")
Back to Journals » Cancer Management and Research » Volume 12
Glucometabolic Reprogramming in the Hepatocellular Carcinoma Microenvironment: Cause and Effect
Authors Tian H, Zhu X , Lv Y , Jiao Y , Wang G
Received 14 April 2020
Accepted for publication 30 June 2020
Published 17 July 2020 Volume 2020:12 Pages 5957—5974
DOI https://doi.org/10.2147/CMAR.S258196
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Seema Singh
Huining Tian,1 Xiaoyu Zhu,2 You Lv,1 Yan Jiao,3 Guixia Wang1
1Department of Endocrinology and Metabolism, The First Hospital of Jilin University, Changchun 130021, Jilin, People’s Republic of China; 2Department of Nephrology, The First Hospital of Jilin University, Changchun 130021, Jilin, People’s Republic of China; 3Department of Hepatobiliary and Pancreatic Surgery, The First Hospital of Jilin University, Changchun 130021, Jilin, People’s Republic of China
Correspondence: Guixia Wang
Department of Endocrinology and Metabolism, The First Hospital of Jilin University, 71 Xinmin Street, Chaoyang District, Changchun 130021, Jilin, People’s Republic of China
Tel +86 158 0438 1103
Fax +86 431 8878 6259
Email [email protected]
Yan Jiao
Department of Hepatobiliary and Pancreatic Surgery, The First Hospital of Jilin University, 71 Xinmin Street, Chaoyang District, Changchun 130021, Jilin, People’s Republic of China
Tel/ Fax +86 13843101157
Email [email protected]
Abstract: Hepatocellular carcinoma (HCC) is a tumor that exhibits glucometabolic reprogramming, with a high incidence and poor prognosis. Usually, HCC is not discovered until an advanced stage. Sorafenib is almost the only drug that is effective at treating advanced HCC, and promising metabolism-related therapeutic targets of HCC are urgently needed. The “Warburg effect” illustrates that tumor cells tend to choose aerobic glycolysis over oxidative phosphorylation (OXPHOS), which is closely related to the features of the tumor microenvironment (TME). The HCC microenvironment consists of hypoxia, acidosis and immune suppression, and contributes to tumor glycolysis. In turn, the glycolysis of the tumor aggravates hypoxia, acidosis and immune suppression, and leads to tumor proliferation, angiogenesis, epithelial–mesenchymal transition (EMT), invasion and metastasis. In 2017, a mechanism underlying the effects of gluconeogenesis on inhibiting glycolysis and blockading HCC progression was proposed. Treating HCC by increasing gluconeogenesis has attracted increasing attention from scientists, but few articles have summarized it. In this review, we discuss the mechanisms associated with the TME, glycolysis and gluconeogenesis and the current treatments for HCC. We believe that a treatment combination of sorafenib with TME improvement and/or anti-Warburg therapies will set the trend of advanced HCC therapy in the future.
Keywords: hepatocellular carcinoma, tumor microenvironment, glycolysis, gluconeogenesis, Warburg effect
Introduction
Liver cancer is the second leading cause of cancer mortality worldwide and the 7th most frequently diagnosed cancer worldwide, with approximately 782,000 deaths and 841,000 new cases diagnosed annually.1 Hepatocellular carcinoma (HCC) is the major type of primary liver cancer (PLC) and accounts for 75–85% of cases.2 The main risk factors for HCC are hepatitis B virus (HBV), hepatitis C virus (HCV), cirrhosis, aflatoxin-contaminated foodstuffs, alcohol abuse, obesity, and type 2 diabetes.1,3–5 Decades ago, Otto Warburg observed that cancer cells rely on glycolysis for the generation of energy even in a normoxic environment, which was known as the “Warburg effect” or “aerobic glycolysis”.6,7 Aerobic glycolysis not only provides energy but also provides intermediates (nucleotides, amino acids, lipids and NADPH) for biosynthesis,8,9 which explains why aerobic glycolysis occurs prior to oxidative phosphorylation (OXPHOS) in proliferation cells such as tumor cells. The distinct proliferation characteristics and glucometabolic reprogramming of tumor create a unique TME different from the overall human environment. The HCC microenvironment consists of various cell types, growth factors, proteolytic enzymes, extracellular matrix (ECM) proteins and cytokines, which are widely known to contribute to hypoxia, acidosis and immune suppression.10 The “suitable” environment provided by the tumor microenvironment (TME) contributes to tumor proliferation, angiogenesis, invasion and metastasis. Aerobic glycolysis and TME can interact with each other and create a vicious spiral.
However, as the major metabolic organ in the body, liver plays an important role in glucose homeostasis by regulating synthesis and decomposition of glycogen. During fasting, approximately 80% of endogenous glucose is produced by liver through gluconeogenesis.11,12 Gluconeogenesis is actually a reverse pathway of glycolysis and can inhibit glycolysis through downstream gluconeogenesis enzymes, such as phosphoenolpyruvate carboxykinase1 (PCK1) and fructose-1,6-bisphosphatase 1 (FBP1).13,14 In addition, gluconeogenesis uses lactate as one of the substrates to consume harmful byproducts of glycolysis. This glucose-metabolizing feature offers a unique opportunity to treat HCC. Nevertheless, the decrease of PCK1 and FBP1 expression in HCC compared to normal liver tissue lead to the suppression of gluconeogenesis and elevation of glycolysis.15,16 As an emerging hallmark of tumors, studies regarding glucose metabolism reprogramming used to focus on glycolysis. However, the correlation between gluconeogenesis and tumors is rarely reported but may provide insight for the treatment of HCC. In this review, we summarized the interaction between glucometabolic reprogramming and the HCC microenvironment. Furthermore, we discussed HCC treatment targeting the improvement of the TME, suppression of glycolysis and elevation of gluconeogenesis aiming to find promising metabolism-related therapeutic targets of HCC.
Hypoxic Microenvironment
Hypoxia is a typical microenvironment feature in nearly all solid tumors, and it contributes to their rapid and uncontrolled proliferation.17 Hypoxia-inducible factors (HIFs) are key transcription factors produced by tumor cells under hypoxia to cope with the hypoxic microenvironment. Furthermore, HIFs contribute to invasive growth, survival, metastasis, treatment resistance and poor prognosis of HCC.18 The HIF family includes three subtypes: HIF-1, HIF-2, and HIF-3. Among them, HIF-1 and HIF-2 are considered to be the most important factors for cells to respond to hypoxia. HIF-1 and HIF-2 consist of an oxygen-sensitive subunit HIF-α and a constitutively expressed HIF-β subunit.19,20 Both HIF-1α and HIF-2α are reported correlating with tumors. Studies have shown that HIF-1α regulates vascular endothelial growth factor (VEGF) during the acute phase of hypoxia, while VEGF is mainly regulated by HIF-2α during long-term hypoxia.21 HIF-2α is overexpressed in primary and metastatic tumors22 and is positively correlated with tumor angiogenesis.23 However, studies on HIF-2α and liver cancer are rare, and HIF-1α is the primary factor in liver cancer hypoxia. In the presence of oxygen, HIF-1α is hydroxylated by prolyl hydroxylases (PHDs), leading to its rapid proteasomal degradation. Under hypoxic conditions, PHDs are no longer active to hydroxylate HIF-1α. HIF-1α will be stabilized and translocated to the nucleus.24
Accumulation of HIF-1α can influence tumor survival and proliferation by regulating tumor glycometabolism in the following four ways. First, HIF-1α can increase the uptake of glucose by upregulating the expression of glucose transporters (GLUT) such as GLUT1. Second, HIF-1α promotes the expression of glycolytic enzymes and accelerates the conversion of glucose to pyruvate. Third, HIF-1a can phosphorylate pyruvate dehydrogenase (PDH) by inducing the expression of pyruvate dehydrogenase kinase (PDK) and inactivate the PDH to prevent the conversion of pyruvate to acetyl CoA. Fourth, HIF-1a upregulates the expression of lactate dehydrogenase A (LDHA) to stimulate the production of lactic acid.25
In addition to the effect the of the glycometabolism of HCC, HIF-1α can also influence HCC survival by regulating the oxidative stress level.26 Oxidative stress can mediate mitochondrial apoptosis and the immune response in liver cancer.27 Reactive oxygen species (ROS), byproducts of oxygen metabolism, are the main causes of oxidative stress, and their concentration changes play dual functions in the regulation of HCC process.28,29 Low levels of ROS may induce DNA mutation by oxidative DNA damage, which eventually increases the likelihood of HCC development.30,31 Overexpression of ROS can inhibit HCC by inducing apoptosis of hepatoma cells and inhibiting metastasis through ROS/Akt/NF-kB pathway, and suppressing liver cancer stem cell via ROS/β-Catenin/FOXO3a Signaling.32–34 In a hypoxic environment, the low oxygen content and the lack of oxygen as electron recipient lead to the imbalance of electron flow through the mitochondrial electron chain, which contributes to the accumulation of ROS and causes irreversible cellular damages in tumors.35,36 However, HIF-1α can promote HCC progression by preventing ROS accumulation through the following pathways. First, HIF-1α prevents pyruvate from entering TCA cycle by inactivating PDH through PDKs and the conversion of pyruvate to lactate by upregulating LDHA expression.21,37,38 HIF-1α therapy ensures that circulating tricarboxylic acid cycle (TCA) substrates cannot enter mitochondrial oxidation.36,39 Second, HIF-1α can reduces ROS accumulation by inhibiting ROS production sites in the electron transport chain (ETC), such as complexes 1 and 4.40,41 Third, HIF-1α decreases the number of mitochondrial cristae and the mitochondrial mass through HEY1/PINK1 pathway, and degrading mitochondria by inducing BNIP3 to restrict ROS production and promote ROS elimination.42 Glutamine is a key source of carbon, secondary only to glucose.43 Decomposition of glutamine will replenish the TCA cycle and provide abundant carbon and nitrogen for hepatocyte growth and proliferation.44,45 Some of the carbon can be used to produce NADPH to achieve redox equilibrium.46 At the same time, glutamic acid produced by glutamine decomposition will directly synthesize the antioxidant glutathione and neutralize ROS.47 Through various pathways, the tumor cells will eventually maintain ROS at an appropriate level that is conducive to their own growth and proliferation. At present, most chemotherapy drugs and radiotherapy kill tumor cells by inducing ROS production.48 Hence, interfering with or reversing of hypoxia and its effects or identifying a suitable way to increase ROS level in tumor cells can reduce the drug resistance of tumors and improve the therapeutic effect.32,49,50 In turn, tumor cell aerobic glycolysis can influence HIF-1α by upregulating glutamine. The TCA cycle is the hinge of metabolism of glucose, fat and amino acid. Glutamine is the most abundant nonessential amino acid in blood serum. The “Warburg effect” of tumors leads to the conversion of pyruvate into lactic acid and the lack of carbon source for TCA cycle. Glutamine is not only a nitrogen source for amino acids and nucleotide synthesis but also the main carbon source for TCA cycle and macromolecule biosynthesis.51 Many tumor cells require much more glutamine than normal cells. Tumor cells take up a large amount of glutamine and then convert it into other metabolic intermediates, meeting the energy requirements for rapid proliferation.52 Glutamine can regulate the stability of HIF-1a in response to hypoxia and support the survival of HCC cells by upregulating proline and hydroxyproline levels.53 The increase in the level of glutamine further exacerbates tumor hypoxia. Thus, targeting glutamine could be a new strategy in oncotherapy.
Acid-Base Microenvironment
Acid-base characteristic of the TME is widely recognized as the acidification of extracellular pH (pHe), which is so-called tumor acidosis (Figure 1). Tumor cells have a lower pHe of ~6.7–7.1 and a higher intracellular pH (pHi) ≥7.4 rather than a higher pHe of ~7.4 and a lower pHi of ~7.2 in normal cells.54 Recently scientists have proposed using high-resolution pH mapping to monitor pHe in HCC, which could be a biomarker for metabolic changes and monitoring tumor aggressiveness and therapeutic outcome.55–57 Tumor acidosis is the consequence of lactate and H+ ions accumulation, which are produced by glycolysis and oxidative metabolism. Most tumor cells, also called as glycolytic tumor cells, prefer glycolysis rather than OXPHOS, leading to increases in lactate and H+ production. However, some tumor cells still use oxidative metabolism and are called oxidative tumor cells.58 The accumulation of lactate in HCC microenvironment is mainly due to the increases of intracellular lactate production and extracellular transport. The interconversion of pyruvate and lactate plays a critical role in intracellular lactate production, which is primarily catalyzed by the lactate dehydrogenase (LDH) family.59 LDH enzymes with high M-subunits (encoded by LDHA) promote the conversion from pyruvate to lactate.59 In contrast to LDHA, LDH enzymes with high H-subunits are encoded by lactate dehydrogenase B (LDHB) and promote the conversion from lactate to pyruvate.59 Moreover, pyruvate dehydrogenase kinase (PDK) can prevent pyruvate from entering mitochondria for OXPHOS.60 Upregulation of LDHA and PDK synergistically promotes the production of lactate in HCC.60,61 Monocarboxylate transporter (MCT) expression on tumor cell membranes is associated with lactate passive transport and prevents glycolytic tumor cells from intracellular lactate accumulation.59,62 MCT1 and MCT4 are major proteins expressed in tumors. MCT1 is a high-affinity lactate transporter that participates in exogenous lactate uptake by endothelial cells and oxidative tumor cells.62 MCT4 is a low-affinity lactate transporter that promotes lactate release from glycolytic tumor cells.63 Although lactate could be incepted as a fuel, lactate accumulation in the TME still exists due to a large amount of lactate release. As a production of oxidative metabolism, CO2 can be hydrated to H2CO3 and then dissociates to HCO3− + H+.64 Furthermore, the production of H+ can also be associated with the metabolism of amino acids and the hydrolysis of ATP. In addition to lactate/H+ symporter MCTs, H+ can also be actively transported by H+-ATPases and Na+/H+ exchangers (NHEs).65 However, carbonic anhydrases (CAs) colocalize with Na+/HCO3− cotransporters (NBCs)66 transporting Na+ and HCO3− into tumor cells and maintaining a mildly alkaline level of pHi and a dynamic balance of Na+.65 V-ATPase, CAIX and CAXII are selectively overexpressed in HCC.67
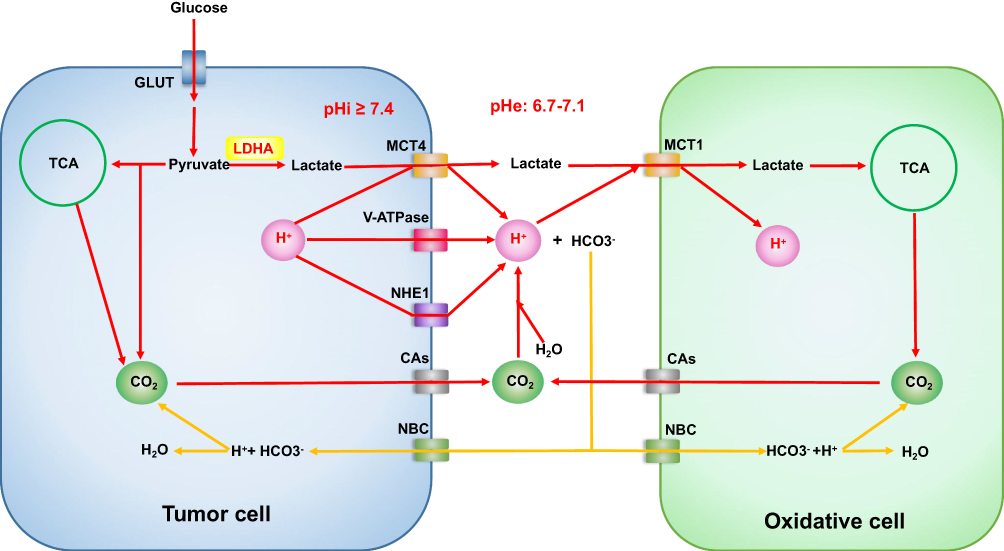 |
Figure 1 Overview of tumor acid-base microenvironment. OXPHOS of the oxidative cells is a compensatory mechanism of tumor acidosis, but the acidosis cannot be reversed. Abbreviations: GLUT, glucose transporters; TCA, tricarboxylic acid cycle; LDH-A, lactate dehydrogenase A; MCT4, monocarboxylate transporter4; MCT1, monocarboxylate transporter1; V-ATPase, vacuolar ATPase; NHE1, sodium-hydrogen exchanger1; CAs, carbonic anhydrases; NBC, sodium bicarbonate cotransporters. |
TME acidosis could influence HIFs reprogramming by increased O2 consumption. An upregulation of HIF-1α under acidosis has been reported in glioma and HEK293 cell.68,69 Additionally, HIF-2α has been reported to play a critical role in regulating metabolic adaptation to acidosis in liver cancer and glioma.68,69 Mild extracellular acidosis could restructure mitochondria and promote mitochondria fusion. Excess H+ and lactate decrease immunological cell function by inhibiting glycolysis and IFN-γ production. Moreover, TME acidosis has been reported to contribute to angiogenesis, invasion and metastasis.65
Immune Microenvironment
Tumor immune microenvironment of liver cancer is mainly associated with T cells, NK cells and tumor-associated macrophages (TAMs). In HCC, dysfunction of the above immune cells leads to reductions in inflammation and the immune response, which contributes to tumor progression.70 Metabolism reprogramming in these cells is closely associated with their functional change in the TME.
T cells play a critical role in antitumor immunity. Different subsets of T cells incline to different types of metabolism pathways. Naïve T cells have two types of subsets: CD4+ T cells and CD8+ T cells. Both of them express a resting mode for OXPHOS, which is accompanied by low lactate levels and low nutrient uptake.71,72 After activation, both CD4+ and CD8+ naïve T cells differentiate into long-lived memory T cells (TM cells) and short-lived effector T cells (TE cells).73–75 CD4+ T cells can differentiate into two affected subsets: helper T cells (Th cells) and regulatory T cells (Treg cells).75–77 CD8+ T cells can also differentiate into cytotoxic T cells (CTL) and Treg cells upon activation.78 Metabolic reprogramming occurs during the process of activation. Long-lived TM cells and Treg cells tend to go through fatty acid oxidation (FAO).79–83 FAO in TM cells can fuel OXPHOS and enhance mitochondrial capacity, which could be a sign of rapid response to infection or cancer recurrence.84 TE cell expansion can be accomplished in just a few days for the immune response and most TE cells die after antigen clearance. However, short-lived TE cells require a fast energy supply.85,86 Both elevated aerobic glycolysis and OXPHOS have been observed in TE cells (except Treg cells) activation.85 Many scientists pointed out that aerobic glycolysis/OXPHOS levels are upregulated in these activated TE cells compared to resting T cells, which indicates a “switch” from OXPHOS to aerobic glycolysis.82,85,87-90 Metabolism reprogramming is associated with T cells fate, in which mitochondria play a critical role.91 The function of mitochondria is correlated with the number and polarization of mitochondria, the number and length of mitochondrial cristae, and mitochondrial dynamics.85 TE cells with a dominant metabolic pathway of aerobic glycolysis have punctate mitochondria, which accelerate cell proliferation.79,92 TM cells with a dominant metabolic pathway of OXPHOS maintain fused networks,79,92 which maximize OXPHOS activity. Control of Treg cells suppress function by mitochondria is closely related to mitochondrial complex III.81 Crosstalk among these cells keeps the dynamic balance of immune response in vivo. TE cells play a role as immune guards, and TM cells can be supplements for TE cells to react rapidly after restimulation. Tregs can inhibit overreaction of TE cells and are essential for protection from autoimmunity and excessive inflammation.93 However, the disruption of this balance leads to the immune escape during tumorigenesis.
Tumorigenesis starts from mutations of tumor-related genes and an uncontrolled cell cycle. In addition to uncontrolled proliferation, tumor progression and migration require the suppression of self-programmed death and evasion of immunosurveillance.94 Immune tolerance and immune evasion play a critical role in poor HCC prognosis and are always concomitant with energy, exhaustion and senescence of T cells, especially TE cells.95 In the TME, T cells’ metabolic pathways can be influenced by glucose competition, extracellular lactate accumulation and interaction between tumor cells and T cells. As a result of glucose competition, T cells cannot obtain enough glucose, which inhibits T cell proliferation and function.87,95 The glucose transporter GLUT1 of T cells is downregulated in tumors, which is essential for glucose uptake and aerobic glycolysis. Macintyre et al suggested that GLUT1 deficiency prevented CD4+ T cells activation and effector functions, and that TE cell expansion and IFN-γ production decreased.96 Stromal cells (tumor cells and cancer-associated fibroblasts) contribute to lactate levels, which lead to cell invasiveness and metastasis.92 It has been demonstrated that tumor cells can inhibit T cell proliferation by expressing indoleamine 2.3-dioxygenase and inhibit T cell function by deriving lactate and blocking lactate export in T cells.97,98 As a byproduct of the “Warburg effect”, lactate production and acidification can lead to immune evasion by diminishing the IFN-γ production of T cells through the NF-kB pathway.99 In contrast, Treg cell proportions can be found increased in the HCC microenvironment.100 It has been reported that tumor-infiltrating Treg cells express higher levels of GLUT1 and glycolysis-related genes than TE cells on the cell surface, which leads to a higher uptake of glucose and an increased level of aerobic glycolysis.101 Treg cells play a synergistic action with tumor cells for glucose competition, which induces T cell senescence and exhaustion by starving effector T cells.102 In the TME, immune tolerance and evasion are only displayed in local tumor rather than the whole body, which is tightly associated with the interaction between tumors and T cells by “immune checkpoints”.103
Immune checkpoints can be negative regulators of the immune response by inhibiting effector lymphocytes (Figure 2). When TE cells are activated, generation of IFN-γ could enhance the antigen presentation and promote T cell maturity. Nevertheless, scientists found that this process could upregulate expression of immune checkpoints, such as programmed death-1 (PD-1). This upregulation could provide negative feedback for the immune response in the normal microenvironment, preventing damage from a hyperimmune response and maintaining peripheral tolerance. However, tumors can take advantage of this mechanism of immune evasion. PD-1 and cytolytic T lymphocyte-associated antigen-4 (CTLA-4), the most focused checkpoints for T cells, are correlated with glucose metabolism.104 PD-1, programmed death-1, is also known as CD279, PDCD1, SLEB2, hPD-1, hPD-l, hSLE1 [NCBI Gene ID: 5133]. A high level of PD-1 expression can be a characteristic of exhausted T cells.105 After being activated by its ligands, such as programmed death-ligand (PD-L1) (CD274, B7-H, B7H1, hPD-L1, PDCD1L1, PDCD1LG1 [NCBI Gene ID: 29,126]), PD-1 can send inhibitory signals to T cells. Expression of myocyte-specific enhancer factor 2D (MEF2D) by HCC cells can upregulate their PD-L1 expression and enhance their combination with PD-1.106 PD-1 can inhibit aerobic glycolysis in T cells in 3 ways: i, PD-1 inhibits expression of GLUT1 leading to reductions in glucose uptake and transmission; ii, PD-1 inhibits a rate-limiting enzyme of aerobic glycolysis, hexokinase2 (HK2); and iii, PD-1 reduces mitochondrial number and induces mitochondrial dysfunction by reducing the number and length of mitochondrial cristae.85,89 In addition, PD-1 induces FAO by upregulating the rate-limiting enzyme of FAO, carnitine palmitoyltransferase (CPT1A).89 Metabolic reprogramming from aerobic glycolysis to FAO makes the dynamics lean toward long-lived TM cells.89 PD-L1 expression in tumor cells is associated with vascular formation in HCC patients.107 PD-1/PD-L1 can be a target for the treatment of HCC. Blocking PD-1 can reinvigorate exhausted CD8+ T cells and program them into durable memory CD8+ T cells.108 However, this reinvigoration CD8+ T cells can be re-exhausted in a high antigen concentration environment.108 Conversely, CTLA-4 inhibits aerobic glycolysis rather than enhancing FAO.89 Blockade of PD-1 and CTLA-4 can reverse the inhibition of aerobic glycolysis and effector function in T cells.85,89,92,95,109 Different from off-targets of the classical immune checkpoint blockers, Treg cells are sensitive to anti-CTLA-4 antibodies and can induce antibody-dependent cell-mediated cytotoxicity.110,111 T cell immunoglobulin and ITM domain (TIGIT) is also involved in the regulation of CD8+ T cell metabolism by downregulating GLUT1 and HK1/HK2.112
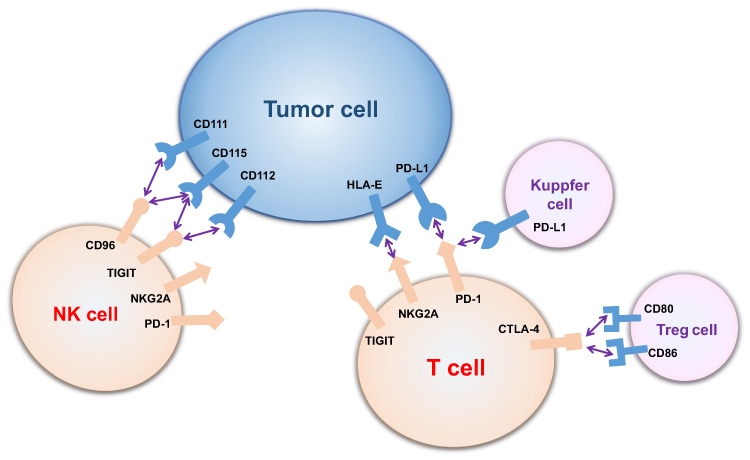 |
Figure 2 Interaction between tumor cells and immune cells by immune checkpoints. T cells and NK cells express various immune checkpoints, which can bind to ligands on tumor cells, Treg cells and Kupffer cells and be inhibited (CD96 binds to CD111 and CD115; TIGHT binds to CD115 and CD112; NKG2A binds to HLA-E; PD-1 binds to PD-L1; and CTLA-4 binds to CD80 and CD86). Abbreviations: PD-1, programmed death-1; CTLA-4, cytolytic T lymphocyte-associated antigen-4; NKG2A, natural killer cell group 2A; TIGIT, T-cell immunoglobulin and ITIM domain; HLA-E, human leukocyte antigen-E. |
Similar to T cells, activated NK cells preferentially go through aerobic glycolysis by maintaining proliferation and effector function and memory NK cells more likely to use FAO to fuel OXPHOS. NK cells can be divided into two different phenotypic and functional subsets depending on the expression levels of CD56 receptor: CD56dim cells and CD56bright cells.113 CD56dim cells are generally deemed to be cytotoxic cells with less GLUT1 expression, whereas the CD56bright cells are considered to be IFN-γ producers with higher GLUT1 expression.113 Either IL-2 or IL-12/15 cytokine combinations can activate NK cells and increase OXPHOS levels for energy supply.114 Sine´ad E. Keating and his colleagues found an interesting phenomenon in which CD56bright cells showed higher GLUT1 expression and levels of aerobic glycolysis than CD56dim cells, leading to a higher level of activation. Downregulation of aerobic glycolysis in CD56bright cells restricts IFN-γ production.113 Lactate accumulation and acidification can impair activation and IFN-γ production of NK cells by diminishing nuclear factor of activated T cells (NFAT).99 Moreover, liver-resident natural killer (LrNK) in the TME displayed a downregulation of NKG2D and impaired cytotoxicity and cytokine production, which could be recovered by IL-15.115 Zhou et al found that LrNK contributes to the tolerogenic microenvironment of the liver by inhibiting TE cells. LrNK inhibits TE cells proliferation and production of INF-γ and TNF-α through a PD-1-PD-L1 axis.116
Immune checkpoints: natural killer cell group 2A (NKG2A)/CD94, TIGIT and CD96 were found to lead to NK cells exhaustion and to predict poor prognosis in HCC.117–119 Scientists found that CD49a+ NK cells, which expressed higher levels of immune checkpoints molecules PD-1, TIGIT and CD96, were correlated with a poor prognosis in HCC patients.120 However, few studies have assessed the correlation between immune checkpoints and metabolic reprogramming in NK cells. NK cell education is the process of NK-cell subsets to obtain functional competence.121 Caroline Pfeifer found that educated NK cells presented with distinct self-inhibitory receptors and went through distinct glycolytic profile and functions.121 Educated NK cells presented with NKG2A (NKG2A-educated NK cells) showed no obvious upregulation in GLUT1 expression, glycolysis or functionality compared with educated NK cells presented with killer cell immunoglobulin (KIR) (KIR-educated cells).121 Furthermore, compared with KIR-educated NK cells NKG2A educated NK cells could better survive glycolysis blockade, allowing NKG2A-educated NK cells to adapt to the hypoxic and low-glucose environment of the tumor.121 Upregulation of the NKG2A ligand on tumor cells further promotes immune evasion from NK cells in the TME.121 In addition, other experiments have proven that anti-NKG2A mAb could block immune evasion by unleashing not only NK cells but also T cells.122
TAM infiltration takes part in tumor invasion and metastasis.123 Macrophages exhibit two diverse phenotypes: M1-classic activation and M2-alternative activation. The M1 type is characterized by pro-inflammatory (IL-1, TNF-α) cytokines and IFN-γ production and can phagocytize tumor cells and induce tumor cell apoptosis. The M2 type is characterized by the production of anti-inflammatory cytokines (IL-6, IL-10, TGF-β) and can induce angiogenesis and tumor cell generation. TAMs were attracted and activated by tumor secretory factors (VEGF, PDGF, TGF-β, CCL2, and M-CSF).10 TAMs mostly exist in the form of M2 in the TME, which could be closely associated with the byproduct of glycolysis in HCC. Lactate secreted by hepatoma cells induces VEGF and arginase1 (Arg1) via HIF-1a to promote M2-like polarization of TAMs.124 As liver-specific macrophages, Kupffer cells survive anoxia by glycolysis and produce PD-L1 ligand to suppress TE cells.125 The dominant TAMs in orthotopic HCC exhibit Kupffer cell (KC) properties and are known as KC-like TAMs (kclTAM).126 TAMs could be “helpers” and target tumorigenesis and development.
Signaling Pathways Involved in HCC Glucometabolic Reprogramming
Tumor cells prefer to choose glycolysis over OXPHOS in hypoxic or even normoxic environment, relying on HIF-1α and c-MYC synergies.127 c-MYC also plays as important a role in glycolysis in HCC as HIF-1α. The importance of collaboration between c-MYC and HIF-1α was demonstrated to activate the Warburg effect by inhibiting IDH1-AS1 in multiple tumors under normal oxygen.127 c-MYC was reported to participate overexpression of MTR4 in HCC, which drives the expression of glycolytic genes such as GLUT1 and PKM2.128 PFK2 in turn was found to up-regulate c-MYC expression in glioma.129 A positive feedback loop between MYC and PFK2 was demonstrated to sustain tumor cell aerobic glycolysis in a Drosophila tumor model.130 c-MYC could be a promising target for HCC treatment, especially in advanced stages.131,132 Moreover, some cytokines and signal pathways can also directly or indirectly affect the glycolysis of tumor cells by increasing the stability and transcription activity of HIF-1α. The major regulatory mechanisms of HIF-1α involved in HCC glucometabolic reprogramming are described in detail below and shown in Figure 3.
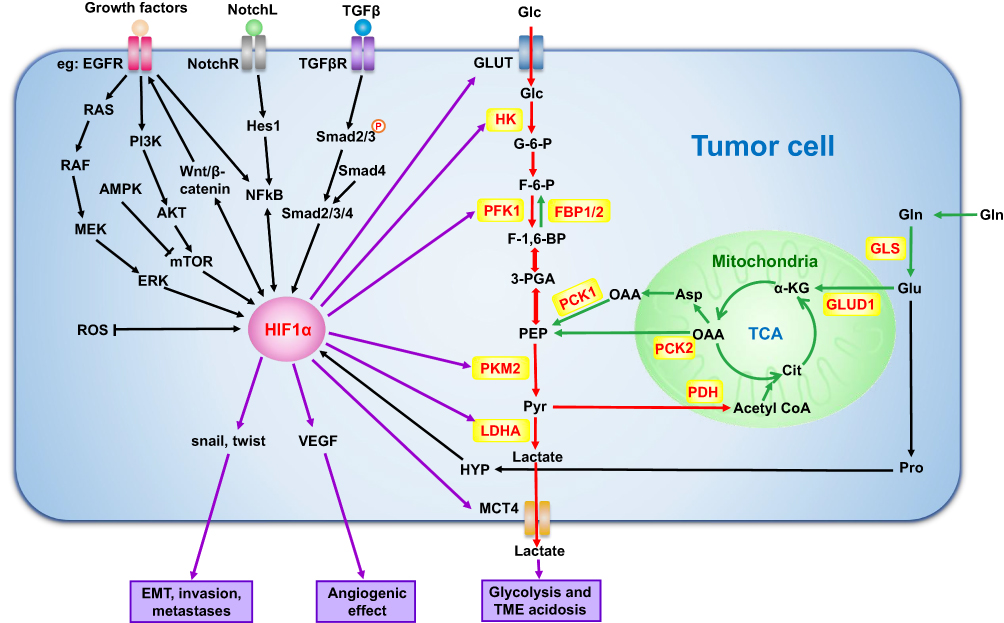 |
Figure 3 Mechanisms of glycolysis regulation by HIF-1α and gluconeogenesis in tumor cells. HIF-1α can directly regulate glycolysis-related enzymes to affect tumor cell glucose metabolism. It can also affect tumor growth and metastasis by regulating the level of active oxygen, EMT and angiogenesis. In addition, cytokines, signaling pathways and glutamine, which play an important role in tumor, can also regulate the biological activity of tumor cells by influencing HIF-1α. Black arrows: pathways regulating HIF-1α; Red arrows: glycolysis-related processes; Green arrows: gluconeogenesis-related processes. Abbreviations: GLS, glutaminase; GLUD1, glutamate dehydrogenase; αKG, α-ketoglutarate; TCA, tricarboxylic acid; GLUT1, glucose transporter1; PFK1, phosphofructokinase; PKM2, pyruvate kinase M2; LDHA, lactate dehydrogenase A; MCT4, monocarboxylate transporter4; VEGF, vascular endothelial growth factor; EMT, epithelial-to-mesenchymal transition; HIF-1α, hypoxia-inducible factor-1α; ICN, the intracelluIar domain of Notch; Glc, glucose; G-6-P, glucose-6-phosphate; F-6-P, fructose-6-phosphate; F-1,6-BP, fructose-1,6-bisphosphate; 3-PGA, glyceraldehyde-3-phosphate; PEP, phosphoenolpyruvate; Pyr, pyruvate; Cit, citrate; OAA, oxaloacetic acid; Asp, asparagine; Glu, glutamate; Gln, glutamine; Pro, proline; HYP, hydroxyproline; NotchL, Notch ligand; NotchR, Notch receptor. |
Phosphatidylinositol-3-kinase (PI3K)/AKT signaling promotes the proliferation of hepatoma cell and EMT in HCC, which contributes to HCC growth, migration and invasion.3,133,134 They transmit cell surface receptor signals and affect a variety of tissue-dependent cellular functions. The PI3K/AKT/mTOR signaling pathway not only directly mediates aerobic glycolysis but also regulates HIF-1α.135 Impairing insulin signaling by inhibiting PI3K/AKT pathway could promote gluconeogenesis in the liver.136 Molecules can treat HCC by inhibiting PI3K/AKT activation, such as MiR-612.137
The Wnt/β-catenin pathway has been reported to occur in both early and late stages of HCC138,139 and suppress mitochondrial respiration and promotes glycolysis.140 HIF-1α can stimulate Wnt/β-catenin signaling via the coactivator BCL9 in HCC.141 Wnt signaling further drives HCC proliferation through MYC, frizzled (FZD), Glypican-3 (GPC3), EGFR and CTNNBIP1.138,142-144 Moreover, activation of the Wnt/β-catenin pathway increases the EMT-associated activity of HIF-1α and enhances the proliferation, EMT, invasion and metastasis of HCC.145,146 Molecules that activate the Wnt/β-catenin pathway can provide therapeutic targets and predictors for molecular precision therapy of HCC, such as LINC00346 and Linc00210 (long noncoding RNAs), and PBOV1 and PROX1 (oncogene).142,143,147,148 PROX1 was also found to be a target for treating HCC sorafenib tolerance.147
Transforming growth factor-β1 (TGF-β1) is a common cytokine that regulates a variety of cellular processes.149 In advanced tumor, TGF-β1 acts as an oncogenic factor and induces tumor proliferation, EMT invasion and metastasis.149,150 TGF-β1 contributes to the metabolic reprogramming of tumor cells by upregulating the expression of key enzymes of the glycolytic pathway via the Smad, p38 MAPK and PI3K/AKT signaling pathways.31 TGF-β1 promotes tumor progression by reducing mitochondrial respiration and enhancing glutamine anaplerosis and the pentose phosphate pathway (PPP) cycle.151 TGF-β1 and its mediated signaling pathway can still induce HIF- α to participate in the process of metabolic reprogramming under normoxic conditions.152
The EGFR/MEK/ERK/HIF-1α/VEGFA cycle regulates glucose metabolism and promotes HCC proliferation, angiogenesis and metastasis.153,154 VEGFs and their cognate receptors (VEGFRs) are critical in the regulation of vessel formation in angiogenesis.155 HIF-1α can induce angiogenesis by binding to the VEGF gene promoter and upregulating VEGF expression.156 In addition, TGF-β1 can induce VEGF expression via Smad and HIF-2α.152
The Notch signaling pathway plays a critical role in crosstalk between glucometabolic reprogramming and HCC microenvironment. High expression of Notch1 indicates a poor prognosis in HCC.157 Notch/Hes1 signaling could induce glycolysis by inactivation of p53 and activation of the NF-kB pathway.126,158 As a target gene of NF-kB, the transcriptional activity of HIF-1α was significantly increased by activated Notch1.158 In turn, HIF-1α was reported to upregulate the expression and function of Notch in HCC.159,160 Notch can promote the proliferation of hepatoma cells through the PI3K-Akt, mTOR and Ras pathways.126 Moreover, Notch inhibits hepatoma cells apoptosis by downregulation of ROS production via the NICD1/Hey1/PINK1 pathway and inactivation of p53.36,126 Notch promotes EMT, invasion and metastasis in HCC through NICD/snail and Wnt3a pathway.157,161-163 However, there is a dispute that blocking Notch promotes HCC progression and metastasis by accelerating proliferation of kclTAMs via Wnt signaling and IL-10 production through c-MYC.164 More evidence is needed in the future.
Unlike the above pathways, AMP-activated protein kinase (AMPK) is the main activation pathway of the anti-Warburg effect in HCC. Activation of AMPK inhibits glycolysis and promotes OXPHOS, which restricts the proliferation of hepatoma cells.165–167 Activation of AMPK/mTOR by glycochenodeoxycholate can also promote HCC invasion and migration by activating autophagy.168 Moreover, upregulation of AMPK reduces the expression of hepatocellular cancer stem cell markers in long-term sorafenib therapy, which provides a new target for overcoming the chemotherapy resistance of HCC.169 AMPK treatment options such as upregulation of HSF1, NOD2 and PEDF or inhibition of 6PGD and GSK-3β could have potential in HCC treatment.165–167,170,171 Among them HSF1 also participates in the promotion of gluconeogenesis.172 Treatments that both inhibit glycolysis and promote gluconeogenesis at the same time are expected to be promising HCC treatment solutions.
Discussion
Along with the improvement of tumor cognition, including disorder of cell cycle, gene mutations and immune evasion, the development of oncotherapy has gone through 3 stages: chemotherapy, targeted therapy and immunotherapy. HCC shows hidden clinical symptoms in the early stage of the disease; thus, the diagnosis often occurs in the advanced stage or metastasis, which is prone to recurrence. Sorafenib is almost the only systemic treatment options for patients with advanced HCC.173 However, sorafenib treatment of advanced HCC is prone to drug resistance and cannot achieve the desired therapeutic effect, which is closely related to the TME.173 Recently, an increasing number of scientists have focused on the effect of TME in tumorigenesis and development, which could be the fourth stage of tumor cognition and therapy. The Warburg effect is the foundation of tumorigenesis, proliferation, migration and metastasis, and it contributes to a unique TME. In this review, we focused on metabolism reprogramming in three aspects of TME: hypoxia, acid-base status and immune microenvironment. Lately, anti-Warburg therapies which not only focus on the characteristics of the TME or directly inhibit glycolysis but also inhibit glycolysis by increasing gluconeogenesis, have become a popular area of research.
Treatments for the Hypoxia Microenvironment
Hypoxia is considered to be a major obstacle to tumor treatment.174 At present, the main idea of hypoxia treatment for HCC is to directly provide/generate oxygen at the tumor site to increase the partial oxygen pressure or indirectly reduce the level of HIF-1α and interfere with a HIF-1α-related signaling pathway to decrease hypoxia effects.17,18 Increasing local oxygen pressure and reversing hypoxia can use nanotechnology to introduce O2 into the tumor or generate oxygen at the tumor site by increasing the decomposition of endogenous hydrogen peroxide and light-triggered water splitting.18,175 However, most of these approaches are in the early stage of development and need more time to evaluate their availability in HCC therapy. Many anticancer drugs aimed at HIF-1α have been reported. Heat shock protein 70 (Hsp70), benzopyranyl 1,2,3-triazole and BIX01294 reduce HIF-1α levels by promoting ubiquitination and proteasome degradation of HIF-1α.176–178 Drugs that inhibit the expression and accumulation of HIF-1α transcriptional activity and protein accumulation include cardenolides and ezn-2208.177,179 Moreover, some inhibitors act on HIF-1α-related signaling pathways, such as glyceollins and apigenin, which inhibit the PI3K/AKT pathway to downregulate HIF-1α.180,181 Semaxanib causes low HIF-1 DNA-binding activity by inhibiting PI3K activity and AKT phosphorylation.182 HIF-1a inhibitors can not only improve the effect of the hypoxia microenvironment on HCC progression but also increase the sensitivity of hepatocytes to targeted therapy. Simvastatin can inhibit HIF-1α/PPAR-γ/PKM2-mediated glycolysis in hepatocytes and resensitize it to sorafenib.183 In addition to treatment, HIF-1-related genes have also been used in the establishment of a novel integrated scoring system, which could contribute to the precise treatment of HCC patients.184 Due to complicated regulations and overlap mechanisms, the clinical trials of HIF-1α inhibitors targeting tumor hypoxia have failed to achieve significantly satisfactory results.
Treatments for the Acid-Base Microenvironment
There are two distinct approaches in tumor acidosis: I. modulating pH to restore chemosensitivity and correct detective immune mechanisms and II. Utilizing an acidic TME to enhance the effect of drugs. The pH can be adjusted in 3 ways: reducing acid production, increasing acid consumption and providing an outside pH buffer. Elevating gluconeogenesis can both reduce acid production and increase acid consumption by inhibiting glycolysis and using lactate as a substrate.185,186 As a promising treatment, the mechanisms and options for increasing gluconeogenesis to treat HCC will be explored in detail in “Treatments for aerobic glycolysis”. Targeted therapy of proton pump/transporters could also reduce acid production. Omeprazole, a proton pump inhibitor (PPI), has been used to analyze the role of V-ATPase in HCC and proved to have a wide range of antitumor effects at the preclinical and clinical levels.187 Since HCC showed partial drug resistance to a CAXII inhibitor compound in an anoxic TME, modifications of compound 25 need to be studied to improve its antitumor effect.67 As mentioned above, outside provision of a pH buffer could be anti-acidifying strategies in HCC. Oral or transarterial chemoembolization (TACE) pH buffer can restrict local invasive growth and metastasis by reducing intratumoral and peritumoral acidosis rather than altering the pH of healthy tissues or blood.188–190 Patients with large HCC showed a marked enhancement of the anticancer activity after TACE with bicarbonate local infusion into tumor.190 Sodium bicarbonate, with a pKa of 6.1, is sufficient to meet the above requirements.188 However, published clinical trials have indicated that pH buffer with a pKa of approximately 7 is a more ideal treatment.189 Alternatively, some drugs have shown an enhanced effect in tumor acidic microenvironment. As protonable weak bases, PPIs can be selectively aggregated and activated in acidic region.191 An acidic TME can not only be the target of PPI but also promote PPI activation. Drugs with the same characteristics as PPIs can be considered for cancer combination therapy. More fundamental studies and clinical trials need to be performed.
Treatments for the Immune Microenvironment
Two treatment strategies aiming at liver cancer immunotherapy include the enhancement of normal immune mechanisms and the correction of detective immune mechanisms.
For enhancement immunotherapy, cytokines (IL-2, IFNs), cancer vaccines and cell therapy (CAR-T) have been approved by the FDA. Although some of the above methods have achieved a certain effect in liver cancer therapy,192 high-frequency negative trials with high toxicity pushed scientists to find other ways.193 Immune checkpoints inhibitors targeting the TME but with lower toxicity have begun to emerge. Immune checkpoint therapy could satisfy the following three principles at the same time: normalizing tumor immunity, targeting the TME and reset of immune response in TME. Treatment with the CTLA-4 inhibitor, tremelimumab, led to a transient complete viral response in 25% of HCC patients with HCV infection (ClinicalTrials.gov Identifier: NCT01008358).194 PD-1 and PD-L1 inhibitors showed lower levels of immune-related adverse events (irAEs) than CTLA-4 inhibitors, with an incidence of 27% versus 72% for all grades and 6% versus 24% for grade 3 or higher in HCC. The irAEs of PD-1 or PD-L1 inhibitors are not related to dose; however, the effect is dose-dependent for CTLA-4 inhibitors.195,196 The PD-1 inhibitor MEDI4736 resulted in lower hepatotoxicity than CTLA-4 antibody in HCC patients.193 Although antitumor activity of PD-1 antibody is promising, less than 20% of HCC patients respond to it.104 Clinical trials targeting at the effect of PD-1 blockade combined with other treatments have been launched. A trial examining the combination of PD-1 blockade and incomplete thermal ablation in patients with advanced HCC has just been completed, the results of which will provide us with a more in-depth understanding of the efficacy once they are published (ClinicalTrials.gov Identifier: NCT03939975). A clinical trial investigating CTLA-4 and PD-L1 combination blockade after transarterial chemoembolization (DEB-TACE) in intermediate-stage HCC patients is underway (ClinicalTrials.gov Identifier: NCT03638141). Given the complexity of liver cancer and the irAEs of immunotherapy alone, more than 16 clinical trials are ongoing in an attempt to explore the therapeutic effects and side effects of combined locoregional immunotherapy.197 Lately, it has been revealed that exhaustion of CD8+ T cells is not the major cause for tumor immune evasion, but rather, it is a lack of stem-like CD8 T cells, providing a fresh perspective to this field of research. Stem-like CD8 T cells can differentiate into CTLs and maintain tumor immune response within the comfortable environment provided by antigen-presenting cells.198 This new discovery may be able to explain why the immune checkpoint treatment is only 20–30% efficient. However, tumor immune evasion has a large and complicated network, which cannot be explained by one single mechanism. The role of stem-like CD8 T cells in tumor immune evasion requires further exploration in the future.
Currently, scientists believe that NK cells are equally as important as T cells and can be used in conjunction with T cells for tumor immunotherapy.122 The antiviral activity of hepatic T cells has been found to be controlled by LrNK via the PD-1-PD-L1 axis.116 Moreover, anti-NKG2A mAb was revealed play an important role in unleashing both T and NK cells.122 The results of the above experiments suggest that PD-1-PD-L1 and NKG2A blockade are important targets for tumor treatment. However, no clinical trials investigating NKG2A blockade in HCC patients have yet been conducted.
Treatments for Aerobic Glycolysis
Strategies targeting for the “anti-Warburg effect” have been primarily considered for key transporter and enzymes involved in glycolysis.199 However, increasing gluconeogenesis could suppress glycolysis at the same time, which became the new target for the “anti-Warburg effect”.
The selective inhibitors of GLUTs include benzamides and rapafucins, which can inhibit GLUTs and inhibit glucose uptake to prevent or reduce the proliferation of tumor cells. Benzamides can directly bind to GLUT1 and inhibit GLUT1 function without affecting GLUT1 protein levels. Benzamides have no obvious toxicity to normal tissues.200 New GLUT inhibitors such as rapafucins are being explored.201 As the healthy tissues also need glucose, it is necessary to select tumor-specific GLUT inhibitors and make appropriate assessments to reduce the toxicity to normal cells.
HK2 is the first rate-limiting enzyme for glucose metabolism.202 Studies have shown that blockade of HK2 in human hepatoma cells can inhibit the occurrence of tumor and increase cell death.203 2-DG is a well-known HK2 inhibitor, that has been reported to inhibit hexokinase by competing with glucose.204,205 Lonidamin is a mitochondrial HK inhibitor that suppresses the activity of HK1 and HK2.206 Others such as 3-bromopyruvic acid (3-BrPA), ketoconazole and posaconazole can also affect tumor metabolism and growth by blocking HK.207,208
Phosphofructokinase 1 (PFK1) is the second rate-limiting enzyme of glycolysis, and tumor formation can be impaired by the O-GlcNAcylation of PFK1 at serine 529.209 Metformin can target the HIF-1α/PFKFB3/PFK1 pathway in hepatoma cells and reduce hepatoma cell proliferation by inhibiting glycolysis.210 Pyruvate kinase (PK) is the third rate-limiting enzyme in glycolysis. It has multiple subtypes, among which PKM2 is upregulated in a variety of cancers. Shikonin is an inhibitor of PKM2 that can reduce the glycolytic rate of tumors. However, its toxicity and poor solubility limit its application.211 Recent studies have found that metformin can also induce tumor cell death and increase sensitivity to chemotherapy drugs by inhibiting PKM2 in osteosarcoma.212
LDHA is located at the bifurcation point of glycolysis and oxidative phosphorylation. Inhibition of LDHA may be a promising antitumor strategy. The piperidindione derivatives miR-30a-5p, miR-41 and GNE-140 have been indicated to inhibit LDHA in breast and pancreatic cancer.213,214 However, glycolysis inhibitors cannot induce cell death to achieve long-term tumor remission and its safety needs further verification. At present, the effect of glycolysis inhibitors alone is not very significant. In the future, it will be necessary to further study the mechanism or a combination of multiple methods for treatment to achieve the purpose of controlling tumors.
The main substrates of hepatic gluconeogenesis are lactate, pyruvate, glycerol and glycosylated amino acids (such as glutamate). Hepatic glycosylation relies on the initial gluconeogenesis enzymes phosphoenolpyruvate carboxykinase (PEPCK), downstream fructose-1, 6-bisphosphatase (FBP) and the final-step glucose-6-phosphatase (G6PC).215 PEPCK has two isoforms: a cytosolic isoform, PCK1, and a mitochondrial isoform, PCK2. Unlike the well-known PCK1, it was recently demonstrated that PCK2 contributed to gluconeogenesis with less efficiency than PCK1.16 FBP also has two isoforms: liver isoform, FBP1 and muscle isoform, FBP2.216 Studies of increasing gluconeogenesis are mainly focused on PCK1 and FBP1.
PCK1 and PCK2 are downregulated in HCC and suggest a poor prognosis.16 Bian found that Nur77 could stabilize PCK1 by attenuating its sumoylation and ubiquitination and then suppress HCC.14 PCK1 was also found to inhibit hepatoma cell proliferation by downregulating cell cycle progression through the AMPK pathway.217
FBP1 appears to be a tumor suppressor and poor prognostic marker in HCC. Gene set enrichment analysis with 594 cases of HCC demonstrated that lower FBP1 expression was correlated with advanced tumor stage, poor overall survival and higher tumor recurrence rates.13 Two double-negative feedback loops have been indicated for FBP1 expression in HCC. The first loop is FBP1 and enhancer of zeste homolog 2 (EZH2): EZH2 can inhibit FBP1 and FBP1 physically competed for EZH2 binding in turn.15 The second loop is FBP1 and polycomb repressive complex 2 (PRC2). PCR2 can downregulate FBP1, and conversely, FBP1 can interfere with PCR2 functions.218 Histone deacetylases and FX11 inhibitor stabilize FBP1 in HCC to inhibit of tumor growth and invasion.219,220 However, more clinical trials are needed.
Conclusion
Tumor aerobic glycolysis is closely associated with TME. They promote each other to provide a suitable growth environment for tumor. Combination treatment of sorafenib with TME improvement and/or anti-Warburg therapies represents the future of advanced HCC therapy. Treatment options that elicit responses with “anti- Warburg effects” are more promising for HCC therapy, such as those that promote the elevation of gluconeogenesis. However, these treatments still need clinical trials for verification.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. doi:10.3322/caac.21492
2. Wang P, Song X, Cao D, et al. Oncogene-dependent function of BRG1 in hepatocarcinogenesis. Cell Death Dis. 2020;11(2):91. doi:10.1038/s41419-020-2289-3
3. Zhang G, Tang X, Liang L, et al. DNA and RNA sequencing identified a novel oncogene VPS35 in liver hepatocellular carcinoma. Oncogene. 2020;39(16):3229–3244. doi:10.1038/s41388-020-1215-6
4. Vitale A, Trevisani F, Farinati F, Cillo U. Treatment of hepatocellular carcinoma in the Precision Medicine era: from treatment stage migration to therapeutic hierarchy. Hepatology. 2020. doi:10.1002/hep.31187
5. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69(1):7–34. doi:10.3322/caac.21551
6. Warburg O, Wind F, Negelein E. The metabolism of tumors in the body. J Gen Physiol. 1927;8(6):519–530. doi:10.1085/jgp.8.6.519
7. Sanderson SM, Locasale JW. Revisiting the Warburg effect: some tumors hold their breath. Cell Metab. 2018;28(5):669–670. doi:10.1016/j.cmet.2018.10.011
8. Wang T, Marquardt C, Foker J. Aerobic glycolysis during lymphocyte proliferation. Nature. 1976;261:4.
9. Pavlova NN, Thompson CB. The emerging hallmarks of cancer metabolism. Cell Metab. 2016;23(1):27–47. doi:10.1016/j.cmet.2015.12.006
10. Novikova MV, Khromova NV, Kopnin PB. Components of the hepatocellular carcinoma microenvironment and their role in tumor progression. Biochemistry (Mosc). 2017;82(8):861–873. doi:10.1134/S0006297917080016
11. Sharabi K, Tavares CD, Rines AK, Puigserver P. Molecular pathophysiology of hepatic glucose production. Mol Aspects Med. 2015;46:21–33. doi:10.1016/j.mam.2015.09.003
12. Goldstein I, Hager GL. Transcriptional and chromatin regulation during fasting - the genomic era. Trends Endocrinol Metab. 2015;26(12):699–710. doi:10.1016/j.tem.2015.09.005
13. Hirata H, Sugimachi K, Komatsu H, et al. Decreased expression of fructose-1,6-bisphosphatase associates with glucose metabolism and tumor progression in hepatocellular carcinoma. Cancer Res. 2016;76(11):3265–3276. doi:10.1158/0008-5472.CAN-15-2601
14. Bian XL, Chen HZ, Yang PB, et al. Nur77 suppresses hepatocellular carcinoma via switching glucose metabolism toward gluconeogenesis through attenuating phosphoenolpyruvate carboxykinase sumoylation. Nat Commun. 2017;8:14420. doi:10.1038/ncomms14420
15. Liao K, Deng S, Xu L, et al. A feedback circuitry between polycomb signaling and fructose-1, 6-bisphosphatase enables hepatic and renal tumorigenesis. Cancer Res. 2020;80(4):675–688. doi:10.1158/0008-5472.CAN-19-2060
16. Liu MX, Jin L, Sun SJ, et al. Metabolic reprogramming by PCK1 promotes TCA cataplerosis, oxidative stress and apoptosis in liver cancer cells and suppresses hepatocellular carcinoma. Oncogene. 2018;37(12):1637–1653. doi:10.1038/s41388-017-0070-6
17. Jing X, Yang F, Shao C, et al. Role of hypoxia in cancer therapy by regulating the tumor microenvironment. Mol Cancer. 2019;18(1):157. doi:10.1186/s12943-019-1089-9
18. Sahu A, Kwon I, Tae G. Improving cancer therapy through the nanomaterials-assisted alleviation of hypoxia. Biomaterials. 2020;228:119578. doi:10.1016/j.biomaterials.2019.119578
19. Briggs KJ, Koivunen P, Cao S, et al. Paracrine induction of HIF by glutamate in breast cancer: EglN1 senses cysteine. Cell. 2016;166(1):126–139. doi:10.1016/j.cell.2016.05.042
20. Shang RZ, Qu SB, Wang DS. Reprogramming of glucose metabolism in hepatocellular carcinoma: progress and prospects. World J Gastroenterol. 2016;22(45):9933–9943. doi:10.3748/wjg.v22.i45.9933
21. Papandreou I, Cairns RA, Fontana L, Lim AL, Denko NC. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. 2006;3(3):187–197. doi:10.1016/j.cmet.2006.01.012
22. Rohwer N, Cramer T. Hypoxia-mediated drug resistance: novel insights on the functional interaction of HIFs and cell death pathways. Drug Resist Updat. 2011;14(3):191–201. doi:10.1016/j.drup.2011.03.001
23. Qing G, Simon MC. Hypoxia inducible factor-2alpha: a critical mediator of aggressive tumor phenotypes. Curr Opin Genet Dev. 2009;19(1):60–66. doi:10.1016/j.gde.2008.12.001
24. Dabral S, Muecke C, Valasarajan C, et al. A RASSF1A-HIF1alpha loop drives Warburg effect in cancer and pulmonary hypertension. Nat Commun. 2019;10(1):2130. doi:10.1038/s41467-019-10044-z
25. Zhang X, Li Y, Ma Y, et al. Yes-associated protein (YAP) binds to HIF-1alpha and sustains HIF-1alpha protein stability to promote hepatocellular carcinoma cell glycolysis under hypoxic stress. J Exp Clin Cancer Res. 2018;37(1):216. doi:10.1186/s13046-018-0892-2
26. Shi DY, Xie FZ, Zhai C, Stern JS, Liu Y, Liu SL. The role of cellular oxidative stress in regulating glycolysis energy metabolism in hepatoma cells. Mol Cancer. 2009;8:32. doi:10.1186/1476-4598-8-32
27. Zhang X, Wang Y, Li X, Yang A, Li Z, Wang D. The anti-carcinogenesis properties of erianin in the modulation of oxidative stress-mediated apoptosis and immune response in liver cancer. Aging (Albany NY). 2019;11(22):10284–10300. doi:10.18632/aging.102456
28. Lee D, Xu IM, Chiu DK, et al. Induction of oxidative stress through inhibition of thioredoxin reductase 1 is an effective therapeutic approach for hepatocellular carcinoma. Hepatology. 2019;69(4):1768–1786. doi:10.1002/hep.30467
29. Huang Q, Zhan L, Cao H, et al. Increased mitochondrial fission promotes autophagy and hepatocellular carcinoma cell survival through the ROS-modulated coordinated regulation of the NFKB and TP53 pathways. Autophagy. 2016;12(6):999–1014. doi:10.1080/15548627.2016.1166318
30. Shen J, Chen M, Lee D, et al. Histone chaperone FACT complex mediates oxidative stress response to promote liver cancer progression. Gut. 2020;69(2):329–342. doi:10.1136/gutjnl-2019-318668
31. Lu J, Tan M, Cai Q. The Warburg effect in tumor progression: mitochondrial oxidative metabolism as an anti-metastasis mechanism. Cancer Lett. 2015;356(2 Pt A):156–164. doi:10.1016/j.canlet.2014.04.001
32. Chang H, Li J, Qu K, et al. CRIF1 overexpression facilitates tumor growth and metastasis through inducing ROS/NFkappaB pathway in hepatocellular carcinoma. Cell Death Dis. 2020;11(5):332. doi:10.1038/s41419-020-2528-7
33. Geng X, Geng Z, Li H, Zhang Y, Li J, Chang H. Over-expression of TFB2M facilitates cell growth and metastasis via activating ROS-Akt-NF-kappaB signalling in hepatocellular carcinoma. Liver Int. 2020;40(7):1756–1769. doi:10.1111/liv.14440
34. Zheng X, Li C, Yu K, et al. Aquaporin-9, mediated by IGF2, suppresses liver cancer stem cell properties via augmenting ROS/beta-catenin/FOXO3a signaling. Mol Cancer Res. 2020;18(7):992–1003. doi:10.1158/1541-7786.MCR-19-1180
35. Fuhrmann DC, Brune B. Mitochondrial composition and function under the control of hypoxia. Redox Biol. 2017;12:208–215. doi:10.1016/j.redox.2017.02.012
36. Kung-Chun Chiu D, Pui-Wah Tse A, Law CT, et al. Hypoxia regulates the mitochondrial activity of hepatocellular carcinoma cells through HIF/HEY1/PINK1 pathway. Cell Death Dis. 2019;10(12):934. doi:10.1038/s41419-019-2155-3
37. Lu CW, Lin SC, Chen KF, Lai YY, Tsai SJ. Induction of pyruvate dehydrogenase kinase-3 by hypoxia-inducible factor-1 promotes metabolic switch and drug resistance. J Biol Chem. 2008;283(42):28106–28114. doi:10.1074/jbc.M803508200
38. Dang CV. The interplay between MYC and HIF in the Warburg effect. Ernst Schering Found Symp Proc. 2007;(4):35–53.
39. Brahimi-Horn MC, Giuliano S, Saland E, et al. Knockout of Vdac1 activates hypoxia-inducible factor through reactive oxygen species generation and induces tumor growth by promoting metabolic reprogramming and inflammation. Cancer Metab. 2015;3:8. doi:10.1186/s40170-015-0133-5
40. Fukuda R, Zhang H, Kim JW, Shimoda L, Dang CV, Semenza GL. HIF-1 regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell. 2007;129(1):111–122. doi:10.1016/j.cell.2007.01.047
41. Tello D, Balsa E, Acosta-Iborra B, et al. Induction of the mitochondrial NDUFA4L2 protein by HIF-1alpha decreases oxygen consumption by inhibiting Complex I activity. Cell Metab. 2011;14(6):768–779. doi:10.1016/j.cmet.2011.10.008
42. Zhang H, Bosch-Marce M, Shimoda LA, et al. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J Biol Chem. 2008;283(16):10892–10903. doi:10.1074/jbc.M800102200
43. Dai W, Xu L, Yu X, et al. OGDHL silencing promotes hepatocellular carcinoma by reprogramming glutamine metabolism. J Hepatol. 2020;72(5):909–923. doi:10.1016/j.jhep.2019.12.015
44. DeBerardinis RJ, Mancuso A, Daikhin E, et al. Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc Natl Acad Sci U S A. 2007;104(49):19345–19350. doi:10.1073/pnas.0709747104
45. Levy PL, Duponchel S, Eischeid H, et al. Hepatitis C virus infection triggers a tumor-like glutamine metabolism. Hepatology. 2017;65(3):789–803. doi:10.1002/hep.28949
46. Shanware NP, Bray K, Eng CH, et al. Glutamine deprivation stimulates mTOR-JNK-dependent chemokine secretion. Nat Commun. 2014;5:4900. doi:10.1038/ncomms5900
47. Li B, Cao Y, Meng G, et al. Targeting glutaminase 1 attenuates stemness properties in hepatocellular carcinoma by increasing reactive oxygen species and suppressing Wnt/beta-catenin pathway. EBioMedicine. 2019;39:239–254. doi:10.1016/j.ebiom.2018.11.063
48. Fulda S, Galluzzi L, Kroemer G. Targeting mitochondria for cancer therapy. Nat Rev Drug Discov. 2010;9(6):447–464. doi:10.1038/nrd3137
49. Lippmann J, Petri K, Fulda S, Liese J. Redox modulation and induction of ferroptosis as a new therapeutic strategy in hepatocellular carcinoma. Transl Oncol. 2020;13(8):100785. doi:10.1016/j.tranon.2020.100785
50. Pibiri M, Sulas P, Camboni T, Leoni VP, Simbula G. Alpha-lipoic acid induces endoplasmic reticulum stress-mediated apoptosis in hepatoma cells. Sci Rep. 2020;10(1):7139. doi:10.1038/s41598-020-64004-5
51. Huang X, Gan G, Wang X, Xu T, Xie W. The HGF-MET axis coordinates liver cancer metabolism and autophagy for chemotherapeutic resistance. Autophagy. 2019;15(7):1258–1279. doi:10.1080/15548627.2019.1580105
52. Long Y, Tsai WB, Wang D, et al. Argininosuccinate synthetase 1 (ASS1) is a common metabolic marker of chemosensitivity for targeted arginine- and glutamine-starvation therapy. Cancer Lett. 2017;388:54–63. doi:10.1016/j.canlet.2016.11.028
53. Tang L, Zeng J, Geng P, et al. Global metabolic profiling identifies a pivotal role of proline and hydroxyproline metabolism in supporting hypoxic response in hepatocellular carcinoma. Clin Cancer Res. 2018;24(2):474–485. doi:10.1158/1078-0432.CCR-17-1707
54. Webb BA, Chimenti M, Jacobson MP, Barber DL. Dysregulated pH: a perfect storm for cancer progression. Nat Rev Cancer. 2011;11(9):671–677. doi:10.1038/nrc3110
55. Savic LJ, Schobert IT, Peters D, et al. Molecular imaging of extracellular tumor pH to reveal effects of locoregional therapy on liver cancer microenvironment. Clin Cancer Res. 2020;26(2):428–438. doi:10.1158/1078-0432.CCR-19-1702
56. Coman D, Peters DC, Walsh JJ, et al. Extracellular pH mapping of liver cancer on a clinical 3T MRI scanner. Magn Reson Med. 2020;83(5):1553–1564. doi:10.1002/mrm.28035
57. Kwon SY, Kim DY, Min JJ, Bae WK. Potential visualization of sorafenib-induced acidosis using 11C-acetate PET/CT in patients with hepatocellular carcinoma. Clin Nucl Med. 2018;43(1):31–32. doi:10.1097/RLU.0000000000001879
58. Mookerjee SA, Goncalves RLS, Gerencser AA, Nicholls DG, Brand MD. The contributions of respiration and glycolysis to extracellular acid production. Biochim Biophys Acta. 2015;1847(2):171–181. doi:10.1016/j.bbabio.2014.10.005
59. Draoui N, Feron O. Lactate shuttles at a glance: from physiological paradigms to anti-cancer treatments. Dis Model Mech. 2011;4(6):727–732. doi:10.1242/dmm.007724
60. Shen YC, Ou DL, Hsu C, et al. Activating oxidative phosphorylation by a pyruvate dehydrogenase kinase inhibitor overcomes sorafenib resistance of hepatocellular carcinoma. Br J Cancer. 2013;108(1):72–81. doi:10.1038/bjc.2012.559
61. Guo Y, Li X, Sun X, et al. Combined aberrant expression of NDRG2 and LDHA predicts hepatocellular carcinoma prognosis and mediates the anti-tumor effect of gemcitabine. Int J Biol Sci. 2019;15(9):1771–1786. doi:10.7150/ijbs.35094
62. Vegran F, Boidot R, Michiels C, Sonveaux P, Feron O. Lactate influx through the endothelial cell monocarboxylate transporter MCT1 supports an NF-kappaB/IL-8 pathway that drives tumor angiogenesis. Cancer Res. 2011;71(7):2550–2560. doi:10.1158/0008-5472.CAN-10-2828
63. Sonveaux P, Vegran F, Schroeder T, et al. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J Clin Invest. 2008;118(12):3930–3942. doi:10.1172/JCI36843
64. Supuran CT. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat Rev Drug Discov. 2008;7(2):168–181. doi:10.1038/nrd2467
65. Viklund J, Avnet S, De Milito A. Pathobiology and therapeutic implications of tumor acidosis. Curr Med Chem. 2017;24(26):2827–2845. doi:10.2174/0929867323666161228142849
66. Svastova E, Witarski W, Csaderova L, et al. Carbonic anhydrase IX interacts with bicarbonate transporters in lamellipodia and increases cell migration via its catalytic domain. J Biol Chem. 2012;287(5):3392–3402. doi:10.1074/jbc.M111.286062
67. Kuchuk O, Tuccitto A, Citterio D, et al. pH regulators to target the tumor immune microenvironment in human hepatocellular carcinoma. Oncoimmunology. 2018;7(7):e1445452. doi:10.1080/2162402X.2018.1445452
68. Filatova A, Seidel S, Bogurcu N, Graf S, Garvalov BK, Acker T. Acidosis acts through HSP90 in a PHD/VHL-independent manner to promote HIF function and stem cell maintenance in glioma. Cancer Res. 2016;76(19):5845–5856. doi:10.1158/0008-5472.CAN-15-2630
69. Nadtochiy SM, Schafer X, Fu D, Nehrke K, Munger J, Brookes PS. Acidic pH is a metabolic switch for 2-hydroxyglutarate generation and signaling. J Biol Chem. 2016;291(38):20188–20197. doi:10.1074/jbc.M116.738799
70. Mossanen JC, Kohlhepp M, Wehr A, et al. CXCR6 inhibits hepatocarcinogenesis by promoting natural killer T- and CD4(+) T-cell-dependent control of senescence. Gastroenterology. 2019;156(6):1877–1889 e1874. doi:10.1053/j.gastro.2019.01.247
71. Fox CJ, Hammerman PS, Thompson CB. Fuel feeds function: energy metabolism and the T-cell response. Nat Rev Immunol. 2005;5(11):844–852. doi:10.1038/nri1710
72. Beyoglu D, Imbeaud S, Maurhofer O, et al. Tissue metabolomics of hepatocellular carcinoma: tumor energy metabolism and the role of transcriptomic classification. Hepatology. 2013;58(1):229–238. doi:10.1002/hep.26350
73. Henning AN, Roychoudhuri R, Restifo NP. Epigenetic control of CD8(+) T cell differentiation. Nat Rev Immunol. 2018;18(5):340–356. doi:10.1038/nri.2017.146
74. Pace L, Goudot C, Zueva E, et al. The epigenetic control of stemness in CD8(+) T cell fate commitment. Science. 2018;359(6372):177–186. doi:10.1126/science.aah6499
75. Sallusto F, Cassotta A, Hoces D, Foglierini M, Lanzavecchia A. Do memory CD4 T cells keep their cell-type programming: plasticity versus fate commitment? T-cell heterogeneity, plasticity, and selection in humans. Cold Spring Harb Perspect Biol. 2018;10(3):a029421. doi:10.1101/cshperspect.a029421
76. Becattini S, Latorre D, Mele F, et al. T cell immunity. Functional heterogeneity of human memory CD4(+) T cell clones primed by pathogens or vaccines. Science. 2015;347(6220):400–406. doi:10.1126/science.1260668
77. Wang K, Yaghi OK, Spallanzani RG, et al. Neuronal, stromal, and T-regulatory cell crosstalk in murine skeletal muscle. Proc Natl Acad Sci U S A. 2020;117(10):5402–5408.
78. Choi JY, Eskandari SK, Cai S, et al. Regulatory CD8 T cells that recognize Qa-1 expressed by CD4 T-helper cells inhibit rejection of heart allografts. Proc Natl Acad Sci U S A. 2020;117(11):6042–6046. doi:10.1073/pnas.1918950117
79. Buck MD, O’Sullivan D, Klein Geltink RI, et al. Mitochondrial dynamics controls T cell fate through metabolic programming. Cell. 2016;166(1):63–76. doi:10.1016/j.cell.2016.05.035
80. Pearce EL, Walsh MC, Cejas PJ, et al. Enhancing CD8 T-cell memory by modulating fatty acid metabolism. Nature. 2009;460(7251):103–107. doi:10.1038/nature08097
81. Weinberg SE, Singer BD, Steinert EM, et al. Mitochondrial complex III is essential for suppressive function of regulatory T cells. Nature. 2019;565(7740):495–499. doi:10.1038/s41586-018-0846-z
82. Scharping NE, Menk AV, Moreci RS, et al. The tumor microenvironment represses T cell mitochondrial biogenesis to drive intratumoral T cell metabolic insufficiency and dysfunction. Immunity. 2016;45(2):374–388. doi:10.1016/j.immuni.2016.07.009
83. Beier UH, Angelin A, Akimova T, et al. Essential role of mitochondrial energy metabolism in Foxp3(+) T-regulatory cell function and allograft survival. FASEB J. 2015;29(6):2315–2326. doi:10.1096/fj.14-268409
84. Poznanski SM, Barra NG, Ashkar AA, Schertzer JD. Immunometabolism of T cells and NK cells: metabolic control of effector and regulatory function. Inflamm Res. 2018;67(10):813–828. doi:10.1007/s00011-018-1174-3
85. Ogando J, Saez ME, Santos J, et al. PD-1 signaling affects cristae morphology and leads to mitochondrial dysfunction in human CD8(+) T lymphocytes. J Immunother Cancer. 2019;7(1):151. doi:10.1186/s40425-019-0628-7
86. Wang S, Campos J, Gallotta M, et al. Intratumoral injection of a CpG oligonucleotide reverts resistance to PD-1 blockade by expanding multifunctional CD8+ T cells. Proc Natl Acad Sci U S A. 2016;113(46):E7240–E7249. doi:10.1073/pnas.1608555113
87. Chang CH, Curtis JD, Maggi LB, et al. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell. 2013;153(6):1239–1251. doi:10.1016/j.cell.2013.05.016
88. Sukumar M, Liu J, Ji Y, et al. Inhibiting glycolytic metabolism enhances CD8+ T cell memory and antitumor function. J Clin Invest. 2013;123(10):4479–4488. doi:10.1172/JCI69589
89. Patsoukis N, Bardhan K, Chatterjee P, et al. PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat Commun. 2015;6:6692. doi:10.1038/ncomms7692
90. Geiger R, Rieckmann JC, Wolf T, et al. L-arginine modulates T cell metabolism and enhances survival and anti-tumor activity. Cell. 2016;167(3):829–842 e813. doi:10.1016/j.cell.2016.09.031
91. Bengsch B, Johnson AL, Kurachi M, et al. Bioenergetic insufficiencies due to metabolic alterations regulated by the inhibitory receptor PD-1 are an early driver of CD8(+) T cell exhaustion. Immunity. 2016;45(2):358–373. doi:10.1016/j.immuni.2016.07.008
92. Franchina DG, He F, Brenner D. Survival of the fittest: cancer challenges T cell metabolism. Cancer Lett. 2018;412:216–223. doi:10.1016/j.canlet.2017.10.014
93. Pacella I, Piconese S. Immunometabolic checkpoints of treg dynamics: adaptation to microenvironmental opportunities and challenges. Front Immunol. 2019;10:1889. doi:10.3389/fimmu.2019.01889
94. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. doi:10.1016/j.cell.2011.02.013
95. Chang CH, Qiu J, O’Sullivan D, et al. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell. 2015;162(6):1229–1241. doi:10.1016/j.cell.2015.08.016
96. Macintyre AN, Gerriets VA, Nichols AG, et al. The glucose transporter Glut1 is selectively essential for CD4 T cell activation and effector function. Cell Metab. 2014;20(1):61–72. doi:10.1016/j.cmet.2014.05.004
97. Fischer K, Hoffmann P, Voelkl S, et al. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood. 2007;109(9):3812–3819. doi:10.1182/blood-2006-07-035972
98. Ma C, Kesarwala AH, Eggert T, et al. NAFLD causes selective CD4(+) T lymphocyte loss and promotes hepatocarcinogenesis. Nature. 2016;531(7593):253–257. doi:10.1038/nature16969
99. Brand A, Singer K, Koehl GE, et al. LDHA-associated lactic acid production blunts tumor immunosurveillance by T and NK cells. Cell Metab. 2016;24(5):657–671. doi:10.1016/j.cmet.2016.08.011
100. Fridman WH, Zitvogel L, Sautes-Fridman C, Kroemer G. The immune contexture in cancer prognosis and treatment. Nat Rev Clin Oncol. 2017;14(12):717–734. doi:10.1038/nrclinonc.2017.101
101. Pacella I, Procaccini C, Focaccetti C, et al. Fatty acid metabolism complements glycolysis in the selective regulatory T cell expansion during tumor growth. Proc Natl Acad Sci U S A. 2018;115(28):E6546–E6555. doi:10.1073/pnas.1720113115
102. Liu X, Mo W, Ye J, et al. Regulatory T cells trigger effector T cell DNA damage and senescence caused by metabolic competition. Nat Commun. 2018;9(1):249. doi:10.1038/s41467-017-02689-5
103. Renner K, Singer K, Koehl GE, et al. Metabolic hallmarks of tumor and immune cells in the tumor microenvironment. Front Immunol. 2017;8:248. doi:10.3389/fimmu.2017.00248
104. Sangro B, Chan SL, Meyer T, Reig M, El-Khoueiry A, Galle PR. Diagnosis and management of toxicities of immune checkpoint inhibitors in hepatocellular carcinoma. J Hepatol. 2020;72(2):320–341. doi:10.1016/j.jhep.2019.10.021
105. Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell. 2015;27(4):450–461. doi:10.1016/j.ccell.2015.03.001
106. Xiang J, Zhang N, Sun H, et al. Disruption of SIRT7 increases the efficacy of checkpoint inhibitor via MEF2D regulation of programmed cell death 1 ligand 1 in hepatocellular carcinoma cells. Gastroenterology. 2020;158(3):664–678 e624. doi:10.1053/j.gastro.2019.10.025
107. Itoh S, Yoshizumi T, Yugawa K, et al. Impact of immune response on outcomes in hepatocellular carcinoma: association with vascular formation. Hepatology. 2020. doi:10.1002/hep.31206
108. Pauken KE, Sammons MA, Odorizzi PM, et al. Epigenetic stability of exhausted T cells limits durability of reinvigoration by PD-1 blockade. Science. 2016;354(6316):1160–1165. doi:10.1126/science.aaf2807
109. Poggio M, Hu T, Pai CC, et al. Suppression of exosomal PD-L1 induces systemic anti-tumor immunity and memory. Cell. 2019;177(2):414–427 e413. doi:10.1016/j.cell.2019.02.016
110. Arce Vargas F, Furness AJS, Litchfield K, et al. Fc effector function contributes to the activity of human anti-CTLA-4 antibodies. Cancer Cell. 2018;33(4):649–663 e644. doi:10.1016/j.ccell.2018.02.010
111. Walker LS, Sansom DM. Confusing signals: recent progress in CTLA-4 biology. Trends Immunol. 2015;36(2):63–70. doi:10.1016/j.it.2014.12.001
112. He W, Zhang H, Han F, et al. CD155T/TIGIT signaling regulates CD8(+) T-cell metabolism and promotes tumor progression in human gastric cancer. Cancer Res. 2017;77(22):6375–6388. doi:10.1158/0008-5472.CAN-17-0381
113. Keating SE, Zaiatz-Bittencourt V, Loftus RM, et al. Metabolic reprogramming supports IFN-gamma production by CD56bright NK cells. J Immunol. 2016;196(6):2552–2560. doi:10.4049/jimmunol.1501783
114. Lotze MT, Buchser WJ, Liang X. Blocking the interleukin 2 (IL2)-induced systemic autophagic syndrome promotes profound antitumor effects and limits toxicity. Autophagy. 2012;8(8):1264–1266. doi:10.4161/auto.20752
115. Easom NJW, Stegmann KA, Swadling L, et al. IL-15 overcomes hepatocellular carcinoma-induced NK cell dysfunction. Front Immunol. 2018;9:1009. doi:10.3389/fimmu.2018.01009
116. Zhou J, Peng H, Li K, et al. Liver-resident NK cells control antiviral activity of hepatic T cells via the PD-1-PD-L1 axis. Immunity. 2019;50(2):403–417 e404. doi:10.1016/j.immuni.2018.12.024
117. Sun C, Xu J, Huang Q, et al. High NKG2A expression contributes to NK cell exhaustion and predicts a poor prognosis of patients with liver cancer. Oncoimmunology. 2017;6(1):e1264562. doi:10.1080/2162402X.2016.1264562
118. Zhang Q, Bi J, Zheng X, et al. Blockade of the checkpoint receptor TIGIT prevents NK cell exhaustion and elicits potent anti-tumor immunity. Nat Immunol. 2018;19(7):723–732. doi:10.1038/s41590-018-0132-0
119. Sun H, Huang Q, Huang M, et al. Human CD96 correlates to natural killer cell exhaustion and predicts the prognosis of human hepatocellular carcinoma. Hepatology. 2019;70(1):168–183. doi:10.1002/hep.30347
120. Sun H, Liu L, Huang Q, et al. Accumulation of tumor-infiltrating CD49a(+) NK cells correlates with poor prognosis for human hepatocellular carcinoma. Cancer Immunol Res. 2019;7(9):1535–1546. doi:10.1158/2326-6066.CIR-18-0757
121. Pfeifer C, Highton AJ, Peine S, et al. Natural killer cell education is associated with a distinct glycolytic profile. Front Immunol. 2018;9:3020. doi:10.3389/fimmu.2018.03020
122. Andre P, Denis C, Soulas C, et al. Anti-NKG2A mAb is a checkpoint inhibitor that promotes anti-tumor immunity by unleashing both T and NK cells. Cell. 2018;175(7):1731–1743 e1713. doi:10.1016/j.cell.2018.10.014
123. Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell. 2010;141(1):39–51. doi:10.1016/j.cell.2010.03.014
124. Colegio OR, Chu NQ, Szabo AL, et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature. 2014;513(7519):559–563. doi:10.1038/nature13490
125. Oh KW, Currin RT, Lemasters JJ. Kupffer cells mediate increased anoxic hepatocellular killing from hyperosmolarity by an oxygen- and prostaglandin-independent mechanism. Toxicol Lett. 2000;117(1–2):95–100. doi:10.1016/S0378-4274(00)00247-2
126. Giovannini C, Bolondi L, Gramantieri L. Targeting Notch3 in hepatocellular carcinoma: molecular mechanisms and therapeutic perspectives. Int J Mol Sci. 2016;18(1):56. doi:10.3390/ijms18010056
127. Xiang S, Gu H, Jin L, Thorne RF, Zhang XD, Wu M. LncRNA IDH1-AS1 links the functions of c-Myc and HIF1alpha via IDH1 to regulate the Warburg effect. Proc Natl Acad Sci U S A. 2018;115(7):E1465–E1474. doi:10.1073/pnas.1711257115
128. Yu L, Kim J, Jiang L, et al. MTR4 drives liver tumorigenesis by promoting cancer metabolic switch through alternative splicing. Nat Commun. 2020;11(1):708. doi:10.1038/s41467-020-14437-3
129. Liang J, Cao R, Zhang Y, et al. PKM2 dephosphorylation by Cdc25A promotes the Warburg effect and tumorigenesis. Nat Commun. 2016;7:12431. doi:10.1038/ncomms12431
130. Wong KKL, Liao JZ, Verheyen EM. A positive feedback loop between Myc and aerobic glycolysis sustains tumor growth in a Drosophila tumor model. Elife. 2019;8. doi:10.7554/eLife.46315
131. Weng Q, Chen M, Yang W, et al. Integrated analyses identify miR-34c-3p/MAGI3 axis for the Warburg metabolism in hepatocellular carcinoma. FASEB J. 2020;34(4):5420–5434. doi:10.1096/fj.201902895R
132. Yin Y, Sun M, Zhan X, et al. EGFR signaling confers resistance to BET inhibition in hepatocellular carcinoma through stabilizing oncogenic MYC. J Exp Clin Cancer Res. 2019;38(1):83. doi:10.1186/s13046-019-1082-6
133. Peng M, Wei-Guo T, Jin-Wu H, et al. HSP4 triggers epithelial-mesenchymal transition and promotes motility capacities of hepatocellular carcinoma cells via activating AKT. Liver Int. 2020;40(5):1211–1223.
134. Dou C, Zhou Z, Xu Q, et al. Hypoxia-induced TUFT1 promotes the growth and metastasis of hepatocellular carcinoma by activating the Ca(2+)/PI3K/AKT pathway. Oncogene. 2019;38(8):1239–1255. doi:10.1038/s41388-018-0505-8
135. Cheng SC, Quintin J, Cramer RA, et al. mTOR- and HIF-1alpha-mediated aerobic glycolysis as metabolic basis for trained immunity. Science. 2014;345(6204):1250684. doi:10.1126/science.1250684
136. Liao YJ, Lee TS, Twu YC, et al. Glycine N-methyltransferase deficiency in female mice impairs insulin signaling and promotes gluconeogenesis by modulating the PI3K/Akt pathway in the liver. J Biomed Sci. 2016;23(1):69. doi:10.1186/s12929-016-0278-8
137. Liu Y, Lu LL, Wen D, et al. MiR-612 regulates invadopodia of hepatocellular carcinoma by HADHA-mediated lipid reprogramming. J Hematol Oncol. 2020;13(1):12. doi:10.1186/s13045-019-0841-3
138. Kim E, Lisby A, Ma C, et al. Promotion of growth factor signaling as a critical function of beta-catenin during HCC progression. Nat Commun. 2019;10(1):1909. doi:10.1038/s41467-019-09780-z
139. de La Coste A, Romagnolo B, Billuart P, et al. Somatic mutations of the beta-catenin gene are frequent in mouse and human hepatocellular carcinomas. Proc Natl Acad Sci U S A. 1998;95(15):8847–8851. doi:10.1073/pnas.95.15.8847
140. Lecarpentier Y, Schussler O, Hebert JL, Vallee A. Multiple targets of the canonical WNT/beta-catenin signaling in cancers. Front Oncol. 2019;9:1248. doi:10.3389/fonc.2019.01248
141. Xu W, Zhou W, Cheng M, et al. Hypoxia activates Wnt/beta-catenin signaling by regulating the expression of BCL9 in human hepatocellular carcinoma. Sci Rep. 2017;7:40446. doi:10.1038/srep40446
142. Zhang N, Chen X. A positive feedback loop involving the LINC00346/beta-catenin/MYC axis promotes hepatocellular carcinoma development. Cell Oncol (Dordr). 2020;43(1):137–153. doi:10.1007/s13402-019-00478-4
143. Fu X, Zhu X, Qin F, et al. Linc00210 drives Wnt/beta-catenin signaling activation and liver tumor progression through CTNNBIP1-dependent manner. Mol Cancer. 2018;17(1):73. doi:10.1186/s12943-018-0783-3
144. Li N, Wei L, Liu X, et al. A frizzled-like cysteine-rich domain in glypican-3 mediates wnt binding and regulates hepatocellular carcinoma tumor growth in mice. Hepatology. 2019;70(4):1231–1245. doi:10.1002/hep.30646
145. Huang JL, Fu YP, Gan W, et al. Hepatic stellate cells promote the progression of hepatocellular carcinoma through microRNA-1246-RORalpha-Wnt/beta-Catenin axis. Cancer Lett. 2020;476:140–151. doi:10.1016/j.canlet.2020.02.012
146. Zhang Q, Bai X, Chen W, et al. Wnt/beta-catenin signaling enhances hypoxia-induced epithelial-mesenchymal transition in hepatocellular carcinoma via crosstalk with hif-1alpha signaling. Carcinogenesis. 2013;34(5):962–973. doi:10.1093/carcin/bgt027
147. Liu Y, Ye X, Zhang JB, et al. PROX1 promotes hepatocellular carcinoma proliferation and sorafenib resistance by enhancing beta-catenin expression and nuclear translocation. Oncogene. 2015;34(44):5524–5535. doi:10.1038/onc.2015.7
148. Guo Y, Wu Z, Shen S, et al. Nanomedicines reveal how PBOV1 promotes hepatocellular carcinoma for effective gene therapy. Nat Commun. 2018;9(1):3430. doi:10.1038/s41467-018-05764-7
149. Batlle E, Massague J. Transforming growth factor-beta signaling in immunity and cancer. Immunity. 2019;50(4):924–940. doi:10.1016/j.immuni.2019.03.024
150. Liu Z, Wang Y, Dou C, et al. Hypoxia-induced up-regulation of VASP promotes invasiveness and metastasis of hepatocellular carcinoma. Theranostics. 2018;8(17):4649–4663. doi:10.7150/thno.26789
151. Soukupova J, Malfettone A, Hyroššová P, et al. Role of the transforming growth factor-β in regulating hepatocellular carcinoma oxidative metabolism. Sci Rep. 2017;7(1). doi:10.1038/s41598-017-12837-y.
152. Chae KS, Kang MJ, Lee JH, et al. Opposite functions of HIF-alpha isoforms in VEGF induction by TGF-beta1 under non-hypoxic conditions. Oncogene. 2011;30(10):1213–1228. doi:10.1038/onc.2010.498
153. Lin J, Cao S, Wang Y, et al. Long non-coding RNA UBE2CP3 enhances HCC cell secretion of VEGFA and promotes angiogenesis by activating ERK1/2/HIF-1alpha/VEGFA signalling in hepatocellular carcinoma. J Exp Clin Cancer Res. 2018;37(1):113. doi:10.1186/s13046-018-0727-1
154. Xuan Z, Zhao L, Li Z, et al. EPS8L3 promotes hepatocellular carcinoma proliferation and metastasis by modulating EGFR dimerization and internalization. Am J Cancer Res. 2020;10(1):60–77.
155. Zhou HJ, Xu Z, Wang Z, et al. SUMOylation of VEGFR2 regulates its intracellular trafficking and pathological angiogenesis. Nat Commun. 2018;9(1):3303. doi:10.1038/s41467-018-05812-2
156. Krzywinska E, Kantari-Mimoun C, Kerdiles Y, et al. Loss of HIF-1alpha in natural killer cells inhibits tumour growth by stimulating non-productive angiogenesis. Nat Commun. 2017;8(1):1597. doi:10.1038/s41467-017-01599-w
157. Zhang L, Chen J, Yong J, Qiao L, Xu L, Liu C. An essential role of RNF187 in Notch1 mediated metastasis of hepatocellular carcinoma. J Exp Clin Cancer Res. 2019;38(1):384. doi:10.1186/s13046-019-1382-x
158. Moriyama H, Moriyama M, Ozawa T, et al. Notch signaling enhances stemness by regulating metabolic pathways through modifying p53, NF-kappaB, and HIF-1alpha. Stem Cells Dev. 2018;27(13):935–947. doi:10.1089/scd.2017.0260
159. Landor SK, Lendahl U. The interplay between the cellular hypoxic response and Notch signaling. Exp Cell Res. 2017;356(2):146–151. doi:10.1016/j.yexcr.2017.04.030
160. Yang SL, Ren QG, Zhang T, et al. Hepatitis B virus X protein and hypoxia-inducible factor-1alpha stimulate Notch gene expression in liver cancer cells. Oncol Rep. 2017;37(1):348–356. doi:10.3892/or.2016.5211
161. Jin M, Wang J, Ji X, et al. MCUR1 facilitates epithelial-mesenchymal transition and metastasis via the mitochondrial calcium dependent ROS/Nrf2/Notch pathway in hepatocellular carcinoma. J Exp Clin Cancer Res. 2019;38(1):136. doi:10.1186/s13046-019-1135-x
162. Chen Z, Zuo X, Pu L, et al. Hypomethylation-mediated activation of cancer/testis antigen KK-LC-1 facilitates hepatocellular carcinoma progression through activating the Notch1/Hes1 signalling. Cell Prolif. 2019;52(3):e12581. doi:10.1111/cpr.12581
163. Zhou J, Zheng X, Feng M, et al. Upregulated MMP28 in hepatocellular carcinoma promotes metastasis via Notch3 signaling and predicts unfavorable prognosis. Int J Biol Sci. 2019;15(4):812–825. doi:10.7150/ijbs.31335
164. Ye YC, Zhao JL, Lu YT, et al. NOTCH signaling via WNT regulates the proliferation of alternative, CCR2-independent tumor-associated macrophages in hepatocellular carcinoma. Cancer Res. 2019;79(16):4160–4172. doi:10.1158/0008-5472.CAN-18-1691
165. Chen H, Wu D, Bao L, et al. 6PGD inhibition sensitizes hepatocellular carcinoma to chemotherapy via AMPK activation and metabolic reprogramming. Biomed Pharmacother. 2019;111:1353–1358. doi:10.1016/j.biopha.2019.01.028
166. Jin X, Moskophidis D, Mivechi NF. Heat shock transcription factor 1 is a key determinant of HCC development by regulating hepatic steatosis and metabolic syndrome. Cell Metab. 2011;14(1):91–103. doi:10.1016/j.cmet.2011.03.025
167. Ma X, Qiu Y, Sun Y, et al. NOD2 inhibits tumorigenesis and increases chemosensitivity of hepatocellular carcinoma by targeting AMPK pathway. Cell Death Dis. 2020;11(3):174. doi:10.1038/s41419-020-2368-5
168. Gao L, Lv G, Li R, et al. Glycochenodeoxycholate promotes hepatocellular carcinoma invasion and migration by AMPK/mTOR dependent autophagy activation. Cancer Lett. 2019;454:215–223. doi:10.1016/j.canlet.2019.04.009
169. Bort A, Sanchez BG, Mateos-Gomez PA, Vara-Ciruelos D, Rodriguez-Henche N, Diaz-Laviada I. Targeting AMP-activated kinase impacts hepatocellular cancer stem cells induced by long-term treatment with sorafenib. Mol Oncol. 2019;13(5):1311–1331. doi:10.1002/1878-0261.12488
170. Li C, Huang Z, Zhu L, et al. The contrary intracellular and extracellular functions of PEDF in HCC development. Cell Death Dis. 2019;10(10):742. doi:10.1038/s41419-019-1976-4
171. Fang G, Zhang P, Liu J, et al. Inhibition of GSK-3beta activity suppresses HCC malignant phenotype by inhibiting glycolysis via activating AMPK/mTOR signaling. Cancer Lett. 2019;463:11–26. doi:10.1016/j.canlet.2019.08.003
172. Qiao A, Jin X, Pang J, Moskophidis D, Mivechi NF. The transcriptional regulator of the chaperone response HSF1 controls hepatic bioenergetics and protein homeostasis. J Cell Biol. 2017;216(3):723–741. doi:10.1083/jcb.201607091
173. Garten A, Grohmann T, Kluckova K, Lavery GG, Kiess W, Penke M. Sorafenib-induced apoptosis in hepatocellular carcinoma is reversed by SIRT1. Int J Mol Sci. 2019;20(16):4048. doi:10.3390/ijms20164048
174. Singh L, Aldosary S, Saeedan AS, Ansari MN, Kaithwas G. Prolyl hydroxylase 2: a promising target to inhibit hypoxia-induced cellular metabolism in cancer cells. Drug Discov Today. 2018;23(11):1873–1882. doi:10.1016/j.drudis.2018.05.016
175. Liu Y, Jiang Y, Zhang M, Tang Z, He M, Bu W. Modulating hypoxia via nanomaterials chemistry for efficient treatment of solid tumors. Acc Chem Res. 2018;51(10):2502–2511. doi:10.1021/acs.accounts.8b00214
176. Luo W, Zhong J, Chang R, Hu H, Pandey A, Semenza GL. Hsp70 and CHIP selectively mediate ubiquitination and degradation of hypoxia-inducible factor (HIF)-1alpha but not HIF-2alpha. J Biol Chem. 2010;285(6):3651–3663. doi:10.1074/jbc.M109.068577
177. Albadari N, Deng S, Li W. The transcriptional factors HIF-1 and HIF-2 and their novel inhibitors in cancer therapy. Expert Opin Drug Discov. 2019;14(7):667–682. doi:10.1080/17460441.2019.1613370
178. Oh SY, Seok JY, Choi YS, Lee SH, Bae JS, Lee YM. The histone methyltransferase inhibitor BIX01294 inhibits HIF-1alpha stability and angiogenesis. Mol Cells. 2015;38(6):528–534. doi:10.14348/molcells.2015.0026
179. Sapra P, Kraft P, Pastorino F, et al. Potent and sustained inhibition of HIF-1alpha and downstream genes by a polyethyleneglycol-SN38 conjugate, EZN-2208, results in anti-angiogenic effects. Angiogenesis. 2011;14(3):245–253. doi:10.1007/s10456-011-9209-1
180. Lee SH, Jee JG, Bae JS, Liu KH, Lee YM. A group of novel HIF-1alpha inhibitors, glyceollins, blocks HIF-1alpha synthesis and decreases its stability via inhibition of the PI3K/AKT/mTOR pathway and Hsp90 binding. J Cell Physiol. 2015;230(4):853–862. doi:10.1002/jcp.24813
181. Fang J, Xia C, Cao Z, Zheng JZ, Reed E, Jiang BH. Apigenin inhibits VEGF and HIF-1 expression via PI3K/AKT/p70S6K1 and HDM2/p53 pathways. FASEB J. 2005;19(3):342–353. doi:10.1096/fj.04-2175com
182. Tang W, Zhao G. Small molecules targeting HIF-1alpha pathway for cancer therapy in recent years. Bioorg Med Chem. 2020;28(2):115235. doi:10.1016/j.bmc.2019.115235
183. Feng J, Dai W, Mao Y, et al. Simvastatin re-sensitizes hepatocellular carcinoma cells to sorafenib by inhibiting HIF-1alpha/PPAR-gamma/PKM2-mediated glycolysis. J Exp Clin Cancer Res. 2020;39(1):24. doi:10.1186/s13046-020-1528-x
184. Deng F, Chen D, Wei X, et al. Development and validation of a prognostic classifier based on HIF-1 signaling for hepatocellular carcinoma. Aging (Albany NY). 2020;12:3431.
185. Yang J, Jin X, Yan Y, et al. Inhibiting histone deacetylases suppresses glucose metabolism and hepatocellular carcinoma growth by restoring FBP1 expression. Sci Rep. 2017;7:43864. doi:10.1038/srep43864
186. Tang Y, Zhang Y, Wang C, et al. Overexpression of PCK1 gene antagonizes hepatocellular carcinoma through the activation of gluconeogenesis and suppression of glycolysis pathways. Cell Physiol Biochem. 2018;47(1):344–355. doi:10.1159/000489811
187. Taylor S, Spugnini EP, Assaraf YG, Azzarito T, Rauch C, Fais S. Microenvironment acidity as a major determinant of tumor chemoresistance: proton pump inhibitors (PPIs) as a novel therapeutic approach. Drug Resist Updat. 2015;23:69–78. doi:10.1016/j.drup.2015.08.004
188. Estrella V, Chen T, Lloyd M, et al. Acidity generated by the tumor microenvironment drives local invasion. Cancer Res. 2013;73(5):1524–1535. doi:10.1158/0008-5472.CAN-12-2796
189. Silva AS, Yunes JA, Gillies RJ, Gatenby RA. The potential role of systemic buffers in reducing intratumoral extracellular pH and acid-mediated invasion. Cancer Res. 2009;69(6):2677–2684. doi:10.1158/0008-5472.CAN-08-2394
190. Chao M, Wu H, Jin K, et al. A nonrandomized cohort and a randomized study of local control of large hepatocarcinoma by targeting intratumoral lactic acidosis. Elife. 2016;5. doi:10.7554/eLife.15691
191. Lu ZN, Tian B, Guo XL. Repositioning of proton pump inhibitors in cancer therapy. Cancer Chemother Pharmacol. 2017;80(5):925–937. doi:10.1007/s00280-017-3426-2
192. Li D, Li N, Zhang YF, et al. Persistent polyfunctional chimeric antigen receptor T cells that target glypican 3 eliminate orthotopic hepatocellular carcinomas in mice. Gastroenterology. 2020.
193. Harding JJ, El Dika I, Abou-Alfa GK. Immunotherapy in hepatocellular carcinoma: primed to make a difference? Cancer. 2016;122(3):367–377. doi:10.1002/cncr.29769
194. Sangro B, Gomez-Martin C, de la Mata M, et al. A clinical trial of CTLA-4 blockade with tremelimumab in patients with hepatocellular carcinoma and chronic hepatitis C. J Hepatol. 2013;59(1):81–88. doi:10.1016/j.jhep.2013.02.022
195. Wang PF, Chen Y, Song SY, et al. Immune-related adverse events associated with anti-PD-1/PD-L1 treatment for malignancies: a meta-analysis. Front Pharmacol. 2017;8:730. doi:10.3389/fphar.2017.00730
196. Xu H, Tan P, Zheng X, et al. Immune-related adverse events following administration of anti-cytotoxic T-lymphocyte-associated protein-4 drugs: a comprehensive systematic review and meta-analysis. Drug Des Devel Ther. 2019;13:2215–2234. doi:10.2147/DDDT.S196316
197. Greten TF, Mauda-Havakuk M, Heinrich B, Korangy F, Wood BJ. Combined locoregional-immunotherapy for liver cancer. J Hepatol. 2019;70(5):999–1007. doi:10.1016/j.jhep.2019.01.027
198. Jansen CS, Prokhnevska N, Master VA, et al. An intra-tumoral niche maintains and differentiates stem-like CD8 T cells. Nature. 2019;576(7787):465–470. doi:10.1038/s41586-019-1836-5
199. Martinez-Outschoorn UE, Peiris-Pages M, Pestell RG, Sotgia F, Lisanti MP. Cancer metabolism: a therapeutic perspective. Nat Rev Clin Oncol. 2017;14(1):11–31. doi:10.1038/nrclinonc.2016.60
200. Fortunato S, Bononi G, Granchi C, Minutolo F. An update on patents covering agents that interfere with the cancer glycolytic cascade. ChemMedChem. 2018;13(21):2251–2265. doi:10.1002/cmdc.201800447
201. Guo Z, Cheng Z, Wang J, et al. Discovery of a potent GLUT inhibitor from a library of rapafucins by using 3D microarrays. Angew Chem Int Ed Engl. 2019;58(48):17158–17162. doi:10.1002/anie.201905578
202. Shi T, Ma Y, Cao L, et al. B7-H3 promotes aerobic glycolysis and chemoresistance in colorectal cancer cells by regulating HK2. Cell Death Dis. 2019;10(4):308. doi:10.1038/s41419-019-1549-6
203. DeWaal D, Nogueira V, Terry AR, et al. Hexokinase-2 depletion inhibits glycolysis and induces oxidative phosphorylation in hepatocellular carcinoma and sensitizes to metformin. Nat Commun. 2018;9(1):446. doi:10.1038/s41467-017-02733-4
204. Zhang Z, Li TE, Chen M, et al. MFN1-dependent alteration of mitochondrial dynamics drives hepatocellular carcinoma metastasis by glucose metabolic reprogramming. Br J Cancer. 2020;122(2):209–220. doi:10.1038/s41416-019-0658-4
205. Terranova T, Feo F, Gravela E, Gabriel L. [the effect of 2-desoxyglucose on energy metabolism and protein synthesis of tumor cells and normal cells]. Z Krebsforsch. 1964;66:41–45. doi:10.1007/BF00525559
206. Yin X, Choudhury M, Kang JH, et al. Hexokinase 2 couples glycolysis with the profibrotic actions of TGF-beta. Sci Signal. 2019;12(612):eaax4067. doi:10.1126/scisignal.aax4067
207. Agnihotri S, Mansouri S, Burrell K, et al. Ketoconazole and posaconazole selectively target HK2-expressing glioblastoma cells. Clin Cancer Res. 2019;25(2):844–855. doi:10.1158/1078-0432.CCR-18-1854
208. Kunjithapatham R, Geschwind JF, Rao PP, Boronina TN, Cole RN, Ganapathy-Kanniappan S. Systemic administration of 3-bromopyruvate reveals its interaction with serum proteins in a rat model. BMC Res Notes. 2013;6:277. doi:10.1186/1756-0500-6-277
209. Yi W, Clark PM, Mason DE, et al. Phosphofructokinase 1 glycosylation regulates cell growth and metabolism. Science. 2012;337(6097):975–980. doi:10.1126/science.1222278
210. Hu J, Zeng Z, Xia Q, et al. Metformin attenuates hepatoma cell proliferation by decreasing glycolytic flux through the HIF-1alpha/PFKFB3/PFK1 pathway. Life Sci. 2019;239:116966. doi:10.1016/j.lfs.2019.116966
211. Su Q, Luo S, Tan Q, et al. The role of pyruvate kinase M2 in anticancer therapeutic treatments. Oncol Lett. 2019;18(6):5663–5672. doi:10.3892/ol.2019.10948
212. Shang D, Wu J, Guo L, Xu Y, Liu L, Lu J. Metformin increases sensitivity of osteosarcoma stem cells to cisplatin by inhibiting expression of PKM2. Int J Oncol. 2017;50(5):1848–1856. doi:10.3892/ijo.2017.3950
213. Li L, Kang L, Zhao W, et al. miR-30a-5p suppresses breast tumor growth and metastasis through inhibition of LDHA-mediated Warburg effect. Cancer Lett. 2017;400:89–98. doi:10.1016/j.canlet.2017.04.034
214. Boudreau A, Purkey HE, Hitz A, et al. Metabolic plasticity underpins innate and acquired resistance to LDHA inhibition. Nat Chem Biol. 2016;12(10):779–786. doi:10.1038/nchembio.2143
215. Grasmann G, Smolle E, Olschewski H, Leithner K. Gluconeogenesis in cancer cells - Repurposing of a starvation-induced metabolic pathway? Biochim Biophys Acta Rev Cancer. 2019;1872(1):24–36. doi:10.1016/j.bbcan.2019.05.006
216. Duda P, Janczara J, McCubrey JA, Gizak A, Rakus D. The reverse Warburg effect is associated with Fbp2-dependent Hif1alpha regulation in cancer cells stimulated by fibroblasts. Cells. 2020;9(1):205. doi:10.3390/cells9010205
217. Tuo L, Xiang J, Pan X, et al. PCK1 negatively regulates cell cycle progression and hepatoma cell proliferation via the AMPK/p27(Kip1) axis. J Exp Clin Cancer Res. 2019;38(1):50. doi:10.1186/s13046-019-1029-y
218. Leithner K. Epigenetic marks repressing gluconeogenesis in liver and kidney cancer. Cancer Res. 2020;80(4):657–658. doi:10.1158/0008-5472.CAN-19-3953
219. Shang F, Liu M, Li B, et al. The anti-angiogenic effect of dexamethasone in a murine hepatocellular carcinoma model by augmentation of gluconeogenesis pathway in malignant cells. Cancer Chemother Pharmacol. 2016;77(5):1087–1096. doi:10.1007/s00280-016-3030-x
220. Srikantan S, Deng Y, Cheng ZM, et al. The tumor suppressor TMEM127 regulates insulin sensitivity in a tissue-specific manner. Nat Commun. 2019;10(1):4720. doi:10.1038/s41467-019-12661-0
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.