A, Gharibshahi L. Size_controlled and optical properties of platinum nanoparticles by gamma radiolytic synthesis. Appl Radiat Isot. 2017;130:211–217. doi:10.1016/j.apradiso.2017.09.012
43. Belapurkar AD, Kapoor S, Kulshreshtha SK, Mittal JP. Radiolytic preparation and catalytic properties of platinum nanoparticles. Mater Res Bull. 2001;36(12):145–151. doi:10.1016/S0025-5408(01)00499-8
44. Pasanphan W, Chunkoh L, Choofong S. Magnetic gadolinium-chitosan composite nanoparticles created by radiolytic synthesis. ICCM Int Conf Compos Mater. 2011;6–11.
45. Luchette M, Korideck H, Makrigiorgos M, Tillement O, Berbeco R. Radiation dose enhancement of gadolinium-based AGuIX nanoparticles on HeLa cells. Nanomedicine Nanotechnology, Biol Med. 2014;10(8):1751–1755. doi:10.1016/j.nano.2014.06.004
46. Treguer M, de Cointet C, Remita H, et al. Dose rate effects on radiolytic synthesis of gold_silver bimetallic clusters in solution. J Phys Chem B. 1998;102(22):4310–4321. doi:10.1021/jp981467n
47. Remita H, Lampre I, Mostafavi M, Balanzat E, Bouffard S. Comparative study of metal clusters induced in aqueous solutions by γ-rays, electron or C6+ ion beam irradiation. Radiat Phys Chem. 2005;72(5):575–586. doi:10.1016/j.radphyschem.2004.03.042
48. Remita H, Remita S. Metal clusters and nanomaterials: contribution of radiation chemistry. In: Wishart JF, Madhava Rao BS, editors. Recent Trends in Radiation Chemistry. World Scientific; 2010:347–383.
49. Belloni J, Mostafavi M, Remita H, Marignier J. Delcourt and M-O. Radiation-induced synthesis of mono- and multi-metallic clusters and nanocolloids. New J Chem. 1998;22(11):1239–1255. doi:10.1039/a801445k
50. Gharibshahi E, Saion E. Influence of dose on particle size and optical properties of colloidal platinum nanoparticles. Int J Mol Sci. 2012;13(12):14723–14741. doi:10.3390/ijms131114723
51. Huang K, Ma H, Liu J, et al. Size-dependent localization and penetration of ultrasmall gold nanoparticles in cancer cells, multicellular spheroids, and tumors in vivo. ACS Nano. 2012;6(5):4483–4493. doi:10.1021/nn301282m
52. Puglia C, Nilsson A, Hernnäs B, Karis O, Bennich P, Mårtensson N. Physisorbed, chemisorbed and dissociated O2 on Pt(111) studied by different core level spectroscopy methods. Surf Sci. 1995;342(13):119–133. doi:10.1016/0039-6028(95)00798-9
53. Liu F, Zhao Z, Qiu L, Zhao L. Applications of XPS on nanoscale material research. J Surf Anal. 2009;15(3):271–273. doi:10.1384/jsa.15.271
54. Fu X, Wang Y, Wu N, Gui L, Tang Y. Surface modification of small platinum nanoclusters with alkylamine and alkylthiol: an XPS study on the influence of organic ligands on the pt 4f binding energies of small platinum nanoclusters. J Colloid Interface Sci. 2001;243(2):326–330. doi:10.1006/jcis.2001.7861
55. López GP, Castner DG, Ratner BD. XPS O 1s binding energies for polymers containing hydroxyl, ether, ketone and ester groups. Surf Interface Anal. 1991;17(5):267–272. doi:10.1002/sia.740170508
56. Forbes LM, O_Mahony AM, Sattayasamitsathit S, Wang J, Cha JN. Polymer end-group mediated synthesis of well-defined catalytically active platinum nanoparticles. J Mater Chem. 2011;21(39):15788. doi:10.1039/c1jm13137k
57. Jansen RJJ, van Bekkum H. XPS of nitrogen-containing functional groups on activated carbon. Carbon N Y. 1995;33(8):1021–1027. doi:10.1016/0008-6223(95)00030-H
58. Deniau G, Azoulay L, Jégou P, Le Chevallier G, Palacin S. Carbon-to-metal bonds: electrochemical reduction of 2-butenenitrile. Surf Sci. 2006;600(3):675–684. doi:10.1016/j.susc.2005.11.021
59. Truica-Marasescu F, Wertheimer MR. Nitrogen-rich plasma-polymer films for biomedical applications. Plasma Process Polym. 2008;5(1):44–57. doi:10.1002/ppap.200700077
60. Forbes LM, Sattayasamitsathit S, Xu PF, et al. Improved oxygen reduction reaction activities with amino acid R group functionalized PEG at platinum surfaces. J Mater Chem A. 2013;1(35):10267–10273. doi:10.1039/c3ta12133j
61. Crispin X, Lazzaroni R, Crispin A, Geskin V, Brédas J, Salaneck W. Understanding the initial stages of polymer grafting on metals: a photoelectron spectroscopy study of acrylonitrile adsorption on transition metal surfaces. J Electron Spectros Relat Phenomena. 2001;121(1):57–74. doi:10.1016/S0368-2048(01)00326-7
62. Silverstein RM, Bassler GC, Morrill TC. Spectrometric Identification of Organic Compounds. John Wiley & Sons, Inc.; 1991.
63. Nakamoto K. Infrared and Raman Spectra of Inorganic and Coordination Compounds. Part B: Applications in Coordination, Organometallic and Bioinorganic Chemistry.
64. Wysokiski R, Kuduk-Jaworska J, Michalska D. Electronic structure, Raman and infrared spectra, and vibrational assignment of carboplatin. Density functional theory studies. J Mol Struct Theochem. 2006;758(23):169–179. doi:10.1016/j.theochem.2005.10.032
65. Glastrup J. Degradation of polyethylene glycol. A study of the reaction mechanism in a model molecule: tetraethylene glycol. Polym Degrad Stab. 1996;52(3):217–222. doi:10.1016/0141-3910(95)00225-1
66. Chibani S, Michel C, Delbecq F, Pinel C, Besson M. On the key role of hydroxyl groups in platinum-catalysed alcohol oxidation in aqueous medium. Catal Sci Technol. 2013;3(2):339–350. doi:10.1039/C2CY20363D
67. Mallat T, Baiker A. Oxidation of alcohols with molecular oxygen on platinum metal catalysts in aqueous solutions. Catal Today. 1994;19(2):247–283. doi:10.1016/0920-5861(94)80187-8
68. Anderson R, Griffin K, Johnston P, Alsters PL. Selective oxidation of alcohols to carbonyl compounds and carboxylic acids with platinum group metal catalysts. Adv Synth Catal. 2003;345(4):517–523. doi:10.1002/adsc.200390060
69. Cox Gad S. Handbook of Pharmaceutical Biotechnology. Cox GS, ed. John Wiley & Sons. Inc; 2007.
70. Wand P, Bartl JD, Heiz U, Tschurl M, Cokoja M. Functionalization of small platinum nanoparticles with amine and phosphines: ligand binding modes and particle stability. J Colloid Interface Sci. 2016;478:72–80. doi:10.1016/j.jcis.2016.06.003
71. Salado-Leza D, Mendoza-Mendoza E, Castillo-Ramírez JA, Escudero-Lourdes C, García-Cerda LA. A simple approach to room-temperature synthesis of cubic Al-doped HfO2 nanoparticles and their toxicity evaluation in normal prostate cells. Mater Lett. 2020;274:128048. doi:10.1016/j.matlet.2020.128048
72. Barabadi H, Najafi M, Samadian H, et al. A systematic review of the genotoxicity and antigenotoxicity of biologically synthesized metallic nanomaterials: are green nanoparticles safe enough for clinical marketing? Med. 2019;55:8.
73. Mortezaee K, Najafi M, Samadian H, Barabadi H, Azarnezhad A, Ahmadi A. Redox interactions and genotoxicity of metal-based nanoparticles: A comprehensive review. Chem Biol Interact. 2019;312((September)):108814. doi:10.1016/j.cbi.2019.108814
74. Jiang H, Goulbourne CN, Tatar A, et al. High-resolution imaging of dietary lipids in cells and tissues by NanoSIMS analysis. J Lipid Res. 2014;55(10):2156–2166. doi:10.1194/jlr.M053363
75. Usami N, Furusawa Y, Kobayashi K, et al. Mammalian cells loaded with platinum-containing molecules are sensitized to fast atomic ions. Int J Radiat Biol. 2008;84(7):603–611. doi:10.1080/09553000802199846
76. Stefanciková L, Porcel E, Eustache P, et al. Cell localisation of gadolinium-based nanoparticles and related radiosensitising efficacy in glioblastoma cells. Cancer Nanotechnol. 2014;5(1):6. doi:10.1186/s12645-014-0006-6
77. Jain S, Coulter JA, Hounsell AR, et al. Cell-specific radiosensitization by gold nanoparticles at megavoltage radiation energies. Int J Radiat Oncol Biol Phys. 2011;79(2):531–539. doi:10.1016/j.ijrobp.2010.08.044
78. Shah NB, Dong J, Bischof JC. Cellular uptake and nanoscale localization of gold nanoparticles in cancer using label-free confocal Raman microscopy. Mol Pharm. 2011;8(1):176–184. doi:10.1021/mp1002587
79. Romer W, Wu T-D, Duchambon P, et al. Sub-cellular localisation of a 15N-labelled peptide vector using NanoSIMS imaging. Appl Surf Sci. 2006;252:6925–6930. doi:10.1016/j.apsusc.2006.02.183
80. Tubiana M, Dutreix J, Wambersie A. Effects cellulaires des rayonnements ionisants. Les courbes de survie cellulaire. In: Hermann, editor. Radiobiologie. Paris: Hermann, éditeurs des sciences et des arts; 1986:73–104.
81. Douglas BG, Fowler JF. The effect of multiple small doses of x rays on skin reactions in the mouse and a basic interpretation. Radiat Res. 1976;66(2):401–426. doi:10.2307/3574407
82. Taggart LE, McMahon SJ, Currell FJ, Prise KM, Butterworth KT. The role of mitochondrial function in gold nanoparticle mediated radiosensitisation. Cancer Nanotechnol. 2014;5(1):5. doi:10.1186/s12645-014-0005-7
83. International Commission on Radiological Protection. Report of the RBE subcommittee to the international commission on radiological protection and the international commission on radiation units and measurements. Health Phys. 1963;9(357386).
84. Porcel E, Li S, Usami N, et al. Nano-Sensitization under gamma rays and fast ion radiation. J Phys Conf Ser. 2012;373:012006. doi:10.1088/1742-6596/373/1/012006
85. Smialek MA, Moore SA, Mason NJ, Shuker DEG. Quantification of radiation-induced single-strand breaks in plasmid DNA using a TUNEL/ELISA-based assay. Radiat Res. 2009;172(5):529–536. doi:10.1667/RR1684.1
86. McMahon SJ, Hyland WB, Muir MF, et al. Nanodosimetric effects of gold nanoparticles in megavoltage radiation therapy. Radiother Oncol. 2011;100(3):412–416. doi:10.1016/j.radonc.2011.08.026
87. Verkhovtsev AV, Korol AV, Solov’yov AV. Electron production by sensitizing gold nanoparticles irradiated by fast ions. J Phys Chem C. 2015;119(20):11000–11013. doi:10.1021/jp511419n
88. Pesic ZD, Hellhammer R, Sulik B, Stolterfoht N. Strong anisotropy in the proton emission following fragmentation of H 2 O molecules by impact with slow, highly charged Xenon ions. J Phys B at Mol Opt Phys. 2009;42(23):235202. doi:10.1088/0953-4075/42/23/235202
89. Stumpf H. On the theory of electronic processes in ionic crystal semiconductors. Phys Der Kondens Mater. 1971;13(2):101–117.
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.