")
Back to Journals » International Journal of Nanomedicine » Volume 16
Gold Nanostars: A Novel Platform for Developing 211At-Labeled Agents for Targeted Alpha-Particle Therapy
Authors Liu Y, Zhou Z, Feng Y, Zhao XG, Vaidyanathan G, Zalutsky MR , Vo-Dinh T
Received 2 July 2021
Accepted for publication 28 August 2021
Published 28 October 2021 Volume 2021:16 Pages 7297—7305
DOI https://doi.org/10.2147/IJN.S327577
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Israel (Rudi) Rubinstein
Yang Liu,1 Zhengyuan Zhou,2 Yutian Feng,2 Xiao-Guang Zhao,2 Ganesan Vaidyanathan,2 Michael R Zalutsky,1,2 Tuan Vo-Dinh1,3,4
1Department of Biomedical Engineering, Duke University, Durham, NC, 27708, USA; 2Department of Radiology, Duke University Medical Center, Durham, NC, 27710, USA; 3Department of Chemistry, Duke University, Durham, NC, 27708, USA; 4Fitzpatrick Institute for Photonics, Duke University, Durham, NC, 27708, USA
Correspondence: Michael R Zalutsky
Department of Radiology, Duke University Medical Center, 311 Research Drive, 161H Bryan Research Building, Durham, NC, 27710, USA
Email [email protected]
Tuan Vo-Dinh
Department of Biomedical Engineering, Duke University, 1427 CIEMAS, Box 90281, Durham, NC, 27708, USA
Tel +1 919 660-8520
Email [email protected]
Aim: To develop an innovative 211At nanoplatform with high radiolabeling efficiency and low in vivo deastatination for future targeted alpha-particle therapy (TAT) to treat cancer.
Methods: Star-shaped gold nanoparticles, gold nanostars (GNS), were used as the platform for 211At radiolabeling. Radiolabeling efficiency under different reaction conditions was tested. Uptake in the thyroid and stomach after systemic administration was used to evaluate the in vivo stability of 211At-labeled GNS. A subcutaneous U87MG human glioma xenograft murine model was used to preliminarily evaluate the therapeutic efficacy of 211At-labeled GNS after intratumoral administration.
Results: The efficiency of labeling GNS with 211At was almost 100% using a simple and rapid synthesis process that was completed in only 1 min. In vitro stability test in serum showed that more than 99% of the 211At activity remained on the GNS after 24 h incubation at 37°C. In vivo biodistribution results showed low uptake in the thyroid (0.44– 0.64%ID) and stomach (0.21– 0.49%ID) between 0.5 and 21 h after intravenous injection, thus indicating excellent in vivo stability of 211At-labeled GNS. The preliminary therapeutic efficacy study demonstrated that 211At labeled GNS substantially reduced tumor growth (P < 0.001; two-way ANOVA) after intratumoral administration.
Conclusion: The new 211At radiolabeling strategy based on GNS has the advantages of a simple process, high labeling efficiency, and minimal in vivo dissociation, making it an attractive potential platform for developing TAT agents that warrants further evaluation in future preclinical studies directed to evaluating prospects for clinical translation.
Keywords: astatine-211, 211At, gold nanostars, GNS, cancer therapy, targeted alpha-particle therapy, TAT
Introduction
Targeted alpha-particle therapy (TAT) is an emerging method for cancer treatment that has the advantages of high linear energy transfer (>50 keV/µm), irreparable double-strand DNA breaks, and independence of dose rate, cell cycle, and cellular oxygenation.1,2 As a result, TAT is conceptually superior to standard-of-care external beam radiation therapy using X-rays or γ-rays particularly for treating hypoxic tumors. Furthermore, since alpha-particles have a range of only a few cell diameters, TAT is an attractive approach for treating disseminated cancers.3 Among the different available alpha-particle emitting radionuclides, astatine-211 (211At) has the unique advantages of an optimal half-life (7.2 h) and absence of long-lived alpha-particle emitting daughter radionuclides, thus preventing off-target radiation toxicity from the redistribution of daughter radionuclides.2,4 Our group performed the first clinical trial for the treatment of glioblastoma (GBM) with 211At-labeled antibodies.5 However, traditional 211At radiolabeling methods including the one used in this clinical study generate a chemical bond between 211At and an aromatic carbon that have the following limitations: (1) the complicated radiochemistry process makes it difficult for clinical applications; (2) low conjugation efficiency results could lead to suboptimal cancer treatment: only 1 in 400 to 1000 antibody molecules can be labeled with an 211At; (3) the 211At from the labeled molecule can dissociate from the prosthetic moiety due to intrinsic low C-At bond strength and in vivo metabolic degradation.6,7 Innovative radiolabeling methods involving a simple process, high yield, and loading capacity are urgently needed for both preclinical studies and clinical translation of TAT agents labeled with 211At.
Our group has developed a novel method to synthesize star-shaped gold nanoparticles, gold nanostars (GNS), for biomedical applications without the need for toxic surfactants.8 The multibranched GNS morphology offered a high surface area for 211At labeling, which helped achieve a very effective delivery platform. Furthermore, the size of GNS can be tailored to optimize the preferential accumulation in tumors by the enhanced permeability and retention effect. In prior studies, we have demonstrated that the developed GNS can accumulate selectively in brain cancer mediated by the enhanced permeability and retention effect.9 The enhanced permeability and retention effect was reported to be due to the tumor leaky vasculature, and this effect was found in most solid tumors.10,11 Brain cancer was selected as the intended target disease for this study because it is one of the most aggressive cancers. Specifically, GBM, with more than 10,000 newly diagnosed patients in the United States each year, is the most common primary brain cancer.12,13 Despite the highest first-year cost (> $120,000) for a cancer type, the prognosis for GBM patients is dismal. The median survival for GBM patients is only 15 months after aggressive treatments, including surgery, chemotherapy, and radiation therapy.14–16 Less than 5% of the patients survive for more than 3 years.17 Despite decades of effort, GBM is still a deadly disease with essentially 100% mortality.18,19 Thus, there is a critical and urgent need to develop new approaches for effective brain cancer treatment.
As a first step towards developing a GNS-based 211At-labeled TAT agent for GBM treatment, we have performed experiments described herein evaluating the potential utility of 211At-labeled GNS for this purpose. Only a few studies have been reported directed at developing nanoparticle-driven carrier systems for use with 211At. Ultrashort, single-walled carbon nanotubes (US-tubes) labeled with 211At via noncovalent van der Waals binding have been evaluated in an ex vivo study.20 However, the radiolabeling efficiency was low, and the in vivo stability of 211At-labeled US-tubes was not studied. Although in vitro cell experiments have been performed with 211At-labeled gold nanospheres, no in vivo investigations of gold nanoparticles have been described to date.21,22
In the current study, we evaluated GNS as a novel platform technology for 211At radiolabeling and for the first time, investigated in vivo stability and biodistribution in normal mice, and therapeutic efficacy of the 211At-GNS in a murine subcutaneous GBM model. Our experimental results showed that 211At labeling of GNS can be achieved by a simple process in a short time with a high labeling efficiency and in vivo stability. In addition, the 211At-labeled GNS demonstrated a substantial therapeutic efficacy by inhibiting tumor growth after intratumoral administration. To the best of our knowledge, this is the first in vivo study investigating gold nanoparticles as an 211At delivery platform for future TAT applications.
Materials and Methods
GNS Synthesis and Characterization
All chemicals were purchased from Sigma-Aldrich unless mentioned otherwise. GNS were synthesized following our developed toxic surfactant-free and seed-mediated method reported in previous studies.23,24 12-nm spherical gold nanoparticles synthesized using the trisodium citrate reduction method were used as seeds. For GNS synthesis, 1 mL of 3 mM AgNO3 in deionized (DI) water and 1 mL of 50 mM ascorbic acid in DI water were rapidly added to a mixture of 100 mL of DI water with 0.25 mM HAuCl4, 1 mM HCl, and 1 mL of the 12-nm gold nanosphere seeds under vigorous stirring. After a 10-min reaction, 1 mL of 1 mM mPEG-SH (MW 6,000) in DI water was added and the mixture was incubated for 1 h at room temperature. The resultant GNS were purified and concentrated by centrifugation at 6000 g for 30 min. Transmission electron microscopy (TEM) of GNS was performed using a Tecnai G2 Twin transmission electron microscope (FEI, Hillsboro, OR) with a 160 kV acceleration voltage. The Vis-NIR spectrum was obtained by using a UV-Vis-NIR spectrometer (UV-3600, Shimadzu, Japan). The hydrodynamic diameter and nanoparticle concentration of the PEGylated GNS were measured with Nanoparticle Tracking Analysis method (Nanosight 500, Malvern, UK).
Radiolabeling and in vitro Stability
Astatine-211 was produced at the Duke University Medical Center cyclotron facility by bombarding a natural bismuth target with a 28.0-MeV alpha-particle beam and subsequently isolated by a dry distillation method.25 For the radiolabeling experiments, 100 µg GNS in DI water were mixed with 7.4 MBq 211At (460 fmol) in different media [DI water, 0.1 M NaOH in DI water, 50 mM ascorbic acid (AA) in DI water, or 0.2 mg/mL N-chlorosuccinimide (NCS) in DI water], and the reaction was allowed to proceed at room temperature for 1, 5 and 30 min. We also evaluated the effect of the GNS amount – ranging from 100 µg (540 fmol) down to 0.01 µg (54 amol) – on radiolabeling efficiency using a 5-min reaction time. The reaction volume was 100 µL, and centrifugation (6,000 g, 10 min) was used to separate free, unreacted 211At remaining in solution and 211At-labeled GNS. The 211At-labeled GNS pellet was resuspended in 100 µL of DI water, and then the radioactivity levels were measured using a dose calibrator. The stability of 211At-labeled GNS was evaluated by incubating it in phosphate-buffered saline (PBS), pH 7.4, and in murine serum at 37°C up to 24 h. The murine serum was isolated by centrifugating the murine whole blood at 3000 g for 10 min. The supernatant (serum) was collected for the in vitro stability measurements. The concentration of the 211At-GNS agent in both the murine serum and PBS was 200 µg/mL (Au) with 7.4 MBq 211At per mL. The same centrifugation protocol (6,000 g, 10 min) was used to separate dissociated 211At in the solution and the 211At-labeled GNS.
In vivo Biodistribution
Freshly prepared PEGylated GNS were labeled with 211At by incubation at room temperature for 5 min in 50 mM ascorbic acid in DI water. Radiolabeled GNS were purified by centrifugation/washing 3 times and resuspended in PBS without ascorbic acid. For the biodistribution experiment, 20 nude mice were randomly divided into 4 groups, and each mouse was intravenously injected with 211At-labeled GNS (0.185 MBq radioactivity and 100 µg gold mass) in 100 µL PBS without ascorbic acid. Mice were euthanized, and organs of interest (liver, spleen, lung, heart, kidney, bladder, stomach, small intestine, large intestine, thyroid, muscle, blood, urine, bone, skin, brain, and tail) were harvested at 0.5, 2, 14, and 21 h after intravenous injection of the radiolabeled GNS. Tissues were weighed and counted along with injection standards for 211At activity using an automated gamma counter (LKB 1282, Wallac, Finland). The measurement geometry of the gamma counter was taken into account by placing samples at the same location as that for the standard. From these counts, the percent injected dose per gram tissue (%ID/g) and percent injected dose per organ (%ID/organ) were calculated using an in-house computer program.
Therapeutic Efficacy Experiment
Human glioma U87MG cells were obtained from Duke Cell Culture Facility and cultured in MEM cell growth medium with 10% fetal bovine serum (Hyclone, MA, USA), 100 units/mL penicillin, and 100 µg/mL streptomycin at 37°C in a 5% CO2 incubator. The use of the U87MG cell line was approved by the ethics committee, and this cell line was authenticated by the short tandem repeat profiling. All animal studies were performed under protocols that had been approved by the Duke University Institutional Animal Care and Use Committee. The guidelines from National Institutes of Health and Duke University Institutional Animal Care and Use Committee were followed. For subcutaneous xenograft generation, 3×106 U87MG human glioma cells in the cell growth medium were subcutaneously injected into each mouse in the flank region. The tumor size was calculated as 0.5×length×width×width. After 18 days, the tumor size reached ~100 mm3 and the intratumoral therapy study was initiated. Groups of 10 mice received either 30 µL of 211At-labeled GNS (1.11 MBq) in PBS or just PBS by intratumoral injection using a syringe pump at a rate of 30 µL/min. The tumor sizes were measured every 3 or 4 days until the end of the study.
Results
The synthesized GNS exhibited a star-like shape and had a maximum absorption at 765 nm (Figure 1). We performed 211At radiolabeling experiments using PEGylated GNS with a hydrodynamic diameter of 50 nm using different reaction conditions and the results are shown in Figure 2. First, we compared the radiolabeling efficiency in four different reaction media. The calculated 211At labeling efficiency was 94.7 ± 0.1% in DI water, 97.4 ± 0.2% in DI water with 0.1 M NaOH, 99.8 ± 0.1% in DI water with 50 mM ascorbic acid, and 42.6 ± 0.3% in DI water with 0.2 mg/mL NCS. The radiolabeling efficiency for the reaction medium with a reducing agent, ascorbic acid, was the highest (99.8%), and almost all the 211At could be attached to GNS after only a 5-min incubation at room temperature. Conversely, the NCS oxidizing agent dramatically decreased the radiolabeling efficiency to 42.6 ± 0.3%. Second, we evaluated the effect of incubation time on radiolabeling efficiency in reaction media consisting of DI water or DI water with 50 mM ascorbic acid. The radiolabeling efficiency in DI water increased from 93.4 ± 0.2% (1 min), to 94.7 ± 0.1% (5 min), and 97.4 ± 0.2% (30 min). The radiolabeling efficiency in DI water with 50 mM ascorbic acid was essentially quantitative by 1 min and remained constant over 30 min. Third, we compared how the nanoparticle-to-211At ratio affected the radiolabeling using DI water with 50 mM ascorbic acid as the reaction medium. The radiolabeling efficiency decreased as the 211At:GNS ratio increased: from 99.8 ± 0.1% (540 fmol GNS, 211At:GNS = 0.85), 99.1 ± 0.1% (54 fmol GNS, 211At:GNS = 8.5), 98.3 ± 0.1% (5.4 fmol GNS, 211At:GNS = 85), 94.1 ± 0.2% (540 amol GNS, 211At:GNS = 850), and 67.4 ± 0.2% (54 amol GNS, 211At:GNS = 8,500). The in vitro stability tests at 37°C indicated that 99.5 ± 0.3% and 99.6 ± 0.2% 211At remained on GNS after 1 h incubation in PBS and serum, respectively. After 24 h incubation, the percentage of 211At on GNS was 98.9 ± 0.2% for PBS and 99.1 ± 0.3% for serum at 37°C.
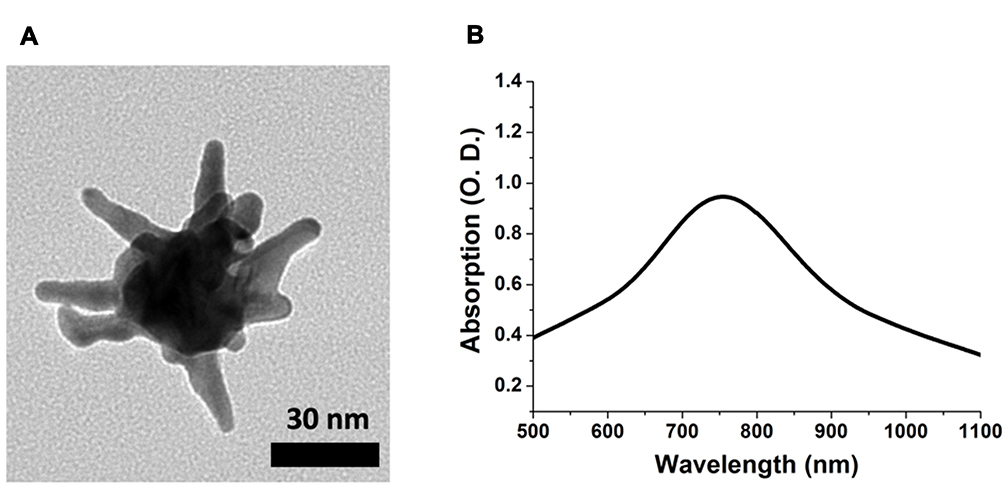 |
Figure 1 TEM image (A) and Vis-NIR absorption spectrum (B) of the synthesized GNS used in this study. |
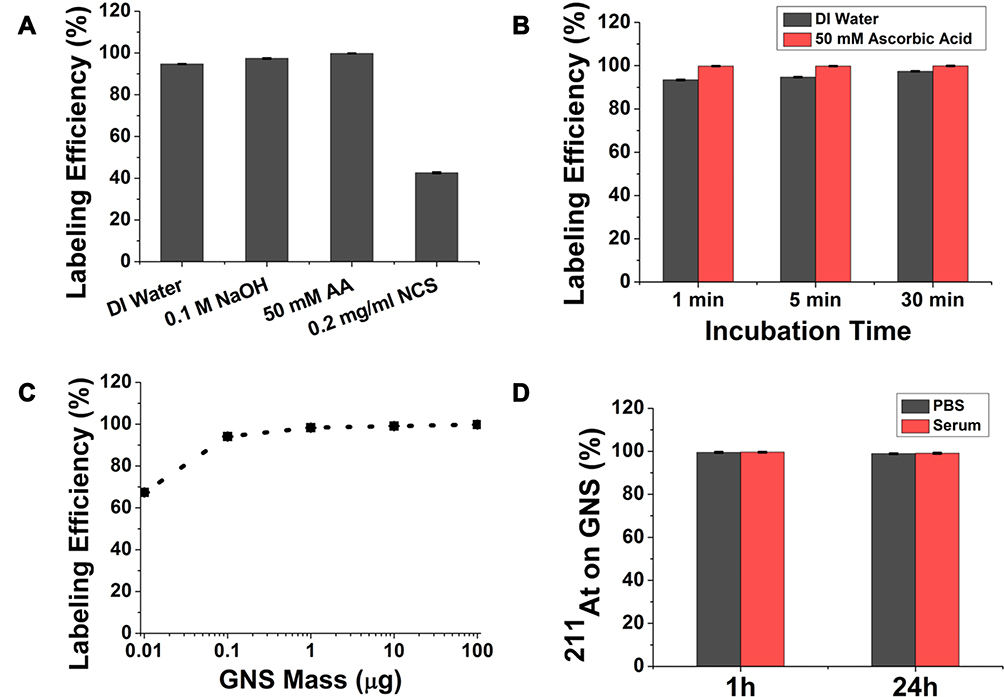 |
Figure 2 Evaluation of radiolabeling GNS with 211At. (A). Radiolabeling efficiency evaluation in different reaction media: DI water, DI water with 0.1 M NaOH, DI water with 50 mM ascorbic acid, DI water with 0.2 mg/mL NCS after a 5 min incubation. (B) Radiolabeling efficiency as a function of incubation time when the reaction was performed using DI water or DI water with 50 mM ascorbic acid as the media. (C) Radiolabeling efficiency evaluation for GNS mass ranging from 0.01–100 µg for 5 min incubation in DI water with 50 mM ascorbic acid. (D) Percentage of 211At remaining on GNS after incubation in PBS and murine serum at 37°C for 1 h and 24 h. All experiments performed in triplicate and error bar shows standard deviation. |
After evaluating the feasibility of labeling GNS with 211At, we performed a comprehensive study to investigate the biodistribution of 211At activity in normal mice after intravenous injection of 211At-labeled GNS. As shown in Figure 3, at 30 min, blood was found to have the highest concentration of 211At activity (44.4 ± 4.5%ID/g) followed by spleen (22.6 ± 1.8%ID/g), lungs (16.9 ± 2.0%ID/g), liver (9.1 ± 2.2%ID/g) and kidneys (8.1 ± 1.1%ID/g). In total, nearly 60%ID was found in the blood pool and ~13%ID was found in the liver. Activity in the blood decreased with time between 0.5 and 21 h after intravenous injection – from 44.4 ± 4.5%ID/g at 0.5 h to 40.6 ± 3.2%ID/g, 22.9 ± 4.17%ID/g and 22.0 ± 4.7%ID/g remaining at 2, 14 and 21 h, respectively. Similar behavior was observed in the lungs. In contrast, the %ID/g in spleen increased with time, from 22.6 ± 1.9%ID/g at 0.5 h, to 37.5 ± 5.9%ID/g at 2 h, 35.3 ± 4.7%ID/g at 14 h, and 105.3 ± 15.8%ID/g at 21 h; a similar trend was observed in the liver. For radiohalogens like 211At, uptake in the thyroid and the stomach can serve as an indicator of in vivo stability because of the proclivity of free halides for these tissues. The %ID in thyroid was low and did not change significantly between 0.5 and 21 h, with values of 0.61 ± 0.21%ID, 0.64 ± 0.28%ID, 0.44 ± 0.18%ID, and 0.61 ± 0.21%ID measured at 0.5, 2, 14 and 21 h, respectively. Likewise, the %ID in the stomach was low at all time points: 0.21 ± 0.06%ID at 0.5h, 0.28 ± 0.08%ID at 2h, 0.43 ± 0.14%ID at 14 h, and 0.49 ± 0.10%ID at 21 h. In summary, the biodistribution results showed that 211At-labeled GNS had a long circulation time in the blood and accumulated in the liver and spleen gradually, with a low degree of deastatination in vivo.
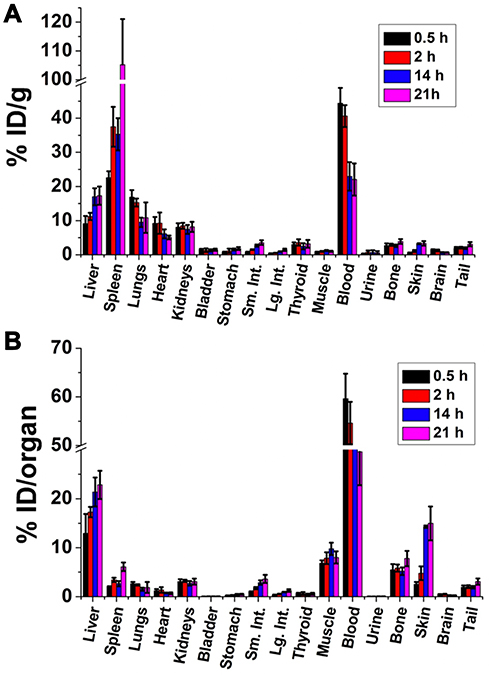 |
Figure 3 Biodistribution of 211At activity after intravenous injection of 211At-labeled GNS in normal mice. The results are shown as percent injected dose per gram tissue (%ID/g) (A), and as percent injected dose per organ (%ID/organ) (B). Error bar shows the standard deviation (n=5). |
As an initial evaluation of the therapeutic potential of 211At-labeled GNS, an experiment was performed in athymic mice with subcutaneous U87MG human glioma xenografts with the labeled drug given by the intratumoral route. As shown in Figure 4, the 211At-labeled GNS substantially inhibited the tumor growth rate. The average tumor volume in the control group increased rapidly from 82 mm3 on Day 1 to 177 mm3, 409 mm3, 798 mm3, and 1456 mm3 on Days 4, 7, 10, and 14, respectively. In contrast, the average tumor volume in the treatment group increased slowly, from 78 mm3 on Day 1 to 95 mm3, 112 mm3, 152 mm3, and 189 mm3 on Days 4, 7, 10, and 14, respectively. The two-way ANOVA test indicated that the difference in tumor volumes between the 211At-labeled GNS treated group and the control group with PBS injection was statistically significant (P < 0.001).
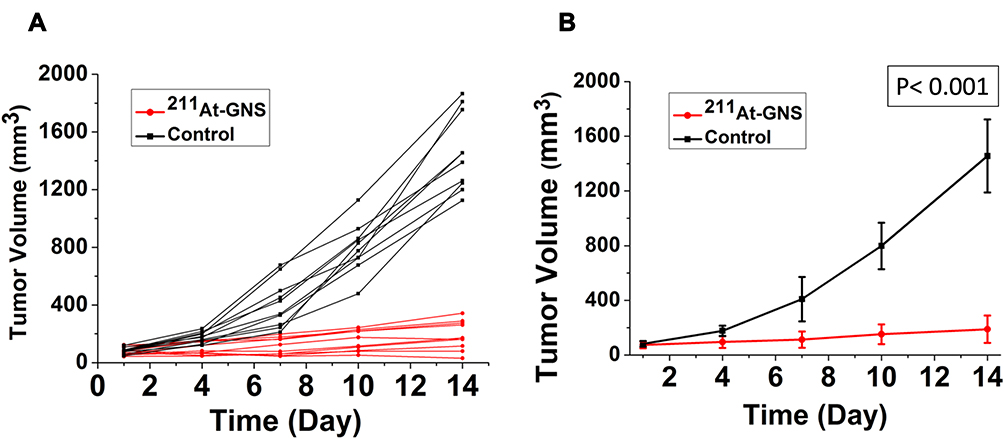 |
Figure 4 Therapeutic efficacy evaluation of 211At TAT using GNS as a novel delivery platform. (A) tumor size change profile for each mouse in the 211At-GNS treatment group (1.11 MBq) and blank control group with PBS injection. (B) Average tumor size change profile for mice in the 211At-GNS treatment group (1.11 MBq) and blank control group with PBS injection. Error bar shows standard deviation (n = 10). P value was calculated using 2-way ANOVA (P< 0.001). |
Discussion
The multibranched GNS morphology of GNS offered a high surface area for loading 211At, which should provide a very effective delivery platform. Consistent with this expectation, our developed GNS had a very promising performance as a nanoplatform for 211At delivery. First, the radiolabeling process is simple and can be completed as fast as 1 min with nearly quantitative labeling efficiency (99.8%). Moreover, the radiolabeling process can be performed without using the organic solvents or a high-performance liquid chromatography system for purification as is generally required for traditional radiolabeling methods based on At-C chemical bond formation. Second, the GNS loading capacity for 211At is high and the radiolabeling efficiency remained at ~94% when an 211At:GNS ratio of 850 was used. The radiolabeling efficiency dropped to 67.4% when we further increased the 211At:GNS ratio to 8,500, indicating that the maximum number of 211At loaded per GNS was approximately 5,700. In contrast, traditional macromolecular radiolabeling methods based on At-C chemical bond formation generally have a low conjugation efficiency: only 1 in 400 to 1,000 antibody molecules can be labeled with an 211At radionuclide.2 Third, the 211At-labeled GNS exhibited high stability and minimal in vivo dissociation. The thyroid and stomach are the two major organs that take up high amounts of free [211At] astatide. We observed a low uptake of 211At activity in the thyroid (0.44–0.61%ID) and the stomach (0.21–0.49%ID) between 0.5 h and 21 h after systemic administration of 211At-labeled GNS, suggesting a low degree of in vivo dehalogenation had occurred.
The observed high in vivo stability of 211At labeled GNS can be explained by the strong chemical bond formed between At and Au.26 The calculated bond energy was reported to be 130 kJ/mol and the oxidation state for 211At for binding to Au was considered to be −1.26 Our radiolabeling results are consistent with this; in the presence of a reducing agent, ascorbic acid, 211At labeling of GNS increased, while in the presence of an oxidizing agent, NCS, 211At labeling of GNS decreased, indicating that the oxidization state of At on GNS is −1. We plan to perform mechanistic studies to better understand our in vivo experimental data in the near future using approaches similar to those used by Teze et al.6
The in vivo stability of the 211At-labeled TAT agent is critical because dissociated 211At can result in off-targeted toxicity and decrease the radiation dose delivered to the tumor. To provide a frame of reference, we compared our in vivo stability results with those from previous studies in mice using clinically applied 211At coupling reagents including N-succinimidyl 3-trimethylstannylbenzoate (m-MeATE) and isothiocyantophenethyl-ureido-closodecaborate(2-) (B10-NCS). Using a murine model, Zalutsky et al reported the stomach uptake for an 211At-labeled antibody prepared using m-MeATE to be 2.94–4.07%ID/g between 0.5 and 24 h after IV injection with a thyroid uptake of 0.36–0.73%ID.27 In a separate biodistribution study using the same m-MeATE reagent for labeling an antibody F(ab’)2 fragment with 211At, Bäck et al reported that the stomach uptake was 4.4–12.3%ID/g during 0.5–21 h after IV injection,28 which is significantly higher than that (0.71–1.79%ID/g) for our GNS delivery platform. Orozco et al utilized the B10-NCS coupling reagent to label an anti-CD45 antibody with 211At and reported a stomach uptake of 1–2%ID/g between 1 h and 7 h, reaching ~5%ID/g at 24 h.29
We next compared our in vivo stability results with those for other 211At TAT agents moving toward clinical trial including meta-[211At]astatobenzylguanidine (211At-MABG), and N-succinimidyl 3-((1,2-bis(tert-butoxycarbonyl)guanidino)methyl)-5-(trimethylstannyl)benzoate (Boc2-iso-SGMTB). Ohshima et al reported that for 211At-MABG, stomach uptake was 2.41–6.71%ID/g and thyroid uptake was 0.61–1.65%ID between 1 and 24 h after injection.30 Choi et al utilized the Boc2-iso-SGMTB linker to label a single-domain antibody fragment (sdAb) with 211At and reported a stomach uptake of 0.5–1.7%ID/g and thyroid uptake of 0.15–0.35%ID over up to 24 h after injection.7 In addition, there are some other 211At-coupling reagents at earlier stages of development that provide data for comparison. For example, Dekempaner evaluated the N-[2-(maleimido)ethyl]-3-(trimethylstannyl)benzamide (MSB) coupling reagent in comparison to Boc2-SGMTB, m-MeATE for labeling an sdAb with 211At.31 The reported stomach uptake for 211At-labeled sdAb using MSB was more than 20%ID/g between 1 and 6 h after injection. Better in vivo stability was observed with sdAb labeled using m-MeATE, but stomach uptake was still more than 10%ID/g even at 1 h after injection. Consistent with the results of Choi et al,7 211At-labeled sdAb prepared using Boc2-SGMTB had the lowest stomach uptake among the three studied methods, with ~2%ID/g between 1 and 6 h after injection.
With regard to the in vivo stability of small organic molecule TAT agents labeled with 211At, Ohshima reported that for [211At]astatophenylalanine, stomach uptake was 0.6–2.99%ID/g during the first 6 h after injection.32 Makvandi et al evaluated the in vivo stability of [211At]MM4 (1-(4-astatophenyl)-8,9-dihydro-2,7,9atriazabenzo[cd]azulen-6(7H)-one) and stomach uptake was more than 10%ID/g within 2 h after injection.33 Liu et al radiolabeled a small-molecule peptide with 211At by using an N-succinimidyl 5-(tributylstannyl)-3-pyridinecarboxylate ester precursor and reported the stomach uptake to be 5.55%ID/g 3 h after intravenous injection.34 However, in a subsequent therapy study using this molecule, gastric toxicity was observed at higher doses.35 In summary, although different animal models were used, uptake of 211At activity in the stomach and thyroid after injection of 211At-labeled GNS is lower than that observed for most other 211At-labeled TAT agents, including those that have either been investigated clinically or will soon enter clinical trials. This suggests that the GNS delivery platform has sufficient in vivo stability for future clinical translation.
It is worth noting that GNS have other properties that may provide advantages compared with more conventional gold nanoparticle configurations, particularly when combination therapeutic strategies are contemplated. GNS have tip-enhanced plasmonics with a dramatically increased electromagnetic field near the sharp tip, making them a superior nanoplatform for certain imaging and therapy approaches. For example, we have used GNS for in vivo surface-enhanced Raman spectroscopy for cancer detection in a sarcoma murine animal model.36 In addition, we have used GNS for both in vitro and in vivo high-resolution imaging with two-photon photoluminescence. The GNS have an extremely high two-photon cross section, which is up to 4 orders of magnitude higher than that for gold nanospheres.37 As a result, we can image a single GNS under two-photon photoluminescence imaging.38 The GNS capability for sensitive optical imaging at the subcellular level makes it an attractive theranostic platform for image-guided therapy and to investigate the therapeutic efficacy of TAT with 211At. We have also used GNS for photothermal therapy and our experimental results demonstrated that GNS are superior to gold nanoshells, which are under clinical trial to treat prostate cancer with photothermal therapy.39 Therefore, GNS are an attractive nanoplatform because they can be used for combining photothermal therapy and TAT with the aim of generating synergistically effective cancer treatments.
A preliminary therapeutic efficacy study on a human GBM flank tumor murine model demonstrated that TAT therapy with 211At-labeled GNS can dramatically decrease tumor growth. With a high linear energy transfer, TAT therapy can generate irreparable DNA double-strand breaks with a therapeutic effect that is independent of tissue oxygen level, which is superior to traditional external beam radiation therapy. External beam radiation therapy is characterized as a low linear energy transfer, reparable DNA single-strand break generating radiation, with the requirement of oxygen to generate radical oxygen species for therapeutic effect. In previous studies, we radiolabeled these same GNS with 124I and performed PET/CT imaging in mice with intracranial tumors and demonstrated that the GNS could accumulate selectively in brain tumors with tumor-to-normal ratios of up to 7.8:1.9 In future studies, we plan to use a similar intracranial GBM murine animal model to evaluate the therapeutic efficacy of 211At-labeled GNS in an orthotopic setting. Finally, the developed GNS nanoplatform can also be applied for TAT to treat other types of cancer and through the use of different targeting ligands.
Conclusion
In summary, we have developed a novel 211At radiolabeling nanoplatform using biocompatible GNS and performed the first in vivo biodistribution and 211At therapy study with gold nanoparticles. Our experimental results demonstrated that the developed nanoplatform has the advantages of a simple radiolabeling process, high radiolabeling efficiency and loading capacity, minimal in vivo dissociation, and potent therapeutic effect. As a result, our novel 211At nanoplatform has a great potential to be applied for future preclinical studies and clinical translation aimed at benefiting cancer patients.
Acknowledgments
The authors would like to acknowledge the support from the US National Institutes of Health (R01EB028078) and the US Department of Defense (W81XWH1910684). The content of the information in this paper does not necessarily reflect the position or the policy of the government, and no official endorsement should be inferred.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Poty S, Francesconi LC, McDevitt MR, et al. Alpha-emitters for radiotherapy: from basic radiochemistry to clinical studies-Part 1. J Nucl Med. 2018;59(6):878–884.
2. Zalutsky MR, Vaidyanathan G. Astatine-211-labeled radiotherapeutics: an emerging approach to targeted alpha-particle radiotherapy. Curr Pharm Des. 2000;6(14):1433–1455.
3. Poty S, Francesconi LC, McDevitt MR, et al. Alpha-emitters for radiotherapy: from basic radiochemistry to clinical studies-Part 2. J Nucl Med. 2018;59(7):1020–1027.
4. Meyer GJ. Astatine. J Labelled Comp Radiopharm. 2018;61(3):154–164.
5. Zalutsky MR, Reardon DA, Akabani G, et al. Clinical experience with alpha-particle emitting 211At: treatment of recurrent brain tumor patients with 211At-labeled chimeric antitenascin monoclonal antibody 81C6. J Nucl Med. 2008;49(1):30–38.
6. Teze D, Sergentu DC, Kalichuk V, et al. Targeted radionuclide therapy with astatine-211: oxidative dehalogenation of astatobenzoate conjugates. Sci Rep. 2017;7(1):2579.
7. Choi J, Vaidyanathan G, Koumarianou E, et al. Astatine-211 labeled anti-HER2 5F7 single domain antibody fragment conjugates: radiolabeling and preliminary evaluation. Nucl Med Biol. 2018;56:10–20.
8. Yuan H, Khoury CG, Hwang H, et al. Gold nanostars: surfactant-free synthesis, 3D modelling, and two-photon photoluminescence imaging. Nanotechnology. 2012;23(7):075102.
9. Liu Y, Carpenter AB, Pirozzi CJ, et al. Non-invasive sensitive brain tumor detection using dual-modality bioimaging nanoprobe. Nanotechnology. 2019;30(27):275101.
10. Maeda H. Tumor-selective delivery of macromolecular drugs via the EPR effect: background and future prospects. Bioconj Chem. 2010;21:797–802.
11. Maeda H. The enhanced permeability and retention (EPR) effect in tumor vasculature: the key role of tumor-selective macromolecular drug targeting. Advan Enzyme Regul. 2001;41:189–207.
12. Ostrom QT, Gittleman H, Fulop J, et al. CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2008-2012. Neuro Oncol. 2015;17(Suppl4):iv1–iv62.
13. Davis ME. Glioblastoma: overview of disease and treatment. Clin J Oncol Nurs. 2016;20(5 Suppl):S2–8.
14. Mariotto AB, Yabroff KR, Shao Y, et al. Projections of the cost of cancer care in the United States: 2010-2020. J Natl Cancer Inst. 2011;103(2):117–128.
15. Stupp R, Mason WP, van den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352(10):987–996.
16. Ningaraj NS, Salimath BP, Sankpal UT, et al. Targeted brain tumor treatment-current perspectives. Drug Target Insights. 2007;2:197–207.
17. Krex D, Klink B, Hartmann C, et al. Long-term survival with glioblastoma multiforme. Brain. 2007;130:2596–2606.
18. Omuro A, DeAngelis LM. Glioblastoma and other malignant gliomas: a clinical review. JAMA. 2013;310(17):1842–1850.
19. Ostrom QT, Bauchet L, Davis FG, et al. The epidemiology of glioma in adults: a “state of the science” review. Neuro-Oncology. 2014;16(7):896–913.
20. Hartman KB, Hamlin DK, Wilbur S, et al. 211AtCl@US-Tube nanocapsules: a new concept in radiotherapeutic-agent design. Small. 2007;3(9):1496–1499.
21. Dziawer L, Koźmiński P, Męczyńska-Wielgosz S, et al. Gold nanoparticle bioconjugates labelled with 211At for targeted alpha therapy. RSC Adv. 2017;7:41024–41032.
22. Dziawer L, Majkowska-Pilip A, Gaweł D, et al. Trastuzumab-modified gold nanoparticles labeled with 211At as a prospective tool for local treatment of HER2-positive breast cancer. Nanomaterials. 2019;9:632.
23. Liu Y, Chongsathidkiet P, Crawford BM, et al. Plasmonic gold nanostar-mediated photothermal immunotherapy for brain tumor ablation and immunologic memory. Immunotherapy. 2019;11(15):1293–1302.
24. Liu Y, Maccarini P, Palmer GM, et al. Synergistic immuno photothermal nanotherapy (SYMPHONY) for the treatment of unresectable and metastatic cancers. Sci Rep. 2017;7(1):8606.
25. Feng Y, Zalutsky MR. Production, purification and availability of 211At: near term steps toward global access. Nucl Med Biol. 2021;100:12–23.
26. Demidov Y, Zaitsevskii A. Adsorption of the astatine species on a gold surface: a relativistic density functional theory study. Chem Phys Lett. 2018;691:126–130.
27. Zalutsky M, Stabin MG, Larsen RH, et al. Tissue distribution and radiation dosimetry of astatine-211-labeled chimeric 81C6, an α -particle-emitting immunoconjugate. Nucl Med Biol. 1997;24:255–261.
28. Bäck T, Andersson H, Divgi CR, et al. 211At radioimmunotherapy of subcutaneous human ovarian cancer xenografts: evaluation of relative biologic effectiveness of an α-Emitter in vivo. J Nucl Med. 2005;46(12):2061–2067.
29. Orozco JJ, Bäck T, Kenoyer A, et al. Anti-CD45 radioimmunotherapy using 211At with bone marrow transplantation prolongs survival in a disseminated murine leukemia model. Blood. 2013;121(18):3759–3767.
30. Ohshima Y, Sudo H, Watanabe S, et al. Antitumor effects of radionuclide treatment using α-emitting meta-211At-astato-benzylguanidine in a PC12 pheochromocytoma model. Eur J Nucl Med Mol Imaging. 2018;45:999–1010.
31. Dekempeneer Y, Bäck T, Aneheim E, et al. Labeling of anti-HER2 nanobodies with astatine-211: optimization and the effect of different coupling reagents on their in vivo behavior. Mol Pharm. 2019;16:3524–3533.
32. Ohshima Y, Suzuki H, Hanaoka H, et al. Preclinical evaluation of new α-radionuclide therapy targeting LAT1: 2-[211At]astato-α-methyl-L-phenylalanine in tumor-bearing model. Nucl Med Biol. 2020;90–91:15–22.
33. Makvandi M, Lee H, Puentes LN, et al. Targeting PARP-1 with alpha-particles is potently cytotoxic to human neuroblastoma in preclinical models. Mol Cancer Ther. 2019;18:1195–1204.
34. Liu W, Ma H, Tang Y, et al. One-step labelling of a novel small-molecule peptide with astatine-211: preliminary evaluation in vitro and in vivo. J Radioanal Nucl Chem. 2018;316:451–456.
35. Liu W, Tang Y, Ma H, et al. Astatine-211 labelled a small molecule peptide: specific cell killing in vitro and targeted therapy in a nude-mouse model. Radiochim Acta. 2021;109(2):119–126.
36. Liu Y, Ashton JR, Moding EJ, et al. A plasmonic gold nanostar theranostic probe for in vivo tumor imaging and photothermal therapy. Theranostics. 2015;5(9):946–960.
37. Gao N, Chen Y, Li L, et al. Shape-dependent two-photon photoluminescence of single gold nanoparticles. J Phys Chem C. 2014;118(25):13904–13911.
38. Liu Y, Huang W, Xiong C, et al. Biodistribution and sensitive tracking of immune cells with plasmonic gold nanostars. Int J Nanomedicine. 2019;14:3403–3411.
39. Rastinehad AR, Anastos H, Wajswol E, et al. Gold nanoshell-localized photothermal ablation of prostate tumors in a clinical pilot device study. Proc Natl Acad Sci USA. 2019;116:18590–18596.
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.