")
Back to Journals » International Journal of Nanomedicine » Volume 15
Gold Nanoparticles Mediated Drug-Gene Combinational Therapy for Breast Cancer Treatment
Authors Shrestha B, Wang L, Zhang H, Hung CY , Tang L
Received 25 April 2020
Accepted for publication 29 July 2020
Published 21 October 2020 Volume 2020:15 Pages 8109—8119
DOI https://doi.org/10.2147/IJN.S258625
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Thomas Webster
Binita Shrestha,1 Lijun Wang,1 Hao Zhang,2 Chiung Yu Hung,2 Liang Tang1
1Department of Biomedical Engineering, The University of Texas at San Antonio, San Antonio, TX, USA; 2Department of Biology, The University of Texas at San Antonio, San Antonio, TX, USA
Correspondence: Liang Tang
Department of Biomedical Engineering, The University of Texas at San Antonio, One UTSA Circle, San Antonio, TX 78249, USA
Tel +1 210-458-7995
Email [email protected]
Background: Cancer is a complex heterogeneous disease to which singular modes of treatment mostly fail to produce a desired therapeutic efficacy. Targeting different cellular pathways using combinational therapies has been gaining popularity in cancer treatment, with the added benefit of reducing dosage and side effects.
Methods: A gold nanoparticle-mediated drug delivery nanoplatform was developed for co-delivery of doxorubicin and polo-like kinase 1 (PLK1) siRNA. Gold nanoparticles were coated with polyethyleneimine to facilitate assembly of PLK1 on the surface. Doxorubicin was loaded on nanoparticles through a pH-sensitive linker with a thiol group at one terminal end for controlled release.
Results: The therapeutic efficiency of this co-delivery system was evaluated in 2D and 3D cultured systems. The reduced IC50 value clearly demonstrated the synergistic effect of combined drug and gene delivery over their individual delivery in a cancer treatment model.
Conclusion: This study may provide an adaptable, facile platform to investigate drug-siRNA combinations for cancer inhibition.
Keywords: nanomedicine, cancer therapy, pH-responsive, co-delivery, PLK1
Introduction
Cancer therapy has been a major public health challenge due to the heterogeneity and complexity of cancer cells. Despite significant advances in cancer biology in the past few decades,1 the efficacy of current cancer treatment strategies is still not ideal. Breast cancer is the most common cancer in women. Due to early diagnosis, the survival rates of breast cancer have improved in recent years; however, it is still the most common form of cancer in women, and the second leading cause of death in women, surpassed by lung and bronchus cancer.2 Most patients receiving single therapies succumb to cancer due to dose-associated toxicity and multidrug resistance.2,3 Targeting multiple cellular pathways for cancer treatment has shown positive results. Combining multiple therapies that target various cellular pathways has demonstrated a synergistic or additive effect in cancer treatment by increasing the genetic barrier, minimizing side effects, and reducing therapeutic doses.4,5 The combination of chemotherapy and gene therapy has shown a synergistic effect against various cancer types, including breast cancer.6,7 Chemotherapy is the first line of treatment for cancer, as indicated by ample chemotherapeutic drugs available for almost all kinds of cancer types. On the other hand, mutations and genetic instability are the basis of the onset of cancer. Therefore, gene therapy has a high potential for use in cancer treatment via the modulation of the genes responsible for cancer. Combining chemotherapy and gene therapy, hence, would be an ideal approach for cancer treatment.
Recent studies have demonstrated the efficacy of combinational therapy over monotherapy in various cancer types. Regardless of therapies being combined, it is crucial that the therapeutic agents are delivered to the same diseased site. For instance, Su et al showed enhanced doxorubicin (DOX) accumulation at the diseased site with the aid of tumor necrosis factor α (TNFα). However, since TNFα and DOX were delivered separately, TNFα leakage into circulation resulted in DOX accumulation in the liver, indicating that co-delivery or simultaneous delivery is desired in chemo-gene combination therapy.8 Nanotechnology-based drug delivery systems can allow for co-delivery or simultaneous delivery of multiple therapeutic agents. Additionally, nanotechnology has enhanced existing drug delivery systems through its high drug loading surface area and its passive targeting capabilities. Multiple types of nanoparticles have been investigated for applications such as drug delivery, imaging, photothermal therapy, and electroporation, and tissue engineering9–14 etc. As such, we developed a gold nanoparticle-based carrier vehicle for the co-delivery of drug and siRNA (Figure 1). DOX, representing chemotherapy, is one of the most common breast cancer therapeutic agents that work by intercalating DNA. Polo-like Kinase 1 (PLK1) siRNA represents gene therapy, down-regulation of PLK1 protein has been reported to inhibit cancer progression.17,18 The nano-delivery system and the synergistic effect of the drug/siRNA co-delivery were systematically characterized and investigated in 2D/3D cultured tumor microenvironments.
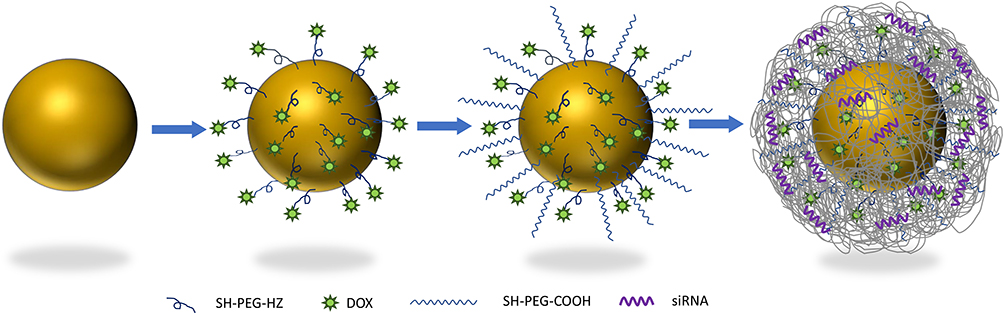 |
Figure 1 Schematic representation of nanocomplex. |
Materials and Methods
Cell Culture
SKBR3 (ATCC® HTB-30™) and HEp-2 (ATCC® CCL-23™) cell lines were obtained from American Type Culture Collection (ATCC). HEp-2 and SKBR3 were cultured in EMEM and Mc-Coy culture media, respectively, at 37°C supplied with 5% CO2 and 95% humidity. Both culture media were supplemented with 5% FBS and 1% penicillin streptomycin.
Synthesis and Characterization of PEI-Coated Gold Nanospheres
Gold nanospheres (GNS) were synthesized using the Turkevich method.19 Briefly, 20 mL of gold chloride solution (Sigma-Aldrich Co., St Louis, MO, USA) was heated under continuous stirring until it started to boil. A total of 2.2 mL of 1% sodium citrate was then added to the solution. As the solution turned ruby red, it was cooled down to room temperature and then filtered. GNS was stored at 4°C until use. GNS was characterized using UV-Vis spectrophotometry, a Zetasizer,
DLS (dynamic light scattering) for particle sizing, and scanning electron microscopy.
Synthesized GNS were then functionalized with SH-PEG-COOH (Mw 5000, JenKem Technologies). For every 1 mL of poly (ethylene glycol) (PEG) (1mM), 9 mL of GNS was added to make the final concentration of 1 nM. The solution was stirred for 2 hours at room temperature and then centrifuged at 7000 g for 20 minutes. The pellet was collected and re-dispersed in water. The pH was adjusted to 4.5–5.5. The carboxyl group was activated using EDC and NHS at the ratio of 4:1 for 30 minutes. To this mixture, 300 µL of 2 mg/mL polyethyleneimine (PEI) (Sigma-Aldrich Co., St Louis, MO, USA) solution was added and further incubated for 2 hours. The pellet was collected after centrifugation at 3000 rpm for 5 minutes and re-dispersed in RNAse and DNAse-free water for storage. PEI-functionalized GNS was characterized using spectrophotometry and DLS.
PEI Quantification and siRNA Assembly
The copper sulfate complexion method reported by Ungaro et al was used for PEI quantification.20 First, 0.05 mg/mL of copper (II) sulfate in acetate buffer was added to a PEI solution in the range of 0–4 mg/mL at a 1:1 ratio. The absorbance at 285 nm was recorded to generate a standard curve. Similarly, 100 µL of 0.05 mg/mL of copper (II) sulfate solution was added to 100 µL of PEI-coated nanoparticles (1nM). The absorbance reading at 285 nm was used to calculate the approximate PEI amount conjugated to GNS. The amount of N present in PEI was further used to calculate N/P ratio for siRNA conjugation. Thus, prepared GNS-PEI was mixed with siRNA at various N/P ratios for 30 minutes at 4 ͦC.
Cytotoxicity Analysis
The cytotoxicity of nanocomplex was evaluated using CellTiter-Glo® assay. Cells were seeded in 24 well plates at the cell density of 1x103 cells. Bare GNS, PEG-GNS, PEI-GNS, and PEI only were added to the cells at the final concentration of 0.2nM. The cell viability was measured after a period of 72 hours for both cell lines. At each time point, the media was replaced by fresh media followed by the addition of CellTiter-Glo® solution at the ratio of 1:1. The plates were kept on the shaker for 2 minutes and left undisturbed for 10 minutes as mentioned in the protocol. A Synergy ™ 2 multi detection microplate reader from BioTek Inc was used to measure chemo-luminescence of the samples. All experiments were performed in triplicate.
Study of siRNA Release and Gene Silencing
5-Carboxyfluorescein (FAM)-tagged siRNA was assembled on the surface of PEI-GNS to study the siRNA release. These nanoparticles were mixed with siRNA 30 minutes before adding to the cells. Cells were seeded in 48 well plates at a density of 0.25x105. The cells were incubated with siRNA-GNS-PEI for 24 hours. siRNA release to the cells was observed using fluorescence microscopy. Lysotracker red stain for acidic organelles was used to evaluate the uptake of nanoparticles through endocytosis (Figure S1). Co-localization of green (FAM) and red signal indicates the presence of nanoparticles within endosomes/lysosomes. To evaluate the silencing efficiency of the nanocomplex, the cells were pre-transfected with plasmid GFP using lipofectamine following the manufacturer protocol. Briefly, cells were seeded at a density of 0.25x106 in 24-well plates. A mixture of pGFP and lipofectamine 3000 was added to cells. After 6 hours, GFP siRNA conjugated nanoparticles at the concentration of 80 nM, and 100nM was added to the cells and further incubated for 24 hours. The cells were then collected, washed, and re-dispersed in PBS with 0.1% FBS for flow cytometry analysis.
PLK1 Silencing
For PLK1 silencing, PLK1 siRNA was assembled to GNS following the previously mentioned protocol. The cells were incubated with PLK1-siRNA conjugated nanocomplexes for 24 hours. The cells were washed with ice-cold PBS and lysed using RIPA buffer containing 1% protease inhibitor. The lysate was centrifuged at 12,000 g for 30 minutes and the supernatant was collected. Total protein concentration was determined using a BCA assay. Western blot analysis was performed to visualize the PLK1 protein in the sample. Briefly, protein samples were mixed with loading buffer and loaded to 12% polyacrylamide gel. The gel was run for 90 minutes at 120 mV. The proteins were transferred to a PVDF membrane using a semi-dry method. After transfer, the PVDF membrane was kept in a blocking buffer overnight. An iBind system was used for antibody binding and washing steps following the manufacturer’s instruction. The membrane was rinsed with PBS with Tween and distilled water followed by the addition of an ECL solution. The membrane was taped to the cassette and developed.
Doxorubicin Conjugation
For the covalent binding of DOX, 4x10 −5 moles of polyethylene-hydrazine (PEG-HYZ) were added to 100 mL of GNS in the dark. After 24 hours, the mixture was centrifuged and washed several times with water to obtain GNS-HYZ. Then, 25 mg of DOX was added to 25 mL of GNS-HYZ followed by the addition of 10 µL sodium cyanoborohydride (5M in 1N Sodium hydroxide).21 The mixture was continuously stirred at 50°C for 24 hours.22 DOX-GNS was extensively dialyzed against PBS for 36 hours. The PBS was exchanged once every 12 hours.
Determination of pH-Sensitive Release
A dialysis cassette was used to determine the drug release kinetics of DOX-GNS. Three buffers with pH 2, pH 5.5, and pH 7.4 to mimic acidic pH, endosomal/lysosomal pH, and physiological pH were used for the evaluation of drug release. For this, 0.1 mL of DOX-GNS was added to each 3 mL buffer solution with varying pH. The cassette was submerged in PBS under continuous stirring. At each time point, 5 mL of PBS was taken and replaced with fresh PBS. The fluorescence intensity of DOX was measured at 485 nm using a multiplate reader. The concentration of DOX was determined by the DOX standard curve. All the experiments were performed in triplicate. The cumulative release of DOX was calculated using the following formula:
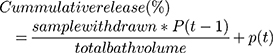
where P is percentage and t is time.
Synthesis of Combinational Nanocomplex
Twenty milliliters of DOX-conjugated GNS was first coated with PEG-COOH (100 µM) for 1 hour. The solution was centrifuged at 5000 rpm for 15 minutes. The pellets were collected and re-dispersed in the RNAse-free water. The pH of the GNS solution was adjusted to 4.5. Three hundred microliters of EDC and NHS in the ratio 4:1 was added to the 10 mL of DOX- PEG-GNS to activate the carboxyl group followed by the addition of 3 mL of PEI (2 mg/mL). The mixture was stirred at room temperature for 2 hours and centrifuged to remove unbound PEI. These nanocomplexes were incubated with PLK1 before cellular experiments.
3D Cell Culture
Cancer cell spheroids were cultured using an aqueous two-phase system as reported by Ham et al.23 Two polymers, PEG and dextran (DEX), were used to obtain two immiscible aqueous phase systems. A total of 0.5 g of PEG was dissolved in 9.5 mL of complete cell culture media. Different percentages of DEX (1%, 3%,5%,7%,9%, 11% and 15%) were prepared. Both PEG and DEX phase was kept at 37°C for 1 hour and were filtered before use. Round bottom 96 well plates were used for 3D culture. These plates were coated with 1% pluronic 1 day before spheroid culture. The cells were then harvested, and the concentration was adjusted to 1x107 cells per mL. These cells were mixed with DEX at a 1:1 ratio. A total of 50 µL of the PEG phase was added to each well in 96 well plates, to which 0.5 µL of cell and DEX mixture was dropped carefully. The plates were then stored at 37°C in presence of 5% CO2 under continuous shaking. The spheroids were imaged every alternate day using a brightfield microscope. For live-cell staining, these spheroids were stained with calcein for 30 minutes and imaged with a fluorescence microscope. The diameter of the spheroids was analyzed by ImageJ software.
To evaluate the therapeutic efficacy of combinational therapy in the 3D culture system, cell viability was determined by CellTiter-Glo® 3D cell viability assay. First, the spheroids were incubated with different nanocomplexes (DOX, DOX-GNS, PLK1-GNS, and DOX-PLK1-GNS) for various time points (24, 48, 72, and 96 hours). Following treatment, the spheroids were incubated with CellTiter-Glo® for 1 hour at room temperature. The luminescence was measured using a BioTek multiplate reader.
Statistics
Student’s t-test was performed to compare two groups whereas two-way ANOVA was used to compare multiple groups followed by post hoc Tukey’s test. GraphPad Prism was used for data analysis. The groups were considered significantly different for P < 0.05 unless otherwise stated. All the data are represented Mean ± SD unless otherwise stated.
Results
Synthesis and Characterization of PEI-GNS
Mono-dispersed GNS and subsequent surface modifications were stable in suspension as demonstrated by the UV-Vis spectrum and zeta potential value (Figure 2A and B). The surface plasmon peak and zeta potential values were 521 nm and −36 ± 5 mV, respectively. These nanoparticles were 12 nm in diameter as determined by SEM (Figure 2C). Following conjugation with SH-PEG-COOH and PEI, the peak absorbance shifted by 1–2 nm towards the upper part of the spectrum consistent with gold nanoparticle functionalization. To further confirm conjugation, zeta potential value was obtained at each conjugation step. The zeta potential value for PEG-GNS, PEI-PEG-GNS (unactivated), and PEI-PEG-GNS (activated) was found to be 0 ± 4 mV, 2 ± 3mV and 44 ± 4 mV, respectively (Figure 2B). An increase in zeta potential value from −36 ± 5 mV to 0 ± 4 mV confirms the conjugation of PEG on the surface of GNS. Further conjugation of cationic polymer PEI increased the zeta potential value to 44 ± 4 mV. The carboxyl group in the PEG linker was activated using EDC-NHS chemistry to conjugate PEI through electrostatic bonding. The minimal positive surface charge was due to nonactivated-COOH and resulted in lower conjugation of PEI. In comparison, activated-COOH resulted in higher efficiency of PEI conjugation as demonstrated by the increase of zeta-potential value. This result confirms that PEI is conjugated via the PEG linker. Hence, this nanocomplex is a core-shell structure where PEI shell is linked to the gold core via PEG linker as demonstrated in the schematic diagram (Figure 1). The hydrodynamic size of the nanoparticles was evaluated using DLS, which was 37 ± 26 nm and 164 ± 14 nm for PEG-GNS and PEI-PEG-GNS, respectively.
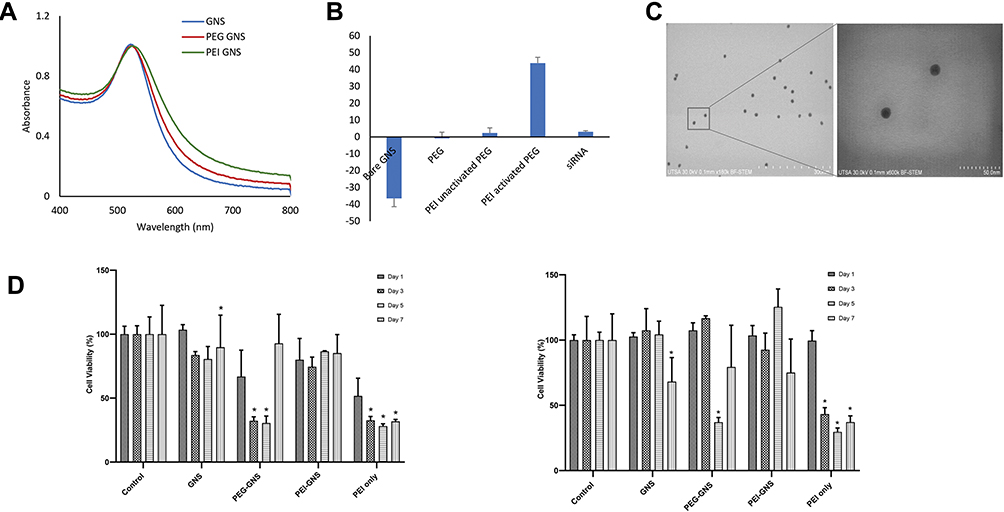 |
Figure 2 GNS characterization. (A) UV-Vis spectrum before and after functionalization. (B) Zeta potential measurement. (C) SEM images. (D) Cell viability assay HEP2 (left) and SKBR3 (right) over the period of 7 days (n=3, Mean ±SEM, *P<0.05). |
Cytotoxicity Evaluation
CellTiter-Glo® assay was used to assess the cytotoxicity of these nanocomplexes at 1,3,5, and 7 days in HEp-2 and SKBR3 cells (Figure 2D). While PEI is widely used for siRNA/DNA delivery, its application has been limited by its significant toxicity. PEI alone demonstrated reduced cell viability in both HEp-2 and SKBR3 cell lines with increased incubation periods. Interestingly, PEG-GNS demonstrated more toxicity in day 3 and 5 HEp-2 cells and day 5 in SKBR3 cells. As PEG is considered to be biocompatible, this toxicity of PEG-GNS can be attributed to the carboxyl group.24 Uncharged COOH crosses the cell membrane easily, as they are lipid-soluble. Inside the cells, COOH becomes charged in the presence of H+ ions and tends to accumulate in cells resulting in acidification. SKBR3 is a metastatic cancer cell line and therefore has a high tendency to extrude H+ ions in comparison to HEp-2 cell line.25 This may have resulted in reduced cell viability in HEp-2 and not in SKBR3. However, no apparent toxicity was observed in both cell lines when treated with PEI-PEG-GNS. This result was further corroborated by necrotic and apoptotic cell analyses performed on Day 1 and Day 7 (Figures S2 and S3). This was because the amount of PEI was minimal to facilitate siRNA assembly without eliciting significant toxicity.
siRNA Release
Binding affinity is an important parameter in gene delivery systems as it affects the gene release efficiency at the targeted site. While it is ideal that the carrier vehicle easily releases its payload at the desired site, it is equally important to prevent non-specific leakage during the transport from the injection site to the target area. Therefore, optimal binding efficiency is desired for any gene delivery application. To optimize the binding affinity, siRNA was assembled to the GNS-PEI surface in various N/P ratios (5:1, 10:1, 15:1, 20:1, and 25:1). The UV-Vis spectrum after siRNA conjugation and zeta potential value corresponding to each N/P ratio is shown in Figure 3A and B, respectively. The amount of PEI in GNS-PEI was determined by the spectrophotometric method using copper conjugation. As shown in Figure 3C, the minimal nucleic acid release occurred at 20:1 and higher N/P ratio whereas excessive release that is comparable to the control group occurred at 5:1 and 10:1 ratio. However, the optimal release of siRNA was observed at the ratio of 15:1. Previous studies demonstrated that the surface charge of nanoparticles affects the cellular uptake of the nanoparticle.26–28 The zeta potential value of the N/P ratio 15:1 showed −14 ±1mV which is optimal for our application. Although a cationic surface charge results in higher nanoparticle internalization, it shows significant toxicity potential. Therefore, a slight negative charge of the functional gold nanoparticle is desirable for drug delivery.
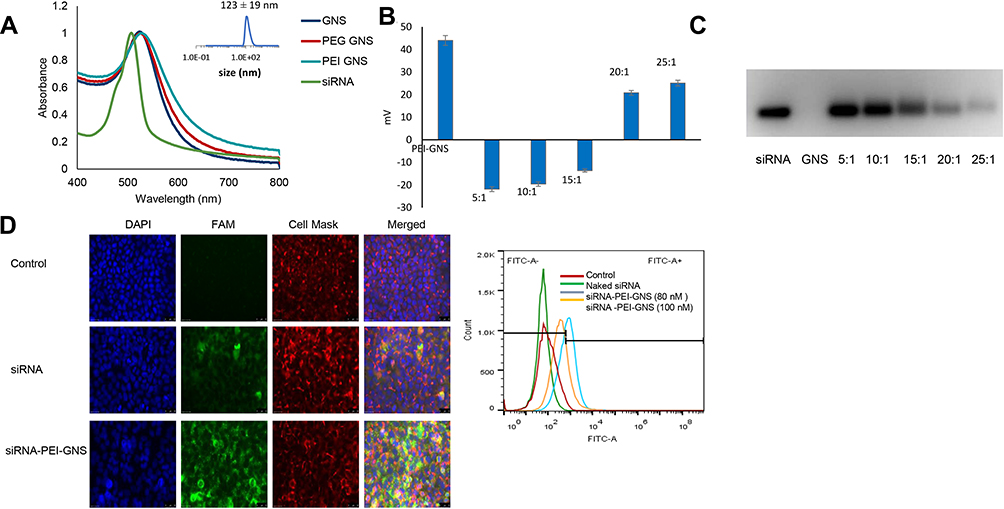 |
Figure 3 siRNA release study. (A) UV-Vis spectrum of siRNA conjugation. (B) Zeta-potential measurement of different N/P ratios. (C) Gel-electrophoresis to optimize siRNA release. (D) Florescence image and flow cytometer analysis was used to examine siRNA release. Naked FAM-siRNA resulted in lower signal intensity whereas FAM-siRNA conjugated to GNS resulted in higher signal intensity. |
FAM-tagged siRNA was used to evaluate siRNA release in the cells for 24 hours. The green fluorescence signal in Figure 3D clearly depicts the degradation of naked siRNA in the cells. However, siRNA complexed with GNS-PEI was held intact during the delivery. Both 80 nM and 100 nM concentration showed siRNA release; however, no significant difference was observed between these two concentrations. Therefore, we selected 100 nM concentration for our further studies.
PLK1 is an enzyme that belongs to Serine/threonine kinases which play an important role in regulating various cell cycle events such as mitotic signaling, centrosome maturation, and DNA damage in almost all cell lines. One of the primary functions of PLK1 is to regulate mitotic checkpoints to ensure genetic stability.29 A wide range of cancers including breast cancer have demonstrated a correlation between PLK1 expression and carcinogenesis.30 As a result, its expression level is used as a measure of tumor severity. Silencing PLK1, therefore, has shown a promising anti-proliferative response. Western blot results of the PLK1 silencing study showed reduced PLK1 protein expression in both HEp-2 and SKBR3 cell lines as shown in Figure 4A. The anti-Plk1 blot of HEp2 cells showed doublet bands. This raised a potential protein degradation concern. However, all treated, and control protein samples were separated on a duplicated Coomassie blue-stained gel that showed uniformed band patterns without any signs of protein degradation. The anti-actin blot showed a sharp band pattern for all samples. Furthermore, the lower band showed consistent density for all samples and it was not inhibited by the siRNA treatment. We concluded that the lower band was a non-specific interacting protein.
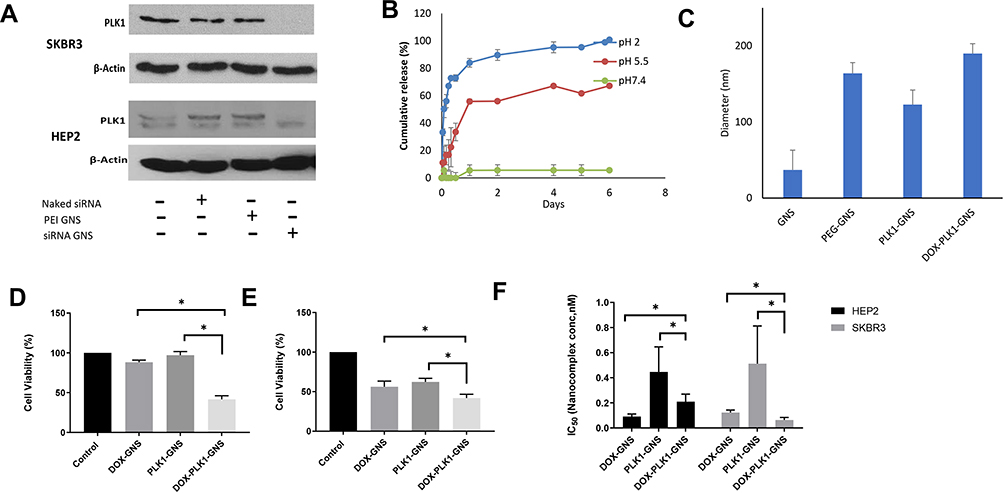 |
Figure 4 (A) Western blot analysis to determine PLK1 protein expression. (B) pH-responsive release of DOX. (C) Size analysis of nanoconjugates following siRNA and DOX conjugation. Cell viability of (D) SKBR3 and (E) HEp2 after 48 hour of incubation with combinational particle. (F) IC50 value for comparison between combinational and single therapy for HEP2 and SKBR3 cell lines. (n=3, Mean ± SEM, *P<0.05). |
Acid Triggered Drug Release Study
Controlled drug release is one of the most desirable features in drug delivery applications, particularly in a complex disease like cancer where non-specific release poses a significant threat to normal cells. The tumor microenvironment is slightly acidic in comparison to its normal environment. This acidic nature is primarily due to glycolytic metabolism, hypoxia, and inefficient blood perfusion in tumors. Stimuli-responsive drug release in the acidic environment provides further assurance for site-specific delivery to cancerous cells. The presence of H+ ions leads to an increased hydrolysis rate which eventually results in the DOX release from the nanocomplex as indicated in Figure 4B. About 67% of DOX was released at pH of 5.5 whereas neutral pH value showed only 5% release over 6 days. Furthermore, 33% DOX release at pH 2.0 and 11% of DOX release at pH 5.5 over the period of 24 hours corroborate sustained release profile at a mildly acidic condition in comparison to higher or lower pH.
Antitumor Effect in Cancer Cells
The antitumor effect of combinational therapy was investigated using cell viability assay and its efficacy was compared to a single therapy. Four different treatment groups (ie, free DOX, DOX-GNS, PLK1-GNS, and DOX-PLK1-GNS) in both HEp-2 and SKBR3 cell lines were systematically investigated. The size analysis of PLK1-GNS was 123±19 nm and after DOX-PLK1-GNS was 190 ± 13 nm (Figure 4C). Cells without any treatment were used as the negative control group. As shown in Figure 4D and E, all treatment groups demonstrated effective cell inhibition as compared to the control group for both cell lines. However, DOX-PLK1 conjugated GNS exhibited significantly higher anti-tumor efficacy among all the treatment groups (P < 0.05). The therapeutic efficacy of nanocomplex was further evaluated in terms of IC50 value (Figure 4F). The IC50 value was calculated using GraphPad Prism software and summarized in Table 1. The IC50 value is represented in terms of GNS concentration. The IC50 value was much lower for DOX-PLK1-GNS than DOX-GNS and PLK1 GNS for both HEp-2 and SKBR3 which indicate the synergistic effect of combinational therapy over chemotherapy or gene therapy alone. This result may have been caused by increased sensitivity of cells against given treatment. PLK1 silencing has reported to sensitize cells against DOX drug. Any change in PLK1 expression level can have a direct effect on various cell cycle regulating events. Deregulated cell cycle events as a result of PLK1 silencing augments DOX treatment, which eventually improves the therapeutic efficacy of combinational therapy. Similarly, 48-hour cell viability of different treatment groups further confirmed the synergistic effect of DOX-PLK1-GNS over other treatment groups as shown in Figure 4D and E
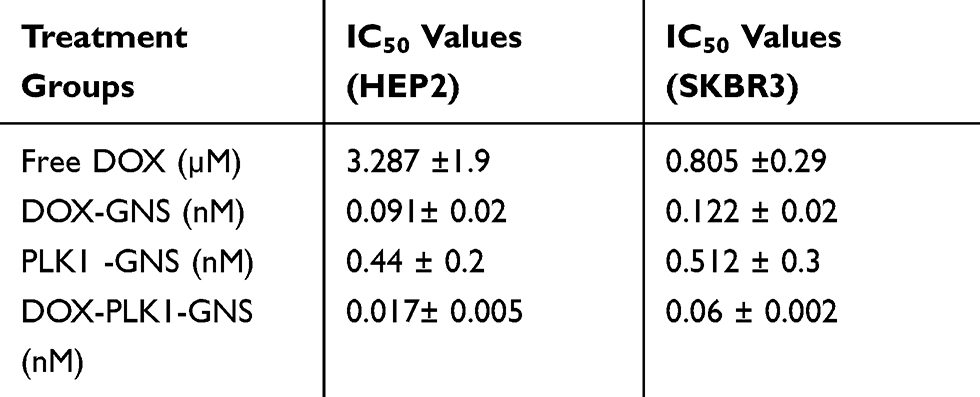 |
Table 1 IC50 Values for Different Treatment Groups |
3D Tumor Spheroids
Although the 2D cell culture system is popular to evaluate the therapeutic efficacy of a drug delivery system, the results may not accurately correlate with an actual model. This is mainly due to the limited capability of 2D system to mimic the complexity of solid tumors in the physiological environment. Limited cell–cell interaction, the large surface area exposed to the given treatment, and cell–matrix interaction in 2D monolayer system affect the predictability of therapeutic outcome in in-vivo studies, which could be very labor-intensive and expensive. 3D spheroids, as a closer approximation of actual solid tumor to mimic an in-vivo study, have been widely used to bridge the gap between 2D system and animal models. 3D spheroids resemble solid tumors in many similar ways such as cell–cell interaction, presence of quiescent and necrotic cell population, limited oxygen and nutrients to the necrotic core, and so on. To culture 3D spheroids, aqueous two-phase system (ATPS) based on two immiscible biocompatible polymers was used.18 Cells were mixed with the dextran (DEX) phase and dropped into PEG solution in round bottom 96 well plate. Bright-field images of spheroids cultured at a different percentage of DEX are shown in Figure S4. Figure 5A shows the size and volume of 3D spheroids increased with the increase in the DEX percentage. Nine percent of DEX resulted in more spherical spheroid formation between 3% and 9%. However, 11% and 15% formed loose aggregates for more than 50% of the wells. Figure 5B indicates normal size distribution and the average diameter of spheres to be 737±12 µm. Live-cell staining was performed to evaluate the viable cells after spheroids were formed (Figure 5C), which confirmed the cell viability by fluorescence detection.
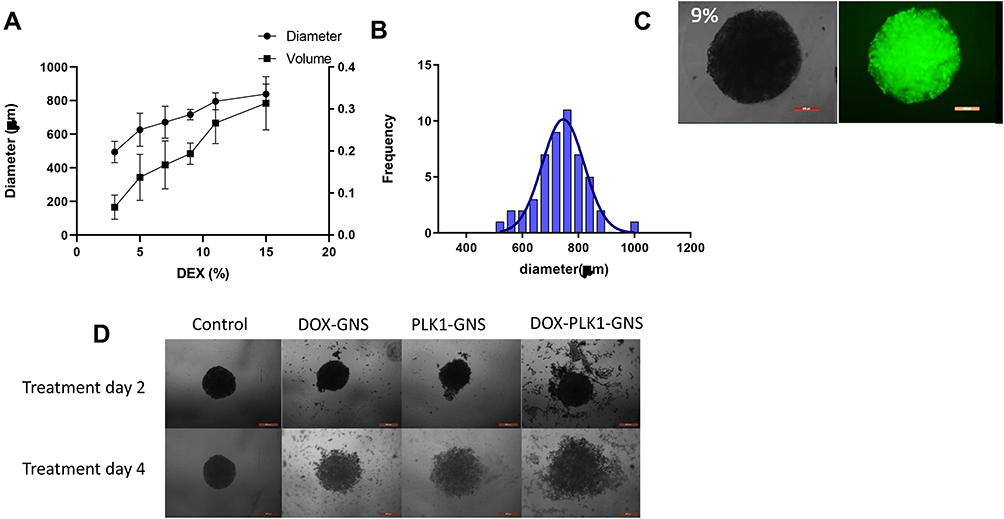 |
Figure 5 Evaluation of therapeutic efficiency in 3D spheroids. (A) Size and volume analysis of 3D spheroids cultured at different percentage of dextran. (B) 3D spheroids demonstrated normal size distribution. (C) Brightfield and florescence images of spheroids. The spheroids were stained with calcein to evaluate the live cells in the spheroids. (D) Bright field images of spheroids treated with DOX-GNS, PLK1-GNS, DOX-GNS and AH-DOX-GNS. |
Synergistic Effect of Combinational Nanoparticles
The spheroids were treated with DOX-GNS, PLK1-GNS, and DOX-PLK1 GNS, respectively, for 24 and 48 hours to evaluate the therapeutic efficacy of combinational vs single therapy. Spheroid incubated with culture media without nanoparticles was used as a negative control. We did not observe any inhibition in cell viability after 24 hours of treatment. Further incubation of spheroids for 48 hours showed some inhibition in cell viability; however, the results were insignificant. The spheroids were then incubated with nanocomplex for 4 days to evaluate the IC50 value. The IC50 value was found to be 0.122 ± 0.02 nM, 0.512 ± 0.3 nM, and 0.06 ± 0.002 nM for DOX-GNS, PLK1-GNS, and DOX-PLK1-GNS, respectively, for SKBR3 spheroids. Reduction in IC50 value for combinational therapy indicated a synergistic effect of combinational therapy. Figure 5D shows the bright-field images of spheroids treated with different treatment groups.
Discussion
As drug discovery and development costs have skyrocketed in the past few decades, it is clinically relevant to explore novel approaches to effectively improve the efficacy of currently available cancer therapeutic reagents. Although there are ample drugs available in the market for a variety of cancers, major challenges such as low bioavailability, multi-drug resistance (MDR), non-specific delivery, and dose-dependent side effects limit their therapeutic efficacy. DOX is a common anti-mitotic drug used for the treatment of a broad range of cancers. However, the full therapeutic potential of DOX is hindered due to its severe dose-associated side effects such as significant cardiac damage. Often, a high injection dose is required to achieve a therapeutic threshold at the diseased site due to the low bioavailability of drugs. This results in MDR development which is another major obstacle in cancer therapy. Therefore, targeted delivery of DOX at the tumor site is highly desirable. On the other hand, siRNA-based therapy is another powerful tool to control cancer cell proliferation and survival. A correlation of PLK1 overexpression and cancer has been established and many studies have reported anti-proliferative response of PLK1 silencing in various types of cancer.27,29,30 Furthermore, PLK1 silencing has reported to sensitize the cells against DOX, thereby reducing the therapeutic systemic dose. In the present study, genes and drugs were co-delivered to improve the therapeutic efficacy of nanocomplex. We hypothesized that PLK1 protein suppression in combination with the anti-mitotic effect of DOX would result in a synergistic effect conferring the improved therapeutic efficiency of combinational therapy over single therapy. The controlled release system is ideal to minimize dose-dependent side effects. Cytocompatibility largely depends on PEI, although considered gold standard for gene delivery, present a significant viability risk to cells. Our designed systems utilize PEI to attach siRNA at a minimal level without triggering toxic effect in the cells. Necrotic and apoptotic cell analysis over the period over 7 days indicated a minimal effect of our nanocomplex design in comparison to the adverse effect of PEI only (Figures S2 and S3). In this study, GNS is the core platform where the PEI is conjugated to the surface via a PEG linker representing a core-shell structure. DOX is simultaneously conjugated to the GNS surface through thiols bonds for combinational therapy. In addition to DOX, our design provides the flexibility to conjugate other drugs or molecules to the GNS surface to develop multi-drug delivery systems. Further, GNS as a core provides the freedom to incorporate photothermal therapy as well as imaging component for diagnostic purposes if desired.
Our results indicated a synergistic effect of chemo-gene combinational therapy in comparison to the single mode of treatment not only in the 2D cell culture system but also in 3D tumor spheroids. We used 3D spheroids as the closest physiological approximation of actual solid tumors because 2D culture often fails to mimic actual physiological tumors due to lack of cell–cell interaction, maximal exposure of cells to the given treatment, and absence of necrotic or quiescent cells. The two-phase aqueous system was used due to its simplicity. The cultured spheroids were slightly bigger than the others reported in previous studies. This was mainly because of the difference in the volume of a cell-DEX mixture (0.25 µL of cell-DEX mixture vs 0.5 µL in this study). We speculated that manual pipetting the cell-DEX mixture to the PEG phase in the well plate may also contribute to the discrepancy in the final spheroid size. Nevertheless, the IC50 value assessment observed for 3D spheres demonstrated the enhanced efficacy of combinational therapy over the single therapy, corroborating with the 2D cell culture study.
Conclusion
To summarize, we developed a universal GNS-based drug delivery vehicle platform which could be further applied to study a combination of various drugs and genes with little modifications. It is a simple and facile platform that can be adapted for further conjugation of other therapeutic drug or clinically relevant biomolecules. The pH-responsive drug release provides an additional assurance to minimize non-specific release and thus minimizes dose-dependent side effects and MDR. This design may represent a paradigm shift to developing a theragnostic platform due to the SPR and imaging capabilities of GNS.
Abbreviations
GNS, Gold nanospheres; PEG, Poly (ethylene glycol); PEI, Polyethyleneimine; PEG-GNS, PEG conjugated GNS; PEI-GNS, PEI conjugated GNS; PEG-HYZ, PEG-hydrazine; DEX, Dextran; DOX-GNS, Dox conjugated GNS; PLK1-GNS, PLK1 conjugated GNS; DOX-PLK1-GNS, DOX and PLK1 conjugated GNS.
Acknowledgments
This work was supported by the US Department of Agriculture (2015-38422-24059) and the National Institutes of Health Grants R01AI135005 (to C.Y.H).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Kleinsmith LJ. Principles of Cancer Biology. Benjamin-Cummings Publishing Company; 2006.
2. Siegiel R, Miller K, Jemal A. Cancer statistics, 2017. CA Cancer J Clin. 2017;67:7–30. doi:10.3322/caac.21387
3. Housman G, Byler S, Heerboth S, et al. Drug resistance in cancer: an overview. Cancers. 2014;6(3):1769–1792. doi:10.3390/cancers6031769
4. Devita Jr VT, Young RC, Canellos GP. Combination versus single agent chemotherapy: a review of the basis for selection of drug treatment of cancer. Cancer. 1975;35(1):98–110. doi:10.1002/1097-0142(197501)35:1<98::AID-CNCR2820350115>3.0.CO;2-B
5. Shrestha B, Tang L, Romero G. Nanoparticles‐mediated combination therapies for cancer treatment. Adv Therap. 2019;2(11):1900076. doi:10.1002/adtp.201900076
6. Sun T-M, Du J-Z, Yao Y-D, et al. Simultaneous delivery of siRNA and paclitaxel via a “two-in-one” micelleplex promotes synergistic tumor suppression. ACS Nano. 2011;5(2):1483–1494. doi:10.1021/nn103349h
7. Gandhi NS, Tekade RK, Chougule MB. Nanocarrier mediated delivery of siRNA/miRNA in combination with chemotherapeutic agents for cancer therapy: current progress and advances. J Control Release. 2014;194:238–256. doi:10.1016/j.jconrel.2014.09.001
8. Su B, Cengizeroglu A, Farkasova K, et al. Systemic TNFα gene therapy synergizes with liposomal doxorubicine in the treatment of metastatic cancer. Mol Ther. 2013;21(2):300–308. doi:10.1038/mt.2012.229
9. Shrestha B, DeLuna F, Anastasio MA, Yong J, Brey EM. Photoacoustic imaging in tissue engineering and regenerative medicine. Tissue Eng Part B Rev. 2020;26(1):79–102. doi:10.1089/ten.teb.2019.0296
10. Betal S, Dutta M, Shrestha B, et al. Cell permeation using core-shell magnetoelectric nanoparticles. Integr Ferroelectr. 2016;174(1):186–194. doi:10.1080/10584587.2016.1196332
11. Betal S, Shrestha B, Dutta M, et al. Magneto-elasto-electroporation (MEEP): in-vitro visualization and numerical characteristics. Sci Rep. 2016;6:32019. doi:10.1038/srep32019
12. Roux, Brianna M., et al. iPSC-derived endothelial networks accelerate vascularization but not bone regeneration. Tissue Engineering ja 2020.
13. Shrestha, Binita, et al. Gold nanorods enable noninvasive longitudinal monitoring of hydrogels in vivo with photoacoustic tomography. Acta Biomaterialia. 2020.
14. Shrestha, Binita, Jacob Brown, and Eric M. Brey. Controlled Delivery of Angiogenic Proteins. Controlled Drug Delivery Systems. CRC Press, 2020;147–162.
15. Spänkuch-Schmitt B, Kaufmann M, Strebhardt K, Strebhardt K. Effect of RNA silencing of polo-like kinase-1 (PLK1) on apoptosis and spindle formation in human cancer cells. J Natl Cancer Inst. 2002;94(24):1863–1877. doi:10.1093/jnci/94.24.1863
16. Reagan-Shaw S, Ahmad N. Silencing of polo-like kinase (Plk) 1 via siRNA causes induction of apoptosis and impairment of mitosis machinery in human prostate cancer cells: implications for the treatment of prostate cancer. FASEB J. 2005;19(6):611–613. doi:10.1096/fj.04-2910fje
17. Kimling J, Maier M, Okenve B, Kotaidis V, Ballot H, Plech A. Turkevich method for gold nanoparticle synthesis revisited. J Phys Chem B. 2006;110(32):15700–15707. doi:10.1021/jp061667w
18. Ungaro F, De Rosa G, Miro A, Quaglia F. Spectrophotometric determination of polyethylenimine in the presence of an oligonucleotide for the characterization of controlled release formulations. J Pharm Biomed Anal. 2003;31(1):143–149.
19. Hermanson GT. Bioconjugate techniques. Academic press; 2013.
20. Aryal S, Grailer JJ, Pilla S, Steeber DA, Gong S. DOXorubicin conjugated gold nanoparticles as water-soluble and pH-responsive anticancer drug nanocarriers. J Mater Chem. 2009;19(42):7879–7884. doi:10.1039/b914071a
21. Ham SL, Atefi E, Fyffe D, Tavana H. Robotic production of cancer cell spheroids with an aqueous two-phase system for drug testing. JoVE. 2015;(98):e52754–e52754.
22. Gutiérrez-Praena D, Pichardo S, Sánchez E, Grilo A, Cameán AM, Jos A. Influence of carboxylic acid functionalization on the cytotoxic effects induced by single wall carbon nanotubes on human endothelial cells (HUVEC). Toxicol in Vitro. 2011;25(8):1883–1888. doi:10.1016/j.tiv.2011.05.027
23. Montcourrier P, Silver I, Farnoud R, Bird I, Rochefort H. Breast cancer cells have a high capacity to acidify extracellular milieu by a dual mechanism. Clin Exp Metastasis. 1997;15(4):382–392. doi:10.1023/A:1018446104071
24. He C, Hu Y, Yin L, Tang C, Yin C. Effects of particle size and surface charge on cellular uptake and biodistribution of polymeric nanoparticles. Biomaterials. 2010;31(13):3657–3666. doi:10.1016/j.biomaterials.2010.01.065
25. Chung T-H, Wu S-H, Yao M, et al. The effect of surface charge on the uptake and biological function of mesoporous silica nanoparticles in 3T3-L1 cells and human mesenchymal stem cells. Biomaterials. 2007;28(19):2959–2966. doi:10.1016/j.biomaterials.2007.03.006
26. Xiao K, Li Y, Luo J, et al. The effect of surface charge on in vivo biodistribution of PEG-oligocholic acid based micellar nanoparticles. Biomaterials. 2011;32(13):3435–3446. doi:10.1016/j.biomaterials.2011.01.021
27. Lu L-Y, Yu X. The balance of Polo-like kinase 1 in tumorigenesis. Cell Div. 2009;4(1):4.
28. Cholewa BD, Liu X, Ahmad N. The role of polo-like kinase 1 in carcinogenesis: cause or consequence? Cancer Res. 2013;73(23):6848–6855. doi:10.1158/0008-5472.CAN-13-2197
29. Takahashi T, Sano B, Nagata T, et al. Polo‐like kinase 1 (PLK1) is overexpressed in primary colorectal cancers. Cancer Sci. 2003;94(2):148–152. doi:10.1111/j.1349-7006.2003.tb01411.x
30. Takai N, Hamanaka R, Yoshimatsu J, Miyakawa I. Polo-like kinases (Plks) and cancer. Oncogene. 2005;24(2):287–291. doi:10.1038/sj.onc.1208272
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.