")
Back to Journals » Drug Design, Development and Therapy » Volume 13
Glucoside Derivatives Of Podophyllotoxin: Synthesis, Physicochemical Properties, And Cytotoxicity
Authors Zi CT, Yang L, Kong QH, Li HM, Yang XZ, Ding ZT, Jiang ZH , Hu JM , Zhou J
Received 16 May 2019
Accepted for publication 8 September 2019
Published 23 October 2019 Volume 2019:13 Pages 3683—3692
DOI https://doi.org/10.2147/DDDT.S215895
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Jianbo Sun
Cheng-Ting Zi,1,2,* Liu Yang,2,* Qing-Hua Kong,2 Hong-Mei Li,2 Xing-Zhi Yang,2 Zhong-Tao Ding,3 Zi-Hua Jiang,4 Jiang-Miao Hu,2 Jun Zhou2
1Key Laboratory of Pu-Er Tea Science, Ministry of Education, College of Science, Yunnan Agricultural University, Kunming, 650201, People’s Republic of China; 2State Key Laboratory of Phytochemistry and Plant Resources in West China, Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, People’s Republic of China; 3Key Laboratory of Medicinal Chemistry for Nature Resource, Ministry of Education, School of Chemical Science and Technology, Yunnan University, Kunming 650091, People’s Republic of China; 4Department of Chemistry, Lakehead University, Thunder Bay ON P7B 5E1, Canada
*These authors contributed equally to this work
Correspondence: Zi-Hua Jiang
Department of Chemistry, Lakehead University, 955 Oliver Road, Thunder Bay ON P7B 5EI, Canada
Tel +1 807 766 7171
Fax +1 807 346 7775
Email [email protected]
Jiang-Miao Hu
State Key Laboratory of Phytochemistry and Plant Resources in West China, Kunming Institute of Botany, Chinese Academy of Sciences, No. 132, Lanhei Road, Kunming 650201, People’s Republic of China
Tel +86 871 6522 3264
Fax +86 871 6522 3261
Email [email protected]
Background: Widespread concern of the side effects and the broad-spectrum anticancer property of podophyllotoxin as an antitumor agent highlight the need for the development of new podophyllotoxin derivatives. Although some per-butyrylated glucosides of podophyllotoxin and 4β-triazolyl-podophyllotoxin glycosides show good anticancer activity, the per-acetylated/free of podophyllotoxin glucosides and their per-acetylated are not well studied.
Methods: A few glucoside derivatives of PPT were synthesized and evaluated for their in vitro cytotoxic activities against five human cancer cell lines, HL-60 (leukemia), SMMC-7721 (hepatoma), A-549 (lung cancer), MCF-7 (breast cancer), and SW480 (colon cancer), as well as the normal human pulmonary epithelial cell line (BEAS-2B). In addition, we investigated the structure–activity relationship and the physicochemical property–anticancer activity relationship of these compounds.
Results: Compound 6b shows the highest cytotoxic potency against all five cancer cell lines tested, with IC50 values ranging from 3.27±0.21 to 11.37±0.52 μM. We have also found that 6b displays higher selectivity than the etoposide except in the case of HL-60 cell line. The active compounds possess similar physicochemical properties: MSA > 900, %PSA < 20, ClogP > 2, MW > 700 Da, and RB > 10.
Conclusion: We synthesized several glucoside derivatives of PPT and tested their cytotoxicity. Among them, compound 6b showed the highest cytotoxicity. Further studies including selectivity of active compounds have shown that the selectivity indexes of 6b are much greater than the etoposide except in the case of HL-60 cell line. The active compounds possessed similar physicochemical properties. This study indicates that active glucoside analogs of podophyllotoxin have potential as lead compounds for developing novel anticancer agents.
Keywords: podophyllotoxin, glucoside, synthesis, cytotoxicity, physicochemical properties
Introduction
Cancer is the second leading cause of death in the worldwide and remains one of the most difficult diseases to combat.1 Developing new anticancer drugs and more effective treatment strategies for cancer is of great importance in medicinal chemistry. Natural products with diverse structures and unique biological activities are valuable sources for drug discovery. Close to 60% clinical drugs are either natural products or structural analogs of natural products with improved pharmacological activity.2–4 Podophyllotoxin (PPT, 1, Scheme 1), a well-known naturally occurring aryltetralin lignan, is mainly isolated from the roots of the North American Podophyllum peltatum Linnaeus, the Tibetan P. emodi Wall, or the Taiwanese species Podophyllum peltatum.5 It shows strong cytotoxic activity against various cancer cell lines and acts at the colchicine-binding site on tubulin.6
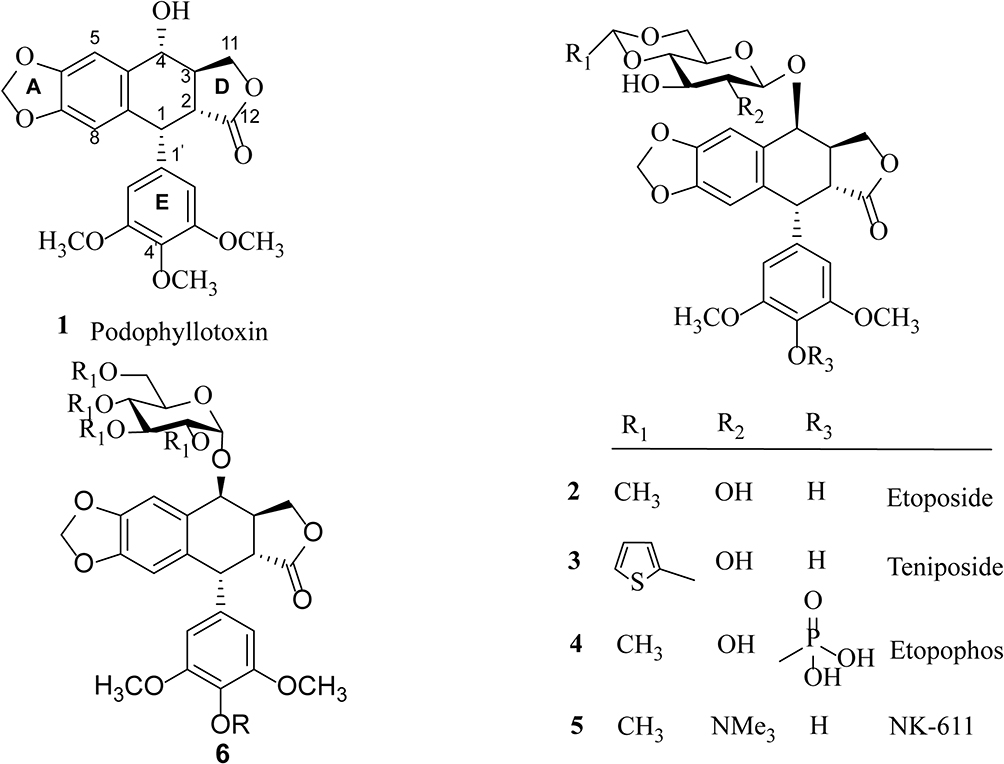 |
Scheme 1 Structure of compounds 1–6: podophyllotoxin (1), etoposide (2), teniposide (3), etopophos (4), NK-611 (5), and podophyllotoxin glucosides (6). |
Due to its high toxicity and poor water solubility, podophyllotoxin has limited application as an anticancer drug. Based on its potent anticancer activity, PPT has served as a lead compound for the discovery and development of new anticancer agents. For example, the two semisynthetic glucosidic cyclic acetals of PPT, etoposide (2) and teniposide (3) (Scheme 1), are in clinical use for the treatment of a variety of cancers, including small-cell lung cancer, non-Hodgkin’s lymphoma, leukemia, Kaposi’s sarcoma, neuroblastoma, and soft-tissue sarcoma.7–10 The mechanism of action for etoposide and teniposide is different from that of PPT in that both etoposide and teniposide block the DNA topoisomerase-II by stabilizing the enzyme–DNA complex.11–14 However, the therapeutic use of etoposide and teniposide is often hindered by problems such as acquired drug resistance, myelosuppression, and their poor water solubility. To overcome the problems of etoposide and teniposide, further structure modifications of PPT have been carried out, which led to the synthesis of other PPT derivatives, such as etopophos (4) and NK-611 (5) (Scheme 1), which reached clinical studies.15 The clinically useful podophyllotoxin-derived glucosides 2–5 possess a 4,6-cyclic acetal moiety and various other substitutions on the sugar residue, suggesting the important role of substituents in modifying the biological activities of these podophyllotoxin derivatives.
In recent years, we have been working on the structural modification of podophyllotoxin and focused on glycosides of podophyllotoxin (such as 6, Scheme 1) and 4β-triazolyl-podophyllotoxin.16–19 Per-butyrylated glucosides of podophyllotoxin16 as well as the glucosides of 4β-triazolyl-podophyllotoxin and their acylated analogues show good cytotoxicity.19,20 The glucosides of podophyllotoxin and their per-acetylated analogs are less well studied.21 In this article, a few glucoside derivatives of PPT were synthesized (Table S1) and evaluated for their in vitro cytotoxic activity against five human cancer cell lines, HL-60 (leukemia), SMMC-7721 (hepatoma), A-549 (lung cancer), MCF-7 (breast cancer), and SW480 (colon cancer). To evaluate the selectivity of these compounds between tumor cells and normal cells, their growth inhibitory effect was tested on normal human pulmonary epithelial cell lines (BEAS-2B). In addition, the physicochemical properties of these compounds were calculated and correlated with their anticancer activity.
Results And Discussion
Chemical Synthesis
There have been several reports on constructing the glucosidic linkages of podophyllotoxin according to known literatures.22–25 The synthesis of glucoside derivatives of podophyllotoxin 6a – 6d following a similar method is reported in the literature and is shown in Scheme 2. 1,2,3,4,6-Penta-O-acetyl-α/β-D-glucopyranose (mainly α-form)26 was treated with ammonia solution (25%) in acetonitrile to give 2,3,4,6-tetra-O-acetyl -α/β-D-glucopyranose (8) as an anomeric mixture (α/β ratio = 6:1) in 46% yield.27,28 Then, compound 8 was allowed to react with podophyllotoxin (1) and 4ʹ-demethylepipodophyllotoxin (9)29 in the presence of trifluoroboran etherate (BF3•Et2O) at −78°C to give the per-acetylated glucoside derivatives of podophyllotoxin 6a and 6b in 58–62% yield.16 Compounds 6a and 6b were treated with sodium methoxide in methanol at room temperature for 2 hrs to yield podophyllotoxin glucosides 6c and 6d in 78–80% yields.30
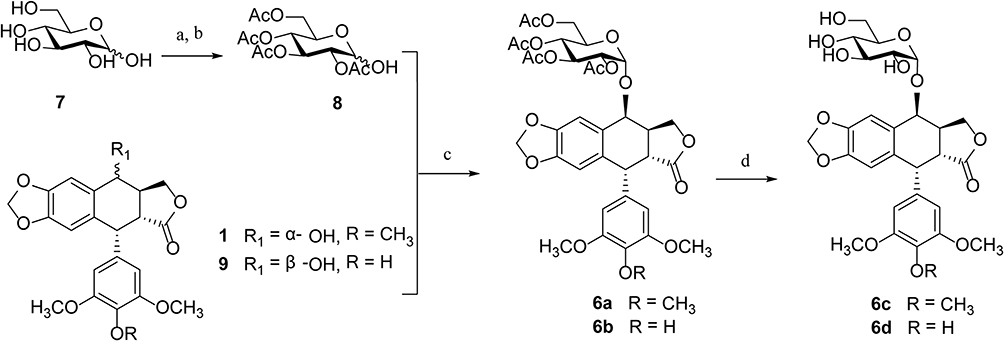 |
Scheme 2 Synthesis of glucoside derivatives of PPT 6a – 6d. Reagents and conditions: (A) Ac2O, sodium acetate, 100°C, 20 mins, ~99%; (B) NH3⋅H2O, CH3CN, rt, overnight, 46%; (C) BF3⋅Et2O, CH2Cl2, −78°C to rt, 58–62%; (D) CH3ONa, CH3OH, 2 hrs, rt, 78–80%. |
All the glucoside derivatives of PPT were characterized by 1H and 13C-NMR, electrospray ionization mass spectrometry (ESI-MS), and high-resolution mass spectrometry (HRESI-MS). The characteristic 1H-NMR and 13C-NMR data of compounds 6a – 6d are shown in Table 1. In the 1H-NMR spectra, the proton at C-4 of 4β-substituted compounds appears as a doublet at 4.72–4.96 ppm, usually with a coupling constant J3-4 < 4.0 Hz, indicating a cis-relationship between C3-H and C4-H.31 The coupling constant of the anomeric proton of the glucose residue (J1”-2”) is typically <4.0 Hz, which confirms that the glycosidic linkage is fan α–linkage.
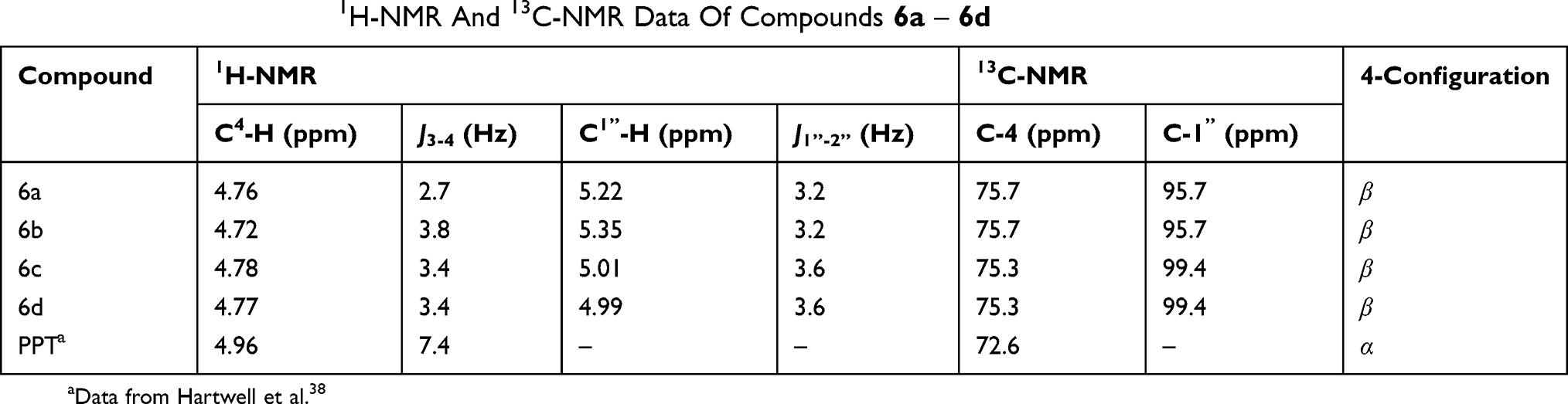 |
Table 1 The Characteristic 1H-NMR And 13C-NMR Data Of Compounds 6a – 6d |
Evaluation Of Biological Activity
The per-butyrylated glucoside derivatives of podophyllotoxin 6e and 6f have been previously documented.16 Per-acetylated glucoside derivatives of podophyllotoxin (6a and 6b) and podophyllotoxin glucosides (6c and 6d) were tested for their cytotoxicity against five human cancer cell lines, including HL-60 (leukemia), SMMC-7721 (hepatoma), A-549 (lung cancer), MCF-7 (breast cancer), and SW480 (colon cancer). Etoposide (2) and cisplatin were taken as control drugs, and their IC50 data are presented in Figure 1 and Table 2. Compounds 6c and 6d having a free glucose residue show weak activity (all having IC50 > 40 μM), while peracetylated glucoside derivatives 6a and 6b show improved activity. Among these derivatives, compound 6b shows the highest cytotoxicity against five cancer cells, with their IC50 values ranging from 3.27±0.21 to 11.37±0.52 μM, which is more potent than the control drug etoposide against the MCF-7 and SW480 cell lines. In our previous study, we reported that the cytotoxic activity of 4β-triazolyl-podophyllotoxin derivatives with a peracetylated glucose residue mostly shows weak activity.19 Furthermore, compound 6b with a hydroxy group at the C-4ʹ position in the E ring is more active than compound 6a which has a methoxyl group at the C-4ʹ position.
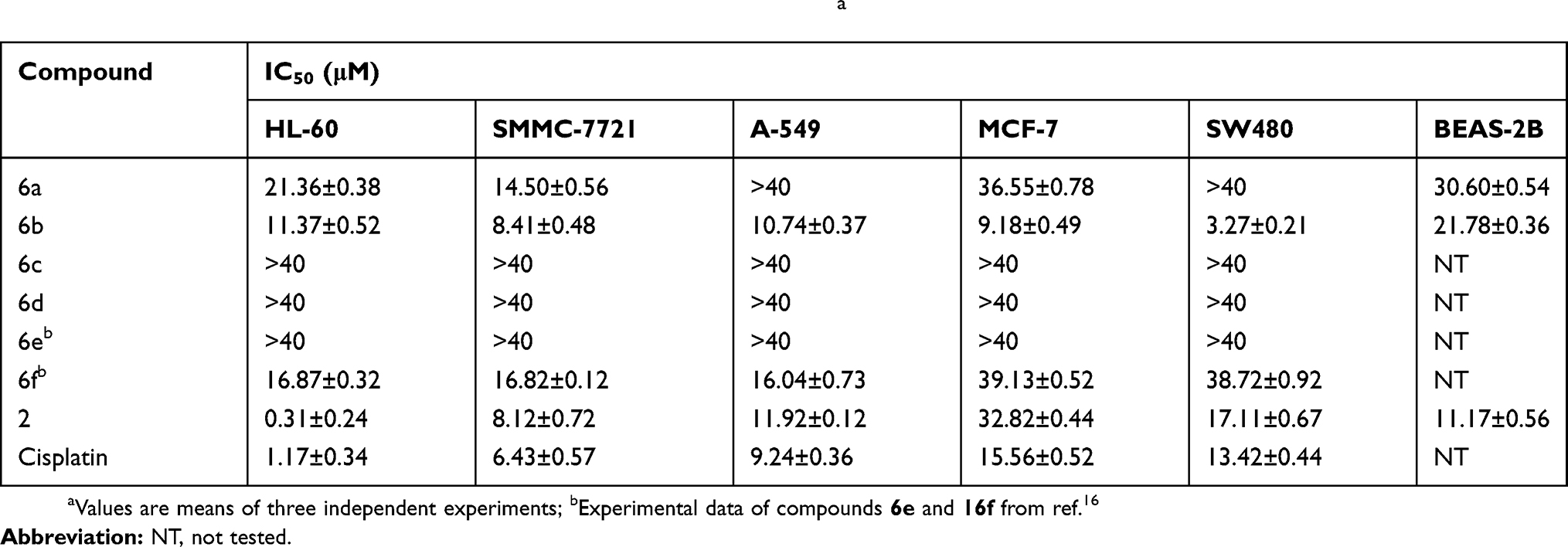 |
Table 2 Cytotoxicity Of Podophyllotoxin Derivatives 6a – 6f In Vitroa |
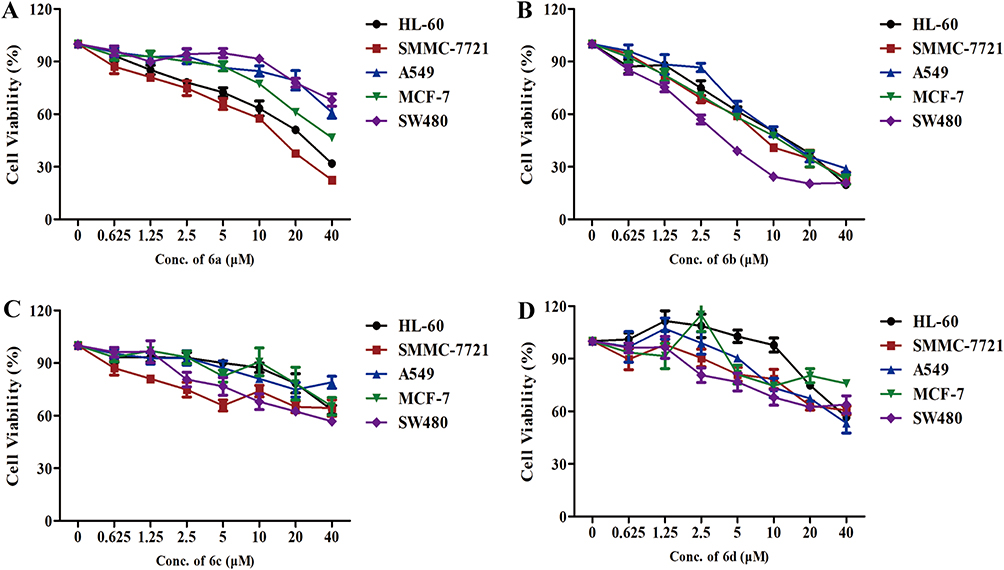 |
Figure 1 Inhibitory effects of podophyllotoxin derivatives on cancer cells. (A–D) The inhibitory effects of compounds 6a – 6d on HL-60 (leukemia), SMMC-7721 (hepatoma), A-549 (lung cancer), MCF-7 (breast cancer), and SW480 (colon cancer) cells, as evaluated by the MTT assay. |
Cancer chemotherapy is often associated with low/nonselectivity of cancer drugs which attack cancer cells as well as normal cells, leading to serious side effects. In order to test their selectivity, compounds 6a and 6b were tested for their growth inhibitory effects on a normal human bronchial epithelial cell line (BEAS-2B) (Table 2). The selectivity index (SI) was expressed as the ratio of the IC50 value of the compound in normal cell line over that in cancer cell line. A larger SI value indicates that the drug displays higher selectivity toward cancer cells over normal cells.32,33 The SI values of compound 6a, 6b and etoposide are presented in Table 3. Compound 6b shows moderate selectivity toward cancer cells with SI values in the range of 1.9–6.7 in all cells tested. Compound 6b displays higher selectivity than etoposide in four of the five cancer cell lines tested except an HL-60 cell line. Among these derivatives, 6b shows the highest potency (IC50 3.27±0.21 μM) and highest selectivity (SI 6.7) in SW480 cell line, suggesting that 6b may be a promising therapeutic agent for colon cancer.
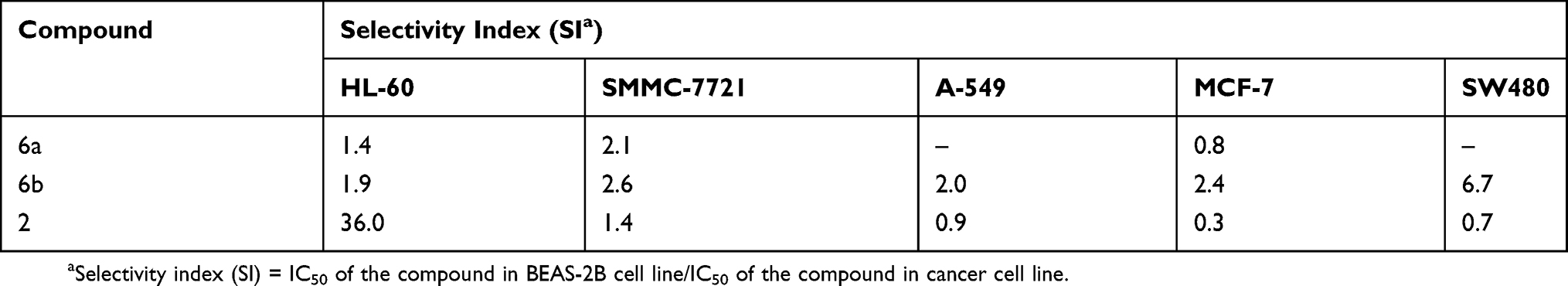 |
Table 3 Selectivity Of The Cytotoxicity Of Compounds 6a, 6b, And Etoposide To Cancer Cells As Compared With BEAS-2B Normal Cells |
Physicochemical Property–Cytotoxicity Relationship
Values Of Partition Coefficient Of The Compounds
The logarithm of the octanol–water partition coefficient investigation (logP) is an important pharmaceutical parameter in evaluating solvency, absorption, and transport of drugs; the preferred logP value is less than 5.11 Compounds etoposide (2) and the most potent compound 6b were measured for values of logP. Solutes were equilibrated between octanol and water. The concentration of compounds in octanol was determined by the HPLC method.12,13 The logP values of compounds 2 and 6b were determined to be 1.44 and 1.78 at 30°C. As shown in Table 4 (see supporting information for the details), compound 6b expressed the logP value and was close to the calculated value of 2.24.
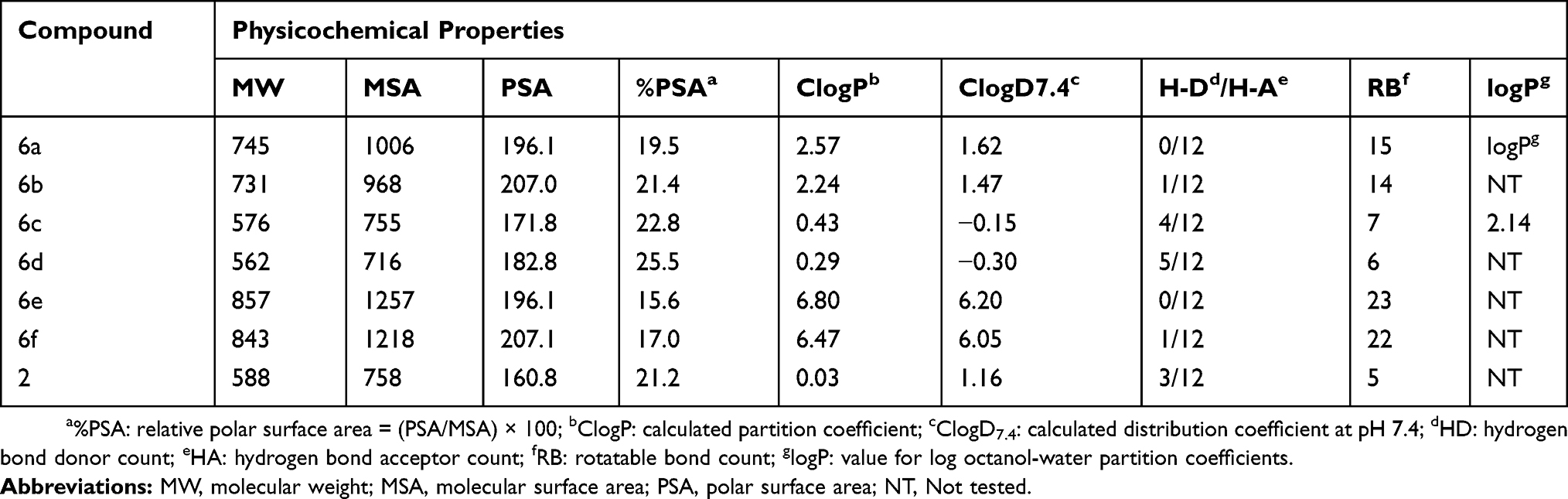 |
Table 4 Physicochemical Properties Of Glucoside Derivatives Of Podophyllotoxin |
Solubility
Poor water solubility is a common problem in developing podophyllotoxin derivatives for therapeutic use. Compounds with glucose residue are slightly soluble in water. The solubility of podophyllotoxin (1) and compounds 6b in aqueous at temperature 25°C are reported 1 has a solubility of 2.2 mg/mL in water, while 6b with a peracetylated glucoside residue has a solubility of 1.7 mg/mL in water (see supporting information for the details). The solubility values obtained for 6b become unfairly soluble in water.
Physicochemical Property
The physicochemical properties of a drug can largely affect the pharmacokinetics and efficacy of a drug. The physicochemical properties of glucoside derivatives of podophyllotoxin 6a – 6d and 6e – 6f19 were calculated and compared with etoposide 2, which include molecular weight (MW), molecular surface area (MSA), polar surface area (PSA), relative polar surface area (%PSA), calculated partition coefficient (ClogP), calculated distribution coefficient at pH 7.4 (ClogD7.4), hydrogen bond donor (HD), hydrogen bond acceptor (HA), and rotatable bond (RB) (Table 4). Noteworthy is that almost all active compounds (having IC50 < 40 μM) are relatively lipophilic (MSA > 900, %PSA < 20, ClogP > 2), and since they have a higher molecular weight (MW > 700 Da) and a larger number of rotatable bonds (RB > 10), with the exception of compound 6e, they are placed at an advantage for further optimization. By contrast, inactive compounds 6c and 6d (having a free glucose residue) have %PSA values >22, ClogD7.4 <0, and a smaller number of rotatable bonds (RB < 10). It is obvious that derivatives with free glucose residues (6c and 6d) are relatively more polar, and this might account for the general lack of activity for these compounds. This result suggests that the peracetylated/perbutyrylated derivatives of podophyllotoxin glucosides may, therefore, be more suitable for further optimization.
Chemical Stability Investigation
The most potent compound 6b was selected for investigations of chemical stability in aqueous phase with comparison of podophyllotoxin (1). The results indicate that compound 6b exhibits better chemical stability under the specific conditions (37°C, pH = 7.0, Figure 2) (see supporting information for the details). Obviously, 6b showed considerable stability with podophyllotoxin.
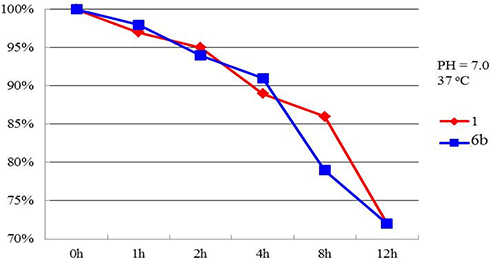 |
Figure 2 Chemical stability investigation of compounds 1 and 6b. |
Experimental
General
All cancer cells (HL-60, SMMC-7721, A-549, MCF-7, and SW480) were obtained from a Shanghai cell bank in China. D-glucose was purchased from Aladdin Chemical Co., Ltd (Guangzhou, China); podophyllotoxin was obtained from Chengdu Proifa Technology Development Co., Ltd (Chengdu, China); boron trifluoride etherate was obtained from J&K Chemical Technology Co., Ltd (Beijing China); 3-(4,5-dimethyl- thiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) was purchased from Sigma-Aldrich (St. Louis, MO, USA). Dichloromethane and acetonitrile were distilled over calcium hydride. All reagents were commercially available and used without further purification unless indicated otherwise. The melting points were measured by an X-4 melting point apparatus and were uncorrected. Optical rotations were obtained with a Jasco P-1020 Automatic Digital Polariscope MS data were obtained in the ESI mode on API Qstar Pulsar instrument; HRMS data were obtained in the ESI mode on LCMS-IT-TOF (Shimadzu, Kyoto, Japan); 1H-NMR and 13C-NMR spectra were recorded on Bruker AVANCE III 400 MHz, or 600 MHz (Bruker BioSpin GmbH, Rheinstetten, Germany) instruments, using tetramethylsilane (TMS) as an internal standard: chemical shifts (δ) are given in ppm, coupling constants (J) in Hz, and the solvent signals were used as references (CDCl3: δC= 77.2 ppm; residual CHCl3 in CDCl3: δH= 7.26 ppm; CD3OD: δC= 49.0 ppm; residual CH3OH in CD3OD: δH= 4.78 ppm). Column chromatography (CC): silica gel (200–300 mesh; Qingdao Makall Group CO., LTD; Qingdao; China). All reactions were monitored using thin-layer chromatography (TLC) on silica gel plates.
Chemistry
Synthesis Of 2,3,4,6-Tetra-O-Acetyl-α/β-D-Glucopyranose (8)
D-glucose (1.8 g, 10 mmol) was suspended in acetic anhydride (9.5 mL, 100 mmol) and anhydrous sodium acetate (0.9 g, 11 mmol) was added, and the resulting mixture was heated at 100°C for 20 mins. The reaction was quenched (saturated aqueous sodium bicarbonate, 20 mL) and diluted with dichloromethane (30 mL); the organic layer was washed with brine (3 × 30 mL) and dried with sodium sulfate. The solvent was evaporated, and the residue dried in vacuo to give the crude 1,2,3,4,6-penta-O-acetyl-D-glucopyranose.
The crude 1,2,3,4,6-penta-O-acetyl-D-glucopyranose was dissolved in acetonitrile (20 mL), and 25% ammonia solution (0.4 mL, 20 mmol) was added dropwise slowly. The mixture was stirred at room temperature for 6 hrs. The solvent was evaporated, and the brown oily residue was passed a short pad of silica column (petroleum ether/ethyl acetate 4:1, v/v) to afford the product 8 (1.6 g, 46% yield for two steps). α/β ratio = 6:1. 1H-NMR (CDCl3, 400 MHz) δ 6.20 (d, 1/7H, J = 8.0 Hz, C1-Hβ), 5.54 (t, 1H, J = 9.6 Hz, C3-H), 5.47 (d, 6/7H, J = 3.2 Hz, C1-Hα), 5.08 (t, 1H, J = 9.6 Hz, C4-H), 4.91 (dd, 1H, J = 3.2 Hz, 10.0 Hz, C2-H), 4.27–4.23 (m, 2H, C6-CH2), 4.14–4.12 (m, 1H, C5-H), 2.10–2.00 (m, 12H, 4 × OCH3); 13C-NMR (CD3Cl, 400 MHz) δ 170.8 (C=O), 170.2 (C=O), 170.1 (C=O), 169.7 (C=O), 95.5 (C-1β), 90.1 (C-1α), 73.2 (C-5β), 72.1 (C-4β), 72.0 (C-2β), 71.9 (C-5α), 69.8 (C-4α), 68.4 (C-2α), 68.3 (C-3β), 67.2 (C-3α), 61.9 (C-6), 20.7 (OCH3), 20.7 (OCH3), 20.6 (OCH3), 20.5 (OCH3); ESIMS: m/z 371 [M + Na]+.
Synthesis Of 4ʹ-Demethylepipodophyllotoxin (9)
4ʹ-Demethylepipodophyllotoxin (9) was prepared according to the literature.29
General Procedure For The Synthesis Of Compounds 6a – 6b
To a mixture of 2,3,4,6-tetra-O-acetyl-α/β-D-glucopyranose (0.2 mmol) and podophyllotoxin/4ʹ-demethylepipodophyllotoxin (0.2 mmol) in dry CH2Cl2 (3 mL) was added of BF3⋅H2O (25 μL, 0.02 mmol) at −78 oC, and the resulting mixture was stirred for 1 hr. Then, triethylamine (0.1 mL) was added to the mixture, and acetic acid (0.1 mL) was added. The solvent was evaporated, and the residue was purified by flash chromatography on silica gel (petroleum ether/ethyl acetate 2:1, v/v) to afford the major product 6a or 6b as white powder.
4-O-(2ʹ’,3ʹ’,4ʹ’,6ʹ’-Tetra-O-Acetyl-α-D-Glucopyranosyl)-Epipodophyllotoxin (6a)
White powder; yield 58%; mp 167–168 oC; 1H-NMR (CDCl3, 400 MHz) δ 6.99 (s, 1H, C6-H), 6.55 (s, 1H, C8-H), 6.23 (s, 2H, C2′, C6′-H), 6.00–5.98 (m, 2H, OCH2O), 5.33 (t, 1H, J = 8.0 Hz), 5.22 (d, 1H, J = 3.2 Hz, C1”-H), 5.08–5.02 (m, 2H), 4.76 (d, 1H, J = 2.7 Hz, C4-H), 4.66–4.65 (m, 1H), 4.30 (d, 1H, J = 2.1 Hz, C1-H), 4.28–4.18 (m, 2H), 4.14–4.03 (m, 1H), 3.79 (s, 3H, C4ʹ-OCH3), 3.76 (s, 6H, C3ʹ, C5ʹ-OCH3), 3.45–3.42 (m, 1H, C3-H), 3.01–2.98 (m, 1H, C2-H), 2.14–2.04 (m, 12H, 4 × OCH3); 13C-NMR (CDCl3, 100 MHz) δ 177.8 (C-12), 170.6 (C=O), 170.0 (C=O), 169.3 (C=O), 169.2 (C=O), 153.4 (C-3ʹ, C-5ʹ), 147.9 (C-6), 146.7 (C-7), 137.5 (C-4ʹ), 136.9 (C-1ʹ), 131.5 (C-9), 128.4 (C-10), 109.7 (C-8), 106.8 (C-5), 105.4 (C-2ʹ, C-6ʹ), 101.3 (OCH2O), 95.7 (C-1ʹ’), 75.7 (C-4), 72.3, 71.0, 69.4, 68.2, 67.7 (C-11), 60.8 (4ʹ-OCH3), 60.7, 56.2 (3ʹ, 5ʹ-OCH3), 44.9 (C-2), 44.2 (C-1), 38.1 (C-3), 20.7 (OCH3), 20.6 (OCH3), 20.6 (OCH3), 20.5 (OCH3); ESIMS: m/z 767 [M + Na]+, HRESIMS: calcd for C36H40O17Na [M + Na]+ 767.2285, found 767.2286.
4-O-(2ʹ’,3ʹ’,4ʹ’,6ʹ’-Tetra-O-Acetyl-α-D-Glucopyranosyl)-4ʹ-Demethylepipodophyllotoxin (6b)
White powder; yield 62%; mp 172–174 oC; 1H-NMR (CDCl3, 400 MHz) δ 6.94 (s, 1H, C6-H), 6.55 (s, 1H, C8-H), 6.45 (s, 2H, C2′, C6′-H), 5.97–5.95 (m, 2H, OCH2O), 5.40 (t, 1H, J = 8.0 Hz), 5.35 (d, 1H, J = 3.2 Hz, C1”-H), 5.10–5.07 (m, 1H), 4.83–4.80 (m, 1H), 4.72 (d, 1H, J = 3.8 Hz, C4-H), 4.41 (d, 1H, J = 2.1 Hz, C1-H), 4.32–4.31 (m, 1H), 4.22–4.21 (m, 1H), 4.09–4.07 (m, 1H), 3.75 (s, 6H, C3ʹ, C5ʹ-OCH3), 3.67–3.64 (m, 1H), 3.45–3.42 (m, 1H, C3-H), 3.00–2.97 (m, 1H, C2-H), 2.15–2.09 (m, 12H, 4 × OCH3); 13C-NMR (CDCl3, 100 MHz) δ 177.7 (C-12), 170.5 (C=O), 170.0 (C=O), 169.3 (C=O), 169.2 (C=O), 152.3 (C-3ʹ, C-5ʹ), 148.0 (C-6), 146.8 (C-7), 140.4 (C-4ʹ), 140.4 (C-1ʹ), 131.1 (C-9), 128.4 (C-10), 109.8 (C-8), 106.8 (C-5), 104.9 (C-2ʹ, C-6ʹ), 101.3 (OCH2O), 95.7 (C-1ʹ’), 75.7 (C-4), 71.0, 69.5, 68.2, 67.8, 67.5 (C-11), 60.7, 56.2 (3ʹ, 5ʹ-OCH3), 45.0 (C-2), 44.3 (C-1), 38.0 (C-3), 20.7 (OCH3), 20.6 (OCH3), 20.6 (OCH3), 20.5 (OCH3); ESIMS: m/z 756 [M + Na]+, HRESIMS: calcd for C35H38O17Na [M + Na]+ 756.2123, found 756.2126.
General Procedure For The Synthesis Of Compounds 6c – 6d
To a solution of 6a/6b (0.1 mmol) in methanol (1.5 mL) was added sodium methoxide (0.03 mmol) at 0 oC, and the resulting mixture was stirred for 2 hrs. The reaction was slowly quenched (anhydrous Amberlite ion-exchange resin IRA-400), and the resin was removed by filtration. The filtrate was concentrated under vacuum, and the residue was purified by flash chromatography on silica gel (chloroform/methanol 9:1, v/v) to afford compound 6c or 6d as white powder.
4-O-(α-D-Glucopyranosyl)-Epipodophyllotoxin (6c)
White powder; yield 80%; mp 190–191 oC; 1H-NMR (CDCl3, 600 MHz) δ 7.07 (s, 1H, C6-H), 6.51 (s, 2H, C2′, C6′-H), 6.48 (s, 1H, C8-H), 5.94–5.93 (m, 2H, OCH2O), 5.01 (d, 1H, J = 3.6 Hz, C1”-H), 4.78 (d, 1H, J = 3.4 Hz, C4-H), 4.47–4.45 (m, 1H), 4.42 (t, 1H, J = 9.6 Hz), 4.36 (d, 1H, J = 2.2 Hz, C1-H), 3.77 (s, 6H, C3ʹ, C5ʹ-OCH3), 3.76 (s, 3H, C4ʹ-OCH3), 3.69–3.63 (m, 2H), 3.55–3.53 (m, 1H), 3.41–3.38 (m, 2H), 3.36–3.33 (m, 1H), 3.24–3.21 (m, 1H, C3-H), 3.16–3.13 (m, 1H, C2-H); 13C-NMR (CDCl3, 150 MHz) δ 178.7 (C-12), 154.7 (C-3ʹ, C-5ʹ), 149.0 (C-6), 148.2 (C-7), 139.5 (C-4ʹ), 137.9 (C-1ʹ), 132.6 (C-9), 131.2 (C-10), 110.1 (C-8), 108.2 (C-5), 106.9 (C-2ʹ, C-6ʹ), 102.5 (OCH2O), 99.4 (C-1ʹ’), 75.3 (C-4), 75.0, 74.3, 73.8, 71.1, 70.0 (C-11), 61.7, 61.1 (4ʹ-OCH3), 56.6 (3ʹ, 5ʹ-OCH3), 46.6 (C-2), 45.4 (C-1), 38.7 (C-3); ESIMS: m/z 575 [M - H]−, HRESIMS: calcd for C28H32O13 [M - H]− 576.1843, found 576.1846.
4-O-(α-D-Glucopyranosyl)-4ʹ- Demethylepipodophyllotoxin (6d)
White powder; yield 78%; mp 201–203 oC; 1H-NMR (CDCl3, 600 MHz) δ 7.07 (s, 1H, C6-H), 6.49 (s, 1H, C8-H), 6.47 (s, 2H, C2′, C6′-H), 5.93–5.92 (m, 2H, OCH2O), 4.99 (d, 1H, J = 3.6 Hz, C1”-H), 4.82–4.81 (m, 1H), 4.77 (d, 1H, J = 4.4 Hz, C4-H), 4.46–4.39 (m, 2H), 4.33 (d, 1H, J = 2.2 Hz, C1-H), 3.78 (s, 6H, C3ʹ, C5ʹ-OCH3), 3.70–3.62 (m, 2H), 3.56–3.53 (m, 1H), 3.41–3.34 (m, 3H), 3.36–3.33 (m, 1H), 3.26–3.23 (m, 1H, C3-H), 3.20–3.18 (m, 1H, C2-H); 13C-NMR (CDCl3, 150 MHz) δ 178.8 (C-12), 149.5 (C-3ʹ, C-5ʹ), 148.9 (C-6), 148.2 (C-7), 135.5 (C-4ʹ), 133.9 (C-1ʹ), 132.9 (C-9), 131.2 (C-10), 110.2 (C-8), 108.1 (C-5), 106.4 (C-2ʹ, C-6ʹ), 102.5 (OCH2O), 99.4 (C-1ʹ’), 75.3 (C-4), 75.0, 74.3, 73.8, 71.1, 69.9 (C-11), 61.7, 56.8 (3ʹ, 5ʹ-OCH3), 46.5 (C-2), 45.6 (C-1), 38.7 (C-3); ESIMS: m/z 561 [M - H]−, HRESIMS: calcd for C27H30O13 [M - H]− 561.1686, found 561.1684.
Cytotoxicity Assay
The following five human cancer lines were used in the cytotoxicity assay: human myeloid leukemia (HL-60), hepatocellular carcinoma (SMMC-7721), lung cancer (A-549), breast cancer (MCF-7), and colon cancer (SW480). All the cells were cultured in RMPI-1640 or DMEM medium (Hyclone, Logan, UT, USA), supplemented with 10% FBS (Hyclone, USA) in 5% CO2 at 37°C. The cytotoxicity assay was performed according to the MTT [3-(4,5-dimethyl- thiazol-2-yl)-2,5-diphenyltetrazolium bromide] method in 96-well microplates.34 Briefly, adherent cells (100 μL) were seeded into each well of a 96-well cell culture plate and allowed to adhere for 12 hrs before drug addition, while suspended cells were seeded just before drug addition, both with an initial density of 1 × 105 cells/mL in 100 μL of the medium. Each tumor cell line was exposed to the test compound at various concentrations in triplicate for 48 hrs. After the incubation, MTT (100 μg) was added to each well, and the incubation continued for 4 hrs at 37°C. The cells were lysed with SDS (200 μL) after removal of 100 μL of the medium. The optical density of lysate was measured at 595 nm in a 96-well microtiter plate reader (Bio-Rad 680). IC50 values were calculated by Reed and Muench’s method.35,36
Calculated Molecular Physicochemical Properties
All structures of podophyllotoxin derivatives were built and energy minimized by the Tripos force field with 0.05 kcal/(mol Å). The Gasteiger–Huchel method was used to calculate charges. Energy minimization was performed by the Powell method with 2000 iterations. Molecular surface area (MSA), polar surface area (PSA), calculated partition coefficient (ClogP), calculated solubility (ClogS), hydrogen bond donor (HD), hydrogen bond acceptor (HA) and rotatable bond (RB) were obtained from MarvinSketch version 5.3.8. (www.chemaxon.org).37
Conclusion
In conclusion, we synthesized a few glucoside derivatives of podophyllotoxin and screened for cytotoxicity against a panel of five human cancer cell lines including HL-60 (leukemia), SMMC-7721 (hepatoma), A-549 (lung cancer), MCF-7 (breast cancer), and SW480 (colon cancer). Derivatives having a free glucose residue show weak activity (IC50 > 40 μM), while the peracetylated derivative 6b shows the highest cytotoxic potency against all five cancer cell lines tested, with IC50 values ranging from 3.27±0.21 to 11.37±0.52 μM. Compound 6b also displays moderate selectivity toward cancer cells over normal human pulmonary epithelial cells (BEAS-2B). The calculated physicochemical properties of these PPT derivatives indicated that more lipophilic compounds are generally more cytotoxic to cancer cells. Our results suggest that some of these compounds have potential as lead compounds for developing novel anticancer agents.
Acknowledgment
We are grateful to the National Nature Science Foundation of China for financial support (No. 21602196); the Yunnan Provincial Science and Technology Department (Nos. 2017ZF003, 2017FD084, and 2017FG001-046); and Yunnan Agricultural University Natural Science Foundation for Young Scientists (No. 2015ZR08).
Disclosure
The authors declare no conflicts of interest in this work.
References
1. Gomes TRH, Fernando MH, Regnia CM. Global trends in nanomedicine research on triple negative breast cancer: a bibliometric analysis. Int J Nanomedicine. 2018;13:2321–2336. doi:10.2147/ijn.s164355
2. Newman DJ, Cragg GM, Snader KM. Natural products as sources of new drugs over the period 1981-2002. J Nat Prod. 2003;66:1022–1037. doi:10.1021/np030096l
3. Newman DJ. Natural products as leads to potential drugs: an old process or the new hope for drug discovery? J Med Chem. 2008;51:2589–2599. doi:10.1021/jm0704090
4. Ojima I. Modern natural products chemistry and drug discovery. J Med Chem. 2008;51:2587–2588. doi:10.1021/jm701291u
5. MacRae WD, Hudson JB, Towers BH. The antiviral action of lignans. Planta Med. 1989;55:531–535. doi:10.1055/s-2006-962087
6. Umesha B, Basavaraju YB, Mahendra C. Synthesis and biological screening of pyrazole moiety containing analogs of podophyllotoxin. Med Chem Res. 2015;24:142–151. doi:10.1007/s00044-014-1100-3
7. You YJ. Podophyllotoxin derivatives: current synthetic approaches for new anticancer agents. Curr Pharm Des. 2005;11:1695–1717. doi:10.2174/1381612053764724
8. Gordaliza M, Garcia PA, Miguel Del Corral JM, Castro MA, Gomez-Zurita MA. Podophyllotoxin: distribution, sources, applications and new cytotoxic derivatives. Toxicon. 2004;44:441–459. doi:10.1016/j.toxicon.2004.05.008
9. Lv M, Xu H. Recent advances in semisynthesis, biosynthesis, biological activities, mode of action, and structure-activity relationship of podophyllotoxins: an update (2008-2010). Mini-Rev Med Chem. 2011;11:901–909. doi:10.2174/138955711796575461
10. Zhang X, Rakesh KP, Shantharam CS, et al. Podophyllotoxin derivatives as an excellent anticancer aspirant for future chemotherapy: a key current imminent needs. Bioorg Med Chem. 2018;26:340–355. doi:10.1016/j.bmc.2017.11.026
11. Lee KH, Imakura Y, Haruna M, et al. Antitumor agents, 107. New cytotoxic 4-alkylamino analogues of 4ʹ-demethyl-epipodophyllotoxin as inhibitors of human DNA topoisomerase II. J Nat Prod. 1989;52:606–613. doi:10.1021/np50063a021
12. Le KH, Beers SA, Mori M, et al. Antitumor agents. 111. New 4-hydroxylated and 4-halogenated anilino derivatives of 4ʹ-demethylepipodophyllotoxin as potent inhibitors of human DNA topoisomerase II. J Med Chem. 1990;33:1364–1368. doi:10.1021/jm00167a013
13. Wang ZQ, Kuo YH, Schnur D, et al. Antitumor agents. 113. New 4 beta-arylamino derivatives of 4ʹ-O-demethylepipodophyllotoxin and related compounds as potent inhibitors of human DNA topoisomerase II. J Med Chem. 1990;33:2660–2666. doi:10.1021/jm00171a050
14. Jardine I. Podophyllotoxins. In: Cassady JM, Douros JD, editors. Anticancer Agents Based on Natural Product Models. New York, NY, USA: Academic Press; 1980:319–351.
15. Reddy DM, Srinivas J, Chashoo G, Saxena AK, Sampath Kumar HM. 4β-[(4-Alkyl)-1,2,3-triazol-1-yl] podophyllotoxins as anticancer compounds: design, synhtesis and biological ecaluation. Eur J Med Chem. 2011;46:1983–1991. doi:10.1016/j.ejmech.2011.02.016
16. Zi CT, Yang D, Dong FW, et al. Synthesis and antitumor activity of novel per-butyrylated glycosides of podophyllotoxin and its derivatives. Bioorg Med Chem. 2015;23:1437–1446. doi:10.1016/j.bmc.2015.02.021
17. Zi CT, Liu ZH, Li GT, et al. Synthesis, and cytotoxicity of perbutyrylated glycosides of 4β-triazolopodophyllotoxin derivatives. Molecules. 2015;20:3255–3280. doi:10.3390/molecules20023255
18. Zi CT, Li GT, Li Y, et al. Synthesis and anticancer activity of 4β-triazole-podophyllotoxin glycosides. Nat Prod Bioprospect. 2015;5:83–90. doi:10.1007/s13659-015-0057-3
19. Zi CT, Xu FQ, Li GT, et al. Synthesis and anticancer activity of glucosylated podophyllotoxin derivatives linked via 4β-triazole rings. Molecules. 2013;18:13992–14012. doi:10.3390/molecules181113992
20. Reddy PB, Paul DV, Agrawal SK, Saxena AK, Kumar HM, Qazi GN. Design, synthesis, and biological testing of 4beta-[(4-substituted)-1,2,3-triazol-1-yl]podophyllotoxin analogues as antitumor agents. Arch Pharm. 2008;341:126–131. doi:10.1002/ardp.200700116
21. Stähelin HF, von Wartburg A. The chemical and biological route from podophyllotoxin glucoside to etoposide: ninth Cain memorial Award lecture. Cancer Res. 1991;51:5–15.
22. Gu XY, Chen L, Wang X, et al. Direct glycosylation of bioactive small molecules with glycosyl iodide and strained olefin as acid scavenger. J Org Chem. 2014;3:1100–1110. doi:10.1021/jo402551x
23. Daley L, Guminski Y, Demerseman P, et al. Synthesis and antitumor activity of new glycosides of epipodophyllotoxin, analogues of etoposide, and NK 611. J Med Chem. 1998;41:4475–4485. doi:10.1021/jm9800752
24. Allevi P, Anastasia M, Ciuffred P, Sanvito AM, Macdonald P. A short and simple synthesis of the antitumor agent etoposide. Tetrahedron Lett. 1992;33:4831–4834. doi:10.1016/S0040-4039(00)61297-2
25. Kuhn M, von Wartburg A. Über ein neues glykosidierungsverfahren synthese von epipodophyllotoxin-β-D-glucopyranosid. 21. Mitt. Über mitosehemmende Naturstoffe. Helv Chim Acta. 1968;51:1631–1641. doi:10.1002/hlca.19680510719
26. Cohen RB, Tsou KC, Rutenburg SH, Seligman AM. The colorimetric estimation and histochemical demonstration of beta-d-galactosidase. J Biol Chem. 1952;195:239–249.
27. Kartha KP, Field RA. Iodine: a versatile reagent in carbohydrate chemistry IV. Per-O-acetylation, regioselective acylation and acetolysis. Tetrahedron. 1997;53:11753–11766. doi:10.1016/S0040-4020(97)00742-4
28. Fiandor J, García-López MT, De Las Heras FG, Méndez-Castrillón PP. A facile regioselective 1-O-deacylation of peracylated glycopyranoses. Synthesis. 1985;12:1121–1123. doi:10.1055/s-1985-31446
29. Hansen HF, Jesen RB, Willumsen AM, et al. New compounds related to podophyllotoxin and congeners: synthesis, structure elucidation and biological testing. Acta Chem Scand. 1993;47:1190–1200. doi:10.3891/acta.chem.scand.47-1190
30. Zi CT, Yang L, Gao W, et al. Click glycosylation for the synthesis of 1,2,3-triazole-linked picropodophyllotoxin glycoconjugates and their anticancer activity. ChemistrySelect. 2017;2:5038–5044. doi:10.1002/slct.201700347
31. Brewer CF, Loike JD, Horwitz SB, Sternlicht H, Gensler WJ. Conformational analysis of podophyllotoxin and its congeners. Structure-activity relationship in microtubule assembly. J Med Chem. 1979;22:215–221. doi:10.1021/jm00190a007
32. de Oliveira PF, Alves JM, Damasceno JL, Oliveira RAM, Dias HJ, Crotti AEM. Cytotoxicity screening of essential oils in cancer cell lines. Rev Bras Farmacogn. 2012;22:88–93.
33. Shi JF, Wu P, Jiang ZH, Wei XY. Synthesis and tumor cell growth inhibitory activity of biotinylated annonaceous acetogenins. Eur J Med Chem. 2014;71:219–228. doi:10.1016/j.ejmech.2013.11.012
34. Malich G, Markovic B, Winder C. The sensitivity and specificity of the MTS tetrazolium assay for detecting the in vitro cytotoxicity of 20 chemicals using human cell lines. Toxicology. 1997;124:179–192. doi:10.1016/s0300-483x(97)00144-3
35. Zamoiskii EA. Evaluation of Reed-Muench method in determination of activity of biological preparations. Zh Mikrobiol Epidemiol Immunobiol. 1956;27:77–83.
36. Stanic MA. simplification of the estimation of the 50 percent endpoints according to the Reed and Muench method. Pathologia Et Microbiologia (basel). 1963;26:298–302.
37. Jeong YC, Anwar M, Moloney MG. Synthesis, antibiotic activity and structure–activity relationship study of some 3-enaminetetramic acids. Bioorg Med Chem Lett. 2014;24:1190–1200. doi:10.1016/j.bmcl.2014.03.013
38. Hartwell JL, Schrecker AW. Components of Podophyllin V. The Constitution of Podophyllotoxin. J Am Chem Soc. 1951;73:2909–2916. doi:10.1021/ja01150a143
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.